

SUMMARY
Aneuploidy, a state of karyotype imbalance, is a hallmark of cancer. Changes in chromosome copy number have been proposed to drive disease by modulating the dosage of cancer driver genes and by promoting cancer genome evolution. Given the potential of cells with abnormal karyotypes to become cancerous, do pathways exist that limit the prevalence of such cells? By investigating the immediate consequences of aneuploidy on cell physiology, we identified mechanisms that eliminate aneuploid cells. We find that chromosome mis-segregation leads to further genomic instability that ultimately causes cell cycle arrest. We further show that cells with complex karyotypes exhibit features of senescence and produce pro-inflammatory signals that promote their clearance by the immune system. We propose that cells with abnormal karyotypes generate a signal for their own elimination that may serve as a means for cancer cell immunosurveillance.
Keywords: aneuploidy, cancer, immune system, genome instability, senescence
INTRODUCTION
In all organisms analyzed to date, aneuploidy, an unbalanced karyotype in which one or more chromosomes are present in excess or reduced copy number, is highly detrimental (Santaguida and Amon, 2015a). Aneuploid budding and fission yeast show proliferation defects under standard growth conditions (Niwa et al., 2006; Torres et al., 2007). In multicellular organisms, chromosomal gain or loss is largely lethal (Hodgkin, 2005; Lindsley et al., 1972; Lorke, 1994). In humans, for example, all monosomies and most trisomies cause embryonic lethality (reviewed in (Hassold and Hunt, 2001)). Only trisomy of the gene poorest chromosome, chromosome 21, is compatible with survival into adulthood. However, even this trisomy leads to high levels of embryonic lethality. Only 12.5 percent of trisomy 21 fetuses survive to birth (reviewed in (Roper and Reeves, 2006)).
The adverse effects of an incorrect karyotype are also observed at the cellular level. Aneuploid mammalian and yeast cells exhibit metabolic alterations (Williams et al., 2008), proliferation defects (Santaguida et al., 2015; Stingele et al., 2012; Tang et al., 2011; Thompson and Compton, 2010; Torres et al., 2007; Williams et al., 2008), genome instability (Blank et al., 2015; Meena et al., 2015; Ohashi et al., 2015; Passerini et al., 2016; Sheltzer et al., 2011; Zhu et al., 2012), proteotoxic stress (Oromendia et al., 2012; Santaguida et al., 2015; Santaguida and Amon, 2015b; Stingele et al., 2012; Tang and Amon, 2013), and aneuploid mammalian cells have been reported to activate p53 (Hinchcliffe et al., 2016; Li et al., 2010; López-García et al., 2017; Sansregret et al., 2017; Thompson and Compton, 2010). In addition to traits observed in a broad range of aneuploidies, aneuploid cells exhibit gene-specific phenotypes where changes in dosage of a particular gene cause a specific phenotype (i.e. (Dodgson et al., 2016)).
The observation that an aneuploid karyotype has detrimental consequences on cellular fitness is consistent with the low prevalence of aneuploid cells in somatic tissues (~2 percent) (Knouse et al., 2014). Aneuploid cells are a rare occurrence even in tissues of mice harboring mutations that cause high levels of chromosome mis-segregation. Mice carrying a hypomorphic mutation in the spindle assembly checkpoint (SAC) component BUB1B (BUB1bH/H allele), exhibit high levels of chromosome mis-segregation in all tissues where this has been analyzed (Baker et al., 2004). Yet, single cell sequencing revealed aneuploid cells to be exceedingly rare in regenerating tissues such as the intestine, skin and blood from these animals (Pfau et al., 2016). Whether aneuploid cells are outcompeted by euploid cells or whether mechanisms exist that eliminate aneuploid cells from tissues is not known.
Paradoxically, despite the adverse effects of an aneuploid karyotype on normal cell physiology, the condition is also a hallmark of cancer, a disease characterized by excessive cell proliferation. 90% of solid tumors harbor whole chromosome gains and/or losses (Gordon et al., 2012; Holland and Cleveland, 2009). Multiple, not mutually exclusive hypotheses have been put forth to explain the prevalence of abnormal karyotypes in cancer. Chromosome copy number alterations have been proposed to drive disease by modulating the dosage of cancer driver genes (Davoli et al., 2013). Aneuploidy also endows cells with phenotypic variability (Beach et al., 2017; Chen et al., 2015; Rutledge et al., 2016), which could help facilitate metastasis or resistance to therapeutic interventions. Indeed aneuploidy has been shown to be associated with metastatic behavior, resistance to chemotherapy and poor patient outcome (Bakhoum et al., 2011; Heilig et al., 2009; Lee et al., 2011; Walther et al., 2008). Finally, the process of chromosome mis-segregation and aneuploidy of many chromosomes have been shown to cause genomic instability (Blank et al., 2015; Crasta et al., 2012; Janssen et al., 2011; Ohashi et al., 2015; Passerini et al., 2016; Sheltzer et al., 2011; Zhu et al., 2012), which could fuel cancer genome evolution.
Given the potential link between aneuploidy and tumorigenesis, it is critical to understand how abnormal karyotypes affect cellular physiology. Here, we examine the immediate consequences of chromosome mis-segregation. We find that following chromosome mis-segregation cells experience replication stress and genomic instability that cause the evolution of cells with highly aberrant karyotypes characterized by complex patterns of whole chromosome and segmental aneuploidies. Such cells cease to divide, undergo senescence and produce pro-inflammatory signals that leads to their elimination by natural killer cells in vitro. Our results indicate that mechanisms exist that eliminate cells with aberrant karyotypes and thus protect organisms from cells with the potential to become cancerous.
RESULTS
Chromosome mis-segregation rarely leads to p53 activation
Previous studies reported that chromosome mis-segregation causes p53 activation and a p53-dependent cell cycle arrest (Li et al., 2010; Thompson and Compton, 2010). The aneuploid state per se or events accompanying chromosome mis-segregation could be responsible for this p53 activation. To distinguish between these possibilities we examined the immediate consequences of chromosome mis-segregation using live cell microscopy.
Several methods have been developed to induce chromosome mis-segregation. For example, compounds that interfere with microtubule dynamics or microtubule – kinetochore attachment cause a SAC dependent delay in mitosis and induce chromosome mis-segregation. Inducing chromosome mis-segregation in this manner was shown to be associated with p53 activation in the subsequent G1 phase (Thompson and Compton, 2010). However, mitotic arrest exceeding ~100 minutes induces a p53-dependent G1 arrest irrespective of whether or not chromosomes are mis-segregated (Uetake and Sluder, 2010). We too observed this phenomenon. We analyzed cells that experienced an extended mitosis induced by the kinesin Eg5 inhibitor monastrol by live cell imaging (Mayer et al., 1999). This analysis showed that the frequency of chromosome mis-segregation and subsequent G1 arrest increased with time spent in mitosis (Figure S1), highlighting that without live imaging it is difficult to compare fates of cells with and without mis-segregation due to the missing information about arrest duration.
To avoid G1 arrest caused by a prolonged mitosis, we generated aneuploid cells by interfering with SAC function rather than by activating the checkpoint. SAC inactivation does not delay cells in mitosis but instead accelerates progression through this cell cycle stage even when chromosomes are not attached to the spindle correctly (Figure S2A) and results in aneuploid progeny.
We examined hTERT immortalized RPE-1 cells stably expressing PCNA-GFP (to determine S phase initiation) and RFP-H2B (to monitor chromosome segregation) grown in the presence of NMS-P715 or reversine. Both compounds inhibit the SAC kinase Mps1 (Colombo et al., 2010; Santaguida et al., 2010). Treatment with NMS-P715 or reversine led to severe chromosome segregation defects. Each chromosome mis-segregated in 6 – 8% of mitoses (Figure S2D–F and (Santaguida et al., 2015)) and virtually all cells harbored lagging chromosomes during anaphase and micronuclei in the following G1 (Figure 1A and Figure S2B, C). Despite severe chromosome mis-segregation, however, mitotic arrest did not occur but cells in fact progressed through mitosis faster than vehicle-control treated cells (Figure S2A). Notably, chromosome mis-segregation did not lead to arrest in the following G1 in the vast majority of aneuploid daughter cells (~80%; Figure 1B). This finding indicates that aneuploidy per se does not cause cell cycle arrest in G1.
Figure 1. p53 activation is not an obligatory consequence of chromosome mis-segregation.

(A) Representative images of hTERT RPE1 cells co-expressing PCNA::GFP and RFP::H2B. Unsynchronized cells were treated with DMSO or 0.5 μM reversine and then immediately filmed for 48h. Cells were filmed every 5 min for 6 hrs to capture mitotic mis-segregation events and then every 20 min for 42 hrs to capture daughter cell S phase timing. Scale bar 5 μm.
(B) Daughter cell fate in NMS-P715-treated hTERT RPE1 cells co-expressing PCNA::GFP and RFP::H2B. Unsynchronized cells were treated with DMSO or 1μM NMS-P715 and immediately filmed as described in (A). Bars represent percentage of daughter cells with the indicated cell fate.
(C–E) Schematic representation of experimental method used to separate cells that arrest in G1 following chromosome mis-segregation from cells that continue to divide (C). RPE-1 cells were synchronized at the G1/S transition by thymidine treatment. Six hours after Thymidine release, cells were treated with 0.5 μM reversine for 12 hours. Six hours later cells were treated with nocodazole. 12 hours later, mitotic cells were removed by shake-off and single cells that detached from the plate were sequenced to determine the karyotype of cells that continue to proliferate after chromosome mis-segregation (cycling). The cells that were not removed by shake-off were placed into fresh medium containing nocodazole. This procedure was repeated three times in order to remove all dividing cells. The cells that remained attached to the plate represented arrested cells (arrested) and their karyotype was determined by single cell sequencing. Heat map in (D) shows chromosome gains and losses in the indicated cell populations. Partially colored boxes represent segmental aneuploidies and are marked as “yes” in the column SA (for segmental aneuploidies). The graph in (E) shows the degree of genome imbalance, defined as the total number of genes that are either gained or lost as a consequence of whole chromosome and segmental aneuploidies (mean ± SEM).
(F) RPE-1 cells were synchronized at the G1/S transition by thymidine treatment. Six hours after Thymidine wash-out, cells were treated with 0.5μM reversine or DMSO (vehicle control) for 12 hours. After drug wash-out, cells were grown for 66 hours (for a total of 72 hours after mitosis) to generate populations of aneuploid dividing cells (aneuploid cycling). Arrested aneuploid cells were generated as described in Figure 1C. The levels of p53, p21 and p16 were determined by Western blot analysis. Actin served as a loading control.
See also Figure S1, S2
Although 80 percent of cells that mis-segregated chromosomes continued to divide, 9% of cells arrested in G1. To determine why these 9% of cells arrested in G1, we developed a method to separate G1-arrested cells from cycling cells following chromosome mis-segregation (Figure 1C). Briefly, we induced chromosome mis-segregation, and then transiently exposed cells to the microtubule poison nocodazole during the cell cycle following chromosome mis-segregation (Figure 1C). Dividing cells arrest in mitosis and can be removed from the plate by shake-off (Figure 1C). We repeated this procedure multiple times to also eliminate cells that progress through the cell cycle more slowly. The only cells that remained adhered to the plate following this procedure were cells that arrested in the interphase immediately following the mitosis during which chromosome mis-segregation was induced. Single cell sequencing revealed highly abnormal karyotypes characterized by multiple chromosome gains and losses in cells that arrested immediately following chromosome mis-segregation (Figure 1D, E). Whereas aneuploid cells that still divided harbored genomic imbalances, which involved less than 5% of their genomes, aneuploid arrested cells exhibited genomic imbalances involving more than 20% of their genomes (Figure 1E). In addition to whole chromosome gains and losses, 42% of cells that had arrested in G1 (5 out of 12 cells) also harbored segmental aneuploidies compared to about 18% of aneuploid cells that were still able to divide (2 out of 11 cells; note that the segmental gain on chromosome 10 was not included in this analysis as it is a characteristic of RPE-1 cells (Zhang et al., 2015)). Aneuploid arrested cells also exhibited signs of cellular senescence. Levels of p53, and the CDK inhibitors p21 and p16 were elevated (Figure 1F).
Inactivation of the DNA damage checkpoint suppresses the G1 arrest following chromosome mis-segregation
Segmental aneuploidies in G1 arrested aneuploid cells could be the result of DNA damage incurring during cytokinesis (Janssen et al., 2011), during accelerated anaphase entry (Figure S2A), or in micronuclei (Crasta et al., 2012). Their presence in G1 arrested aneuploid cells thus raised the possibility that cell cycle arrest was due to DNA damage associated with chromosome mis-segregation. To test this we analyzed γ-H2AX levels following chromosome mis-segregation. Although not detectable by Western blot analysis (Figure S3A), we observed a transient, modest increase in the average number of γ-H2AX foci 3 – 6 hours after chromosome mis-segregation (Figure 2A, S3B, C and (Janssen et al., 2011)). More importantly, preventing activation of the DNA damage response by inhibiting the DNA damage-response kinases ATR, p38 or CHK2 or inactivation of p53 partially suppressed the infrequent G1 delay following chromosome mis-segregation (Figure 2B, C; S3D). These findings suggest that DNA damage occurring during chromosome mis-segregation might be responsible for the G1 arrest following chromosome mis-segregation in a small number of cells. Support for this conclusion comes from the finding that constitutive aneuploidies do not elicit a p53 response. Mouse embryonic fibroblasts (MEFs) trisomic for various different chromosomes do not activate p53 (Tang et al., 2011) nor does p53 depletion suppress their proliferation defects (Sheltzer et al., 2017). It is also possible that stresses associated with aneuploidy, such as oxidative and proteotoxic stress, which scale with the degree of aneuploidy, cause p53 activation. Indeed, we note, that cells that arrest in G1 and activate p53 following chromosome mis-segregation harbor highly aberrant karyotypes (Figure 1D).
Figure 2. DNA damage incurred during chromosome mis-segregation causes p53 activation.
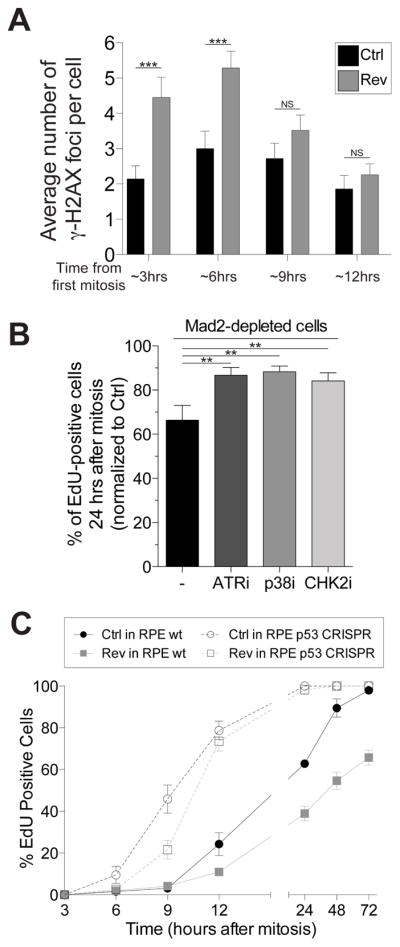
(A) RPE-1 cells were synchronized at the G1/S transition by thymidine treatment. After Thymidine wash-out, cells were treated with 0.5μM reversine or DMSO (vehicle control). The average number of γ-H2AX foci per cell was determined at the indicated times (mean ± SEM). ***: p value < 0.0001; **; NS: not significant, analysis of variance plus Bonferroni’s test. F test of variance: 3 hour time point: 7.54718E-05; 6 hour time point: 0.002922704
(B) RPE-1 cells were exposed to a single round of siRNA-mediated depletion of Mad2 or control (Ctrl) oligo followed by thymidine arrest. 14h after thymidine wash-out (which corresponds roughly to 2 hours after mitosis), control- and Mad2-depleted cells were exposed to the indicated kinase inhibitors and EdU. The percentage of EdU-positive cells was determined 36 hours after thymidine release (~24 hours after mitosis). The graph shows the percentage of EdU-positive cells normalized to control-depleted cells. The following small molecule inhibitors were used: VE821 (ATR inhibitor, working concentration 1 μM), SB203580 (p38 inhibitor, working concentration 10 μM), Chk2 inhibitor II (Chk2 inhibitor, working concentration 10 μM). Graph shows mean ± SEM. ***: p value < 0.0001, analysis of variance plus Bonferroni’s test.
(C) RPE-1 cells, either wild type for p53 or lacking the tumorsuppressor (p53 CRISPR), were synchronized at the G1/S transition by thymidine treatment. After thymidine wash-out, cells were treated with 0.5 μM reversine or DMSO (vehicle control). 14 hours later, the drug was washed-out and cells were exposed to EdU. Percentage of EdU-positive cells was determined at the indicated times. Graph shows mean ± SEM.
See also Figure S3
Aneuploidy causes DNA replication defects
While aneuploidy per se did not impair G1 progression, it was possible that other cell cycle stages were affected by an unbalanced karyotype. Previous studies in budding yeast and human cells showed that many but not all aneuploidies cause DNA replication defects and genomic instability because of changes in gene copy number of factors critical for DNA replication and segregation (Blank et al., 2015; Meena et al., 2015; Ohashi et al., 2015; Passerini et al., 2016; Sheltzer et al., 2011; Zhu et al., 2012). To assess the immediate effects of chromosome mis-segregation on the subsequent S phase, we induced chromosome mis-segregation using reversine (aneuploid) or did not interfere with mitosis (euploid), then synchronized cells in G1 and measured DNA replication fork rate by DNA combing after release from the G1 arrest. Fork rates were significantly slower in cells that had mis-segregated their chromosomes compared to their euploid counterparts (0.59 ± 0.02 kb min−1 versus 0.88 ± 0.03 kb min−; Figure 3A, B). Furthermore, replication fork stalling was increased in aneuploid cells (Figure 3B). Live cell imaging of reversine-treated cells confirmed this result (Figure 3C, D; Movie S1). PCNA foci, a sign of ongoing DNA replication, persisted for longer times in cells that had mis-segregated their chromosomes in the preceding mitosis (Figure 3D). The prolonged presence of PCNA foci in cells was not a consequence of reversine treatment. Cells that were in G1 at the time of reversine treatment (cells that had completed mitosis but did not harbor PCNA foci at the time of reversine addition) were slightly delayed in S phase by reversine treatment (Figure 3D, Table S1; compare G1 population +/− reversine) but this delay was not nearly as dramatic as that of cells that had mis-segregated chromosomes because reversine was added prior to mitosis (Figure 3D; compare G2 population +/− reversine). Not surprisingly, DNA replication defects were accompanied by DNA damage as judged by an increase in 53BP1 foci (Figure 3E, F). Our results indicate that chromosome mis-segregation leads to DNA replication stress in the following S phase which results in DNA damage. We propose that genomic imbalances caused by the aneuploid state as well as replication problems in micronuclei are causes of this replication defect (Passerini et al., 2016).
Figure 3. Chromosome mis-segregation causes replication stress.

(A, B) RPE-1 cells were treated with reversine (0.5μM; aneuploid) or vehicle control (euploid) for 24 hours. The inhibitor was then washed out and cells were arrested in G1 by treatment with Mimosine for 24 hours. After Mimosine wash-out, cells were placed into fresh medium and three hours later pulse labeled with IdU (green) for 60 min and chased with CldU (red) for 60 min. Sample fiber images from euploid and aneuploid cells are shown in (A). Fork rate, fork density and fork stall rate are shown in (B).
(C, D) Unsynchronized RPE-1 cells co-expressing PCNA::GFP and RFP::H2B were treated with DMSO or reversine (2 μM), imaged every 5 min for 5 hrs to capture mitotic mis-segregation events, and then imaged every 20 min for 48h to capture daughter cell S phase timing. Representative images of mother cell anaphase and one daughter cell S phase after treatment with DMSO or reversine are shown in (C). Quantification of the time interval from PCNA focus appearance to dissolution, a measure for S phase duration in living cells, is shown in (D). The analysis was performed on daughter cells whose mother cells divided prior to addition of reversine (reversine exposure occurred during G1) and on daughter cells whose mothers mis-segregated chromosomes in the presence of reversine (reversine exposure occurred during G2; mean ± SD). Scale bar 5 μm.
(E, F) RPE-1 cells were treated with 0.5μM reversine or vehicle control for 24 hours. The drug was then washed out and cells were synchronized in G1 using Mimosine for 24 hours. After Mimosine wash-out, cells were placed into fresh medium and 53BP1 foci were analyzed 4 hours later. Representative images (E) and quantification (F) are shown. Scale bar 5 μm.
See also Movie S1
Chromosome mis-segregation triggers the evolution of complex abnormal karyotypes
What are the consequences of aneuploidy-induced DNA damage? Do such cells permanently arrest in G2 or do some of them proceed into mitosis? Our immunofluorescence studies indicate that the latter occurs. We observed DNA damage to persist into pro-metaphase. Aneuploid pro-metaphase cells harbored increased levels of γ-H2AX foci compared to euploid cells but lower than those seen in cells treated with aphidocolin which interferes with replication of late replicating regions of the genome and results in DNA damage persisting into prometaphase (Figure 4A, B; (Minocherhomji et al., 2015)).
Figure 4. Chromosome mis-segregation results in genomic instability.

(A, B) RPE-1 cells were synchronized at the G1/S transition by thymidine treatment. Six hours after Thymidine release, cells were treated with control vehicle (euploid) or 0.5 μM reversine (aneuploid) for 12 hours. 12 hours after drug wash-out, cells were treated with the CDK1 inhibitor RO-3306 for 12 hours to enrich for G2 cells. Cells were then released in fresh medium containing nocodazole and γ-H2AX foci were analyzed 2 hours later. CREST is used to mark centromeres. As a positive control, cells were treated with aphidicolin 18 hours after thymidine release. Representative images are shown in (A). Quantification of γ-H2AX foci is shown in (B). γ-H2AX is in green, CREST in red, DNA in blue (mean ± SEM). Scale bar 10 μm. **: p value < 0.01, analysis of variance plus Bonferroni’s test.
(C–F) RPE-1 cells stably expressing H2B-GFP were synchronized at the G1/S transition by thymidine treatment. Six hours after thymidine wash-out, cells were treated with control vehicle (euploid) or 0.5 μM reversine (aneuploid) for 12 hours. After drug wash-out, cells were filmed every 5′. Quantification of mitotic aberrations (lagging chromosomes and micronuclei) is shown in (E). Length of mitosis (nuclear envelope breakdown (NEBD) to DNA decondensation) is shown in (F; mean ± SEM).
(G) Cells were grown as described in Figure 4A. After RO-3306 wash-out, cells were released into fresh medium and fixed 90′ later to analyze DNA bridges in anaphase. Representative images of DNA bridges are shown. PICH is in green, BLM in red, CREST in magenta, DNA in blue. Scale bar 10 μm.
To further characterize how chromosome mis-segregation affect genome integrity, we analyzed the cell division following the mitosis during which cells were treated with reversine by live cell microscopy (henceforth second mitosis; Figure 4C). Aneuploid cells exhibited a high degree of mitotic aberrations during the second mitosis - lagging chromosomes during anaphase and micronuclei in the following G1 (Figure 4D, E, Figure S4 and Movie S2). Importantly, these abnormalities were not due to incomplete drug wash out and hence lack of a SAC function. Duration of the abnormal second mitoses was significantly longer than that of unperturbed mitoses, indicating that the spindle assembly checkpoint was active (Figure 4F). Furthermore, we observed ultrafine anaphase DNA bridges – DNA threads that connect under-replicated genomic regions – as revealed by staining with antibodies against the Bloom’s syndrome helicase (BLM) protein and Plk1-interacting checkpoint helicase (PICH) protein (Figure 4G).
Signs of DNA damage, identified as 53BP1 foci, were apparent even in G1 following this second mitosis (Figure 5A, B). Comparison of the karyotypes of cells immediately after chromosome mis-segregation (“First cell cycle” in Figure 5C, D; S5A) and of cells that had undergone an additional mitosis thereafter confirmed that complex karyotypes evolve in aneuploid cells (“Second cell cycle” in Figure 5C, D; S5B). Only 20% of cells displayed greater than two chromosome aberrations immediately after chromosome mi-segregation (Fig 5C, D, in agreement with Figure S2C–F and (Colombo et al., 2010; Hewitt et al., 2010; Santaguida et al., 2015; 2010)), but in the second mitosis, 80% of cells harbored more than 2 chromosome gains or losses (Figure 5C, D). Together our results indicate that chromosome mis-segregation has consequences beyond the production of cells with whole chromosome gains or losses. It sets in motion a process where replication stress and DNA damage drive chromosome segregation errors and mitotic aberrations in the next mitosis.
Figure 5. Chromosome mis-segregation triggers the evolution of complex abnormal karyotypes.

(A, B) Cells were grown as described in Figure 4A. After RO-3306 wash-out, cells were released into fresh medium and fixed 6 hours later to analyze 53BP1 foci in the following G1. Representative images are shown. 53BP1 is in red, γ-H2AX in green, CREST in magenta, DNA in blue. Quantification of 53BP1 foci in G1 is shown in (B). Scale bar 5 μm.
(C, D) Karyotype analysis after the first and second cell cycle following chromosome mis-segregation (see Figure S5 for experimental details). The percentage of cells with more than 2 chromosome changes (C) and the total number of chromosomal changes per cell (D) are shown for aneuploid cells after the first and second cell cycle following chromosome mis-segregation. *: p value < 0.05, Student’s t test.
See also Figure S5
Cells with complex karyotypes senesce
Our results indicate that chromosome mis-segregation leads to the generation of cells with highly aberrant karyotypes. Their ability to undergo mitosis decreased as karyotypes became more aberrant (Figure 6A) indicating that they arrest in interphase. To characterize the cells that had stopped proliferating within three days after they had been induced to mis-segregate their chromosomes, we induced chromosome mis-segregation, let cells divide for three days and then repeatedly used the previously described nocodazole-shake off protocol to remove cells that were still proliferating, which we determined to be approximately 60 percent (Figure 6B). Of the 40 percent of cells that had arrested within three days following chromosome mis-segregation, approximately half had never undergone a cell division and the other half had undergone at least one cell cycle as judged by EdU incorporation (Figure 6C).
Figure 6. Chromosome mis-segregation causes the generation of cells with complex karyotypes that undergo senescence.

(A) RPE-1 cells were treated with reversine or control vehicle for 24 hours. Cells were exposed for 12 hours to nocodazole either after wash-out or 24 hours later, to determine the ability of cells to enter mitosis during the first or second cell cycle following chromosome mis-segregation (mean ± SEM).
(B) Schematic representation of the experimental method used to isolate arrested cells with complex karyotypes (see STAR methods for details). Shown below the cartoon is the percentage of cells that detached from the plate after each shake-off.
(C) Aneuploid arrested cells were isolated as described in STAR methods. Cells were exposed to EdU after reversine wash-out, plated on coverslips after the fourth nocodazole shake-off and fixed 12 hours later to determine the percentage of EdU-positive cells. Graph shows mean ± SEM.
(D, E) Aneuploid arrested cells were isolated as described in STAR methods and their karyotype determined by single cell sequencing. The heat map of chromosome gains and losses of arrested cells with complex karyotypes is shown in (D). Partially colored boxes represent segmental aneuploidies and are marked as “yes” in the column SA (for segmental aneuploidies). The graph in (E) shows the degree of genome imbalance, defined as the total number of genes that are either gained or lost as a consequence of whole chromosome and segmental aneuploidies (mean ± SEM). The “Cycling” sample is the same as presented in Figure 1E and is comprised of aneuploid cells able to divide.
(F) RPE-1 cells were synchronized at the G1/S transition by thymidine treatment. Six hours after thymidine release, cells were treated with DMSO (euploid) or 0.5 μM reversine (aneuploid) for 12 hours. After wash-out, cells were placed into fresh medium and harvested 18 or 66 hours later, which corresponds to 24 and 72 hours after mitosis, respectively. Arrested cells were obtained as described in Figure 6B. The levels of p53, p21, p16 and γ-H2AX were determined by Western blot analysis. Actin served as a loading control. Euploid cells treated with doxorubicin for 6 or 12 hours were used as positive controls. Euploid and aneuploid samples represent cells that were initially treated with DMSO or 0.5 μM reversine, respectively.
(G) Euploid, aneuploid cycling and aneuploid arrested cells were generated as described in STAR methods. The graph shows the quantification of γ-H2AX foci per cell. Euploid cells treated with doxorubicin for 12 hours were used as a positive control. Graph shows mean ± SEM.
(H) Senescence-associated β-galactosidase (β-gal) activity was determined in euploid cells and arrested cells with complex karyotypes obtained as described in Figure 6B.
(I) The percentage of cycling and arrested euploid or aneuploid RPE-1 cells, either wild type or lacking p53, was determined following the scheme shown in Figure 6B. Graph shows the percentage of arrested cells or cells recovered after the indicated mitotic shake-offs.
The cell population obtained in this manner exhibited the same characteristics as cells that arrested in G1 immediately following chromosome mis-segregation. They harbored highly abnormal karyotypes with multiple whole chromosome and segmental aneuploidies (Figure 6D, E) and displayed the hallmarks of senescence, including elevated levels of the senescence markers p53, p21 and p16 (Figure 6F), higher numbers of γ-H2AX foci (Figure 6G), and increased senescence-associated β-galactosidase activity (Figure 6H and (Baker et al., 2004)).
Like cells that arrest in G1 immediately following chromosome mis-segregation, inactivation of p53 partially suppressed their cell cycle arrest. We induced chromosome mis-segregation, removed proliferating cells by nocodazole shake off and then determined the number of cells that detached from the plate as a measure of the percentage of the population that continued to proliferate following chromosome mis-segregation. This analysis revealed that fewer aneuploid cells arrested in interphase when p53 was deleted compared to aneuploid cells with intact p53 (Figure 6I). Our results indicate that approximately 50% of cells arrest in G1 due to p53 activation but other pathways prevent cell proliferation in the other half of G1 cells.
The aneuploid state causes a senescence-associated gene expression signature
Gene expression analysis of the G1 arrested aneuploid cells was consistent with cells being senescent. We observed down-regulation of genes involved in cell cycle progression (Figure S6A–C) and a senescence-associated gene expression profile known as senescence-associated secretory phenotype (SASP; (Freund et al., 2010)) (Figure S6D and Table S2; the leading edge of the enrichment includes the genes listed in Table S3). DNA damage and cell cycle arrest are likely a major cause of the SASP gene expression signature observed in cells with complex karyotypes but other aspects of the aneuploid state likely also contribute. MEFs harboring specific trisomies also show a SASP gene expression pattern (Figure S6E and (Sheltzer et al., 2012)) yet these cells do not experience significant DNA damage nor activate p53 nor undergo cell cycle arrest (Figure S6F; (Tang et al., 2011; Williams et al., 2008)).
Cells with complex karyotypes produce pro-inflammatory signals
Our gene expression analysis not only revealed the existence of a SASP-like gene expression signature in aneuploid cells but also the upregulation of genes that mediate inflammation and an immune response (Figure 7A). The top 7 upregulated gene set categories in arrested cells with complex karyotypes represented gene expression profiles associated with an immune response. Interestingly, with the exception of the “Interferon alpha/beta signaling” gene-set, immune response gene sets observed in aneuploid arrested cells did not match gene sets previously identified in cells in which senescence was induced by DNA damage (Figure 7A; (Krizhanovsky et al., 2008)). Furthermore, we found the cGAS/STING pathway, an innate immune system pathway that is activated in response to cytosolic DNA, to be upregulated in cells with complex karyotypes ((Schneider et al., 2014); Figure S7G, Table S2). Whether these findings indicate that aneuploid arrested cells exhibit a different immune response than cells that senesce due to DNA damage remains to be determined. We conclude that cells that arrest in G1 with highly abnormal karyotypes induce multiple immune response pathways. In agreement with the idea that the aneuploid state produces a pro-inflammatory signal is the recent finding that fibroblasts derived from Down Syndrome individuals activate an interferon response (Sullivan et al., 2016).
Figure 7. Aneuploidy triggers an immune response.

(A) Canonical pathway gene set (c2cp of msigdb) enrichment for up-regulated genes in arrested cells with complex karyotypes. Normalized enrichment scores (NES) for the top 7 up-regulated gene sets are shown. The columns on the right show the normalized enrichment score for these gene categories in gene expression data sets obtained from cells in which senescence was induced by DNA damage (Iannello et al., 2013; Krizhanovsky et al., 2008).
(B) Cytokine levels were determined in supernatants of euploid and arrested cells with complex karyotypes (obtained as described in Figure 6B). Graph shows the cytokine fold change in arrested cells with complex karyotypes normalized to euploid cells (mean ± SEM).
(C) Early passage wild-type and BUB1BH/H mouse embryonic fibroblasts were cultured and CCL2 levels were determined in culture supernatants.
(D–G) MICA/B (D), CD155/PVR (E) and ULBP2 (F) cell surface levels in euploid cells and aneuploid arrested cells (obtained as described in Figure 6B). Euploid cells treated with doxorubicin (100 ng/ml, 48 hours) were used as a positive control for CD155/PVR and ULBP2 expression. Graphs show fluorescence intensity of cells of similar size (gating see Figure S7). IgG2A isotype control was used for MICA/B, ULBP2 and ULBP1 staining in euploid cells (Figure S7B). IgG1 isotype control was used for CD155/PVR and Nectin-2/CD112 in euploid cells (Figure S7A). Mean fluorescence intensities are listed in (G).
(H) Euploid and arrested cells with complex karyotypes were generated as described in Figure 6B and levels of the indicated protein determined by enzyme-linked immunosorbent assay (mean ± SEM).
(I) Euploid and arrested cells with complex karyotypes were generated as described in Figure 6B. Cells were co-cultured with NK92 cells at a target:effector ratio of 1:10 and filmed every 10′ for 36 hours. Representative images from live imaging datasets are shown.
(J) Euploid and aneuploid arrested cells were generated as described in Figure 6B. Cells were seeded and allowed to attach to the plate for 12 hours, after which NK92 cells were introduced. At the indicated times, dead and NK92 cells were removed by gentle shake-off and the remaining cells were counted and are shown as a percentage of cells grown for the same time in the absence of NK92 cells (mean ± SD). ***: p value < 0.0001; *: p value < 0.05; NS: not significant, analysis of variance plus Bonferroni’s test.
(K) Euploid and arrested cells with complex karyotypes and NK92 cells were cultured as in Figure 7J for 24 hours. For experiments with antibodies blocking NKG2D, NK92 cells were pre-incubated with 20 μg/ml anti-NKG2D antibody for three hours before co-culturing them with RPE-1 cells. Dead and NK92 cells were removed and the remaining cells were counted and normalized to cells grown under the same condition but without co-culture (mean ± SEM). ***: p value < 0.0001; NS: not significant, analysis of variance plus Bonferroni’s test.
See also Figure S6, S7 and Movie S3
Consistent with the inflammatory gene expression profile observed in cells with complex karyotypes we found the secretion of cytokines (IL-6, IL-8 and CCL2) to be elevated (Figure 7B). Interestingly, we also observed a subtle elevation in secretion of CCL2 but not other cytokines in early passage mouse embryonic fibroblasts (MEFs) derived from BUB1bH/H (Figure 7C), which do not exhibit signs of senescence. This finding raises the interesting possibility that increased immunogenicity is not just a characteristic of cells with complex karyotypes that ceased to divide but also of cells with aberrant karyotypes that are proliferating.
To further characterize the immunogenic potential of arrested aneuploid cells we examined various cell surface proteins known to trigger recognition by the innate immune system, specifically natural killer (NK) cells. First, we examined expression of MICA and MICB. MICA and MICB are cell surface proteins that belong to the natural killer group 2, member D (NKG2D) ligand family, and activate NK cells in response to proteotoxic stress (Raulet and Guerra, 2009). Although euploid cells expressed MICA and MICB at their cell surface, MICA/B levels were elevated two fold in arrested aneuploid cells (Figure 7D, G; (Chien et al., 2011; Krizhanovsky et al., 2008)). This increase was not due to differences in cell size between aneuploid and euploid cells, as we compared MICA/B mean fluorescence intensity between cell populations of the same size (Figure S7A). We also examined the expression of the NKG2D ligands ULBP1 and ULBP2 at the cell surface. ULBP1 and ULBP2 are induced by cellular stresses and DNA damage (Raulet and Guerra, 2009). Their levels were also elevated in aneuploid arrested cells compared to euploid cells but did not reach levels seen in euploid cells treated with the DNA damaging agent doxorubicin (Figure 7E, G and S7B–D).
DNAM1 is an adhesion molecule at the surface of NK cells which mediates interactions between NK cells and target cells (Raulet and Guerra, 2009). This protein binds to CD112 (also known as Nectin-2) and CD155 (also known as PVR) two surface molecules expressed in response to DNA damage (Raulet and Guerra, 2009). Aneuploid cells expressed CD112 and CD155 at elevated levels (Figure 7F, G and S7E–G). CD155 levels, in particular, were as high in aneuploid arrested cells with complex karyotypes as in euploid cells treated with doxorubicin (Figure 7F, G). Finally, we found phosphorylation of STAT3 (Y705) and SAPK/JNK (T183/Y185) (Figure 7H) to be higher in aneuploid arrested cells which suggests that the inflammatory response caused by arrested cells with complex karyotypes triggers a feed-forward loop in which secreted cytokines propagate the inflammation response by activating other inflammatory pathways such as those dependent on STAT3 and SAPK/JNK (Kyriakis and Avruch, 2001). Together, these findings indicate that aneuploid arrested cells elicit an immune response.
To test whether aneuploid arrested cells were indeed targeted by immune cells we co-cultured euploid cells or aneuploid arrested cells with NK92 cells and followed cell viability by live cell imaging (see STAR methods). NK92 cells did not interact with euploid cells, but effectively killed cells with complex karyotypes (Figure 7I, J and movie S3). NK92-mediated killing of aneuploid cells was not immediate but started 6–12 hours after mixing of cells (Figure 7J and movie S3). Despite the fact that NK92 mediated killing occurred only after a lag, the response was nevertheless NK92 cell-specific. Aneuploid arrested cells did not die when cultured in the absence of NK92 cells (Figure S7H). Further evidence supporting the conclusion that selective killing occurred stems from the observation that the interaction between NK cells and aneuploid arrested cells was at least partially dependent on the NK cell receptor NKG2D. Pre-incubation of NK cells with blocking antibodies against NKG2D decreased their ability to kill aneuploid arrested cells (Figure 7K). We conclude that cells with complex karyotypes elicit an innate immune response aimed at their removal.
DISCUSSION
By investigating the immediate consequences of aneuploidy on cells, we identified mechanisms that eliminate aneuploid cells. Chromosome mis-segregation leads to genomic instability and increased karyotype complexity ((Blank et al., 2015; Meena et al., 2015; Ohashi et al., 2015; Passerini et al., 2016; Sheltzer et al., 2011; Zhu et al., 2012), this study). Cells with complex aberrant karyotypes ultimately cease to divide, exhibit features of senescence and produce pro-inflammatory signals that promote their clearance by the immune system. Together, these findings indicate that multiple mechanisms prevent the accumulation of aneuploid cells in tissues. A senescence program limits their proliferation and the innate immune system facilitates their clearance. The latter mechanism could very well represent a means whereby cancer cells, which are frequently highly aneuploid, are recognized and eliminated by the immune system.
Cells cannot count their chromosomes
It was previously reported that chromosome mis-segregation induced by interference with spindle function causes p53 activation (Thompson and Compton, 2010). This observation led to the interesting proposal that the cells somehow “know” how many chromosomes they have and that a chromosome number that deviates from the euploid karyotype triggers a p53 response. However chromosome mis-segregation brought about by interfering with spindle function results in a mitotic delay, which when it exceeds 100 minutes causes p53 activation in the subsequent G1 phase irrespective of whether or not chromosome mis-segregation occurred (Uetake and Sluder, 2010) and requires the DNA damage binding protein 53BP1 and the deubiquitinating enzyme USP28 (Lambrus and Holland, 2017). To examine the effects of chromosome mis-segregation on cell cycle progression without this complication, we used methods to interfere with chromosome segregation that accelerated rather than delayed mitosis. Live cell imaging of cells induced to mis-segregate chromosomes in this manner showed that the vast majority of cells that mis-segregate chromosomes do not delay or arrest in G1 following chromosome mis-segregation. p53 activation and a p53-dependent cell cycle arrest do not occur in cells harboring constitutive aneuploidies either (Sheltzer et al., 2017; Tang et al., 2011) further supporting the idea that aneuploidy per se does not trigger a p53-dependent G1 arrest.
While the vast majority of cells that mis-segregated chromosomes continued to proliferate, some cells (10–15%, depending on the experimental set up) do arrest in G1. Such cells harbor highly complex karyotypes and show signs of DNA damage. We propose that DNA damage accrued during chromosome mis-segregation is largely responsible for p53 activation and G1 arrest in these cells. In agreement with this hypothesis is the observation that inactivation of DNA damage-responsive kinases or p53 greatly reduces the number of cells that arrest in G1 following chromosome mis-segregation. We further note that a previous study utilizing cells harboring a mutant allele of the SAC target Cdc20 (CDC20AAA) that is resistant to SAC inhibition found that p53 is activated by the DNA damage checkpoint kinase ATM in aneuploid cells (Li et al., 2010). Several sources of DNA damage likely contribute to p53 activation. Lagging chromosomes might become trapped in the cytokinetic furrow (Janssen et al., 2011), which could lead to DNA damage but perhaps not breakage of chromosomes (Houchmandzadeh et al., 1997; Maciejowski et al., 2015). Premature anaphase entry induced by SAC inactivation could lead to anaphase onset in the presence of incompletely replicated or decatenated DNA. DNA damage could also occur when chromosomes contained in micronuclei are exposed to cytoplasmic nucleases, due to ruptured nuclear envelopes (Crasta et al., 2012; Hatch et al., 2013; Janssen et al., 2011). Aneuploidy-associated stresses that include oxidative, metabolic and proteotoxic stress also likely contribute to p53 activation (Kruiswijk et al., 2015), especially in cells with high levels of genomic imbalances such as is seen in aneuploid cells that arrest in G1. We conclude that the aneuploid state per se does not lead to p53 activation and G1 arrest, but that events associated with chromosome mis-segregation such as DNA damage and aneuploidy associated stresses do. Thus, p53 activation is a potential but not an obligatory outcome of chromosome mis-segregation.
Aneuploidy causes chromosome instability
Previous analyses showed that human and yeast cells harboring many different constitutive aneuploidies exhibit DNA replication and repair defects (Blank et al., 2015; Ohashi et al., 2015; Passerini et al., 2016; Sheltzer et al., 2011; Zhu et al., 2012). Here, we show that this effect on DNA replication is immediate. Our studies in yeast and those of others in mammalian cells further point to changes in the levels of DNA replication factors as being responsible for these DNA replication defects. In yeast, specific chromosome gains, cause specific DNA replication and repair defects, indicating that altered expression of specific genes located on the aneuploid chromosome is responsible for the observed defects (Sheltzer et al., 2011; Zhu et al., 2012). In mammalian cells, multiple DNA replication factors such as the ssDNA binding protein RPA and the lagging strand DNA polymerase δ have been shown to be haploinsufficient (Murga et al., 2016; O’Driscoll, 2008). Furthermore, aneuploid cells downregulate the expression of DNA replication proteins, such as the MCM helicases (Passerini et al., 2016).
The aneuploidy-induced aberrant S phase precipitates further chromosome instability. DNA damage incurred during the abnormal S phase persists into mitosis leading to chromosome mis-segregation and other mitotic abnormalities. We note that this finding could provide an explanation for the puzzling observation that cancers harbor highly abnormal karyotypes yet mutations in genes encoding chromosome segregation factors are rare in the disease (Kops et al., 2005). Our data show that a single chromosome mis-segregation event can set in motion the evolution of complex karyotypes that are characteristic of solid tumors.
Aneuploidy causes an innate immune response
Previous studies showed that aneuploidy is a rare occurrence in tissues even when chromosome segregation is compromised (Pfau et al., 2016) raising the possibility that mechanisms exist to eliminate cells with highly aberrant karyotypes in vivo. Our findings indicate that this is indeed the case. Cells with complex karyotypes express higher levels of NKG2D ligands, such as MICA/B and ULBPs, and of the DNAM1 ligands CD112 and CD155. These cell surface molecules mediate NK cells activation, trigger NK-mediated clearance in vitro (Iannello et al., 2013; Krizhanovsky et al., 2008) and have been shown to mediate tumor cell recognition (Raulet and Guerra, 2009).
Several characteristics of aneuploid cells likely contribute to their recognition by NK cells. For example, aneuploid arrested cells experience DNA damage and produce a SASP gene expression signature, which were previously shown to elicit NK cell recognition and to play a crucial role in the removal of cancer cells in vivo (Raulet and Guerra, 2009). Up-regulation of genes regulated by the cGAS/STING pathway could hint to the presence of DNA in the cytoplasm in aneuploid arrested cells (Lan et al., 2014). The aneuploid state per se might also trigger immune recognition. MEFs harboring specific trisomies exhibit a gene expression pattern very similar to that of aneuploid arrested cells yet these cells do not experience significant DNA damage nor undergo cell cycle arrest. Aneuploidy causes a number of stresses such as proteotoxic stress and oxidative stress (Santaguida and Amon, 2015a), which have previously been shown to induce MICA/B and DNAM1-ligand expression, respectively (Raulet and Guerra, 2009). Moreover, it is important to note that the proteome of aneuploid cells is fundamentally altered because changes in gene copy number generally lead to changes in protein levels (Santaguida and Amon, 2015a). Aneuploidy-induced changes in the cell surface proteome could also elicit an immune response.
It has not escaped our attention that our study could shed light on how the immune system recognizes cancer cells. Neoantigens have been proposed to be a major source of cancer immunosurveillance (Schumacher and Schreiber, 2015). Our data raise the possibility that immune recognition of cancer cells is also mediated by their aneuploid state and the physiological changes associated with complex aberrant karyotypes. At some point during disease evolution however, aneuploid cancer cells escape immune detection and once this point has been reached, aneuploidy appears to correlate with immune evasion (Davoli et al., 2017). Understanding which aspect of tumorigenesis transforms aneuploidy from an immunogenic trait into an immune evasive property will be key to understanding cancer immune evasion.
STAR Methods
EXPERIMENTAL MODEL AND SUBJECT DETAILS
RPE-1 hTERT cell lines and MEFs were cultured in DMEM (Invitrogen) supplemented with 10% FBS, 2 mM L-glutamine and 100 U/ml penicillin/streptomycin. Cells were grown at 37°C with 5% CO2 in a humidified environment.
To generate an RPE-1 hTERT cell line co-expressing GFP-PCNA and H2B-RFP, cells were transduced with pBABE-Puro, a vector encoding human histone H2B C-terminally fused to mRFP1.3 (gift from Don Cleveland), and with an MGC collection human PCNA cDNA engineered to harbor an N-terminal eGFP fusion and cloned into pBABE-Hygro. A population of cells expressing both transgenes at moderate levels was selected by fluorescence activated cell sorting (FACS). This cell population was then cultured in DMEM/F12 (Invitrogen) supplemented with 10% FBS and 100U/ml penicillin/streptomycin and grown at 37°C with 5% CO2 in a humidified environment. NK92-MI cells were obtained from ATCC and cultured in Alpha Minimum Essential medium without ribonucleosides and deoxyribonucleosides but with 2 mM L-glutamine and 1.5 g/L sodium bicarbonate, 0.2 mM inositol, 0.1 mM 2-mercaptoethanol, 0.02 mM folic acid, horse serum to a final concentration of 12.5%, fetal bovine serum to a final concentration of 12.5%.
METHOD DETAILS
Drug treatments
Reversine was obtained from Cayman Chemical or Sigma-Aldrich and used at a working concentration of 0.5 μM or 2μM; Monastrol (working concentration 100 μM) from Tocris; SB203580 (working concentration 10 μM) from CellSignalingTechnology; Thymidine (working concentration 5 mM), Aphidicolin (working concentration 400 nM), RO-3306 (working concentration 7.5 μM), Chk2 inhibitor II hydrate (working concentration 10 μM), VE821 (working concentration 1 μM) and Nocodazole (working concentration 330 nM) were obtained from Sigma-Aldrich. NMS-P715 (working concentration 1 μM) was obtained from EMD/Millipore.
Isolation of cells that stopped dividing following chromosome mis-segregation
To enrich for cells that ceased to divide following chromosome mis-segregation, RPE-1 cells were synchronized at the G1/S phase transition by thymidine treatment. Six hours after release from the G1/S phase block, cells were treated with vehicle or 0.5 μM reversine for 12 hours. After wash-out of the drug, cells were placed into fresh medium and either harvested 66 hours later (euploid and aneuploid cycling cells) or exposed to nocodazole. 12 hours later, mitotic cells were removed by shake-off and the remaining cells were placed again into fresh medium containing nocodazole. Because the percentage of dividing cells was very low after the fourth shake-off (see Figure 6B), we performed four nocodazole treatment/shake-offs in all experiments shown.
Cell imaging methods
For fluorescence imaging RPE-1 cells were plated at about 30% of confluence onto coverslips coated with 10 μg/ml Fibronectin (Sigma-Aldrich). Cells were fixed with 4% paraformaldehyde (in PBS) for 15 minutes at room temperature, then treated with 4% BSA-PBS and incubated with the appropriate antibodies diluted in BSA-PBS. The following antibodies were used for immunofluorescence: anti γH2AX (Cell Signaling Technology #9718), anti 53BP1 (Novus #NB100–305), anti PICH (Millipore #04–1540), anti BLM (Santa Cruz #sc-7790), anti-centromeric antibody (Antibodies Inc. #15-234-0001). Alexa 488- and Alexa 546-labeled secondary antibodies were from Invitrogen. DyLight649-conjugated secondary antibody was purchased from Jackson ImmunoResearch Laboratories. DNA was stained with Hoechst. The coverslips were mounted using ProLong Gold Antifade reagent (Life Technologies).
EdU incorporation into DNA was visualized using the Click-iT EdU Alexa Fluor 647 or Alexa Fluor 488 Imaging Kit (Invitrogen) following the manufacturer’s instructions. EdU-positive nuclei were scored by: (i) applying a global intensity threshold (manually adjusted to detect nuclei by DAPI staining and EdU-positive cells at an intensity value of about 1000); (2) applying the Separate Touching Objects tool in Volocity; (3) excluding touching nuclei with a separation guide of 7 μm; and (4) rejecting nuclei with an area of less than 30 μm2. The method was validated manually and found to give accurate cell counts.
Cells were imaged at 25°C on a Zeiss Axio Observer. Z1 inverted microscope (Zeiss. Thornwood, NY) with an ORCA-ER C4742-80 CCD camera (Hamamatsu Corporation. Middlesex, NJ) and an X-Cite Series 120 arc lamp (Life Sciences & Industrial Division. Ontario, Canada) or on a DeltaVision Elite imaging system (Applied Precision) and microscope (model IX-71; Olympus) controlled by SoftWoRx software (Applied Precision) and a 60× objective lens with a CoolSNAP HQ2 camera (Photometrics). Images were acquired as z-sections at 0.3 μm (DeltaVision) and converted into maximal intensity projections using SoftWoRx (Applied Precision) software. Deconvolution was performed using a constrained-iterative algorithm in SoftWoRx.
Quantification of fluorescence intensity as well as quantification of 53BP1 and γH2AX foci was conducted using Volocity (Perkin Elmer) and Python 2.7. γH2AX -positive foci within a cell were determined by analyzing the number of objects within each cell containing fluorescent accumulations of DAPI and γH2AX greater than 0.2 μm2 as measured by Volocity. Foci were scored by: (1) applying the Volocity “find spots” option; (2) filtering the population by applying a pixel intensity threshold of 1500; (3) using the compartmentalize tool in Volocity to analyze only previously identified nuclei; and (4) applying a 0.2 μm2 size filter to remove speckles and noise. This protocol was validated manually and found to reliably detect foci. Images were imported into Photoshop CS5.1 (Adobe Systems, Inc.), and levels were adjusted.
For FISH analyses, RPE-1 cells were plated on 24×60mm coverslips, treated with DMSO or 1μM NMS-P715 for 24 hrs and then fixed and stained with Hoechst and FISH probes (Cytocell Aquarius Satellite Enumeration probe LPE-011G, Cytocell Aquarius Satellite Enumeration probe LPE-006R). Nuclei were imaged on a Deltavision microscope with a 40x objective and number of foci/nucleus were counted.
Video microscopy
Live cell imaging was performed either using an inverted microscope (IX70; Nikon) with a magnification objective of 10x or using a Yokagawa CQ1 spinning disk confocal (40x objective, reversine-treated cells) or Yokogawa CV1000 (20x objective, monastrol-treated cells). All microscopes were equipped with an incubation chamber maintained at 37°C in an atmosphere of 5% CO2. For experiments with NK92 cells, euploid and aneuploid arrested cells were co-cultured in the presence of NK92 cells at a target:effector ratio of 1:10 right before the beginning of filming.
For experiments described in Figure S1, unsynchronized RPE-1 hTERT GFP-PCNA H2B-RFP cells were plated on Greiner SCREENSTAR 96-well plates (#655866), incubated overnight, and then treated with DMSO or 100 μM monastrol and immediately filmed for 6 hrs using a Yokagawa CV1000 microscope with a 20x objective. After 6 hours plates were removed, cells were washed twice with complete medium, and returned to the microscope for an additional 50 hrs of filming. Because cells were unsynchronized, mother cells entered mitosis throughout monastrol treatment and thereby experienced variable mitotic delays. After drug washout, the mother cells exited mitosis and the cell cycle progression of their daughter cells was tracked. Images were acquired every 10 min for the first 8 hrs to capture mother cell mitotic timing and mis-segregation events, and then every 20 min to capture daughter cell cycle progression.
To quantify daughter cell S phase timing after chromosome mis-segregation in the mother cell, unsynchronized RPE1 hTERT GFP-PCNA H2B-RFP cells were plated on Greiner SCREENSTAR 96-well plates (#655866), incubated overnight, and then treated with DMSO, 0.5 μM, or 2 μM reversine. Cells were immediately filmed on a Yokagawa CQ1 with a 40x objective. Images were acquired every 5 minutes for 5 hrs and then every 20 min for a total of 48 hrs. Reversine was not washed out for the duration of the experiment. Because cells were unsynchronized, we determined the time from PCNA focus appearance to disappearance for two types of cells. The first type (G1) were cells that progressed through mitosis before drug treatment and hence were exposed to reversine only during G1 and S phase. The second type (G2) were the daughters of mother cells that progressed through mitosis in the presence of reversine and had mis-segregated their chromosomes.
To track daughter cell cycle fate after mother cells had mis-segregated chromosomes, RPE-1 hTERT GFP-PCNA H2B-RFP cells were plated on 96-well cycloolifin plates overnight, treated with DMSO or 1 μM NMS-P715 and immediately filmed on a Yokagawa CV1000 microscope using a 20x objective. Images were acquired every 10 min for 8 hrs to capture mother cell mitosis and then every 15 min for 2 days to track daughter cell fate.
Protein detection by Western blots
For protein analyses cells were lysed in lysis buffer (50 mM Tris-HCl, pH8.0, 150 mM NaCl, 0.5 % Sodium deoxycholate, 0.1% SDS, 1% NP-40, protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Roche)) and resolved on 15% SDS PAGE gels. The following primary antibodies were used: anti-actin (Sigma-Aldrich #A2228, 1:10,000), anti-p53 (Santa Cruz #sc-126, 1:200), anti-p21 (Santa Cruz #sc-6246, 1:200), anti-p16 (BD #554079, 1:200), anti γH2AX (Cell Signaling Technology #9718, 1:500), anti-GAPDH (Santa Cruz #sc-365062).
RNAseq data processing and analysis
RNAseq analyses were conducted on euploid, and aneuploid cells that proliferate or that had ceased to divide. Total RNA was isolated using the RNeasy Mini Kit (QIAGEN). Quality control of single end reads was performed by aligning reads to the human genome (hg19) with tophat 2.0.9. Alignment rates to various genomic features were counted and summarized using bedtools 2.17.0 and a series of custom scripts. The QC results are all within acceptable ranges.
To quantify gene expression, RNAseq data were aligned and summarized using bowtie version 1.0.1, rsem version 1.2.15, samtools/0.1.19 and a UCSC known genes annotation file from the hg19 assembly. Differential expression analysis was performed with R version 3.2.2 and DESeq_1.20.0. Raw data for the trisomic MEF experiment (GSE12501) and senescence experiment (GSE11954) were downloaded from GEO and reprocessed using R version 3.2.2 with bioconductor packages affy_1.48.0, affyPLM_1.46.0 and gcRMA_2.42.0. For the senescence experiment, markers of growth and senescence were identified by differential expression analysis using limma_3.26.3 with statistical cut-offs of log2 fold change > 1 and adjusted p-value < 0.05.
Gene Set Enrichment Analysis was run with Java application version 2.2.2 obtained from the Broad Institute and custom gene sets (sting_sasp.gmx, grVsen_degs.gmx) or canonical pathway gene sets (c2cp) available at MsigDB (http://software.broadinstitute.org/gsea/msigdb/index.jsp). Genes with low average expression and variance across all samples (expression < 0.5, variance < 0.02) were excluded from the GCT file.
A group of histone genes shows some degree of differential expression in our experiment and these genes are found in many different c2cp gene sets such as REACTOME_RNA_POL_I_TRANSCRIPTION and REACTOME_MEIOTIC_RECOMBINATION. These gene sets show enrichment in our arrested cells with complex karyotypes. Because our experiment depends on polyA tail purification of mRNAs for sequencing and given the controversy surrounding the polyA status of histone mRNAs, we have excluded the gene sets dominated by histone genes from our GSEA summary in order to highlight other biological processes.
DNA Combing
RPE-1 cells were treated with 2 μM AZ3146 or vehicle control for 24 hours. The cells were then washed and arrested in late G1 by treatment with Mimosine for 24 hours. Mimosine was removed and, cells were placed into fresh medium. Three hours later cells were pulsed at 37°C with 25 μM IdU for 60minu tes and chased with 200 μM CldU for 60 minutes. After labeling, cells were harvested and washed twice with PBS. Cells were re-suspended in 50 μl of PBS to a final concentration of 0.5×106 cells/ml. Cells were incubated at 42°C for 2–3 minutes and mixed with equal volume of 1.5 % low melting agarose (made in PBS and pre-warmed to 42°C) and cast into plug molds and allowed to set at 4°C for half an hour. The plugs w ere digested with Proteinase K for a total of 60 hours at 50°C with solution changes eve ry 12 hours. The plugs were washed with TE and the combed fibers were stained as previously described (Iyer et al., 2016). Fibers were visualized using a Zeiss Axioskop 2 Plus epifluorescence microscope with a 100x Plan-NEOFLUAR oil objective and imaged using a SPOT monochrome cooled-CCD camera. Fibers were measured using ImageJ. Pixels were converted to kb using λ DNA as a standard. Analysis of the data was automated using custom-made MATLAB scripts.
For fork stall rate estimation, two unidirectional forks moving in the same direction on a fiber (green-red-unlabeled-green-red or red-green-unlabeled-red-green) were inferred to contain a stalled fork in between them. Specifically, stalled forks were recognized by red-unlabeled-green (RUG) or green-unlabeled-red (GUR) patterns. The frequency of such patterns in the dataset was used to identify the apparent stall rate. In addition, two forks moving away from each other on a fiber (a RGUGR pattern) can be interpreted as forks elongating from a single origin in the middle or two origins whose forks on the inner side have both stalled. To estimate the stall rate across the entire dataset, the probability of forks stalling was estimated from the unambiguous stall events and extrapolated to ambiguous events to determine the net stall rate (Iyer and Rhind, 2017).
Karyotype analysis
Karyotype determination by single cell sequencing was performed as previously described (Knouse et al., 2014). Briefly, single cells were isolated by microaspiration, and genomic DNA was amplified using the GenomePlex Single Cell Whole Genome Amplification Kit (Sigma). Amplified DNA was purified, barcoded, pooled, and sequenced on an Illumina HiSeq2000. Sequencing reads were aligned using BWA (0.6.1). HMMcopy (0.1.1) was used to estimate gene copy number in 500-kb bins. Cells with high variability in copy number across bins were excluded from the analysis.
Karyotype analysis by G-banding was performed by Cell Line Genetics (Madison, WI).
Cytokine measurement
Euploid and cells with complex karyotypes were isolated and placed into fresh medium for 36 hours. Then, medium was harvested and cell number determined using a Cellometer AutoT4 (Nexcelom). Levels of secreted cytokines were determined using the human cytokine array kit (R&D Systems) following the manufacturer’s instructions and normalized to total number of cells.
ELISA measurement
Euploid and cells with complex karyotypes were isolated and the levels of NF-κB p65, phospho- NF-κB p65 (Ser536), phospho-SAPK/JNK (Thr183/Tyr185), phospho-p38 MAPK (Thr180/Tyr182), phospho-Stat3 (Tyr705) and phospho-IκB-α (Ser32) were measured by a solid phase sandwich enzyme-linked immunosorbent assay (PathScan Inflammation Multi-Target Sandwich ELISA Kit, Cell Signaling Technology) following the manufacturer’s instructions.
FACS
Euploid and arrested cells with complex karyotypes were generated as described above and the levels of MICA/B, CD155/PVR, CD112, ULBP1 and ULBP2 were measured by flow cytometry and analyzed using FlowJo software. Forward (FSC-A) scatter was used to determine the cell size distribution of the cell population.
NK-mediated cell death assay
51Chromium release assays are traditionally used to determine the cytotoxicity of immune cells (Brunner et al., 1968). Because of concerns over spillage during the repeated nocodazole shake-offs necessary to generate aneuploidy arrested cell populations, we did not use this assay. Instead we followed cell killing by life-cell microscopy. In this assay, euploid and aneuploidy arrested cells were generated as described above and plated into a 12 well plate at 104 cells/well. 12 hours later, cells were placed in NK92 growth medium for 24 hours and then co-cultured in the presence of NK92 cells at a target:effector ratio of 1:10 for 24 hours. For antibody-blocking experiments, NK92 cells were pre-incubated with 20 μg/ml anti-NKG2D antibody (R&D systems) for three hours. Dead cells, together with NK92 cells, were gently removed. Adherent cells were counted using a Cellometer AutoT4 (Nexcelom) and normalized to cells grown under the same condition but in the absence of NK92 cells.
We note that the kinetics of NK92 cell mediated killing of aneuploidy arrested cells is slower than what is usually seen with the 51Chromium release assay (Brunner et al., 1968). This difference could be biological, that is NK92 cells take longer to become activated by aneuploid arrested cells than by other target cells. We favor the idea that differences in assay sensitivity are responsible for the slow NK92 cell response that we observe. To detect release of 51Chromium from cells membrane perforation has to occur. In contrast, life cell imaging based assessment of cell death requires the complete lysis of cells, which is likely to take longer than membrane perforation.
RNAi
For RNAi experiments, RPE-1 cells were plated at about 25–30% of confluence 12 hours before transfection. Negative control siRNA (with sequence that do not target any gene product) or oligos targeting Mad2 were purchased from Life Technologies (catalogue number 4390846 and 4392420/s8391, respectively) and transfected with Lipofectamine RNAiMAX (Life Technologies) at final concentration of 10 nM following the manufacturer’s instruction.
β-galactosidase staining
Euploid and arrested cells with complex karyotypes were plated into a 6 well plate at 106 cells/well, allowed to attach overnight, and then stained using the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology) following the manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism software. Details of the statistical tests employed are reported in figure legends. Error bars represent SEM unless otherwise indicated. All experiments were performed in two or more replicates and at least 50 cells/condition/replicate were analyzed.
DATA AND SOFTWARE AVAILABILITY
The RNA-seq data sets generated for this study can be accessed at Gene Expression Omnibus (GEO) database with the accession number GSE83647.
Supplementary Material
Representative movie of DMSO and reversine-treated hTERT RPE-1 PCNA::GFP RFP::H2B cells (from dataset shown in Figure S2C). The image acquisition interval is 20 min. Time 0 represents appearance of PCNA foci. The first frame shows anaphase of the mother cell and the next frame is 20 min prior to the appearance of PCNA foci in one of the daughter cells. Time is indicated in minutes on the lower right.
RPE-1 cells stably expressing H2B-RFP were synchronized at the G1/S transition by thymidine treatment. Six hours after thymidine release, cells were treated with control vehicle or 0.5 μM reversine for 12 hours. After drug wash-out, cells were immediately filmed every 5′. Representative movies of DMSO (A) and reversine-treated hTERT RPE-1 (B) cells are shown. Time is indicated in hours:minutes on the upper left.
Representative movies of euploid cells (A) and arrested cells with complex karyotypes (B) co-cultured with NK92 cells at a target:effector ratio of 1:10. Time is indicated in hours:minutes on the upper left.
Daughter cell S phase length in unsynchronized RPE-1 cells co-expressing PCNA::GFP and RFP::H2B treated with DMSO or reversine (0.5 or 2 μM). Table shows S phase length of cells exposed to the indicated agent either in G1 or in G2.
Table S2. Custom gene list for the gene set SASP and the gene set STING_ISG (Related to Figure 6).
Table S3. List of genes included in the leading edge of the enrichment for the gene set SASP in arrested cells with complex karyotypes compared to euploid cells (Related to Figure 6).
Highlights.
p53 activation is a potential but not obligatory outcome of chromosome mis-segregation
Chromosome segregation errors lead to replication stress and DNA damage
Aneuploidy drives genome instability and evolution of complex karyotypes
Aneuploid cells with complex karyotypes are cleared by natural killer cells
Acknowledgments
We thank members of the Amon lab for discussions and reading of the manuscript and Charlie Whittaker of the Barbara K. Ostrom (1978) Bioinformatics and Computing Facility KI in the Swanson Biotechnology Center for help with the gene expression analysis. Work in the Amon lab is supported by grants from the NIH (CA206157 and GM118066) and the Kathy and Curt Marble Cancer Research Fund. A. A. is an investigator of the Howard Hughes Medical Institute, the Paul F. Glenn Center for Biology of Aging Research at MIT and the Ludwig Institute for Cancer Research. S. S. was supported by the American Italian Cancer Foundation (AICF), by a Fellowship in Cancer Research from Marie Curie Actions and the Italian Association for Cancer Research (AIRC) and by a KI Quinquennial Cancer Research Fellowship. K.A.K. is supported by National Institute of General Medical Sciences Training Grant T32GM007753. Work in the Rhind lab was supported by grant GM98815 from NIGMS. Work in the Desai group is supported by grant GM074215 from NIGMS and by the Ludwig Institute for Cancer Research; A.K.R. acknowledges support from UC San Diego’s Cancer Cell Biology Training Grant (NIH-NCI-T32-CA067754). We thank Andy Shiau (Head, Ludwig Small Molecule Discovery group) for providing inhibitors and for access to high-content imaging and tissue culture equipment.
Footnotes
Author Contributions
Conceptualization, S.S. and A.A.; Investigation, S.S, A.R., D.R.I., O.M.S., L.Z., K.A.K., Y.L.W.; Writing, S.S. and A.A.; Funding Acquisition and Supervision, N.R., A.D., A.A. All authors discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Danilova OV, Kaur P, Levy NB, Compton DA. Chromosomal Instability Substantiates Poor Prognosis in Patients with Diffuse Large B-cell Lymphoma. Clin Cancer Res. 2011;17:7704–7711. doi: 10.1158/1078-0432.CCR-11-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach RR, Ricci-Tam C, Brennan CM, Moomau CA, Hsu PH, Hua B, Silberman RE, Springer M, Amon A. Aneuploidy Causes Non-genetic Individuality. Cell. 2017;169:229–242. e21. doi: 10.1016/j.cell.2017.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank HM, Sheltzer JM, Meehl CM, Amon A. Mitotic entry in the presence of DNA damage is a widespread property of aneuploidy in yeast. Mol Biol Cell. 2015;26:1440–1451. doi: 10.1091/mbc.E14-10-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner KT, Mauel J, Cerottini JC, Chapuis B. Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology. 1968;14:181–196. [PMC free article] [PubMed] [Google Scholar]
- Chen G, Mulla WA, Kucharavy A, Tsai HJ, Rubinstein B, Conkright J, McCroskey S, Bradford WD, Weems L, Haug JS, Seidel CW, Berman J, Li R. Targeting the adaptability of heterogeneous aneuploids. Cell. 2015;160:771–784. doi: 10.1016/j.cell.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes & Development. 2011;25:2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo R, Caldarelli M, Mennecozzi M, Giorgini ML, Sola F, Cappella P, Perrera C, Depaolini SR, Rusconi L, Cucchi U, Avanzi N, Bertrand JA, Bossi RT, Pesenti E, Galvani A, Isacchi A, Colotta F, Donati D, Moll J. Targeting the Mitotic Checkpoint for Cancer Therapy with NMS-P715, an Inhibitor of MPS1 Kinase. Cancer Research. 2010;70:10255–10264. doi: 10.1158/0008-5472.CAN-10-2101. [DOI] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012 doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355:eaaf8399. doi: 10.1126/science.aaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodgson SE, Kim S, Costanzo M, Baryshnikova A, Morse DL, Kaiser CA, Boone C, Amon A. Chromosome-Specific and Global Effects of Aneuploidy in Saccharomyces cerevisiae. Genetics. 2016;202:1395–1409. doi: 10.1534/genetics.115.185660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends in Molecular Medicine. 2010;16:238–246. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet. 2012;13:189–203. doi: 10.1038/nrg3123. [DOI] [PubMed] [Google Scholar]
- Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet. 2001;2:280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell. 2013;154:47–60. doi: 10.1016/j.cell.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig CE, Löffler H, Mahlknecht U, Janssen JWG, Ho AD, Jauch A, Krämer A. Chromosomal instability correlates with poor outcome in patients with myelodysplastic syndromes irrespectively of the cytogenetic risk group. Journal of Cellular and Molecular Medicine. 2009;14:895–902. doi: 10.1111/j.1582-4934.2009.00905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt L, Tighe A, Santaguida S, White AM, Jones CD, Musacchio A, Green S, Taylor SS. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J Cell Biol. 2010;190:25–34. doi: 10.1083/jcb.201002133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe EH, Day CA, Karanjeet KB, Fadness S, Langfald A, Vaughan KT, Dong Z. Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone H3.3 Ser31 phosphorylation. Nat Cell Biol. 2016;18:668–675. doi: 10.1038/ncb3348. [DOI] [PubMed] [Google Scholar]
- Hodgkin J. WormBook, The C. elegans Research Community, editor. Karyotype, ploidy, and gene dosage. 2005 doi: 10.1895/wormbook.1.3.1. http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–487. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houchmandzadeh B, Marko JF, Chatenay D, Libchaber A. Elasticity and structure of eukaryote chromosomes studied by micromanipulation and micropipette aspiration. J Cell Biol. 1997;139:1–12. doi: 10.1083/jcb.139.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. 2013;210:2057–2069. doi: 10.1084/jem.20130783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer DR, Rhind N. Replication Fork Slowing And Stalling are Distinct, Checkpoint-Independent Consequences of Replicating Damaged DNA. 2017 doi: 10.1101/122895. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, Das, Rhind . Cold Spring Harb Protoc. 2016. Analysis of DNA Replication in Fission Yeast by Combing. [DOI] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJPL, Medema RH. Chromosome Segregation Errors as a Cause of DNA Damage and Structural Chromosome Aberrations. Science. 2011;333:1895–1898. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, Amon A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proceedings of the National Academy of Sciences. 2014;111:13409–13414. doi: 10.1073/pnas.1415287111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJPL, Weaver BAA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Lambrus BG, Holland AJ. A New Mode of Mitotic Surveillance. Trends Cell Biol. 2017:1–8. doi: 10.1016/j.tcb.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan YY, Londoño D, Bouley R, Rooney MS, Hacohen N. Dnase2a Deficiency Uncovers Lysosomal Clearance of Damaged Nuclear DNA via Autophagy. CellReports. 2014;9:180–192. doi: 10.1016/j.celrep.2014.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AJX, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IPM, Howell M, Kschischo M, Swanton C. Chromosomal Instability Confers Intrinsic Multidrug Resistance. Cancer Research. 2011;71:1858–1870. doi: 10.1158/0008-5472.CAN-10-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, Han S, Van Deursen JM, Zhang P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proceedings of the National Academy of Sciences. 2010;107:14188–14193. doi: 10.1073/pnas.1005960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley DL, Sandler L, Baker BS, Carpenter AT, Denell RE, Hall JC, Jacobs PA, Miklos GL, Davis BK, Gethmann RC, Hardy RW, Steven AH, Miller M, Nozawa H, Parry DM, Gould-Somero M. Segmental aneuploidy and the genetic gross structure of the Drosophila genome. Genetics. 1972;71:157–184. doi: 10.1093/genetics/71.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorke DE. Developmental characteristics of trisomy 19 mice. Acta Anat (Basel) 1994;150(3):159–69. doi: 10.1159/000147614. [DOI] [PubMed] [Google Scholar]
- López-García C, Sansregret L, Domingo E, McGranahan N, Hobor S, Birkbak NJ, Horswell S, Grönroos E, Favero F, Rowan AJ, Matthews N, Begum S, Phillimore B, Burrell R, Oukrif D, Spencer-Dene B, Kovac M, Stamp G, Stewart A, Danielsen H, Novelli M, Tomlinson I, Swanton C. BCL9L Dysfunction Impairs Caspase-2 Expression Permitting Aneuploidy Tolerance in Colorectal Cancer. Cancer Cell. 2017;31:79–93. doi: 10.1016/j.ccell.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell. 2015;163:1641–1654. doi: 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- Meena JK, Cerutti A, Beichler C, Morita Y, Bruhn C, Kumar M, Kraus JM, Speicher MR, Wang ZQ, Kestler HA, d’Adda di Fagagna F, Gunes C, Rudolph KL. Telomerase abrogates aneuploidy-induced telomere replication stress, senescence and cell depletion. EMBO J. 2015:1–14. doi: 10.15252/embj.201490070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015;528:286–290. doi: 10.1038/nature16139. [DOI] [PubMed] [Google Scholar]
- Murga M, Lecona E, Kamileri I, Díaz M, Lugli N, Sotiriou SK, Anton ME, Méndez J, Halazonetis TD, Fernandez-Capetillo O. POLD3 Is Haploinsufficient for DNA Replication in Mice. Molecular Cell. 2016:1–8. doi: 10.1016/j.molcel.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa O, Tange Y, Kurabayashi A. Growth arrest and chromosome instability in aneuploid yeast. Yeast. 2006;23:937–950. doi: 10.1002/yea.1411. [DOI] [PubMed] [Google Scholar]
- O’Driscoll M. Haploinsufficiency of DNA Damage Response Genes and their Potential Influence in Human Genomic Disorders. Curr Genomics. 2008;9:137–146. doi: 10.2174/138920208784340795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi A, Ohori M, Iwai K, Nakayama Y, Nambu T, Morishita D, Kawamoto T, Miyamoto M, Hirayama T, Okaniwa M, Banno H, Ishikawa T, Kandori H, Iwata K. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nature Communications. 2015;6:7668. doi: 10.1038/ncomms8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes & Development. 2012;26:2696–2708. doi: 10.1101/gad.207407.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passerini V, Ozeri-Galai E, de Pagter MS, Donnelly N, Schmalbrock S, Kloosterman WP, Kerem B, Storchova Z. The presence of extra chromosomes leads to genomic instability. Nature Communications. 2016;7:10754. doi: 10.1038/ncomms10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfau SJ, Silberman RE, Knouse KA, Amon A. Aneuploidy impairs hematopoietic stem cell fitness and is selected against in regenerating tissues in vivo. Genes & Development. 2016;30:1395–1408. doi: 10.1101/gad.278820.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nature Publishing Group. 2009;9:568–580. doi: 10.1038/nri2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RJ, Reeves RH. Understanding the basis for Down syndrome phenotypes. PLoS Genet. 2006;2:e50. doi: 10.1371/journal.pgen.0020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge SD, Douglas TA, Nicholson JM, Vila-Casadesús M, Kantzler CL, Wangsa D, Barroso-Vilares M, Kale SD, Logarinho E, Cimini D. Selective advantage of trisomic human cells cultured in non-standard conditions. Nature Publishing Group. 2016;6:22828. doi: 10.1038/srep22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansregret L, Patterson JO, Dewhurst S, López-García C, Koch A, McGranahan N, Chao WCH, Barry DJ, Rowan A, Instrell R, Horswell S, Way M, Howell M, Singleton MR, Medema RH, Nurse P, Petronczki M, Swanton C. APC/C Dysfunction Limits Excessive Cancer Chromosomal Instability. Cancer Discovery. 2017;7:1–17. doi: 10.1158/2159-8290.CD-16-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol. 2015a;16:473–485. doi: 10.1038/nrm4025. [DOI] [PubMed] [Google Scholar]
- Santaguida S, Amon A. Aneuploidy triggers a TFEB-mediated lysosomal stress response. Autophagy. 2015b;11:2383–2384. doi: 10.1080/15548627.2015.1110670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D’Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol. 2010;190:73–87. doi: 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Vasile E, White E, Amon A. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes & Development. 2015;29:2010–2021. doi: 10.1101/gad.269118.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, Brito IL, Hiraoka Y, Niwa O, Amon A. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–1030. doi: 10.1126/science.1206412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Ko JH, Replogle JM, Habibe Burgos NC, Chung ES, Meehl CM, Sayles NM, Passerini V, Storchova Z, Amon A. Single-chromosome Gains Commonly Function as Tumor Suppressors. Cancer Cell. 2017;31:240–255. doi: 10.1016/j.ccell.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, Amon A. Transcriptional consequences of aneuploidy. Proceedings of the National Academy of Sciences. 2012;109:12644–12649. doi: 10.1073/pnas.1209227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele S, Stoehr G, Peplowska K, Cox JUR, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol. 2012;8:1–12. doi: 10.1038/msb.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, Smith KP, Liggett LA, Gomez EB, Galbraith MD, DeGregori J, Espinosa JM. Trisomy 21 consistently activates the interferon response. eLife. 2016;5:1709. doi: 10.7554/eLife.16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Amon A. Gene Copy-Number Alterations: A Cost-Benefit Analysis. Cell. 2013;152:394–405. doi: 10.1016/j.cell.2012.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144:499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol. 2010;188:369–381. doi: 10.1083/jcb.128.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol. 2010;20:1666–1671. doi: 10.1016/j.cub.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther A, Walther A, Houlston R, Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut. 2008;57:941–950. doi: 10.1136/gut.2007.135004. [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–184. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Pavelka N, Bradford WD, Rancati G, Li R. Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS Genet. 2012;8:e1002719. doi: 10.1371/journal.pgen.1002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative movie of DMSO and reversine-treated hTERT RPE-1 PCNA::GFP RFP::H2B cells (from dataset shown in Figure S2C). The image acquisition interval is 20 min. Time 0 represents appearance of PCNA foci. The first frame shows anaphase of the mother cell and the next frame is 20 min prior to the appearance of PCNA foci in one of the daughter cells. Time is indicated in minutes on the lower right.
RPE-1 cells stably expressing H2B-RFP were synchronized at the G1/S transition by thymidine treatment. Six hours after thymidine release, cells were treated with control vehicle or 0.5 μM reversine for 12 hours. After drug wash-out, cells were immediately filmed every 5′. Representative movies of DMSO (A) and reversine-treated hTERT RPE-1 (B) cells are shown. Time is indicated in hours:minutes on the upper left.
Representative movies of euploid cells (A) and arrested cells with complex karyotypes (B) co-cultured with NK92 cells at a target:effector ratio of 1:10. Time is indicated in hours:minutes on the upper left.
Daughter cell S phase length in unsynchronized RPE-1 cells co-expressing PCNA::GFP and RFP::H2B treated with DMSO or reversine (0.5 or 2 μM). Table shows S phase length of cells exposed to the indicated agent either in G1 or in G2.
Table S2. Custom gene list for the gene set SASP and the gene set STING_ISG (Related to Figure 6).
Table S3. List of genes included in the leading edge of the enrichment for the gene set SASP in arrested cells with complex karyotypes compared to euploid cells (Related to Figure 6).