

Abstract
Cardiac arrest and cardiopulmonary resuscitation (CA/CPR) produce brain ischemia that results in cognitive and motor coordination impairments subsequent to injury of vulnerable populations of neurons, including cerebellar Purkinje neurons. To determine the effects of CA/CPR on plasticity in the cerebellum, we used whole cell recordings from Purkinje neurons to examine long-term depression (LTD) at parallel fiber (PF) synapses. Acute slices were prepared from adult male mice subjected to 8 min cardiac arrest at 1, 7, and 30 days after resuscitation. Concurrent stimulation of PF and climbing fibers (CFs) resulted in robust LTD of PF-evoked excitatory postsynaptic currents (EPSCs) in controls. LTD was absent in recordings obtained from mice subjected to CA/CPR, with no change in EPSC amplitude from baseline at any time point tested. AMPA and mGluR-mediated responses at the PF were not altered by CA/CPR. In contrast, CF-evoked NMDA currents were reduced following CA/CPR, which could account for the loss of LTD observed. A loss of GluN1 protein was observed following CA/CPR that was surprisingly not associated with changes in mRNA expression. These data demonstrate sustained impairments in synaptic plasticity in Purkinje neurons that survive the initial injury and which likely contribute to motor coordination impairments observed after CA/CPR.
Keywords: Cerebral ischemia, electrophysiology, synaptic plasticity, NMDA
Introduction
Cardiac arrest occurs in approximately 600,000 people each year and is a major cause of mortality and morbidity, with survivors experiencing motor and cognitive deficits.1–5 The underlying cause of the behavioral impairments is primarily attributed to death of vulnerable populations of neurons including hippocampal CA1 neurons, striatal neurons, and cerebellar Purkinje cells.6–8 Therefore, studies in animal models are often focused on early neuroprotection, with the only successful intervention to date being therapeutic hypothermia.9 Clinical translation of pharmacological neuroprotective strategies have largely failed, making it important to examine the function of surviving neurons after ischemia to identify strategies that enhance plasticity and recovery in the post-ischemic brain. Deficits in hippocampal plasticity have been observed for up to a month after ischemic insults, suggesting the brains surviving networks are unable to compensate for neuronal cell loss.10–12 Targeting such synaptic plasticity deficits may present an opportunity to enhance functional recovery from ischemic brain injury with a broadened therapeutic window. While there are a number of studies demonstrating impaired long-term potentiation in the hippocampus, it remains unclear whether similar plasticity deficits are observed in other ischemia sensitive brain regions.
Purkinje cells in the cerebellum are particularly sensitive to ischemic damage and dysfunction of these neurons likely contributes to the motor deficits observed in cardiac arrest survivors.6,7,13,14 Indeed, Purkinje cell loss is apparent in post-mortem cerebellums in victims who died 5-180 h after suffering a cardiac arrest.6,7 We recently reported a similar time course of Purkinje cell degeneration in our mouse model of cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Following 8 min of cardiac arrest, we observed a 30% loss of Purkinje cells that occurred between one and seven days after resuscitation.15 Purkinje cells in the cerebellum receive and integrate multiple inputs and ion channel dysfunction in Purkinje cells is associated with ataxia and impaired motor learning,16–20 increasing confidence that Purkinje cell injury and dysfunction contribute to post-ischemic motor deficits. The goal of the current study is to determine whether cerebral ischemia causes altered ion channel and receptor function in surviving Purkinje cells, contributing to post-ischemic deficits in plasticity.
The best characterized form of synaptic plasticity in the cerebellum is long-term depression (LTD) at parallel fiber to Purkinje cell synapses. Purkinje cells receive glutamate excitation from two inputs: climbing fiber (CF) projections from the inferior olive and parallel fibers (PFs) from cerebellar granule cells.21–24 LTD occurs following paired activation of PF and CF synapses, resulting in reduced AMPA receptor function at the PF synapse.25–27 LTD requires highly elevated calcium levels and subsequent phosphorylation and internalization of AMPA receptors. Plasticity in the cerebellum is thought to underlie motor learning, and deficits in cerebellar LTD have been associated with impaired cerebellar-dependent learning.28–30 We hypothesize that ischemia causes sustained plasticity impairments in Purkinje cells as a consequence of alterations in glutamatergic signaling. The data presented here show long-lasting impairments in cerebellar LTD in surviving Purkinje cells of mice subjected to CA/CPR which are associated with reduced NMDA receptor function.
Methods
Experimental animals and cardiac arrest model
The Institutional Animal Care and Use Committee (IACUC) at the University of Colorado approved all experimental protocols in accordance with National Institutes of Health guidelines for the care and use of animals in research. Experiments were performed in accordance with ARRIVE guidelines.31,32 Blinding was performed prior to electrophysiological recording. Adult (8–10-week-old) male C57Bl6 (Charles River, Wilmington, MA) were subjected to CA/CPR as previously described12,15,33 during ON light cycle. A total of 119 surgeries were performed, including sham surgeries of which 14 mice did not survive to reach their final end point. There were an additional 10 animals that survived; however, we were unable to obtain high quality recordings.
Briefly, anesthesia was induced with 3% isoflurane and maintained with 1.5–2% isoflurane in oxygen-enriched air via facemask. Temperature probes were inserted in the left ear and rectum to monitor head and body temperature simultaneously. A PE-10 catheter was inserted into the right internal jugular vein for drug administration. Needle electrodes were placed subcutaneously on the chest for continuous EKG monitoring. Animals were endotracheally intubated, connected to a mouse ventilator (MiniVent Ventilator, Harvard Apparatus). Cardiac arrest was induced by injection of 50 µL KCl (0.5M) via the jugular catheter and confirmed by asystole on EKG. The endotracheal tube was disconnected and anesthesia stopped. During cardiac arrest, body temperature was allowed to spontaneously decrease to a minimum of 35.5℃, and head temperature was maintained at 37.5℃. Resuscitation was begun 8 min after induction of cardiac arrest by injection of 0.05–0.10 ml epinephrine solution (16 µg epinephrine/ml 0.9% saline), chest compressions, and ventilation with 100% oxygen at a respiratory rate of 200/min and 25% greater tidal volume. Chest compressions were stopped as soon as spontaneous circulation was restored. Resuscitation was abandoned if spontaneous circulation was not restored within 2 min. Mice were extubated after they recovered an adequate respiratory rate and effort. Sham controls underwent the same procedures as mice undergoing cardiac arrest including anesthesia intubation, placement of jugular catheter, EKG leads and temperature management. Sham controls did not receive KCl or epinephrine injections or chest compressions. The animals were placed in a single housed static recovery cage on a heated water blanket (35℃) for 24 h recovery and at room temp for long-term recovery (up to 30 days). Mice received soft food and subcutaneous saline for three days after surgery and had free access to water and regular chow.
Electrophysiology
Following CA/CPR or sham surgery, mice were anesthetized with isoflurane (3%) and transcardially perfused with ice-cold artificial cerebral spinal fluid (ACSF) containing (in mM): 126 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11 D-glucose, bubbled with 95% O2/5% CO2 to maintain pH of 7.4. Mice were decapitated and brains were rapidly removed. Parasagittal cerebellar sections (300 µm) were cut in ice-cold ACSF using a VT1200S vibratome (Leica, Buffalo Grove, IL) and then incubated at 35℃ for 30 min. Slices were maintained at room temperature until the slices were transferred to a recording chamber which was continuously perfused with ACSF (1.5 ml/min) containing picrotoxin (100 µM). Recordings were performed at 22℃ with the exception of NMDA current recordings that were performed at 32℃. Purkinje neurons were identified by their large soma and location in the Purkinje cell layer. Whole cell recordings were obtained using 2–4 MΩ pipette containing (in mM): 135 Kgluconate, 10 HEPES, 0.05 EGTA, 1 MgCl2, 4 MgATP, 0.3 Na2GTP, pH to 7.3 with KOH or 120 Kgluconate, 9 KCl, 10 KOH, 4 NaCl, 10 HEPES, 0.05 EGTA, 1 MgCl2, 4 Na2ATP, 0.4 Na2GTP. Series resistance was < 20 MΩ and did not change more than 20% during the experiment. Whole cell voltage clamp recordings were performed at −70 mV and compensated at 70%.
LTD recording
Parallel fiber excitatory postsynaptic currents (PF-EPSC) were evoked (0.05 Hz) with a glass stimulus electrode placed in the molecular layer and stimulus intensity adjusted to produce a measurable inward evoked excitatory postsynaptic current (EPSC) (200–400 pA). A stable baseline amplitude was recorded for 5 min before LTD induction. For LTD induction, simultaneous PF and CF stimulation was performed in current clamp configuration at 1 Hz for 5 min. Following induction, PF responses were measured for 25 min in voltage clamp configuration. Data were compressed to 1 min averages and the extent of LTD was measured as % of the baseline EPSC during the last 5 min of the recording.
Glutamate receptor recordings
To measure metabotropic glutamate receptor (mGluR)-mediated currents, a single PF-EPSC was evoked every 20 s. Once a stable baseline amplitude was obtained, the mGluR antagonist, (RS)-α-Methyl-4-carboxyphenylglycine (MCPG, 500 µM) was perfused and the difference with and without MCPG was quantified. For measurements of the CF-EPSCs, a glass electrode containing ACSF was placed in the granule cell layer approximately 50 µm away from the recorded Purkinje cell soma and a single stimulus was applied at 0.05 Hz. For N-methyl-D-aspartate (NMDA) receptor-mediated currents, recordings were performed in Mg-free ACSF containing picrotoxin (100 µM), glycine (25 µM) and 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX, 250 nM). CF-EPSC was identified as a large amplitude, all-or-none large inward depolarization. NBQX (10 µM) was perfused to isolate NMDA-receptor-mediated currents. Once amplitudes plateaued (5–10 min), D-(-)-2-Amino-5-phosphonopentanoic acid (D-AP5,50 µM) was perfused to block NMDA receptors. NMDA receptor-mediated currents were quantified as the difference in plateau amplitude in the presence of NBQX and D-AP5. Alternatively, exogenous application of NMDA (10 mM) using an iontophoretic electrode (100–200 nA, 4 s) was used to activate NMDA receptors and the difference in amplitude with and without D-AP5 present was quantified.
Recording of miniature excitatory postsynaptic currents (mEPSC) was performed in the presence of tetrodotoxin (250 nM) and picrotoxin (100 µM). Purkinje cells were held at −80 mV and gap-free continuous recordings were performed in 3-min sweeps. mEPSC events were identified using Clampfit software with template event detection and mEPSC amplitude and inter-event interval were quantified.
Laser capture microdissection and quantitative real-time PCR
Following CA/CPR or sham surgery, mice were deeply anesthetized with isoflurane (3%) decapitated and brains were rapidly removed. Cerebellums were placed in cryomolds with optimum cutting temperature (OCT) freezing medium and rapidly frozen with 2-methylbutane on dry ice. Cryostat sections were cut (8 µm), placed on slides and stored at −80℃ until laser capture was performed later the same day. Prior to laser capture, slides were ethanol fixed (70%, 30 s), washed in nuclease free water (2 × 30 s) and dehydrated in ethanols (70%/95%/100%, 30 s each) and xylene (5 min). Sections were visualized on a Nikon microscope and Purkinje cells were captured using Arcturus laser capture system. Collection caps were incubated with lysis buffer for 30 min at 42℃ and then stored at −80℃ until RNA isolation was performed. Four caps were collected from each animal and pooled on RNA isolation columns to yield approximately 1000 cells. Picopure RNA kit (Arcturus) was used to isolate RNA and were treated on column with DNase (Qiagen) to remove genomic DNA contamination. cDNA was generated from RNA using iScript reverse transcription kit (Bio-Rad) and used for quantitative real-time PCR (qRT-PCR). Real-time PCR reactions were performed using SsoFast mastermix (Bio-rad) and Taq-man primer/probes (Applied biosystems) for 18S and GluN1. Expression levels were calculated using the ΔΔCT method relative to 18 s and were normalized to controls.
Immunohistochemistry
Mice were transcardially perfused with phosphate-buffered saline (PBS) and 4% paraformaldehyde under isoflurane anesthesia (3%). Brains were removed and fixed overnight and then stored in cryoprotectant solution containing 20% glycerol in 0.1 M Sorenson’s buffer, pH 7.6. Freezing microtome sections (50 µM), sections were cut and stored in well plates in cryostorage solution (30% ethylene glycol, 30% sucrose, and 1% PVP-40 in 0.1 M Sorenson’s buffer) at 4℃. Sections were rinsed with PBS and were blocked and permeabilized for 1 h at room temperature with 0.3% Triton-X, 5% normal donkey serum, and 1% BSA in PBS. Primary antibody incubations with anti-GluN1 1:100 (Thermo Fisher) and anti-calbindin D28K 1:500 (Santa-Cruz Biotech) were performed for 48 h at 4℃. Secondary antibody incubations with Alexa-488 and Alexa-594 (Jackson Immuno) were performed at room temperature for 2 h. Slides were mounted onto slides and visualized using an LSM510 confocal microscope (Zeiss). Z stacks were collected using Zen6 software and analyzed using ImageJ software. Background subtracted mean fluorescence intensity and Pearson’s correlation analysis were performed for each image and were averaged across sections to generate a single value for each animal.
Statistics
For electrophysiology experiments, n indicates number of recordings with no more than two recordings for a given experiment from a single animal. Power analysis using expected variances was performed to determine sample size required for 80% power using StatMate2 (Graphpad). For GluN1 expression, n indicates number of animals. Data are presented as mean ± SEM. Statistical comparisons were made between two groups using student’s test and multiple groups using one-way analysis of variance (ANOVA) followed by Dunnet’s post hoc comparison of groups relative to control. Statistical comparisons were performed using Prism 6.0 (Graphpad). Differences with a p-value of < 0.05 were considered significant.
Results
Impairments in cerebellar LTD following CA/CPR
PF-LTD is an associative form of plasticity that contributes to cerebellar-dependent learning processes. We assessed whether LTD induced by paired activation of parallel fiber and climbing fiber (PF+CF) inputs is altered by cardiac arrest-induced global cerebral ischemia in adult mice. Whole cell voltage clamp recording of PF-EPSCs was recorded in the presence of the GABAA receptor antagonist picrotoxin (100 µM). Following a 5-min stable baseline, the LTD induction stimulus of paired PF+CF stimulation (1 Hz, 5 minutes) was performed in current clamp configuration. Importantly, this configuration allowed for generation of a complex spike during CF stimulation (Figure 1(a), inset). After paired stimulation, there was a significant depression in PF-EPSC amplitude immediately following the stimulus that was sustained for the duration of recordings (25 min); decreasing to 45.6 ± 10.1% of baseline (n = 5). In contrast, Purkinje cell recordings in slices obtained from mice 1 day after CA/CPR exhibited a complete loss of PF-LTD compared to controls (96.1 ± 5.2% of baseline, n = 6, p = 0.002) (Figure 1(b)). A sustained impairment in LTD was observed at 7 days (81.6 ± 10.9%, n = 6) and 30 days (88.0 ± 7.1%, n = 5) after cardiac arrest (p = .0004, compared to control) (Figure 1(c)). These data demonstrate for the first time that CA/CPR causes a rapid and sustained impairment in parallel fiber LTD.
Figure 1.
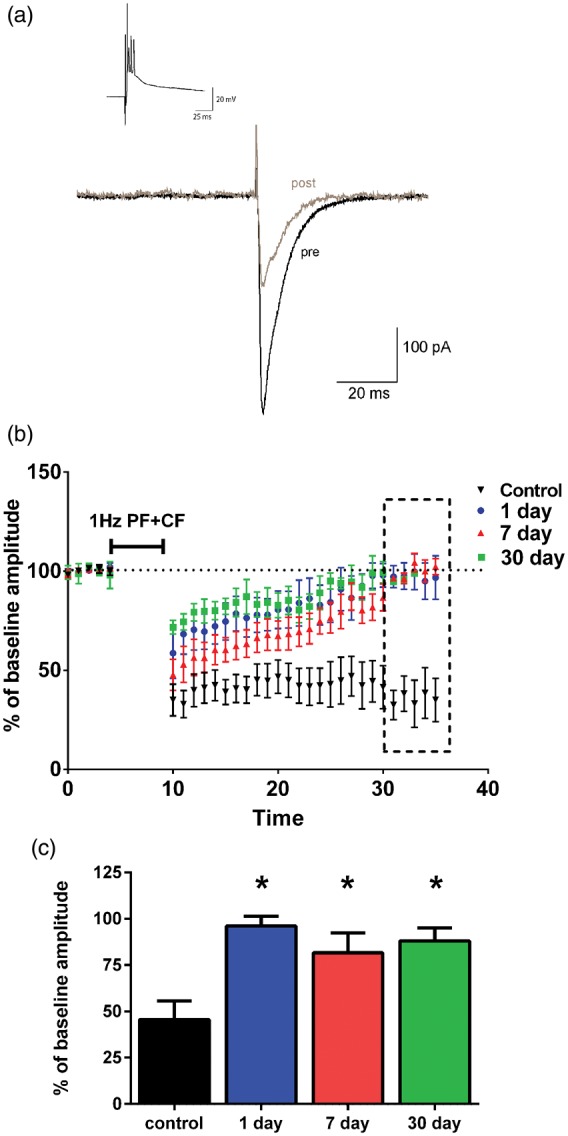
Impaired LTD following CA/CPR. (a) Representative trace of PF-EPSC pre (black trace) and post (gray trace) LTD induction. Inset shows induction stimulus of paired PF+CF that was delivered at 1 Hz for 5 min. (b) Time course of EPSC amplitudes normalized to mean baseline amplitude (% of baseline). LTD induction stimulus was delivered at 5 min after baseline. LTD was quantified during the last 5 min of recording (dotted box). (c) Mean change from baseline amplitude. Controls: n = 5 slices, from 5 mice; 1 day: n = 6 slices from 5 mice; 7 day: n = 6 slices from 6 mice; 30 day: n = 5 slices from 5 mice *indicates groups that are significantly different from controls (one-way ANOVA with Dunnets post hoc; p < 0.05)
Excitatory transmission at parallel fiber unaltered by CA/CPR
We next recorded basal excitatory transmission to determine whether the loss of LTD observed after cardiac arrest was due to alterations in excitatory transmission at the parallel fiber synapse. Recordings were performed at 7 days after CA/CPR as this is a time point where we observe plasticity impairments and the cell death process has stabilized.15 Paired pulse ratio (PPR) was used to determine whether cardiac arrest altered the probability of release from PF-Purkinje cell synapses. Two parallel fiber stimuli were delivered 50 ms apart and the ratio between the amplitude of pulse 1 (P1) and pulse 2 (P2) was calculated. In slices from control animals, paired-pulse facilitation was observed with a ratio of 1.7 ± 0.08 (n = 10) (Figure 2(a)). No difference in PPR was observed in slices from mice subjected to cardiac arrest (1.88 ± 0.13, n = 10) compared to controls (p = 0.28), suggesting no changes in probability of glutamate release at the PF synapse (Figure 2(b)). These data suggest that the LTD impairment observed after CA/CPR was not of presynaptic origin.
Figure 2.
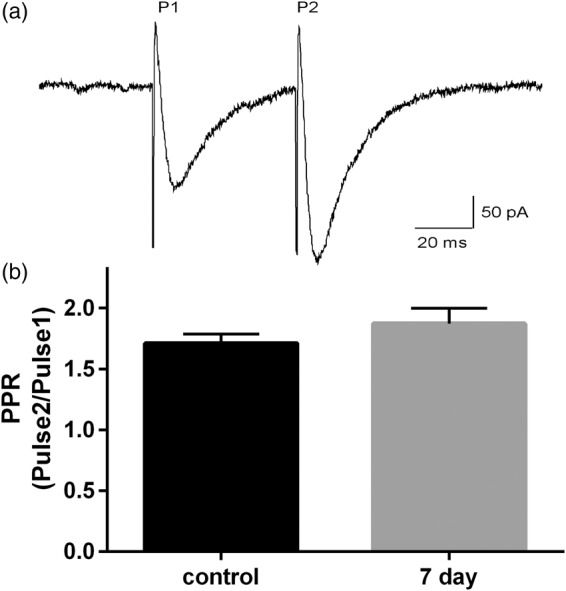
No changes in synaptic release probability at parallel fiber synapse following CA/CPR. (a) Representative tracing showing EPSCs evoked by two parallel fiber stimuli (P1 and P2) delivered 50 ms apart. (b) Paired pulse ratios (PPR) calculated by dividing the amplitude of EPSC from P2/P1. Bar graphs represent mean PPR ± SEM and were not different between control and seven-day CA/CPR. Control: n = 10 slices from 6 mice; 7 day: n = 10 slices from 7 mice. (Student’s t-test p > 0.05).
We next examined postsynaptic glutamate receptor function in Purkinje cells. The induction of LTD requires the activation of AMPA and metabotropic glutamate (mGluR) receptors at the parallel fiber synapse. Miniature EPSCs (mEPSC) were recorded to determine whether there were any ischemia-induced alterations in post-synaptic Purkinje cell AMPA receptor function. Excitatory miniature events were isolated using TTX (250 nM) and picrotoxin (100 µM) and were recorded at −80 mV (Figure 3(a)). Mean mEPSC amplitudes in controls were −19.1 ± 0.74 (n = 9) and were not different following CA/CPR (20.33 ± 0.72, n = 11, p = 0.23 compared to controls) (Figure 3(b)). Analysis of inter-event interval was used to determine mEPSC frequency, which was not changed by CA/CPR (control: 3.48 ± 0.62 Hz; 7 day CA/CPR: 4.11 ± 0.51 Hz; p = 0.43) (Figure 3(c)). These results indicate that CA/CPR does not alter PF-Purkinje cell post-synaptic AMPA receptor function, making changes in these receptors an unlikely mediator of the CA/CPR-induced plasticity deficits.
Figure 3.

No alterations in post-synaptic AMPA receptor function following CA/CPR. (a) Representative recording of mEPSCs in Purkinje cells. Astericks indicate events. (b) Mean mEPSC amplitudes (c) and frequency were not different between control and CA/CPR (student’s t-test p > 0.05). Each n represents the mean amplitude for an individual recording. Control: n = 9 slices from 6 mice; 7 day: n = 11 slices from 7 mice.
Activation of mGluR results in release of endoplasmic reticulum calcium stores and activation of the calcium-permeable non-selective cation channels, TRPC3. Currents resulting from mGluR activation of TRPC3 were recorded to assess mGluR function following CA/CPR. Parallel fiber responses were evoked and the reduction in EPSC amplitude with the mGluR antagonist MCPG (500 µM) was quantified (Figure 4(a)). In control animals, mGluR-mediated currents comprised 9.8 ± 0.2% (n = 5) of the total EPSC amplitude (Figure 4(b)). Following CA/CPR, the mGluR-mediated currents were 18.6 ± 0.5% (n = 7) of the total EPSC amplitude, not different from controls (p = 0.21). Similarly, exogenous application of the mGluR agonist DHPG (50 µM) produced an inward current that was not different between controls (48.7 ± 4.2 pA) and CA/CPR (56.91 ± 6.0 pA, p = 0.29) (Figure 4(c) and (d)). Consistent with these data, we did not observe an alteration in TRPC3 mean fluorescence intensity or Pearson’s correlation of TRPC3/calbindin colocalization at seven days after CA/CPR relative to control (Figure 4(e) to (g)). Therefore, we did not observe any alterations in excitatory transmission at the parallel fiber to Purkinje cell synapse that could account for the loss of LTD observed following CA/CPR.
Figure 4.

No change in metabotropic glutamate receptor-mediated currents following CA/CPR. (a) Representative traces showing PF-EPSC in absence and presence of MCPG (500 µM). (b) No difference in mGluR responses quantified as a percent of baseline EPSC amplitude (student’s t-test p > 0.05). Control: n = 4 slices from 4 mice; 7 day: n = 7 slices from 5 mice. (c) Representative trace showing bath application (solid line) of DHPG (50 µM). (d) No difference in current amplitude induced by DHPG (student’s t-test p > 0.05). Control: n = 6 slices from 4 mice; 7 day: n = 6 slices from 4 mice. (e) Representative image of TRPC3 immunofluorescence in control (left) and seven days after CA/CPR (right). N = 4 mice per group. (f) Mean background-subtracted fluorescence intensity and (g) Pearson’s correlation of TRPC3 and calbindin colocalization was not different between groups.
CA/CPR results in a loss of functional NMDA receptors at the CF synapse
We next examined excitatory transmission at the CF synapse. CF stimulation produces AMPA and NMDA receptor activation and a subsequent complex spike that results from activation of P/Q type voltage-dependent calcium channels (VDCC). NMDA receptor and VDCC channels are both sources of calcium influx that are required for LTD induction. CF-evoked responses were analyzed to assess AMPA and VDCC function. The number of spikelets produced by climbing fiber stimulation was not different between controls (4.2 ± 0.7, n = 5) and mice subjected to CA/CPR (4.1 ± 0.3, n = 8, p = 0.91), suggesting similar AMPA receptor and VDCC activation between the two groups. To measure synaptic AMPA and NMDA receptor currents in adult Purkinje cells, climbing fiber stimulation was performed in the presence of low levels of the AMPA receptor antagonist NBQX (250 nM) allowing reasonable voltage clamp. In controls, subsequent addition of maximal concentration of NBQX (10 µM) inhibits the majority of inward current elicited by CF stimulation. The NBQX-sensitive current was not different between controls (5259 ± 322 pA, n = 8) compared to CA/CPR (4566 ± 493, n = 6, p = 0.24). The current remaining in saturating NBQX (10 µM) is abolished by the NMDA receptor antagonist D-AP5 (50 µM), confirming previous reports that Purkinje cells from adult mice express NMDA receptors at the CF synapse (Figure 5(a)). In mice subjected to CA/CPR, there was a significant reduction in NMDA receptor-mediated currents. In control, the D-AP5 sensitive component of the CF-evoked EPSC was 292.8 ± 40.5 pA (n = 8) and was 110.1 ± 45 pA seven days after CA/CPR (n = 7, p = 0.0072). A similar reduction in NMDA receptor-mediated currents was also present at 1 (80 ± 16.6, n = 5, p = 0.0046) and 30 days (107 ± 40.8, n = 6, p = 0.0066) (Figure 5(b)). The decay kinetics of the NMDA EPSC were similar in all groups (control: 67.1 ± 3.9 ms; 24 h: 78.6 ± 24 ms; 7 day: 59.6 ± 3 ms; 30 day: 55.5 ± 10.3 ms), suggesting that the subunit composition was not changed by CA/CPR (Figure 5(c)). To bypass a possible pre-synaptic contribution to CA/CPR-induced reduction in NMDA receptor function, the current response to local exogenous application of NMDA was measured in mice after CA/CPR and compared to control mice. Iontophoresis of NMDA (10 mM, 4 s) onto the Purkinje cell soma resulted in an inward current that was insensitive to NBQX and blocked by D-AP5 (Figure 5(d)). The NMDA current recorded seven days after CA/CPR was significantly reduced (430.6 ± 21.55 pA vs. 80.73 ± 9.9 pA; n = 4) (Figure 5(e)). These results confirm a postsynaptic loss of NMDA receptor function in Purkinje cells subsequent to global ischemia and was the only alteration in excitatory transmission that was correlated with the loss of LTD.
Figure 5.

CA/CPR-induced reduction in NMDA receptor function. (a) Representative traces from control and CA/CPR of CF-evoked responses in the presence of NBQX (250 nM: black trace; 10 µM: red trace) and D-AP5 (blue trace). (b) D-AP5 sensitive currents were calculated as the difference in current amplitude in NBQX (10 µM) and D-AP5 (50 µM) and are significantly reduced in mice subjected to cardiac arrest compared to control. Control: n = 9 slices from 7 mice; 1 day: n = 5 slices from 4 mice; 7 day: n = 7 slices from 5 mice; 30 day: n = 7 slices from 5 mice. (c) Decay kinetics of D-AP5 sensitive current is not different between groups (one-way ANOVA with Dunnett’s post hoc analysis *p < 0.05). (d) Representative traces of current response to iontophoretic application of 10 mM NMDA for 4 s. (e) NMDA-mediated current was significantly smaller in mice subjected to CA/CPR. Control: n = 4 slices from 3 mice; 7 day: n = 4 slices from 4 mice. (Student’s t-test p < 0.05).
Reduced protein but not mRNA of GluN1 subunit
We next determined whether CA/CPR resulted in altered expression of the requisite NMDA receptor subunit, GluN1. We first assessed transcriptional changes in GluN1 mRNA using qRT-PCR. Cerebellar homogenates are not ideal for the study of Purkinje cell mRNA as granule cells are the most abundant cell type in the cerebellar cortex. Therefore, laser capture microdissection was performed to specifically analyze the expression of NMDA receptors in Purkinje cells. Approximately 1000 Purkinje cells per animal were pooled for RNA isolation. We have previously demonstrated a high purity of Purkinje cells using this method.15 qRT-PCR was performed and expression levels were quantified relative to the housekeeping gene 18 S. Surprisingly, expression of the GluN1 subunit was not changed by ischemia compared to control (control: 1.08 ± 0.06, n = 4; 7 day: 1.2 ± 0.06, n = 7, p = 0.25) (Figure 6(a)). In order to assess NMDA protein expression immunohistochemistry using antibodies, recognizing the GluN1 subunits was performed in cerebellar sections from control and seven days after CA/CPR. There was a visible reduction in GluN1 staining, particularly in Purkinje cells (Figure 6(b)). We observed a reduction in mean GluN1 intensity in sections from CA/CPR relative to control (control: 65974 ± 5051, n = 4; CA/CPR: 47485 ± 4547, n = 4; p = 0.035) (Figure 6(c)). In order to quantify Purkinje cell-specific GluN1 expression, Pearson’s correlation analysis of GluN1 and calbindin immunofluorescence was performed. We observed a significant reduction in GluN1/calbindin colocalization at seven days after CA/CPR (0.09 ± 0.01, n = 4) compared to controls (0.15 ± 0.01, p = 0.011), consistent with a reduction in GluN1 expression in Purkinje cells (Figure 6(d)). Therefore, we see an alteration in GluN1 expression in the absence of mRNA changes.
Figure 6.

Reduced GluN1 protein but not mRNA following CA/CPR. (a) Normalized GluN1 mRNA levels from isolated Purkinje cells is not different following CA/CPR. N = 4 mice per group. (b) Representative confocal z-stack images of calbindin and GluN1 staining in cerebellar sections. (c) Normalized fluorescence intensity of GluN1 staining was reduced following CA/CPR. N = 4 mice per group. (d) Reduced colocalization of calbindin and GluN1 staining at seven days after CA/CPR (student’s t-test p < 0.05).
Discussion
In the present study, we show for the first time long-lasting alterations in synaptic transmission and synaptic plasticity in cerebellar Purkinje cells. Impairments in LTD were observed as early as one day after CA/CPR and were sustained for at least 30 days. The loss of LTD was not associated with deficits in parallel fiber transmission, but rather correlates with a reduction in NMDA receptor function at the climbing fiber synapse. Loss of NMDA receptor function was likely the result of reduced translation or enhanced degradation of the receptor, as we observed changes in GluN1 protein without a concurrent change in mRNA levels.
Until recently it was thought that Purkinje cells did not express NMDA receptors and that glutamate excitation in Purkinje cells was mediated solely through AMPA and metabotropic glutamate receptors. However, recent synaptic physiology experiments performed in adult, rather than juvenile, mice has revealed the presence of NMDA receptors at the climbing fiber synapse that contributes to excitatory currents and the induction of synaptic plasticity.34,35 Our recent work has demonstrated a contribution of NMDA receptors as mediators of Purkinje cell death and that acute inhibition of NMDA receptors protect Purkinje cells from ischemic injury.15 Here we use a mouse model of CA/CPR to examine NMDA receptor function in Purkinje cells in the post-ischemic brain. The ischemia-induced reduction of NMDA receptor function and concurrent loss of LTD observed in this study is consistent with a critical role for NMDA receptors in the induction of this form of synaptic plasticity in the adult brain. Indeed, a recent study by Piochon et al. demonstrated for the first time that induction of LTD in adult Purkinje cells requires functional synaptic NMDA receptors. This study demonstrated that postsynaptic NMDA receptors are required for the induction and maintenance of LTD induced by simultaneous stimulation of climbing fiber and PFs.35 Therefore, the loss of LTD maintenance observed following CA/CPR is consistent with a loss of NMDA receptor function. While ours is the first report of ischemia-induced changes in NMDA receptor function and synaptic plasticity in the Purkinje cell, it is remarkably similar to observations made in Purkinje cells following acute ethanol exposure. A 50% reduction in NMDA receptor-mediated currents occurred in slices exposed to acute ethanol and this reduction was sufficient to impair LTD in Purkinje cells.36 Therefore, the chronic 60–70% reduction in synaptic NMDA receptors current we observed after CA/CPR likely explains the loss of LTD observed in Purkinje cells.
We did not observe any alterations in parallel fiber transmission, further supporting our conclusion that the loss of NMDA receptor function plays a causal role in plasticity impairments. NMDA receptors are localized to the climbing fiber synapse and along with VDCCs are a source of cytosolic calcium that is required for LTD induction. Our data showing a similar complex spike in post-ischemic slices and controls suggest a similar level of VDCC activation. Additional signals required for the induction and expression of LTD are activation of AMPA and metabotropic receptors at the parallel fiber synapse.28 We were unable to detect ischemia-induced changes in miniature EPSCs to assess AMPA receptor or mGluR-mediated currents suggesting that there were no changes in excitatory transmission at the parallel fiber that could account for the loss of LTD observed following CA/CPR. While it is possible that there are biochemical changes at the parallel fiber synapse that cannot be detected with electrophysiology, our data strongly point towards an NMDA-receptor-dependent LTD impairment.
The observation that synaptic plasticity is impaired following global cerebral ischemia is consistent with reports in other ischemia-sensitive neuronal populations. Similar to what we report here, in CA1 neurons, long-term potentiation (LTP) is impaired following CA/CPR12 for 30 days following ischemia. Hippocampal LTP requires calcium influx through the NMDA receptor and produces an enhancement in synaptic transmission rather than a depression. Data regarding NMDA receptor function in the CA1 are conflicting, with some studies showing no change in NMDA receptor function and others reporting reduced function and expression following cerebral ischemia. In a four-vessel occlusion model of cerebral ischemia, there have been reports of reduced expression and function of NMDA receptors for up to seven days after injury.37,38 In contrast, our previous data demonstrated no change in AMPA: NMDA ratio in CA1 neurons following CA/CPR suggesting the mechanism for loss of plasticity in the hippocampus is likely different than the mechanism described here in Purkinje cells. The mechanism of the observed reduction in NMDA receptor current in Purkinje cells remains unclear, as no significant reductions in the requisite GluN1 subunit mRNA were observed after CA/CPR. This is somewhat surprising given that the loss of function lasts for 30 days after the insult and that others have reported reduced mRNA in the CA1 that is associated with reduced functionality.38 In contrast, we observed less immunofluorescence for GluN1 in Purkinje cells at seven days after CA/CPR. This disconnects between mRNA and protein expression of GluN1 suggest a non-transcriptional regulation of NMDA receptor expression such as reduced translation or enhanced degradation that requires further investigation. It is also possible that there are additional mechanisms that contribute to the loss of NMDA receptor function that are independent of expression, such as posttranslational modifications. Regardless of the underlying mechanisms, these data support growing evidence that in addition to causing cell death, cerebral ischemia can have profound effects on synaptic plasticity in ischemia sensitive brain regions. It is likely that these plasticity impairments hinder the ability of the brains surviving networks to allow for recovery and function and may serve as a promising therapeutic target that is independent of cell death.
In summary, we have shown long-lasting deficits in cerebellar LTD that are correlated with reduced NMDA receptor function. Impairments in LTD are likely to exacerbate motor coordination and motor learning deficits resulting from cardiac arrest and may reduce the capacity of the cerebellar network to undergo recovery. Over the last decade, advances in functional imaging and lesion-symptom mapping have expanded the role of the cerebellum to include non-motor functions such as cognitive and emotional processing.39–42 Therefore, impairments in cerebellar function may also contribute to cognitive impairments in cardiac arrest survivors. Further, while inhibiting the NMDA receptor in the early acute phase after ischemia may be beneficial for preventing Purkinje cell degeneration, these strategies would likely impair function when administered at delayed time points. Our data suggest that instead strategies to enhance plasticity and repair in the post-ischemic brain should target chronic reduced NMDA receptor function. Indeed, there is evidence that serine, a co-agonist for NMDA receptor, can serve as a therapeutic to promote recovery in the post-ischemic brain. Further studies are needed to determine whether NMDA receptor potentiators promote functional recovery through enhanced synaptic plasticity, and whether such an approach is beneficial in the cerebellum.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NINDS K01 NS086969 (NQ); NINDS R01 NS080851 (PSH); NINDS R01NS046072 (RJT).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Nidia Quillinan designed experiments, collected and analyzed data and wrote manuscript. Guiying Deng collected and analyzed data. Kaori Shimizu collected and analyzed data. Ivelisse Cruz-Torres collected and analyzed data and edited manuscript. Christian Schroeder collected and analyzed data and edited manuscript. Richard J Traystman edited manuscript. Paco S Herson designed experiments and edited manuscript.
References
- 1.Writing Group M, Mozaffarian D, et al. Executive summary: heart disease and stroke statistics – 2016 update: a report from the American Heart Association. Circulation 2016; 133: 447–454. [DOI] [PubMed] [Google Scholar]
- 2.Lim C, Alexander MP, et al. The neurological and cognitive sequelae of cardiac arrest. Neurology 2004; 63: 1774–1778. [DOI] [PubMed] [Google Scholar]
- 3.Khot S, Tirschwell DL. Long-term neurological complications after hypoxic-ischemic encephalopathy. Sem Neurol 2006; 26: 422–431. [DOI] [PubMed] [Google Scholar]
- 4.Peskine A, Rosso C, et al. Neurological sequelae after cerebral anoxia. Brain injury 2010; 24: 755–761. [DOI] [PubMed] [Google Scholar]
- 5.Venkatesan A, Frucht S. Movement disorders after resuscitation from cardiac arrest. Neurol Clin 2006; 24: 123–132. [DOI] [PubMed] [Google Scholar]
- 6.Ng T, Graham DI, et al. Changes in the hippocampus and the cerebellum resulting from hypoxic insults: frequency and distribution. Acta Neuropathol 1989; 78: 438–443. [DOI] [PubMed] [Google Scholar]
- 7.Horn M, Schlote W. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol 1992; 85: 79–87. [DOI] [PubMed] [Google Scholar]
- 8.Kofler J, Hattori K, et al. Histopathological and behavioral characterization of a novel model of cardiac arrest and cardiopulmonary resuscitation in mice. J Neurosci Meth 2004; 136: 33–44. [DOI] [PubMed] [Google Scholar]
- 9.Dell'anna AM, Scolletta S, et al. Early neuroprotection after cardiac arrest. Curr Opin Crit Care 2014; 20: 250–258. [DOI] [PubMed] [Google Scholar]
- 10.Sick T, Pérez-Pinzón M, Feng Z. Impaired expression of long-term potentiation in hippocampal slices 4 and 48 h following mild fluid-percussion brain injury in vivo. Brain Res 1998; 785: 287–292. [DOI] [PubMed] [Google Scholar]
- 11.Mori K, Yoshioka M, et al. An incomplete cerebral ischemia produced a delayed dysfunction in the rat hippocampal system. Brain Res 1998; 795: 221–226. [DOI] [PubMed] [Google Scholar]
- 12.Orfila J, Shimizu K, et al. Increasing SK channel activity reverses ischemia-induced impairment of LTP. Eur J Neurosci 2014; 40: 3179–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brasko J, Rai P, et al. The AMPA antagonist NBQX provides partial protection of rat cerebellar Purkinje cells after cardiac arrest and resuscitation. Brain Res 1995; 699: 133–138. [DOI] [PubMed] [Google Scholar]
- 14.Sato M, Hashimoto H, Kosaka F. Histological changes of neuronal damage in vegetative dogs induced by 18 minutes of complete global brain ischemia: two-phase damage of Purkinje cells and hippocampal CA1 pyramidal cells. Acta Neuropathol 1990; 80: 527–534. [DOI] [PubMed] [Google Scholar]
- 15.Quillinan N, Grewal H, et al. Region-specific role for GluN2B-containing NMDA receptors in injury to Purkinje cells and CA1 neurons following global cerebral ischemia. Neuroscience 2015; 284: 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilbert PF, Thach WT. Purkinje cell activity during motor learning. Brain Res 1977; 128: 309–328. [DOI] [PubMed] [Google Scholar]
- 17.Llinás R, Welsh JP. On the cerebellum and motor learning. Curr Opin Neurobiol 1993; 3: 958–965. [DOI] [PubMed] [Google Scholar]
- 18.Thach WT. A role for the cerebellum in learning movement coordination. Neurobiol Learn Memory 1998; 70: 177–188. [DOI] [PubMed] [Google Scholar]
- 19.Herson PS, Virk M, et al. A mouse model of episodic ataxia type-1. Nat Neurosci 2003; 6: 378–383. [DOI] [PubMed] [Google Scholar]
- 20.Aiba A, Kano M, et al. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 1994; 79: 377–388. [PubMed] [Google Scholar]
- 21.Eccles JC, Llinás R, Sasaki K. The excitatory synaptic action of climbing fibres on the Purinje cells of the cerebellum. J Physiol 1966; 182: 268–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eccles JC, Sasaki K, Strata P. The profiles of physiological events produced by a parallel fibre volley in the cerebellar cortex. Exp Brain Res 1966; 2: 18–34. [DOI] [PubMed] [Google Scholar]
- 23.Konnerth A, Llano I, Armstrong CM. Synaptic currents in cerebellar Purkinje cells. Proc Natl Acad Sci U S A 1990; 87: 2662–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perkel DJ, Hestrin S, et al. Excitatory synaptic currents in Purkinje cells. Proc R Soc Lond 1990; 241: 116–121. [DOI] [PubMed] [Google Scholar]
- 25.Wang YT, Linden DJ. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron 2000; 25: 635–647. [DOI] [PubMed] [Google Scholar]
- 26.Matsuda S, Launey T, et al. Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J 2000; 19: 2765–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee HK, Barbarosie M, et al. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 2000; 405: 955–959. [DOI] [PubMed] [Google Scholar]
- 28.Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev 2001; 81: 1143–1195. [DOI] [PubMed] [Google Scholar]
- 29.Hartell NA. Parallel fiber plasticity. Cerebellum 2002; 1: 3–18. [DOI] [PubMed] [Google Scholar]
- 30.Hansel C, Jeu M, et al. αCaMKII is essential for cerebellar LTD and motor learning. Neuron 2006; 51: 835–843. [DOI] [PubMed] [Google Scholar]
- 31.Kilkenny C, Browne W, et al. Animal research: reporting in vivo experiments: the ARRIVE guidelines. J Gene Med 2010; 12: 561–563. [DOI] [PubMed] [Google Scholar]
- 32.Kilkenny C, Browne W, et al. Animal research: reporting in vivo experiments – the ARRIVE guidelines. J Cereb Blood Flow Metab 2011; 31: 991–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hutchens MP, Traystman RJ, et al. Normothermic cardiac arrest and cardiopulmonary resuscitation: a mouse model of ischemia-reperfusion injury. J Visual Exp 2011; 54: pii3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piochon C, Irinopoulou T, et al. NMDA receptor contribution to the climbing fiber response in the adult mouse Purkinje cell. J Neurosci 2007; 27: 10797–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piochon C, Levenes C, et al. Purkinje cell NMDA receptors assume a key role in synaptic gain control in the mature cerebellum. J Neurosci 2010; 30: 15330–15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He Q, Titley H, et al. Ethanol affects NMDA receptor signaling at climbing fiber-Purkinje cell synapses in mice and impairs cerebellar LTD. J Neurophysiol 2013; 109: 1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, Hsu J, et al. Transient global ischemia alters NMDA receptor expression in rat hippocampus: correlation with decreased immunoreactive protein levels of the NR2A/2B subunits, and an altered NMDA receptor functionality. J Neurochem 1997; 69: 1983–1994. [DOI] [PubMed] [Google Scholar]
- 38.Hsu J, Zhang Y, et al. Decreased expression and functionality of NMDA receptor complexes persist in the CA1, but not in the dentate gyrus after transient cerebral ischemia. J Cereb Blood Flow Metab 1998; 18: 768–775. [DOI] [PubMed] [Google Scholar]
- 39.Schmahmann JD. Disorders of the cerebellum: ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J Neuropsychiatr Clin Neurosci 2004; 16: 367–378. [DOI] [PubMed] [Google Scholar]
- 40.Fabbro F, Tavano A, et al. Long-term neuropsychological deficits after cerebellar infarctions in two young adult twins. Neuropsychologia 2004; 42: 536–545. [DOI] [PubMed] [Google Scholar]
- 41.Strick PL, Dum RP, Fiez JA. Cerebellum and nonmotor function. Ann Rev Neurosci 2009; 32: 413–434. [DOI] [PubMed] [Google Scholar]
- 42.Lagarde J, Hantkie O, et al. Neuropsychological disorders induced by cerebellar damage. Ann Phys Rehabil Med 2009; 52: 360–370. [DOI] [PubMed] [Google Scholar]