

Abstract
Nucleosomes regulate the transcription output of the genome by occluding the underlying DNA sequences from DNA-binding proteins that must act on it. Knowledge of the precise locations of nucleosomes in the genome is thus essential towards understanding how transcription is regulated. Current nucleosome-mapping strategies involve digesting chromatin with nucleases or chemical cleavage followed by high-throughput sequencing. In this review, we compare the traditional Micrococcal nuclease (MNase)-based approach with a chemical cleavage strategy, with discussion on the important insights each has uncovered about the role of nucleosomes in shaping transcriptional processes.
Descriptive keywords: nucleosomes, chromatin, MNase, chemical mapping, transcriptional regulation
Nucleosome positioning plays a critical role in the regulation of transcription
Eukaryotic genomes are packaged hierarchically into a nucleoprotein complex called chromatin. The basic unit of chromatin is the nucleosome, which comprises 147 base pairs (bp) of DNA wrapped 1.65 super helical turns around a small disk-shaped octamer of histone proteins [1, 2]. Nucleosomes are repeated throughout the genome, each separated by unwrapped linker DNA of lengths varying from a few bp to over 100 bp. Approximately 20 bp of linker DNA may associate with a fifth histone, such as H1, thus forming a stable complex of the histone octamer core, H1, and ~165 bp of DNA known as the chromatosome [3].
Nucleosomes sterically occlude their wrapped DNA from proteins that must bind to it for DNA-related processes, including gene regulation and transcription. Access to occluded DNA often requires ATP-dependent nucleosome remodeling factors to unwrap nucleosomal DNA or slide nucleosomes to new locations along DNA [4]. However, remodeling factors are not always required as proteins can still bind nucleosomal targets through “DNA breathing,” in which a stretch of DNA lifts off the histone core and allows transient access to occluded DNA regions [5–7]. In vitro experiments demonstrate that breathing occurs every ~250 milliseconds on the outer stretches of the nucleosome (~20 bp), making these sites relatively accessible [5–7]. At ~40 bp into the nucleosome, however, the breathing frequency decreases to every 10 minutes and even slower at sites close to the nucleosome dyad [8]. It is evident then that the precise location of a nucleosome relative to a target site can influence factor binding by orders of magnitude. In the context of transcriptional regulation, a consequence of this competition over binding sites is that the outcome can have a dramatic effect on gene expression. As an illustration, in vivo work has shown that repositioning nucleosomes by as little as a few base pairs can alter the accessibility of once-hidden TATA boxes, leading to changes in transcription activation [9, 10]. Another example comes from recent studies in yeast: insertion of nucleosome-disfavoring poly(dA) tracts near TF binding sites influences gene expression in a manner that is tunable by the location, length, and composition of the poly(dA) tract [11, 12]. In addition to competing with TFs for DNA accessibility, nucleosomes can function as physical barriers to an elongating RNA Polymerase II (RNAPII) (see Glossary) [13–19]. In in vitro transcription studies on chromatin templates, nucleosomes decrease the rate of transcription elongation by RNAPII [20]. Both in vitro and in vivo studies suggest that nucleosomes contribute to RNAPII pausing. Significantly, in vitro data shows that RNAPII often pauses at the nucleosome dyad, where the histone-DNA contacts are the strongest [15, 16], although in vivo experiments suggest that RNAPII pausing occurs predominantly near the nucleosome entry site [17–19]. Thus, nucleosome positioning along DNA plays a critical role in regulating the transcription output of the genome.
Recent technological advances have enabled nucleosome landscapes to be determined for many different organisms and cell types. The most widely used nucleosome mapping method is Micrococcal nuclease (MNase) digestion of chromatin followed by high-throughput sequencing of the resulting DNA fragments (MNase-seq) [21, 22]. Additional enzymatic methods have been put forward for mapping nucleosomes, including DNase-seq [23], NOME-seq [24], and ATAC-seq [25, 26]. Another approach to determining nucleosome positioning maps is with targeted hydroxyl radical cleavage directed by genetically modified histones (chemical mapping) [27], methidiumpropyl-EDTA-Fe(II) (MPE-seq) [28], or ionizing radiation (RICC-seq) [29]. All existing mapping methods take advantage of the biophysical properties of nucleosomes and involve either enzymatic digestion or chemical cleavage of chromatin. These mapping techniques have collectively contributed to our understanding of nucleosome organization throughout eukaryotic genomes. Due to space constraints, we focus the present review on a comparison between the traditional use of MNase and the chemical mapping approach and discuss the insights they have revealed about the role of nucleosomes in transcriptional regulation.
MNase-seq: a nuclease protection assay to generate global nucleosome maps
MNase digestion yields nucleosome-protected DNA
Most nucleosome mapping experiments are based on the protection of nucleosomal DNA against Micrococcal nuclease (MNase) digestion. MNase is an endo-exonuclease that preferentially degrades the accessible linker DNA between nucleosomes, while most of the nucleosome-bound DNA is left intact (Figure 1a). Brief digestion of chromatin with MNase yields a ‘ladder’ of discrete DNA fragments by agarose gel electrophoresis, where each rung of the ladder corresponds to DNA protected by integer units of nucleosomes (Figure 1b). Thus, the fastest migrating fragment (~147–200 bp) represents DNA wrapped by a single nucleosome (or “mononucleosome”), with fragment sizes increasing by multiples of this length. This characteristic ladder pattern provided early evidence that the regular repeating unit of chromatin consisted of histones wrapped by about 200 bp of DNA [30–33]. As MNase digestion proceeds, the fragment sizes decrease (as indicated by the gradual shortening of the ladder) and, accordingly, the proportion of mononucleosomes is increased. This process can be divided into three major stages [34]. First, monomers produced by initial MNase cleavage are nucleosomes still attached to linker DNA. They tend to be ~200 bp for the most part, but sizes can vary from as little as 154 bp (in Aspergillus fungus [35]) to 260 bp (in sea urchin [36]) due to the wide range of linker lengths. Second, MNase processively trims residual linkers until the fragment lengths are reduced to ~165 bp, as it reaches the boundary of the chromatosome. Third, digestion of the chromatosome eventually results in the concomitant loss of histone H1 and leaves behind the 147 bp nucleosome core particle [33]. At the upper limit of digestion, nucleosomal DNA may be broken down further into subnucleosomal sizes.
Figure 1.
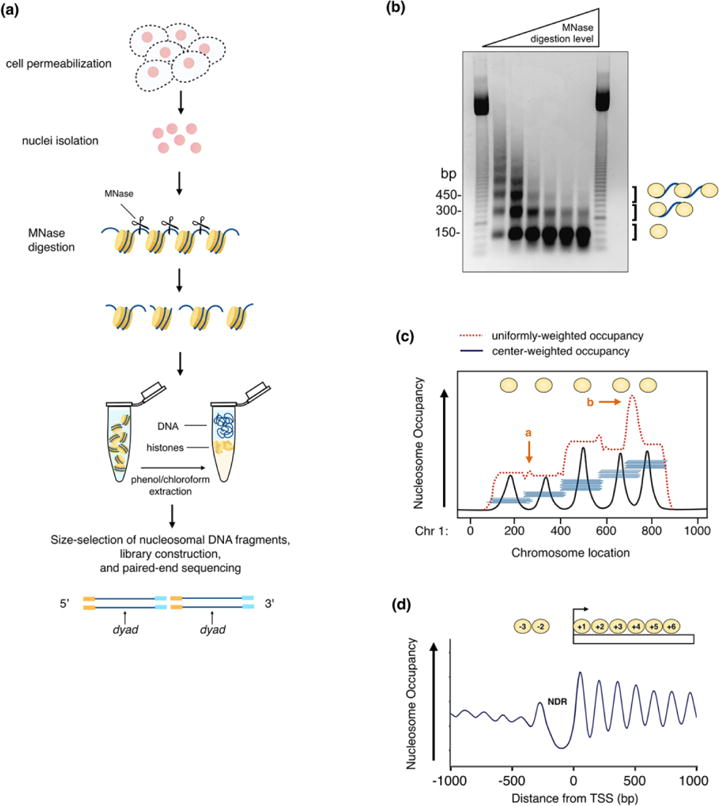
Micrococcal nuclease (MNase) digestion is commonly used to map nucleosomes positions genome-wide. (a) Schematic of MNase mapping experiment. Cells are permeabilized and nuclei are isolated. MNase digests the accessible linker DNA and frees mononucleosomes from chromatin. (MNase is depicted as a pair of scissors.) Undigested DNA fragments are stripped of histones and other DNA-binding proteins by high salt and/or proteinase K digestion, followed by phenol-chloroform extraction. Mononucleosomal DNA is size-selected from an agarose gel (Figure 1b), purified for library construction, followed by paired-end sequencing. Nucleosome dyad positions are inferred from the midpoints of sequenced reads. (b) MNase digestion fragments visualized by agarose gel electrophoresis. With increased MNase digestion (either in time or concentration), the average fragment size gets smaller and the characteristic DNA ladder shortens. Meanwhile, the lowest band (~150 bp), which represents the mononucleosome population, is further enriched. This band is subsequently selected for library construction and paired-end sequencing. (c) Sequenced reads (horizontal blue lines) are mapped back to the genome to reveal the position of the protected DNA fragment on each chromosome. Shown is a comparison of “uniformly-weighted nucleosome (reads) occupancy” versus (red line) “center-weighted nucleosome (reads) occupancy” (black line). Using a center-weighted function prevents spikes (indicated with orange arrows) in the reads occupancy curve due to (a) overlap of reads arising from two neighboring, non-overlapping nucleosomes or (b) two alternatively positioned, overlapping nucleosomes due to population mixture, thus providing better definition of nucleosome centers. (d) Center-weighted nucleosome occupancy around the transcription start sites (TSS) of genes displays a nucleosome-depleted region (NDR). Nucleosome phasing diminishes further downstream from the well-positioned +1 nucleosome.
Determination of nucleosome positions using MNase
Nucleosome protection against MNase has been widely exploited to map individual nucleosome positions in vivo. In a standard MNase experiment, chromatin in either isolated nuclei or permeabilized cells is treated with MNase. Following, mononucleosomal sized fragments are excised and gel-purified for further analysis. Historically, MNase-digested fragments were hybridized to DNA microarrays to obtain low-resolution nucleosome maps of small genomic windows, and later, to high-density microarrays for maps that span the entire genome [37]. Today, MNase digestion is more frequently combined with high-throughput paired-end sequencing (MNase-seq) to determine nucleosome positions genome-wide. Sequence tags are aligned to the reference genome to reveal the positions of nucleosome-protected fragments on each chromosome. With paired-end tags, the location of both boundaries are provided and the nucleosome center position can be inferred by defining the midpoint of each paired read (Figure 1a) [38].
To construct the nucleosome map, DNA fragments of around 147 bp lengths are often selected. The nucleosome occupancy score at any genomic location is often defined as the number of reads that cover the given position. However, this uniformly-weighted occupancy may generate spurious peaks due to closely positioned neighboring nucleosomes or overlapping nucleosomes from alternative nucleosome positionings that arise from a population mixture or nucleosome dynamics; uniformly-weighted occupancy can thus lead to miscalls of nucleosome centers (Figure 1c). To better infer the locations of nucleosome dyads, a Gaussian kernel is often applied to weight each read towards the center to compute the center-weighted nucleosome occupancy. The peaks from center-weighted occupancy often provide better accuracy for mapping dyads than the uniformly-weighted score.
Technical variations of the standard MNase protocol have been described in the literature, including formaldehyde-crosslinking chromatin to preserve DNA-histone interactions before MNase treatment and immunoprecipitating MNase-digested chromatin with antibodies against specific regulatory factors or histone modifications and variants (MNase-ChIP-seq) [37]. Additionally, one study introduced a modified MNase footprinting protocol to simultaneously map the occupancies of nucleosomes, subnucleosomes, and TFs [39].
Canonical nucleosome patterns uncovered by MNase-based studies
MNase has been used to define nucleosome landscapes in a growing list of genomes [21], including yeast [40–46], fly [47–49], worm [50], mouse [51–54], and human [55–57]. Remarkably, these studies have uncovered common patterns of nucleosome organization that are shared across all species.
First, gene promoters exhibit a prominent nucleosome-depleted region (“NDR”) directly upstream of the transcriptional start site (TSS) (Figure 1d). This NDR has an average length of 150 bp, which is enough to accommodate a single nucleosome [40]. Nucleosome-depletion is thought to enable the binding of trans regulatory factors, as many functional cis-regulatory sequences, including TF-binding sites and TATA boxes, are found in the NDRS [40, 58]. In all organisms, the NDR is flanked by two well-positioned nucleosomes, the first upstream nucleosome (−1 nucleosome) and the first downstream nucleosome (+1 nucleosome), followed by an array of regularly spaced nucleosomes that package the gene body. (In yeast, the first nucleosome upstream of the NDR is commonly referred to as the −1 nucleosome. However, in human, this nucleosome is often referred to as the “−2 nucleosome” since the −1 classification is given to the subset of nucleosomes found in the NDR of some genes [56]). The +1 nucleosome is the most strongly positioned while downstream nucleosomes gradually lose their phasing with increasing distance from the TSS [31]. Positioning of the +1 nucleosome is shown to vary in a species- or cell-type-dependent manner [59]. For example, in yeast, the boundary of the +1 nucleosome is consistently positioned just over the TSS, with genes typically starting ~10 bp inside of the +1 nucleosome [43]. In flies, however, the +1 nucleosome is shifted further downstream (~40–60 bp) of the TSS [49]. Differences in the +1 position may be correlated with intrinsic differences in transcriptional regulatory mechanisms.
Consistent with the role of nucleosomes in regulating gene expression, global nucleosome mapping by MNase have demonstrated that promoters of highly expressed genes contain pronounced NDRs [57]. Conversely, genes that are expressed at low levels tend to have promoters that are more occupied by nucleosomes [57], although stable NDRs are present even in the promoters of genes that are infrequently transcribed [49]. These findings have generally supported the idea that nucleosomes compete for binding sites in the regulatory elements and generally repress gene expression [44, 56, 57, 60].
In addition to promoters, transcription terminations sites (TTS) are also characterized by NDRs [43]. This NDR is preceded by a well-positioned nucleosome that demarcates the 3′ end of the gene. Similar to the promoter, the TTS of highly active genes typically feature increased nucleosome depletion, while the TTS of less active genes are more occupied by nucleosomes [44, 61]. Altogether, MNase-based mapping studies have established a “textbook” picture of nucleosome organization in which genes are bookended by well-positioned nucleosomes and NDRs.
Potential problems with the MNase approach
Although MNase-derived maps have enriched our understanding of nucleosome patterning in eukaryotes, there are several factors that limit their accuracy. First, MNase has a strong preference for cutting A/T sequences [62], which could bias recovered sets of nucleosome DNA sequences and result in misrepresentation of nucleosomes in A/T rich regions. Second, MNase does not cut precisely at the nucleosome boundary, which leads to variability in determining the exact positions of nucleosome centers [34]. Third, MNase may digest away nucleosomes that are more prone to nuclease digestion (often referred to in the literature as “fragile nucleosomes”) and are only detected at relatively low MNase concentrations or short digestion times. Recent experimental evidence in different species suggests that fragile nucleosomes occupy gene promoters and the TTS, thereby challenging the classical picture of the MNase-derived NDRs discussed above [39, 44, 63–68]. The reasons for MNase sensitivity appear to vary, indicating that several classes of fragile nucleosomes might exist. For our purposes, we define fragile nucleosomes as nucleosomes that demonstrate high sensitivity to MNase digestion. Because the exact cause of MNase sensitivity of individual nucleosomes is unknown, this qualitative definition of fragile nucleosomes may include some nucleosomes in A/T-rich genomic regions due to the aforementioned MNase sequence bias. Worth noting, while differential MNase digestion can detect fragile nucleosomes, the identity of MNase digestion products—whether they are nucleosomal or non-nucleosomal— cannot be determined by MNase-seq alone and requires further validation by histone ChIP-seq analysis [67, 69–71]. It is also difficult to quantify the relative abundance of fragile nucleosomes to stable nucleosomes after pooling different experimental datasets. Moreover, limited MNase digestion results in nucleosomal DNA fragments with long linkers still attached, decreasing the accuracy of called nucleosome center positions.
Chemical Mapping: determination of nucleosome positions genome-wide by site-directed hydroxyl radical cleavage
Site-directed chemical cleavage of nucleosome DNA in vitro
To overcome the limitations of MNase, a site-directed chemical cleavage method was developed to determine nucleosome positions in vitro [72]. This method relies on hydroxyl radical cleavage of the nucleosome center. Briefly, nucleosomes were reconstituted in vitro using a genetically modified histone octamer and known nucleosomal DNA templates. The histone octamer contains a histone H4 where the serine at position 47 is mutated to a cysteine (H4S47C) (Figure 2a). This position symmetrically flanks the nucleosome dyad axis and is in close proximity to the DNA backbone. By derivatizing a sulfhydryl-binding, iron-chelating reagent (EDTAcyst-NPS) to the cysteine, Fe2+ ions are anchored at the same location—at sites symmetrically flanking the nucleosome dyad axis [72]. With the addition of hydrogen peroxide, chelated Fe2+ ions undergo a Fenton reaction to create hydroxyl radicals that ultimately cleave the DNA backbone at sites symmetrically flanking the nucleosome center [73–75]. Therefore, unlike MNase digestion, chemical cleavage takes advantage of the structure of the nucleosome to cleave within the nucleosome rather than between nucleosomes.
Figure 2.
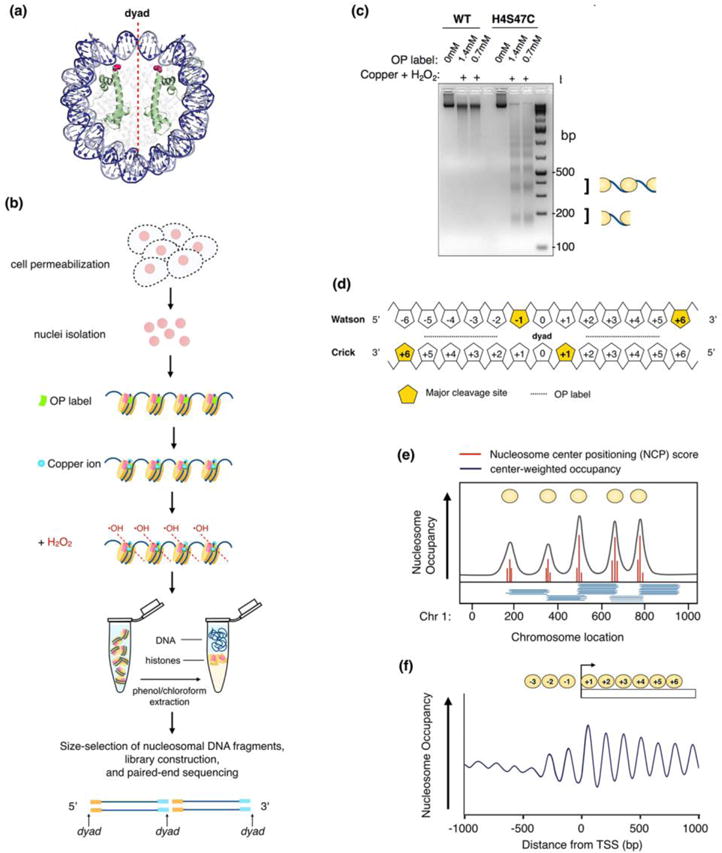
Chemical mapping depends on site-directed hydroxyl radical cleavage of nucleosomes containing modified histone H4 to determine nucleosome positions genome-wide. (a) Structure of a nucleosome, highlighting histone H4 (green) and residue serine 47 (magenta spheres), which is mutated to a cysteine where the sulfhydryl-reactive, copper-chelating reagent (OP label) covalently binds. The nucleosome structure shown here is from [2] and selectively colored as in [27]. (b) Experimental design to chemically map nucleosomes. As in Figure 1a, nuclei are isolated from permeabilized H4S47C-containing cells and incubated with a sulfhydryl-binding, copper-chelating reagent (OP label). The label binds the cysteine of H4S47C and anchors copper ions to DNA at sites flanking the nucleosome dyad. With the addition of hydrogen peroxide (H2O2), hydroxyl radicals cleave where the copper is bound at sites flanking the dyad. Cleaved fragments are treated with high salt and/or proteinase K and subjected to phenol-chloroform extraction. After size selection and DNA purification, chemical cleavage products representing the monolinker between adjacent nucleosomes are prepared for library construction followed by paired-end sequencing. The 5′ and 3′ ends of each read correspond to positions around nucleosome dyad. (c) Resulting DNA fragments from chemical cleavage are visualized by agarose gel electrophoresis. Chemical mapping results in a characteristic DNA banding pattern, which occurs only when the reaction includes the sulfhydryl-reactive OP label (shown in two different concentrations), copper, H2O2 and the H4S47C (from [85]). The monolinker step (lowest band) is excised and purified for library construction, followed by paired-end sequencing. (d) A diagram showing locations of dominant hydroxyl radical cleavages relative to the nucleosome dyad (base pair 0). The cleavage sites are the first nucleotide in the sequencing reads. Relative location of OP label is indicated with perforated line. (e) Sequenced reads are mapped back to a reference genome. The raw cleavage counts are deconvoluted to calculate the nucleosome center positioning (NCP) scores to quantify nucleosome center occupancy at every genomic location. The center-weighted nucleosome occupancy score is defined the summation of Gaussian weighted NCP scores in ±73 bp. (f) Center-weighted nucleosome occupancy from the chemical map reveals a −1 nucleosome in the MNase-derived “NDR” at the TSS (see Figure 1d).
Chemical mapping of nucleosome positions genome-wide in yeast
To apply site-directed chemical cleavage toward mapping nucleosome positions in vivo, Widom and colleagues engineered yeast strains with the H4S47C mutant [27, 76]. (For consistency, genome-wide nucleosome mapping using site-directed chemical cleavage will be referred to simply as “chemical mapping” in this review.) In short, permeabilized H4S47C-containing yeast cells are labeled with a sulfhydryl-reactive, copper-chelating reagent N-(1,10-Phenanthrolin-5-yl)iodoacetamide (“OP” label) (Figure 2b). Next, copper ions and hydrogen peroxide are added to mediate •OH-dependent strand scission of nucleosome centers [77, 78]. On an agarose gel, cleaved products exhibit an MNase-like ladder, wherein the lowest rung corresponds to half of two nucleosome neighbors connected by linker DNA (Figure 2c). Subsequently, DNA products in this size range are isolated and purified, followed by library construction and paired-end sequencing. The 5′ and 3′ ends of the reads denote the center-to-center distance between two adjacent nucleosomes. Therefore, in contrast to MNase-seq, mapping the chemical cleavage sites provides the positions of two nucleosome centers rather than one.
The chemical method generates a genome-wide map of cleavages on both strands of the DNA. The major cleavage sites occur at −1 and +6 bp from the dyad (position 0) for each strand (Figure 2d). However, cleavages also occur at nearby positions, and the pattern may depend on the base composition around dyad [27]. Furthermore, alternatively positioned nucleosomes in a local region may cause convolution of cleavage signals. To resolve these issues, a deconvolution algorithm was built based on cleavage patterns around dyad to decode the exact locations of nucleosomes genome-wide at base pair resolution [27, 79]. This algorithm outputs the nucleosome center positioning (NCP) score that quantifies the dyad occupancy at every genomic location (Figure 2e). Nucleosome occupancy can be calculated by summing up all NCP scores (uniformly weighted) or all Gaussian kernel weighted NCP scores in ±73 bp of the given location.
Chemical mapping has improved resolution over MNase-based mapping
Genome-wide nucleosome maps in yeast Saccharomyces cerevisiae and Schizosaccharomyces pombe generated with chemical mapping are generally consistent with maps from MNase-based methods [27, 76]. However, the chemical map reveals new aspects of in vivo nucleosome organization for the yeast genome.
Most notably, a key insight was made about the role of the genomic DNA sequences in nucleosome positioning. It had been shown previously that nucleosomes are enriched for specific DNA dinucleotide motifs, mostly in the form of ~10-bp periodicity of AA/TT/AT/TA dinucleotides where the DNA backbone (minor groove) faces inward toward the histone core and ~10-bp periodicity of GG/CC/GC/CG dinucleotides where the major groove faces inward [80]. Such sequences motifs are thought to make DNA more bendable and therefore more favorable for nucleosome formation. These periodic dinucleotide signals are collective features of nucleosomes when they are aligned at their centers, and thus the strength of periodicity, which is affected by alignment quality, is used to gauge accuracy in mapping nucleosome centers [80]. Nucleosomes in both MNase- and chemically-defined maps display periodic AA/TT/AT/TA dinucleotide signals [27, 81], but the signals in the chemical map are far stronger. The degraded dinucleotide signal in the MNase map is due in part to the incomplete and unequal cutting efficiency of nucleosome boundaries by MNase and, consequently, the poor alignment of nucleosome centers. On this basis, chemical mapping of nucleosomes in S. cerevisiae and S. pombe [27, 76, 82] supports previous results showing that DNA sequence features such as the ~10-bp periodicity facilitate the fine-tuning of nucleosome positioning in vivo [59, 81, 83].
Due to its single base pair resolution, chemical mapping provides fine details in global chromatin structure and its role in transcription regulation. For example, chemical data show that yeast has a linker length distribution pattern peaked at ~10n+5 bp (for integer n) [27, 76]. In other words, given a nucleosome somewhere in the genome, the next nucleosome down the chain likely starts at the opposite face of the DNA helix, creating an intrinsic zigzag configuration. Second, the chemical map in budding yeast indicates RNAPII has enhanced pause sites across the full length of nucleosomes. During transcription elongation, RNAPII initially pauses at periodic locations inside the nucleosome where the DNA backbone faces out away from the histones and then backtracks by 5 bp and pauses at locations where DNA backbone faces inwards towards the histones (with maximal steric clash between histone and RNAPII) [27]. Lastly, chemical mapping has recently been used to identify alternative nucleosome structures, such the hemisome at the yeast centromere [84].
Chemical mapping in mammalian cells
In an effort to extend chemical mapping capabilities into mammalian genomes, a recent study established a chemical mapping strategy for mouse embryonic stem (ES) cells [85]. The first step was the substitution of multiple endogenous H4 with H4S47C. In yeast, targeted mutation of H4S47 is achieved through standard gene replacement strategies. To meet the challenge of genetically replacing 13 sequence variable histone H4 genes in mouse, the study looked to chemical mapping experiments in fission yeast, which showed that substitution of two of the three H4 genes sufficed to produce levels of chemical cleavage comparable to when all three copies of H4 are replaced [76]. In light of this result from fission yeast, a strategy was implemented to substitute a majority of endogenous H4 proteins in mouse ES cells with mutant H4S47C. First, small hairpin RNAs (shRNAs) against a common region shared by all H4 genes were designed. Second, an RNAi-resistant H4S47C transgene was synthesized. To generate stable H4S47C-expressing mouse ES cells, the H4-shRNA and H4S47C transgene were simultaneously expressed using PiggyBac transgenesis. The simultaneous introduction of H4-shRNA and H4S47C minimizes the deleterious effects of endogenous H4 knockdown. A procedure for chemical mapping in mouse ES cells was then developed based on the yeast chemical mapping protocol. Major modifications included changes in cell permeabilization reagents and reductions in the concentration and incubation time with the OP label. Under optimized conditions, H4S47C-targeted nucleosome cleavage resulted in a characteristic DNA ladder. To generate a genome-wide map of nucleosomes in mouse ES cells, DNA in the monolinker band was isolated for library construction and paired-end sequencing.
Insights from chemical mapping in mouse embryonic stem cells
The chemical map in mouse ES cells uncovered surprising features of genome-wide nucleosome organization in the mouse genome. First, many MNase-defined NDRs including gene promoters and TTS are abundantly occupied by nucleosomes in the chemical map (Figure 2f). This observation is in contrast to the longstanding view of promoter nucleosome architecture provided by MNase-derived maps [86]. Second, while MNase data show that relatively higher nucleosome occupancy in the promoter and TTS is associated with decreased gene expression, the chemical map presents the opposite pattern— high levels of nucleosome occupancy are associated with elevated gene expression. The discrepancy between the two maps may arise from MNase’s inability to reliably map fragile nucleosomes, which exhibit greater susceptibility to digestion than stable nucleosomes. Several lines of evidence argue for the existence of fragile nucleosomes. First, nucleosome-sized DNA fragments over the NDR are recovered with limited MNase digestion, suggesting that some nucleosomal reads are lost under prolonged digestion conditions. Second, short read fragments (50–80 bp) from extensive MNase digestions [53] are enriched in the NDRs, with a high portion of their edges anchored between an AA/AT/TA/TT dinucleotide, coinciding with preferred rotational motif sites inside of nucleosome DNAs. Third, recent MNase titration studies in mammalian genomes identified MNase-sensitive nucleosomes upstream of the TSS [28, 68, 87]. Fourth, methidiumpropyl-EDTA sequencing (MPE-seq), which uses hydroxyl free radicals to preferentially cleave the linker DNA between nucleosomes without the sequence bias of MNase, coupled with histone ChIP-seq confirmed the occupancy of histones H3 and H2B at the TSS [28]. Collectively, results in mouse ES cells illustrate that fragile nucleosomes exist in the mouse genome and chemical mapping is capable of detecting them.
In contrast to previous genome-wide MNase maps, chemically-derived nucleosome maps show that target sites of vertebrate insulator protein CTCF and pluripotent TFs Oct4, Sox2, Nanog, and Klf4 are occupied by nucleosomes [85]. Furthermore, factor binding is positively correlated with nucleosome occupancy such that sites most bound by these factors display relatively higher nucleosome density. Consistent with previous ChIP-seq [88] and MPE-seq [28] experiments in mouse ES cells (including our analysis of available MPE-seq data [85]), the chemical map provides in vivo evidence for the pioneering function of these factors.
Chemical mapping is broadly applicable to investigations on how the nucleosome landscape regulates transcription. For instance, to examine the regulatory role of nucleosomes in transcription elongation by RNAPII, the chemical map in mouse ES cells was integrated with data from global run-on sequencing (GRO-seq) [89]. First, a statistically significant and positive correlation was observed between the occupancy of the +1 nucleosome and RNAPII pausing in the proximal promoter, implicating a role for the nucleosome as a transcriptional barrier in vivo. By extending this analysis into the gene body, RNAPII pausing was found to coincide with nucleosome enrichment at the 5′ and 3′ exon-intron junctions. Specifically, the chemical map illustrates that splice sites are preferentially positioned near the nucleosome dyad where the strongest DNA-histone contacts occur [20]. These findings disagree with MNase-based studies, which suggests nucleosomes are well positioned at exon centers and splices sites are frequently located in linker regions [90, 91]. The chemical nucleosome patterning at exons better explains the two RNAPII pausing events around exons. Thus, the study contends that nucleosomes regulate exon recognition by stalling RNAPII at exon boundaries [92].
Potential limitations to chemical mapping
A basic limitation to chemical mapping is the requirement of the genetically engineered H4S47C. For genomes that encode a high copy number of H4 genes, often residing within multiple loci, substitution of all H4 alleles through standard gene targeting is experimentally challenging. Nevertheless, the chemical mapping strategy used in mouse ES cells provides a practical solution to this limitation, involving transgenic replacement of most of the endogenous H4 with H4S47C (as described above). Another caveat of chemical mapping is the possibility of labeling non-histone cysteine-containing proteins in addition to H4S47C, which may contribute to non-specific cleavage of DNA and inaccurate nucleosome maps. As a result, higher coverage of sequencing is required to accurately discern nucleosome center positions from noise, which inevitably requires higher costs for larger genomes. In addition, it remains unknown whether transgenic H4S47C is evenly distributed throughout the genome and small molecule cysteine-reactive label has equal access to different chromatin regions. These factors potentially affect the relative efficiency of DNA cleavage and influence the accuracy of chemical mapping.
Concluding remarks: the road ahead for nucleosome mapping
Nucleosomes modulate the accessibility of DNA to other DNA-binding proteins. Consequently, where nucleosomes are positioned in relation to genetic elements can have a profound impact on transcription outcomes. Integration of nucleosome mapping strategies with recent advances in high-throughput sequencing technologies have revolutionized the depth to which nucleosome positions are mapped genome-wide, which, in turn, have increased our understanding of how nucleosomes regulate gene expression [21, 22]. In this review, we focused our attention on two nucleosome-mapping methods. First, MNase-seq is a nuclease protection assay that can determine nucleosome positions, but its accuracy is limited by sequence bias and over-digestion of fragile nucleosomes. Second, chemical mapping offers a means to determine nucleosome center positions without sequence bias but requires genetic manipulation of H4 genes. While parallels drawn between MNase-seq and chemical maps can verify each other to a large extent, chemical maps with improved accuracy have begun to revise prevailing narratives governing how to think about the functions of nucleosomes with regard to transcription regulation. Foremost, insights from the chemical map revealed that classical NDRs in promoters and functional TF binding sites are populated by fragile nucleosomes, leaving open the question what are these fragile nucleosomes and what are their roles in transcription regulation (see Outstanding Questions)? Studies in human cells reveal that transcriptionally active promoters, enhancers, and insulator regions are enriched for nucleosome core particles containing histone variants H3.3 and H2A.Z [88], which have been shown to form labile nucleosomes structures with increased sensitivity to MNase digestion and high salt conditions [88, 93]. Future studies will be necessary to understand the nature of the fragile nucleosome species, their structural identities, and possible interactions with chromatin-remodeling complexes and transcriptional machinery. Chemical mapping will be useful toward this purpose. Second, the nucleosome-TF co-occupancy model proposed by the chemical map suggests that not all TFs compete for exclusive binding on nucleosomal targets, as perhaps more may function as pioneer factors. As recent studies have shown that pioneer factors are critical for cellular reprogramming and misregulation of their expression or function is associated with various forms of cancer [94, 95], it will be important to understand the mechanisms by which pioneer factors influence the nucleosome landscape. For example, canonical pioneer factor FoxA has recently been shown to displace linker histone H1 and increase MNase accessibility of nucleosomes at liver-specific enhancers [96]. It is possible that CTCF, Oct4, Nanog, Sox2, or Klf4 may function through a similar mechanism in ES cells. In fact, recent in vitro data have shown that Oct4, Sox2, and Klf4 can bind directly to nucleosomal target DNA [97]. We anticipate that chemical mapping will be an excellent tool in discovering other pioneer factors.
Future steps towards advancing our understanding of how nucleosome positions and dynamics regulate gene expression include identifying cellular factors that drive the in vivo positioning of nucleosomes. (For review on determinants of nucleosome positioning, we direct readers to [59].) To achieve this goal, chemical mapping can be implemented in organisms or cell populations carrying deletions of candidate positioning factors. Further, we anticipate that chemical mapping will be used to delineate nucleosome landscapes in a multitude of organisms and cell types. Most excitingly, we look forward to its applications in human cells and in the study of how nucleosomes reposition during cellular differentiation and disease progression.
TRENDS BOX.
Genome-wide nucleosome positioning maps provide deep insight into the regulatory role of nucleosomes in transcriptional processes.
Micrococcal nuclease (MNase) digests linker DNA in between nucleosomes while nucleosome-protected DNA remains intact. Sequencing the protected DNA allows for the determination of nucleosome positions genome-wide.
The chemical mapping method relies on site-directed hydroxyl radical cleavage of nucleosomes carrying modified histones to determine the positions of nucleosomes in the genome.
MNase-defined NDRs of cis regulatory elements are nucleosome enriched in the chemical map of mouse ES cells. Emerging evidence shows that such regions are occupied by “fragile nucleosomes”, which are lost due to over-digestion by MNase Results in mouse ES cells illustrate that fragile nucleosomes exist in the mouse genome and chemical mapping is capable of detecting them.
OUTSTANDING QUESTIONS BOX.
What is the nature of fragile nucleosomes and what is their role in gene regulation?
How do pioneer factors bind to nucleosome targets? What other pioneer factors are there? What are their roles in the transcriptional control of cell fate?
What is the relationship between nucleosome positioning and splicing?
Acknowledgments
We thank K. Durbin and A. Sebeson for assisting with this manuscript. L.N.V. was funded by the CMBD training grant NIH T32 GM08061 and the NSF Graduate Research Fellowship. J.-P.W. and X.W. were supported by a grant from NIGMS R01GM107177.
Glossary
- Fenton reaction
is a reaction in which transition metal ions, such as iron or copper, are oxidized by hydrogen peroxide, forming a hydroxyl radical and a hydroxide ion in the process. In the case of chemical mapping, cuprous ions are oxidized as follows: Cu+ + H2O2—> Cu2+ + •OH + OH−
- Global run-on sequencing (GRO-seq)
measures elongating RNAPII activity genome-wide.
- Nucleosome dyad
is the center of the nucleosomal DNA fragment.
- Nucleosome occupancy
is the fraction of cells from a population in which a given base pair is wrapped in a nucleosome. Unlike nucleosome positioning, occupancy is not concerned with where the nucleosome is positioned so long as the base pair is covered by one.
- Nucleosome positioning
commonly refers to the exact base pair position of a nucleosome on DNA with respect to a reference point, such as the start location or the dyad.
- Phasing (with regard to nucleosomes)
refers to an array of nucleosomes that are approximately aligned and show rhythmic patterning in nucleosome occupancy when the DNA is aligned a genomic landmark, (e.g., TSS, TTS, etc.)
- RNA polymerase II (RNAPII)
is an enzyme that transcribes DNA into mRNA.
- Well-positioned nucleosome
describes a nucleosome that is dominantly positioned in the exact same location in all cells, shown in the data as a sharp and well-separated nucleosome occupancy peak. Conversely, in the literature, the term fuzzy nucleosomes describes poorly positioned nucleosomes or nucleosomes that have many alternatively positioning nearby, shown as a very wide and noisy occupancy peak in the data
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luger K, et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423(6936):145–50. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 3.Luger K, Hansen JC. Nucleosome and chromatin fiber dynamics. Curr Opin Struct Biol. 2005;15(2):188–96. doi: 10.1016/j.sbi.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Tsukiyama T. The in vivo functions of ATP-dependent chromatin-remodelling factors. Nat Rev Mol Cell Biol. 2002;3(6):422–9. doi: 10.1038/nrm828. [DOI] [PubMed] [Google Scholar]
- 5.Li G, et al. Rapid spontaneous accessibility of nucleosomal DNA. Nat Struct Mol Biol. 2005;12(1):46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 6.Li G, Widom J. Nucleosomes facilitate their own invasion. Nat Struct Mol Biol. 2004;11(8):763–9. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- 7.Polach KJ, Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol. 1995;254(2):130–49. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 8.Tims HS, et al. Dynamics of nucleosome invasion by DNA binding proteins. J Mol Biol. 2011;411(2):430–48. doi: 10.1016/j.jmb.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lomvardas S, Thanos D. Nucleosome sliding via TBP DNA binding in vivo. Cell. 2001;106(6):685–96. doi: 10.1016/s0092-8674(01)00490-1. [DOI] [PubMed] [Google Scholar]
- 10.Martinez-Campa C, et al. Precise nucleosome positioning and the TATA box dictate requirements for the histone H4 tail and the bromodomain factor Bdf1. Mol Cell. 2004;15(1):69–81. doi: 10.1016/j.molcel.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 11.Raveh-Sadka T, et al. Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat Genet. 2012;44(7):743–50. doi: 10.1038/ng.2305. [DOI] [PubMed] [Google Scholar]
- 12.Sharon E, et al. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat Biotechnol. 2012;30(6):521–30. doi: 10.1038/nbt.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teves SS, et al. Transcribing through the nucleosome. Trends Biochem Sci. 2014;39(12):577–86. doi: 10.1016/j.tibs.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 14.Petesch SJ, Lis JT. Overcoming the nucleosome barrier during transcript elongation. Trends Genet. 2012;28(6):285–94. doi: 10.1016/j.tig.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bondarenko VA, et al. Nucleosomes can form a polar barrier to transcript elongation by RNA polymerase II. Mol Cell. 2006;24(3):469–79. doi: 10.1016/j.molcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Kireeva ML, et al. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell. 2002;9(3):541–52. doi: 10.1016/s1097-2765(02)00472-0. [DOI] [PubMed] [Google Scholar]
- 17.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469(7330):368–73. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwak H, et al. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 2013;339(6122):950–3. doi: 10.1126/science.1229386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber CM, et al. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol Cell. 2014;53(5):819–30. doi: 10.1016/j.molcel.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 20.Kulaeva OI, et al. Mechanism of transcription through a nucleosome by RNA polymerase II. Biochim Biophys Acta. 2013;1829(1):76–83. doi: 10.1016/j.bbagrm.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teif VB. Nucleosome positioning: resources and tools online. Brief Bioinform. 2016;17(5):745–57. doi: 10.1093/bib/bbv086. [DOI] [PubMed] [Google Scholar]
- 22.Zentner GE, Henikoff S. High-resolution digital profiling of the epigenome. Nat Rev Genet. 2014;15(12):814–27. doi: 10.1038/nrg3798. [DOI] [PubMed] [Google Scholar]
- 23.Bell O, et al. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12(8):554–64. doi: 10.1038/nrg3017. [DOI] [PubMed] [Google Scholar]
- 24.Kelly TK, et al. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012;22(12):2497–506. doi: 10.1101/gr.143008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buenrostro JD, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schep AN, et al. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 2015;25(11):1757–70. doi: 10.1101/gr.192294.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brogaard K, et al. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012;486(7404):496–501. doi: 10.1038/nature11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishii H, et al. MPE-seq, a new method for the genome-wide analysis of chromatin structure. Proc Natl Acad Sci U S A. 2015;112(27):E3457–65. doi: 10.1073/pnas.1424804112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Risca VI, et al. Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping. Nature. 2017;541(7636):237–241. doi: 10.1038/nature20781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hewish DR, Burgoyne LA. Chromatin sub-structure. The digestion of chromatin DNA at regularly spaced sites by a nuclear deoxyribonuclease. Biochem Biophys Res Commun. 1973;52(2):504–10. doi: 10.1016/0006-291x(73)90740-7. [DOI] [PubMed] [Google Scholar]
- 31.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184(4139):868–71. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 32.Noll M. Subunit structure of chromatin. Nature. 1974;251(5472):249–51. doi: 10.1038/251249a0. [DOI] [PubMed] [Google Scholar]
- 33.Noll M, Kornberg RD. Action of micrococcal nuclease on chromatin and the location of histone H1. J Mol Biol. 1977;109(3):393–404. doi: 10.1016/s0022-2836(77)80019-3. [DOI] [PubMed] [Google Scholar]
- 34.Clark DJ. Nucleosome positioning, nucleosome spacing and the nucleosome code. J Biomol Struct Dyn. 2010;27(6):781–93. doi: 10.1080/073911010010524945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonzalez R, Scazzocchio C. A rapid method for chromatin structure analysis in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res. 1997;25(19):3955–6. doi: 10.1093/nar/25.19.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keichline LD, Wassarman PM. Developmental study of the structure of sea urchin embryo and sperm chromatin using micrococcal nuclease. Biochim Biophys Acta. 1977;475(1):139–51. doi: 10.1016/0005-2787(77)90348-3. [DOI] [PubMed] [Google Scholar]
- 37.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10(3):161–72. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zentner GE, Henikoff S. Surveying the epigenomic landscape, one base at a time. Genome Biol. 2012;13(10):250. doi: 10.1186/gb-2012-13-10-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henikoff JG, et al. Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci U S A. 2011;108(45):18318–23. doi: 10.1073/pnas.1110731108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan GC, et al. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309(5734):626–30. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- 41.Lee W, et al. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet. 2007;39(10):1235–44. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- 42.Albert I, et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446(7135):572–6. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- 43.Mavrich TN, et al. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008;18(7):1073–83. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weiner A, et al. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res. 2010;20(1):90–100. doi: 10.1101/gr.098509.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Z, Pugh BF. High-resolution genome-wide mapping of the primary structure of chromatin. Cell. 2011;144(2):175–86. doi: 10.1016/j.cell.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitehouse I, et al. Chromatin remodelling at promoters suppresses antisense transcription. Nature. 2007;450(7172):1031–5. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- 47.Gilchrist DA, et al. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell. 2010;143(4):540–51. doi: 10.1016/j.cell.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen K, et al. A global change in RNA polymerase II pausing during the Drosophila midblastula transition. Elife. 2013;2:e00861. doi: 10.7554/eLife.00861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mavrich TN, et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453(7193):358–62. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valouev A, et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18(7):1051–63. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Z, et al. The nucleosome map of the mammalian liver. Nat Struct Mol Biol. 2011;18(6):742–6. doi: 10.1038/nsmb.2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teif VB, et al. Genome-wide nucleosome positioning during embryonic stem cell development. Nat Struct Mol Biol. 2012;19(11):1185–92. doi: 10.1038/nsmb.2419. [DOI] [PubMed] [Google Scholar]
- 53.Carone BR, et al. High-resolution mapping of chromatin packaging in mouse embryonic stem cells and sperm. Dev Cell. 2014;30(1):11–22. doi: 10.1016/j.devcel.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scruggs BS, et al. Bidirectional Transcription Arises from Two Distinct Hubs of Transcription Factor Binding and Active Chromatin. Mol Cell. 2015;58(6):1101–12. doi: 10.1016/j.molcel.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 56.Schones DE, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132(5):887–98. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Valouev A, et al. Determinants of nucleosome organization in primary human cells. Nature. 2011;474(7352):516–20. doi: 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gaffney DJ, et al. Controls of nucleosome positioning in the human genome. PLoS Genet. 2012;8(11):e1003036. doi: 10.1371/journal.pgen.1003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Struhl K, Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol. 2013;20(3):267–73. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22(9):1798–812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, et al. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol. 2009;16(8):847–52. doi: 10.1038/nsmb.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chung HR, et al. The effect of micrococcal nuclease digestion on nucleosome positioning data. PLoS One. 2010;5(12):e15754. doi: 10.1371/journal.pone.0015754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xi Y, et al. Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res. 2011;21(5):718–24. doi: 10.1101/gr.117101.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vera DL, et al. Differential nuclease sensitivity profiling of chromatin reveals biochemical footprints coupled to gene expression and functional DNA elements in maize. Plant Cell. 2014;26(10):3883–93. doi: 10.1105/tpc.114.130609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knight B, et al. Two distinct promoter architectures centered on dynamic nucleosomes control ribosomal protein gene transcription. Genes Dev. 2014;28(15):1695–709. doi: 10.1101/gad.244434.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jeffers TE, Lieb JD. Nucleosome fragility is associated with future transcriptional response to developmental cues and stress in C. elegans. Genome Res. 2017;27(1):75–86. doi: 10.1101/gr.208173.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kubik S, et al. Nucleosome Stability Distinguishes Two Different Promoter Types at All Protein-Coding Genes in Yeast. Mol Cell. 2015;60(3):422–34. doi: 10.1016/j.molcel.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 68.Mieczkowski J, et al. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun. 2016;7:11485. doi: 10.1038/ncomms11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chereji RV, et al. MNase-Sensitive Complexes in Yeast: Nucleosomes and Non-histone Barriers. Mol Cell. 2017;65(3):565–577 e3. doi: 10.1016/j.molcel.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chereji RV, et al. Genome-wide profiling of nucleosome sensitivity and chromatin accessibility in Drosophila melanogaster. Nucleic Acids Res. 2016;44(3):1036–51. doi: 10.1093/nar/gkv978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mueller B, et al. Widespread changes in nucleosome accessibility without changes in nucleosome occupancy during a rapid transcriptional induction. Genes Dev. 2017;31(5):451–462. doi: 10.1101/gad.293118.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Flaus A, et al. Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc Natl Acad Sci U S A. 1996;93(4):1370–5. doi: 10.1073/pnas.93.4.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fenton HJH. LXXIII.—Oxidation of tartaric acid in presence of iron. Journal of the Chemical Society, Transactions. 1894;65:899–910. [Google Scholar]
- 74.Tullius TD, et al. Hydroxyl radical footprinting: a high-resolution method for mapping protein-DNA contacts. Methods Enzymol. 1987;155:537–58. doi: 10.1016/0076-6879(87)55035-2. [DOI] [PubMed] [Google Scholar]
- 75.Ebright YW, et al. Incorporation of an EDTA-metal complex at a rationally selected site within a protein: application to EDTA-iron DNA affinity cleaving with catabolite gene activator protein (CAP) and Cro. Biochemistry. 1992;31(44):10664–70. doi: 10.1021/bi00159a004. [DOI] [PubMed] [Google Scholar]
- 76.Moyle-Heyrman G, et al. Chemical map of Schizosaccharomyces pombe reveals species-specific features in nucleosome positioning. Proc Natl Acad Sci U S A. 2013;110(50):20158–63. doi: 10.1073/pnas.1315809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sigman DS, et al. Oxygen-dependent cleavage of DNA by the 1,10-phenanthroline. cuprous complex. Inhibition of Escherichia coli DNA polymerase I. J Biol Chem. 1979;254(24):12269–72. [PubMed] [Google Scholar]
- 78.Que BG, et al. Degradation of deoxyribonucleic acid by a 1,10-phenanthroline-copper complex: the role of hydroxyl radicals. Biochemistry. 1980;19(26):5987–91. doi: 10.1021/bi00567a007. [DOI] [PubMed] [Google Scholar]
- 79.Xi L, et al. A locally convoluted cluster model for nucleosome positioning signals in chemical map. J Am Stat Assoc. 2014;109(505):48–62. doi: 10.1080/01621459.2013.862169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang JP, Widom J. Improved alignment of nucleosome DNA sequences using a mixture model. Nucleic Acids Res. 2005;33(21):6743–55. doi: 10.1093/nar/gki977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Segal E, et al. A genomic code for nucleosome positioning. Nature. 2006;442(7104):772–8. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin H, et al. Categorical spectral analysis of periodicity in nucleosomal DNA. Nucleic Acids Res. 2016;44(5):2047–57. doi: 10.1093/nar/gkw101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Radman-Livaja M, Rando OJ. Nucleosome positioning: how is it established, and why does it matter? Dev Biol. 2010;339(2):258–66. doi: 10.1016/j.ydbio.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Henikoff S, et al. The budding yeast Centromere DNA Element II wraps a stable Cse4 hemisome in either orientation in vivo. Elife. 2014;3:e01861. doi: 10.7554/eLife.01861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Voong LN, et al. Insights into Nucleosome Organization in Mouse Embryonic Stem Cells through Chemical Mapping. Cell. 2016;167(6):1555–1570 e15. doi: 10.1016/j.cell.2016.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hughes AL, Rando OJ. Mechanisms underlying nucleosome positioning in vivo. Annu Rev Biophys. 2014;43:41–63. doi: 10.1146/annurev-biophys-051013-023114. [DOI] [PubMed] [Google Scholar]
- 87.Deng T, et al. HMGN1 modulates nucleosome occupancy and DNase I hypersensitivity at the CpG island promoters of embryonic stem cells. Mol Cell Biol. 2013;33(16):3377–89. doi: 10.1128/MCB.00435-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jin C, et al. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet. 2009;41(8):941–5. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Williams LH, et al. Pausing of RNA polymerase II regulates mammalian developmental potential through control of signaling networks. Mol Cell. 2015;58(2):311–22. doi: 10.1016/j.molcel.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tilgner H, et al. Nucleosome positioning as a determinant of exon recognition. Nat Struct Mol Biol. 2009;16(9):996–1001. doi: 10.1038/nsmb.1658. [DOI] [PubMed] [Google Scholar]
- 91.Schwartz S, et al. Chromatin organization marks exon-intron structure. Nat Struct Mol Biol. 2009;16(9):990–5. doi: 10.1038/nsmb.1659. [DOI] [PubMed] [Google Scholar]
- 92.Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet. 2014;15(3):163–75. doi: 10.1038/nrg3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lombrana R, et al. High-resolution analysis of DNA synthesis start sites and nucleosome architecture at efficient mammalian replication origins. EMBO J. 2013;32(19):2631–44. doi: 10.1038/emboj.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Iwafuchi-Doi M, Zaret KS. Pioneer transcription factors in cell reprogramming. Genes Dev. 2014;28(24):2679–92. doi: 10.1101/gad.253443.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Iwafuchi-Doi M, Zaret KS. Cell fate control by pioneer transcription factors. Development. 2016;143(11):1833–7. doi: 10.1242/dev.133900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Iwafuchi-Doi M, et al. The Pioneer Transcription Factor FoxA Maintains an Accessible Nucleosome Configuration at Enhancers for Tissue-Specific Gene Activation. Mol Cell. 2016;62(1):79–91. doi: 10.1016/j.molcel.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Soufi A, et al. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell. 2015;161(3):555–68. doi: 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]