

Abstract
The outcome of therapy with CAR-modified T-ells is strongly influenced by the subset origin of the infused T-cells. However, since polyclonally activated T-cells acquire a largely CD45RO+CCR7− effector memory phenotype after expansion, regardless of subset origin, it is impossible to know which subsets contribute to the final T-cell product. To determine the contribution of naïve (Tnaive), memory stem (TSCM), central memory (TCM), effector memory (TEM) and terminally differentiated effector (TEMRA) T-cell populations to the CD3 and CD28-activated CAR-T-cells that we use for therapy, we followed the fate and function of individually sorted CAR-T-cell subsets after activation with CD3 and CD28 antibodies (CD3/28), transduction and culture alone or after reconstitution into the relevant subset-depleted population. We show that all subsets are sensitive to CAR transduction and each developed a distinct T-cell functional profile during culture. Naïve-derived T-cells showed the greatest rate of proliferation but had more limited effector functions and reduced killing than memory-derived populations. When cultured in the presence of memory T-cells, naïve-derived T-cells show increased differentiation, reduced effector cytokine production, and a reduced re-proliferative response to CAR stimulation. CD3/28-activated T-cells expanded in IL-7 and IL-15 produced greater expansion of TSCM- and TCM-derived T-cells compared to IL-2. Our strategy provides a powerful tool to elucidate the characteristics of CAR T-cells, regardless of the protocol used for expansion, reveals the functional properties of each expanded T-cell subset and paves the way for a more detailed evaluation of the effects of manufacturing changes on the subset contribution to in vitro expanded T-cells.
Introduction
Adoptive T-cell immunotherapy with CAR-modified T-cells (CAR-T-cells) targeting tumor antigens have been incorporated into cancer treatment with promising efficacy in distinct settings (1–4). CARs are genetically engineered immunoreceptors comprising a single-chain antibody fragment (scFv) linked to cytosolic endodomains from costimulatory receptors and/or the T-cell receptor (TCR)ζ chain (5–7). The structure of the CAR, including the affinity of the scFv, the type of spacer and costimulatory endodomains, the design of the clinical protocol and the disease targeted profoundly affect the fate and function of CAR T-cells, as does the manufacturing protocol that determines the character of the T-cell product infused. (2–4, 8–23). Data regarding the best T-cell subset from which to derive CAR T-cells for infusion are inconclusive and controversial and most patients receive CD4+ and CD8+ T-cells whose subset derivation is unknown (2–4, 11–20). The ultimate objective of T-cell therapy is to transfer a long-lived T-cell population with the capacity to differentiate into potent tumor-specific effectors and to self-renew (8, 24). Short-lived effector T-cells (TEFF) possess potent effector function in vitro, however, appear less attractive for adoptive immunotherapy because of their limited proliferation and engraftment in vivo (25–27). Memory T-cells subsets have been shown to expand substantially in vivo and are long-lived with their self-renewal capacity being inversely proportional to their differentiation state (28). Recently, it has been reported that antigen-experienced memory T-cell subsets directly promote the phenotypic and functional differentiation of naïve T-cells, which as a consequence lost anti-tumor potential when transferred in vivo (29).
Expression of the lymph node homing molecules CCR7 and the leucocyte common antigen (CD45) isoforms RA and RO distinguishes memory from naïve T-cells and allows the dissection of the memory/effector T-cell compartment at least into four main subsets (30, 31): Memory stem T-cells (TSCM), central memory T-cells (TCM), effector memory (TEM) and terminally differentiated effector T-cells (TEMRA) (24, 30, 31). TCM co-express CCR7 and CD45RO, having lost CD45RA during naïve → memory transition (32). Upon antigenic restimulation TCM lose CCR7 expression and differentiate into TEM (32, 33) and finally into TEMRA, which are considered to be terminally differentiated. TEMRA lack both CCR7 and CD45RO and re-express CD45RA (34). A 4th memory subset TSCM resides phenotypically within the naïve-like T-cell compartment (CD45RO−CD45RA+CCR7+), distinct from naïve T-cells by their expression of CD95 (Fas) (24, 31). Each T-cell subset has distinct engraftment capacities and function following adoptive transfer in preclinical trials (31–33, 35). In particular, TCM are thought to have superior engraftment and persistence compared to more differentiated memory T-cell subsets (24, 28, 30–33, 35–39). The recently described TSCM subset may represent the earliest stage of memory T-cell differentiation and may have the ability to transfer stem cell-like T-cells for improved long-term efficacy (40, 41).
To identify the characteristics and subset derivation of CAR T-cells polyclonally expanded on CD3 and CD28 antibody-coated plates as used in our clinical studies (2–4, 11–20), we sorted each T-cell subset and followed its fate and function after activation, CAR-transduction and culture alone and after reconstitution into the corresponding subset-depleted, polyclonally activated bulk peripheral blood mononuclear cells (PBMC). In a proof-of-concept study, we demonstrate that each T-cell subset is sensitive to CAR transduction, and each displays a specific functional profile. Naïve T-cell-derived populations showed the most rapid expansion and dominated the cultures by the end of the culture period, but had reduced effector functions and killing compared to memory subsets. Furthermore, Tnaive-derived cells cultured in the presence of memory T-cells differentiate more than when cultured alone, and show coincidentally reduced effector cytokine production and ability to proliferate in response to CAR stimulation. TSCM show the most rapid expansion of all subsets, but due to their low frequency at the start of culture, they represented only a minor fraction at the end. Finally we found that compared to IL-2, a combination of IL-7 and IL-15, increased the yield of TSCM and TCM derived T-cells within the bulk cultures. Irrespective of the protocol used for expansion, our comprehensive approach reveals the characteristics of CAR T-cells polyclonally expanded from PBMC, demonstrates the functional properties of each expanded T-cell subset and underlines the importance of culture conditions to influence the desired T-cell populations. To this end, our strategy allows a meticulous assessment of the effects of manufacturing changes on the subset contribution to in vitro expanded T-cells.
Materials and Methods
Cell culture
All PBMC and T-cell cultures and all assays were performed using complete media (45% RPMI 1640 (Hyclone, CA, USA), 45% Click’s (Irvine, CA, USA)) supplemented with 5mM L-glutamine (Invitrogen, Carlsbad, CA), penicillin (100IU/ml), and streptomycin (Hyclone, Logan, UT), containing 10% fetal bovine serum (Hyclone), in humidified incubators at 37°C and 5% CO2.
Blood samples
For all experiments, blood samples were collected with informed consent from healthy volunteers on protocols approved by the Baylor College of Medicine Institutional Review Board (IRB). Peripheral blood mononuclear cells were isolated from blood by lymphoprep density gradient centrifugation (Stem Cell Technologies, Vancouver, BC).
T cell isolation and sorting
PBMCs were enriched for CD3+ T-cells with the pan T-cell “untouched” Isolation Kit II (Miltenyi Biotech) following the manufacturer’s instructions. Cells were labeled with fluorescent antibodies to CD3, CD45RO, CD95, CCR7 (all purchased from Biolegend) and CD45RA (Beckman Coulter). FACSorting was performed for TCM (CCR7+CD45RA−), TEM (CCR7−CD45RA−), TEMRA (CCR7−CD45RA+), TSCM (CD45RA+CCR7+→CD95+) and Tnaive (CD45RA+CCR7+→CD95−) (24, 30, 42) on a BD FACSAriaII SORP (BD Biosciences). Postsorting analysis of purified subsets revealed greater than 98% purity.
Cell lines
NIH 293T, JEKO and HDLM-2 cell lines (ATCC city state) were cultured in IMDM (BioWhittaker, Walkersville, MD) supplemented with 10% FCS (Hyclone, Logan, UT) and 5 mM L-glutamine (Thermo Fisher, Waltham, MA). 2nd generation GD2.CAR (14g2a.CD28ζ CAR) transduced T-cells were directed to the disialoganglioside (GD2) to assess specific in vitro re-expansion and effector function. Expanded 2nd generation CD19.CAR+ (FMC63.CD28ζ CAR) and CD19.CAR-NGFR+ (FMC63.CD28ζ-I-NGFR CAR) T-cells were analyzed for their ability to produce cytokines in response to CAR-antigen-expressing target cells, which constituted JEKO (CD19+) and HDLM-2 (CD19−) cell lines.
Constructs
We used CAR constructs directed to the disialoganglioside, GD2, and the B-cell marker, CD19, that have been targeted in many clinical studies. 2nd generation GD2.CAR (14g2a.CD28-ζ) and the CD19.CAR (FMC63.CD28ζ) cloned into SFG vectors have previously been described (43–45). The CD19.CAR construct was used alone and or cloned upstream of an IRES-NGFR or IRES mOrange to facilitate selection and detection.
Retroviral supernatant
Transient retroviral supernatants were produced by co-transfection of NIH 293T cells with the MoMLV gag/pol expression plasmid PeqPam3(-env), the RD114(-env) expression plasmid RDF, and SFG vectors at a ratio of 3:2:3, respectively, with a total of 10μg DNA. The transfection was facilitated with GeneJuice reagent (Calbiochem). The supernatant was harvested 2 and 3 days after transfection, filtered (using a 0.45μm filter), snap-frozen, and stored at −80°C in 5mL aliquots.
Cell stimulation and expansion
PBMC or FACSorted T-cells were activated on CD3 (from the OKT3 hybridoma: ATCC #CRL 8001, Manassas, VA) and CD28 (Becton Dickinson BD, Franklin Lakes, NJ) antibody-coated plates at (500 μL of 1μg/mL each in the presence of recombinant human (rh) IL-2 (50IU/mL) or rhIL-7 and rhIL-15 (each at 10ng/mL). In some experiments, CD3/28 antibody-activated subsets were transduced on day 2 after activation, then reconstituted into CD3/28-activated TSUBSET-depleted PBMC on day 3 of culture at their initial frequency or cultured separately. The medium and cytokines were changed every 3 days during culture or when passaging the T-cells for splitting during expansion.
Transduction
T cells or T-cell subsets were transduced 2 days after activation: Non-tissue-treated 24-well plates were coated with 500μL 1mg/mL retronectin (Takara Biochemicals, Shiga, Japan) overnight. After removing the retronectin, 1.5 – 2mL of retroviral supernatant was added per well and centrifuged at 2000 × g for 1 hr at 25°C to allow vector adherence. The supernatant was removed prior to the addition of CD3/28 activated peripheral blood derived T-cells or FACSorted T-cell subsets. Cells were incubated for 2 days on the virus-coated plate in the presence of 50IU/ml rhIL-2 (Proleukin, Chiron Corp., Emeryville, CA, USA) or 10ng/mL rhIL-7 and 10ng/mL rhIL-15 (Peprotech), then transferred to tissue-culture-treated plates. Where indicated, one day after transduction, each TSUBSET was reconstituted at its initial frequency into the corresponding subset-depleted population.
Flow cytometry, antibodies and intracellular staining
Following antigen specific stimulation, cytokine production (IFNγ, TNFα, IL-2) was determined by intracellular fluorescence staining. All antibodies were purchased from Biolegend, unless indicated otherwise. GD2+ JF cell lines (serving as stimulator cells for 2nd generation GD2.CAR T-cells) and CD19+ JEKO cell lines (serving as stimulator cells for 2nd generation CD19.CAR T-cells) were added to T-cells at a stimulator to responder ratio of 10:1. HDLM-2 cells lack GD2 and CD19 were used as controls and respective background responses have been subtracted from CAR-specific cytokine production. For effector cytokine detection, cultured T-cells were re-stimulated for 6 hrs in the presence of 1μg/mL brefeldin A (BFA; Sigma-Aldrich). After harvesting, phenotype staining was performed using monoclonal antibodies for CD3 (OKT3), CD4 (SK3) and CD8 (RPA-T8). To define memory subsets, T-cells were extracellularly stained for CCR7 (G043H7), CD45RA (HI100), CD45RO (UCHL1) and CD95 (DX2). In particular experiments a αNGFR (C40-1457, BD) antibody was used to stain for NGFR expression on CD19.CAR-NGFR+ T-cells. Transduction efficacies were assessed by staining for the CAR expression on the T-cell surface utilizing antibodies targeting GD2 (IA7) (46) or CD19.CAR (goat anti-human-IgG (H+L)) (Jackson ImmunoResearch, West Grove, PA). To exclude dead cells, live/dead-discrimination staining-dye (Invitrogen) was added. Subsequently, cells were permeabilized with Perm2 solution (BD Biosciences) and stained for IFNγ (4S.B3), TNFα (MAb11), and IL-2 (MQ1-17H12). Cells were analyzed on a LSR-II flow cytometer and FlowJo Version 10 software (Tree Star). Lymphocytes were gated based on the FSC versus SSC profile and subsequently gated on FSC (height) versus FSC to exclude doublets.
Assessment of cytotoxic activity
A modified vital assay was used for cytotoxicity testing as described previously (24, 47, 48). Briefly, the JEKO cell line served as a CD19-positive target cell for 2nd generation CD19.CAR T-cells, while HDLM-2 cells that lack CD19 were used as control targets. JEKO cells were labeled with 10μM carboxyfluorescein succinimidyl-ester (CFDA-SE; Molecular Probes). As controls, CD19− HDLM-2 were labeled with 5μM dimethyldodecylamine oxide-succinimidyl-ester (DDOA; Invitrogen). Cells were co-cultured in duplicate for 16 hrs in T-cell / target cell ratio of 1:1 and 10:1. Labeled cells were analyzed as duplicates using LSR-II flow cytometer. Samples without T-cells, containing only targets and non-targets (CD19+ JEKO cell lines or CD19− HDLM-2) displayed as internal controls. Cells were gated using live/dead-discrimination staining-dye negative cells (Invitrogen). The mean percent survival of CD19+ JEKO target cells was calculated relative to CD19− HDLM-2 controls. Percentage target cell lysis was calculated as follows: mean percent survival of targets in cultures containing defined numbers of effector T-cells in comparison to control cells without T-cells.
Statistical analysis and calculations
Statistical analysis was performed with GraphPad Prism version 6. Data were analyzed using repeated measures 1-way ANOVA, 2-way ANOVA or paired t-test as indicated, after verifying Gaussian distribution with the Kolmogorov-Smirnov test. p-values ≤ 0.05 were considered significant: *p<0.05, **p<0.01, ***p<0.001. T-cell subset fold expansion expresses T-cell subset numbers (assessed by FACS) in relation to total cell numbers of the indicated days post-initial stimulation and day 1.
Results
Comparative proliferation of isolated T-cell subsets cultured alone
To analyze the characteristics of each T-cell subset transduced with a GD2.CAR we compared their proliferation and phenotype following CD3/28 antibody activation (in the presence of IL-7 and IL-15) and cytokine production following stimulation via the CAR using the CAR antigen (diasialoganglioside: GD2). First CD3+ T-cells were enriched from PBMC by magnetic bead sorting, then TCM (CCR7+CD45RA−), TEM (CCR7−CD45RA−), TEMRA (CCR7−CD45RA+), TSCM (CD45RA+CCR7+→CD95+) and Tnaive (CD45RA+CCR7+→CD95−) were isolated by polychromatic fluorescence-activated cell sorting (FACSorting) (Fig. 1A). The average frequency of each T-cell subset from pre-sorted CD3+ T-cells was 21.5±6.2% for TCM, 17.8±8.8% for TEM, 9.8±5.5% for TEMRA, 0.9±0.2% for TSCM and 50.5±9.2% for Tnaive (Fig. 1B). To determine if all T-cell subsets isolated from PBMC were sensitive to retroviral transduction, we transduced each subset with a 2nd generation CAR (14g2a.CD28-ζ) targeting GD2 (43) on day 2 after stimulation.
FIGURE 1. Strategy for T-cell subset sorting, activation and transduction.
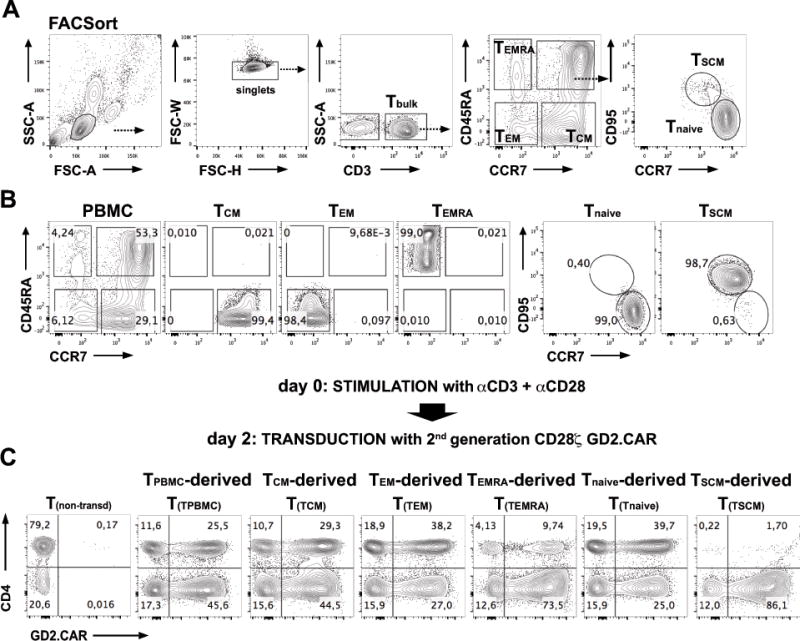
(A) Illustrates the strategy for sorting T-cell subsets from PBMC according to their expression of CD3+, CD45RA+, CCR7+ and CD95+. Prior to FACSorting, CD3+ T-cells were enriched from PBMC by untouched MACS separation. They were then sorted into the following subsets: TCM (CCR7+CD45RA−), TEM (CCR7−CD45RA−), TEMRA (CCR7−CD45RA+), TSCM (CD45RA+CCR7+→CD95+) and Tnaive (CD45RA+CCR7+→CD95−). (B) FACSort purified T-cell populations were isolated to greater than 98% purity. (C) Strategy for polyclonal activation and retroviral transduction of PBMC-derived T(TPBMC), TCM-derived (T(TCM)), TEM-derived (T(TEM)), TEMRA-derived (T(TEMRA)), TSCM-derived (T(TSCM)) and Tnaive-derived (T(Tnaive)) cells. FACSort purified TSUBSET were activated with CD3 and CD28 antibodies and cultured in the presence of IL-7 and IL-15 at 10ng/mL each and transduced on day 2 with a 2nd generation GD2.CAR (14g2a.CD28-ζ CAR). Shown are examples of GD2.CAR transduction efficacies in separately cultured PBMC-, TCM-, TEM-, TEMRA-, TSCM- and Tnaive-derived populations on day 7 post activation. Transduction efficacies were assessed by fluorescence staining for the GD2.CAR expression on the T-cell surface using the 1A7 antibody targeting 14g2a.
As presented in Figure 1C, the GD2.CAR could be expressed in all T-cell subsets with median transduction efficacies of 60.2±10.2%, 60.1±7.2%, 66.6±7.9%, 61.2±7.1% and 64.6±6.1% for CD4+ TPBMC, TCM, TEM, TEMRA, Tnaive and 65.6±20.7%, 70.8±15.4%, 75.3±12.3%, 61.7±16.8%, 66.6±19.2% and 82.7±7.9% in CD8+ TPBMC, TCM, TEM, TEMRA, TSCM, Tnaive, respectively (summarized in Fig. 2A). We observed a trend towards higher transduction efficacies in CD8+ compared with CD4+ T-cells (62.5% in CD4+ T-cells and 70.4% in CD8+ T-cells) (Fig. 2A). As shown in Figure 2B, we detected a preferential expansion of CD8+ T-cells compared to CD4+ T-cells within TPBMC, TCM, TEM, TEMRA, TSCM, but not in Tnaive populations. As previously reported for TSCM-derived cells (24), we observed almost exclusive proliferation of CD8+ TSCM, whereas CD4+ TSCM expansion could not be detected. However, all other expanded T-cell subsets showed a balanced CD4+ / CD8+ ratio (Fig. 2A, B). When cultured individually, all T-cell subsets proliferated in response to CD3/28 stimulation, confirming their viability after separation and all transduced T-cell subsets showed rapid expansion with the highest expansion at day 14 from TSCM-derived (311±63 fold) followed by Tnaive-derived (210±24 fold), TCM-derived (150±65 fold), and lower expansion of TEM-derived (88±31 fold), and TEMRA-derived (61±22 fold) cells (Fig. 2C). All transduced subsets differentiated during culture so that by day 14, CCR7−CD45RO+ TEM cells predominated in TEMRA- and TEM –derived subsets, whereas a substantial proportion of CCR7+CD45RO+ TCM cells was present in TCM-, Tnaive- and TSCM-derived cultures (Fig. 2D), demonstrating that naïve T-cells develop a memory phenotype in response to CD3/28 stimulation in vitro.
FIGURE 2. Fold Expansion and phenotype of separately cultured GD2.CAR-modified TSUBSETS in response to GD2 stimulation.
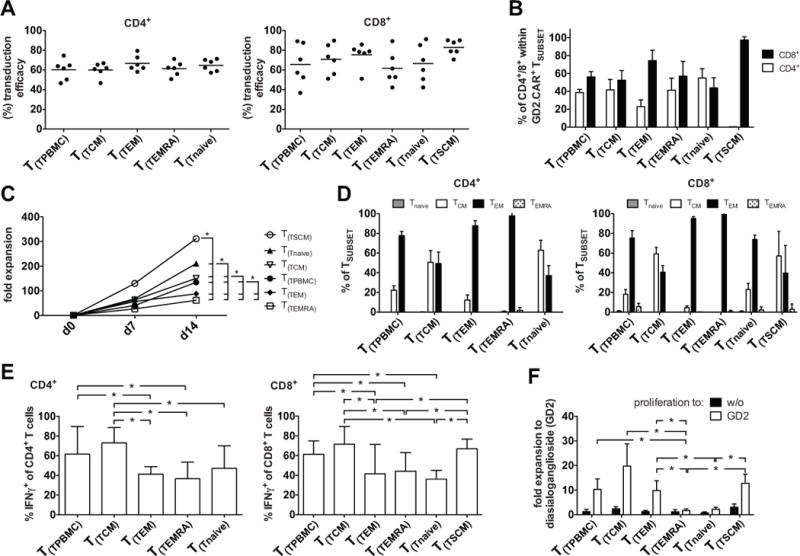
(A) FACSort purified TSUBSET were cultured according to Fig. 1. GD2 transduction efficacies from 6 donors: left graph: CD4+; right graph: CD8+ TSUBSET-derived (T(TSUBSET)) populations on day 14 after CD3/28-activation. (B) CD4+/CD8+ proportion within GD2.CAR+ TSUBSETS. (C) TSUBSET expansion in response to CD3/28 stimulation (measured as fold increase) analyzed for each TSUBSET on days 7 and 14. (D) Quantification of CCR7 and CD45RO expression 14 days after CD3/28-activation in each TSUBSET-derived population: TCM (CCR7+CD45RO+), TEM (CCR7−CD45RO+), TEMRA (CCR7−CD45RO−) and Tnaive (CCR7+CD45RO−). Left graph: CD4+; right graph: CD8+ TSUBSET-derived populations. (E) CD3/28-expanded TSUBSET-derived populations were re-stimulated via the CAR using a GD2-positive neuroblastoma cell line, JF at a stimulator to responder ratio of 10:1 14 days after the initial CD3/28 activation and a non-CAR control stimulation (GD2− Hodgkin lymphoma: HDLM-2). Shown is background-subtracted (negative control: HDLM-2) IFNγ production to JF by CD4+ (left graph) and CD8+ (right graph) TSUBSET-derived populations and was determined by intracellular staining 6 hrs after co-culture. Cells were permeabilized and stained for IFNγ, CD3, CD4 and CD8. (F) TSUBSET expansion (measured as fold increase) was analyzed for each cell subset between days 14 and 21 after stimulation with plate-bound GD2 on day 14. Mean data from 6 healthy donors are presented, and error bars represent SEM. Data were analyzed using repeated measures 1-way ANOVA, after verifying Gaussian distribution with the Kolmogorov-Smirnov test. p-values ≤ 0.05 were considered significant: *p<0.05.
Cytokine production and proliferation of TSUBSETS in response to CAR stimulation
To examine CAR-specific effector functions, we stimulated each transduced subset on day 14 with the GD2-positive JF neuroblastoma cell line and a GD2-negative Hodgkin lymphoma (HDLM2) and measured IFNγ cytokine expression in intracellular cytokine assays (Fig. 2E). GD2-specific IFNγ production from CD4+ and CD8+ TCM-, TPBMC- and TSCM-derived cells was greater than from Tnaive-, TEMRA- and TEM-derived cells. No IFNγ was produced by non-transduced T-cells in response to JF cells (data not shown). To compare T-cell subset proliferation in response to CAR stimulation each transduced subset was stimulated with plate bound GD2 (used to avoid tumor-derived activating or inhibitory signals) in the absence of exogenous cytokines on day 14 of culture. Intriguingly, TEMRA- and Tnaive-derived cells failed to proliferate, whereas TSCM-, TCM- and TEM-derived cells showed robust re-expansion in response to CAR antigen over 7 days of culture (Fig. 2F). However, Tnaive-derived cells did proliferate (18-fold expansion) when IL-7 and IL-15 were added to the re-expansion culture (data not shown) indicating greater dependence on exogenous cytokines than memory-derived T-cells.
In summary, all tested T-cell subsets were susceptible to CAR transduction and expanded when activated via the TCR with greatest fold expansion by TSCM- > Tnaive- > TCM- > TEM- > TEMRA-derived cells. While lower in TEM- and TEMRA-derived cells, all subsets produced IFNγ following CAR-stimulation and acquired a memory T-cell phenotypic profile. TSCM-, TCM- and TEM-derived cells showed a greater capacity to expand in response to CAR stimulation than TEMRA- and Tnaive-derived cells.
Naïve derived T-cells expand preferentially within polyclonally activated PBMC but show reduced effector cytokine production and killing
To evaluate the characteristics of each T-cell subset when reconstituted into the bulk subset-depleted PBMC population, each T-cell subset was purified from PBMC by FACS (Fig. 3A, B) then activated with CD3 and CD28 antibodies and transduced with a retroviral vector encoding both a 2nd generation CD19.CD28ζ CAR and a truncated nerve growth factor receptor (NGFR) (CD19.CAR-NGFR) separated by an IRES to allow flow-cytometric detection following expansion. In parallel, each subset-depleted PBMC population was CD3/28-activated and transduced with the same 2nd generation CD19.CAR, but without NGFR (Fig. 3C, D). One day after transduction, each TSUBSET was reconstituted at its initial frequency into the corresponding subset-depleted population. This design allowed us to distinguish the depleted bulk transduced PBMC from the reconstituted transduced T-cell subset by flow cytometric detection of NGFR (Fig. 3D). All T-cell subsets were sorted to greater than 98% purity as shown in Figure 3B and 3C and transduced with frequencies exceeding 80% (Fig. 3E). CD19.CAR transduction did not affect the survival or expansion of any T-cell subset compared to non-transduced subsets cultured alone in parallel (data not shown).
FIGURE 3. Experimental design to assess the fate of TSUBSETS polyclonally expanded within PBMC.

(A) Illustrates the strategy for FACSort-purification of T-cell populations: TCM, TEM, TEMRA, Tnaive and TSCM were FACSort-depleted from PBMC (B: TSUBSETS; C: PBMC-TCM, PBMC-TEM, PBMC-TEMRA, PBMC-Tnaive, PBMC-TSCM). After isolation each TSUBSET and each single subset-depleted PBMC population was activated with CD3 and CD28 antibodies in the presence of IL-7 and IL-15, then transduced on day 2 with a CD19.CAR construct encoding a marker gene (NGFR), while TSUBSET–depleted cells were transduced with the CD19.CAR alone (CD19.CD28ζ CAR). Each activated and transduced TSUBSET was reconstituted into the appropriate activated and transduced TSUBSET –depleted PBMC at their initial frequencies one day after transduction to allow tracking of the subset within the bulk population after reconstitution (details in methods). (D) Representative FACS plots are shown for the flow cytometric detection of each reconstituted CD19.CAR-NGFR+ TSUBSET-derived (T(TSUBSET)) population (CD4+ T(TSUBSET) indicated in red dots; CD8+ T(TSUBSET) indicated in blue dots) and CD19.CAR+ PBMC-TSUBSET (P-TSUBSET, gray dots) by the assessment of the CD19.CAR and the CD19.CAR-NGFR+, respectively. (E; upper graph) CD19.CAR-NGFR transduction efficacies for CD4+ and CD8+ TCM, TEM, TEMRA, TSCM and Tnaive-derived (T(TSUBSET)) population and (E; lower graph) CD19.CAR transduction efficacies for CD4+ and CD8+ PBMC-TSUBSET (P-TSUBSET).
Differences in the rate of proliferation of each transduced subset cultured alone or within the transduced bulk population could not be detected (Fig. 4A). However, the rate of expansion of T-cell subsets within the bulk culture was much greater for Tnaive- (CD3+ 234±36 fold expansion) and TSCM-derived cells (CD3+ 621±189 fold expansion) than for TEMRA- and TEM-derived cells (CD3+ 44±6 and 54±9 fold expansion), while TCM-derived cells showed an intermediate rate of expansion (CD3+ 204±48 fold expansion) (Fig. 4A), so that by day 14 Tnaive-derived cells dominated the cultures (76±19% of CD3+ T-cells) with TCM-derived cells comprising a mean of 30±12% of CD3+ T-cells (Fig. 4B, C). CD4+ and CD8+ TEMRA- and TEM-derived cells could barely be detected because of the other more rapidly expanding subsets (Fig. 4B, C). Hence, bulk CD3/28-activated T-cells were derived almost entirely from naïve CD8+ T-cells and naïve and TCM CD4+ T-cells, with little or no contribution from TEM and TEMRA subsets (Fig. 4B, C). Of note, despite their high rate of expansion when cultured in bulk PBMC, TSCM-derived cells represented less than 2% of the bulk cultures on day 14 because of their low starting frequency.
FIGURE 4. Fate of TSUBSETS polyclonally expanded within PBMC: naïve-derived T-cells expand preferentially within polyclonally activated PBMC.

CAR-modified TSUBSET were reconstituted into CD19.CAR modified, subset-depleted PBMC according to Figure 3. (A) Fold expansion of CD19.CAR-NGFR+ TSUBSET-derived (T(TSUBSET)) population cultured alone or after reconstitution into the PBMC-TSUBSET: CD4+ (upper graph), CD8+ (middle graph) and whole CD3+ (lower graph). (B) Analysis on day 14 post initial polyclonal activation of each T(TSUBSET) (CD19.CAR-NGFR+) within PBMC-TSUBSET expanded T-cells (CD19.CAR+) showing higher frequencies of CD4+ (red, upper graph), CD8+ (blue, middle graph) and whole CD3+ (lower graph) T(Tnaive) and T(TCM) cells compared to other T(TSUBSET). Data were analyzed using repeated measures 1-way ANOVA, after verifying Gaussian distribution with the Kolmogorov-Smirnov test. p-values ≤ 0.05 were considered significant: *p<0.05. (C) Frequency of each TSUBSET within CD3+ PBMC (ex vivo) and frequency at day 14 of CD4+ (red, upper graph) and CD8+ (blue, lower graph) CD19.CAR-NGFR+ T(TSUBSET) within PBMC-TSUBSET expanded T-cells. Mean data from 6 healthy donors are presented, and error bars represent SEM.
By day 14 of culture, T-cells with a TEM phenotype (CCR7−CD45RO+) dominated all subset derived-cultures (Supplemental Fig. 1A, B). However a substantial proportion of T-cells with a TCM phenotype could be detected within expanded TCM- (CD4+ 20.4±2.9%; CD8+ 13.5±2.6%), Tnaive- (CD4+ 45.2±6.9%; CD8+ 18.5±2.4%) and CD8+ TSCM-derived cells (19.4±2.7%) as well as in bulk PBMCs-derived cells (CD4+ 12.4±2.8%; CD8+ 15.7±3.9%).
To evaluate cytokine production in response to CAR stimulation by T-cell subset-derived cells expanded within bulk PBMC, we performed intracellular flow and co-stained with antibodies to NGFR (the marker for the subset) and to the spacer/hinge region of the CD19.CAR (Fig. 5A, B) to distinguish subset- and bulk-derived T-cells. Figures 5C–D show IFNγ, TNFα and IL-2 production from each T-cell subset (CD4+: red- CD8+ blue bars) and from the T-cell subset-depleted bulk population (CD4+: black- CD8+ white bars) in response to a CD19+ mantle cell lymphoma (JEKO) and a CD19− Hodgkin lymphoma (HDLM2). Tnaive-derived cells produced relatively low levels of all cytokines compared to bulk-derived T-cells and to other subsets, which showed similar capacities to produce cytokines in response to CAR stimulation, even when their frequencies within the co-cultures were low (Fig. 5C, D and E).
FIGURE 5. Cytokine production by reconstituted T-cell subsets in response to CAR-stimulation.

CAR-modified T-cell subsets were reconstituted into CD19.CAR modified, subset-depleted PBMC according to Figure 3. On day 14, cultures were stimulated with a CD19− (HDLM-2) or CD19+ JEKO lymphoma cells for 6 hrs at a stimulator to responder ratio of 10:1. Cytokine production was measured by intracellular cytokine staining. (A) Shows a representative FACSplot in which IFNγ and TNFα production by Tnaive–derived cells in response to HDLM-2 (upper plots) or JEKO (lower plots) is shown. TSUBSETS could be distinguished from the depleted fraction by their expression of NGFR (B). The fraction of all subsets producing IFNγ, TNFα, IL-2 is shown for CD4+ and CD8+ T-cells in C, D and E. Cytokine production by transduced and non-transduced PBMC is also shown. Black bars represent PBMC-TSUBSET–derived CD4+ T-cells, red bars represent TSUBSET–derived CD4+ T-cells, white bars represent PBMC-TSUBSET–derived CD8+ T-cells, blue bars represent TSUBSET–derived CD8+ T-cells. Data from TSUBSET–derived (CD19.CAR-NGFR+) and PBMC-TSUBSET–derived (CD19.CAR+) populations within one approach were analyzed using paired t-test, after verifying Gaussian distribution with the Kolmogorov-Smirnov test. p-values ≤ 0.05 were considered significant: *p<0.05. Mean data from 3 healthy donors are presented, and error bars represent SEM.
We next tested the killing capacity of each subset cultured alone and its respective subset-depleted and subset-reconstituted PBMC cell preparation (Fig. 6A). Figure 6B shows the killing capacity of each T-cell subset cultured alone, T-cell subset-depleted bulk population and T-cell subset-reconstituted bulk population in response to a CD19+ mantle cell lymphoma (JEKO) and a CD19− Hodgkin lymphoma (HDLM2). Intriguingly, killing of CD19+ JEKO was significantly reduced in cultures where TCM and TEM were depleted from the bulk PBMC population (Fig. 6B upper panel). By contrast T-cell subset-reconstituted populations showed little variance in their killing (Fig. 6B middle panel). Comparison of the T-cell subsets cultured alone revealed a significantly lower killing capacity of Tnaive-derived cells (Fig. 6B lower panel).
FIGURE 6. Cytotoxic killing capacity of each TSUBSET–derived population cultured alone and its respective TSUBSET-depleted and TSUBSET-reconstituted PBMC cell preparation.

Assessment of cytotoxic activity by flow cytometric vital assay. CAR-modified T-cell subsets were cultured alone or reconstituted into CD19.CAR modified, subset-depleted PBMC according to Figure 3. (A) CFDA-SE+ CD19+ JEKO cell lines (serving as stimulator APC for 2nd generation CD19.CAR T-cells) were used as targets, whereas DDOA+ CD19− HDLM-2 cells lack CD19 were used as control targets. Cells were co-cultured for 16 hrs in T-cell / target cell ratio of 1:1 and 10:1. Samples without T-cells, containing only APC (CD19+ JEKO cell lines or CD19− HDLM-2) displayed as internal control. The mean percent survival of CD19+ JEKO incubated targets was calculated relative to CD19− HDLM-2 controls. (B) Shows the killing capacity of each TSUBSET-depleted bulk population (upper panel), TSUBSET-reconstituted bulk population (middle panel) and TSUBSET cultured alone (lower panel). Data from TSUBSET–derived population cultured alone, TSUBSET-depleted and TSUBSET-reconstituted PBMC cell preparations were analyzed using paired t-test, after verifying Gaussian distribution with the Kolmogorov-Smirnov test. p-values ≤ 0.05 were considered significant: *p<0.05. Mean data from 3 healthy donors are presented, and error bars represent SEM.
In summary, Tnaive-derived cells expand preferentially within polyclonally activated PBMC, express a memory T-cell surface marker profile but show reduced effector cytokine production and killing compared to memory-derived subsets.
Naïve derived T-cells show increased differentiation when cultured in the presence of bulk (memory) T-cells and show reduced effector cytokine production and re-expansion capacity in response to CAR stimulation
To evaluate the phenotypic and functional characteristics of each T-cell subset cultured in the presence of the whole PBMC preparation compared to T-cells cultured alone, we followed the experimental setup presented earlier, comprising the transduction of each T-cell subset using a retroviral vector encoding both a 2nd generation CD19.CD28ζ CAR and a truncated NGFR (CD19.CAR-NGFR) and in parallel transduction of each subset-depleted PBMC population with the same 2nd generation CD19.CAR, but without NGFR. Initially, we followed the fate of individually sorted CAR-transduced T-cell subsets after activation, transduction and culture alone or after reconstitution into the relevant subset-depleted population (Fig. 7A). Each T-cell subset differentiated during culture and by day 7 of culture T-cells with TCM and TEM phenotypes (CCR7+CD45RO+ and CCR7−CD45RO+, respectively) dominated all subset-derived cultures (Fig. 7A, B). We defined the TCM proportion of each T-cell subset as a measure of differentiation. When comparing the phenotype of each transduced subset cultured alone or within the transduced bulk PBMC population we found significantly higher proportions of phenotypically less-differentiated TCM cells in separately cultured TCM- and Tnaive-derived cells (day 14: TCM-derived cells cultured in bulk: CD4+ 13.1±2.6%; CD8+ 12.9±1.4% versus TCM-derived cells cultured alone: CD4+ 21.7.1±9.9%; CD8+ 21.2±5.4% and Tnaive-derived cells cultured in bulk: CD4+ 14.8±4.7%; CD8+ 11.2±1.1% versus Tnaive-derived cells cultured alone: CD4+ 39.8±7.8%; CD8+ 29.1±6.6%) (Figure 7A, B). A substantial proportion of T-cells with a TCM phenotype could also be detected within expanded CD8+ TSCM-derived cells (day 14: TSCM-derived cells cultured in bulk: 15.3±1.7% versus TSCM-derived cells cultured alone: 22.6±0.9%)(Fig. 7A, B). Of note, T-cell cultures derived from less-differentiated TSCM-, TCM- and Tnaive had more stable CAR expression in long-term cultures up to day 21, whereas TEM- and TEMRA-derived cultures showed partial loss of their CAR (Supplemental Fig. 2).
FIGURE 7. T-cells with TCM and TEM phenotypes dominate all TSUBSET–derived populations during culture.

CAR-modified T-cell subsets were cultured alone or reconstituted into CD19.CAR modified, subset-depleted PBMC according to Figure 3. (A) Representative FACS plots illustrating flow cytometric detection of CCR7 and CD45RO expression on days 7, 14 and 21 post initial activation of TSUBSET-derived (T(TSUBSET)) populations (cultured alone) and of TSUBSET-derived (T(TSUBSET)) populations within the relevant PBMC-TSUBSET preparation (cultured within PBMC): (B) Results for TCM phenotype (CCR7+CD45RO+) at indicated time-points (days 7, 14 and 21) for all subsets of CD4+CAR+ and CD8+CAR+ T-cells. Data from TSUBSET–derived (CD4+CD19.CAR-NGFR+ cultured alone: black bars; CD8+CD19.CAR-NGFR+ cultured alone: white bars versus CD4+CD19.CAR+ cultured within PBMC preparations: red bars; CD8+CD19.CAR+ cultured within PBMC preparations: blue bars) were analyzed using paired t-test, after verifying Gaussian distribution with the Kolmogorov-Smirnov test. p-values ≤ 0.05 were considered significant: *p<0.05. Mean data from 3 healthy donors are presented, and error bars represent SEM.
When comparing intracellular cytokine production in response to CAR stimulation (using the CD19+ mantle cell lymphoma: JEKO) of each transduced subset cultured alone or within the transduced bulk PBMC population, Tnaive-derived cells produced less IFNγ when cultured within the transduced bulk PBMC population than when cultured alone, whereas all other subsets showed similar capacity of IFNγ production, regardless of whether cells were cultured in the presence of the whole PBMC preparation or alone (Supplemental Fig. 3). Tnaive-derived cells produced relatively low levels of IFNγ compared to other T-cell subsets that produced similar cytokine levels in response to CAR stimulation (Supplemental Fig. 3).
Finally, we compared T-cell subset proliferation in response to CD19.CAR stimulation. Each transduced subset cultured alone or T-cell subset-derived cells expanded within bulk PBMC was stimulated on day 14 of culture using CD19+ JEKO cells in the presence or absence of exogenous cytokines (IL-7 and IL-15) (Fig. 8A) and fold T-cell subset expansion was measured 7 days later. To assess the expansion of each subset cultured in the presence of the whole PBMC preparation, we used antibodies to NGFR (T-cell subset) and to the spacer/hinge region of the CD19.CAR before and after CAR stimulation (Fig. 8A). In the absence of exogenous cytokines TEMRA- and Tnaive-derived cells failed to proliferate, whereas TSCM-, TCM- and TEM-derived cells expanded to a similar degree regardless of whether cultured alone or within the transduced bulk population (Fig. 8B). When culture with cytokines, only TEMRAs did not expand. As expected the expansion of the remaining subsets was much greater than without cytokines (Fig. 8C), and notably, Tnaive-derived cells showed significantly greater expansion when cultured alone than when cultured within the transduced bulk population (65-fold for CD4+ and 36-fold for CD8+ alone compared to 43-fold for CD4+ and 15-fold for CD8+ cultured in bulk) (Fig. 8C). In contrast, all other subsets showed slightly greater expansion when cultured in bulk (Fig. 8C).
FIGURE 8. Proliferation of CD19.CAR-modified TSUBSETS cell cultured alone or expanded within bulk PBMC in response to CAR stimulation.

CAR-modified TSUBSET were cultured alone or reconstituted into CD19.CAR modified, subset-depleted PBMC according to Figure 3. (A) Each CD3/28-expanded transduced TSUBSET-derived population cultured alone or within bulk PBMC were stimulated via the CAR using vital CD19+ JEKO cells in the presence or absence of exogenous cytokines (IL-7 and IL-15) at a stimulator to responder ratio of 10:1 14 days after the initial CD3/28 activation. The frequency of each subset was measured by co-staining with antibodies to NGFR and to the spacer/hinge region of the CD19.CAR at assay initiation and post 7 days of consecutive expansion. (B and C) TSUBSET expansion (measured as fold increase) was analyzed for each cell subset between days 14 and 21 after stimulation with CD19+ JEKO cells on day 14 in the absence (B) or presence (C) of exogenous cytokines (IL-7 and IL-15). Data from TSUBSET–derived (CD19.CAR-NGFR+ cultured alone versus cultured within PBMC preparations) were analyzed using paired t-test, after verifying Gaussian distribution with the Kolmogorov-Smirnov test. p-values ≤ 0.05 were considered significant: *p<0.05. Mean data from 3 healthy donors are presented, and error bars represent SEM.
In summary, Tnaive-derived cells differentiate more when cultured in the presence of memory T-cells than when cultured alone, and show coincidentally reduced effector cytokine production and re-expansion capacity in response to CAR stimulation. Overall, Tnaive-derived cells show reduced effector cytokine production compared to memory-derived subsets, and lack the ability to respond to stimulation in the absence of cytokines.
The yield of TSCM- and TCM-derived T-cells is increased by culture in IL-7 with IL-15, compared to culture in IL-2
Because the Tnaive-derived CAR-cells that dominated our cultures showed inferior CAR-mediated effector function, and may have limited longevity, we aimed to increase the frequencies of TCM- and TSCM-derived cells during expansion of the cultures. We therefore compared the effects of the two cytokine regimens on relative T-cell subset expansion. To determine how IL-2 could influence the outgrowth of TCM and TSCM within bulk cultures, we reconstituted FACSort-purified TCM and TSCM as described earlier (Fig. 9A) followed by polyclonal activation with CD3 and CD28 antibodies in the presence of either IL-2 (50IU/mL) or IL-7 and IL-15 (each at 10ng/mL) and transduction with a retroviral vector encoding mOrange. We reconstituted each subset into its respective subset depleted population on day 3, then assessed the fate of TCM and TSCM by tracing their path in the course of expansion (Fig. 9B). The medium and cytokines were changed every 3 days during culture and expansion. The frequency and absolute numbers of each subset was assessed weekly for up to day 35 after the initial stimulation (Fig. 9C). Although the trend to higher frequencies of CD4+ or CD8+ TCM- and TSCM-derived cells induced by culture in IL-7 and IL-15 (Fig. 9C), did not reach significance, we plan to enhance this trend using cytokine modifications and to use our separation approach to validate the changes. Owing to the higher overall fold expansion rate of T-cells expanded in IL-7 and IL-15 the absolute numbers of TCM- and TSCM-derived cells were significantly higher than in IL-2-grown cultures (Fig. 9D). Furthermore, T-cells cultured in IL-2 showed an earlier contraction defined by reduction of cell numbers from day 20, whereas T-cells cultured in IL-7 and IL-15 expanded long-term with no signs of culture contraction up to day 35 (Fig. 9D). These results confirm that IL-7 and IL-15 better supported the expansion of both TCM and TSCM within bulk cultures (24, 41).
FIGURE 9. A combination of IL-7 and IL-15 increases the yield of TSCM and TCM derived T-cells compared to IL-2.

TCM and TSCM were FACSort-depleted from PBMC, activated with CD3 and CD28 antibodies in the presence of either IL-2 (50IU/mL) or IL-7 and IL-15 (each at 10ng/mL) and transduced on day 2 with a retroviral vector encoding mOrange. mOrange+ TCM and TSCM were reconstituted on day 3 to the respective TSUBSET-depleted PBMC. The medium and cytokines were changed every 3 days during culture or when passaging the T-cells for splitting during expansion. (A) Shows the frequency of TSCM and TCM in PBMC. (B) Representative plot showing the frequency of mOrange-positive TCM-derived (T(TCM)) and TSCM-derived (T(TSCM)) populations in reconstituted cultures on day 11. Cells were stained for CD3, CD4 and CD8. (C) Frequencies of reconstituted CD4+ TCM (upper graph), CD8+ TCM (middle graph) and CD8+ TSCM (lower graph) during expansion from day 2 to 35 of culture in either IL-2 or IL-7 and IL-15 (TCM-derived: T(TCM); TSCM-derived: T(TSCM)). (D) Expansion within the bulk population of reconstituted CD4+ TCM (upper graph), CD8+ TCM (middle graph) and CD8+ TSCM (lower graph) measured as fold increase of mOrange+ T-cells over 35 days of culture in IL-2 or IL-7/15 (TCM-derived: T(TCM); TSCM-derived: T(TSCM)). Data were analyzed using repeated measures 2-way ANOVA, after verifying Gaussian distribution with the Kolmogorov-Smirnov test. p-values ≤ 0.05 were considered significant: *p<0.05. Mean data from 4 healthy donors are presented.
Discussion
The central purpose of our study was to understand the contribution of naïve and effector/memory T-cell subsets to the bulk in vitro-stimulated CAR T-cell products that are eventually infused into patients on our clinical trials. To this end, we developed an approach to assess the fate of each T-cell subset by tracing its path in the course of expansion after transduction with either a GD2.CAR or a CD19.CAR and cultured alone or after reconstitution into the relevant subset-depleted PBMCs that had been activated and transduced in parallel (e.g. TSCM into TSCM-depleted PBMC). In our proof-of-concept study we show that all subsets were similarly susceptible to transduction, and the rate of TSUBSET expansion was unchanged by transfer into the subset-depleted population. The greatest rate of expansion was observed in TSCM-derived T-cells, but since TSCM represented less than 1% (0.9±0.2%) of the starting population, their descendants comprised less than 2% of the final product on day 14. Naïve-derived T-cells showed the second greatest rate of expansion, and this population dominated the cultures by day 14, comprising up to 89% of CD19.CAR-modified T-cells. TCM showed the 3rd greatest rate of proliferation and comprised up to 44% of the final population. TEM- and TEMRA-derived populations proliferated relatively poorly and were poorly represented in the final population (<5% and <3%, respectively). Despite the greatest proliferative response to TCR stimulation, the dominant naïve-derived T-cell majority showed relatively poor cytokine production, killing and poor proliferative responses to CAR stimulation. Further, Tnaive-derived cells differentiated to a greater extent when cultured in the presence of memory T-cells compared to when cultured alone, and show reduced re-expansion capacity in response to CAR stimulation. IL-7 and IL-15 induced greater total fold expansion of CD3/28-activated T-cells and produced higher frequencies of TCM– and TSCM–derived cells than IL-2.
Successful CAR-T cell therapy depends on multiple cell and patient-dependent factors. Among these, the T-cell subset origin of the final T-cell product infused may be critical to the final outcome by contributing to post infusion T-cell expansion, effector function and long-term persistence (28, 36, 39). However, while the effector memory phenotype of the final CAR-T cell product infused is often described, the T-cell subset of origin is rarely investigated, so that functional dissection of the infusion product (TEFF cell products) remains a prerequisite to better understand and correlate therapy outcome with T-cell product input. While individual subsets have been expanded in isolation, neither their expansion and function after reconstitution into the bulk population, nor the subset contribution to the final T-cell product have been reported. Of note, we analyzed the T-cell subset contribution only of PBMCs activated on CD3 and CD28-coated plates in the presence of either IL-7 and IL-15 or IL-2, which are the methods we have used to expand CAR-T-cells on our clinical trials. Many other methods are used by other groups, including CD3/28-coated beads, soluble CD3 and CD28 or PHA, and these may produce different results. Regardless, our strategy presents a means to evaluate the effects on T-cell subsets resulting from any changes in culture conditions.
There is intense discussion of the optimal phenotypic T-cell profile and optimal T-cell subset for immunotherapeutic use with regards to longevity, engraftment and anti-tumor effector function (24, 28, 30, 33, 36–39). Berger et al., showed that cytomegalovirus (CMV)-specific TEFF clones derived from TCM exhibited superior engraftment capacities compared to clones derived from TEM, and were long-lived and established persistent memory T-cell in a primate model in which TCR stimulation was provided by endogenous CMV (32). Other groups have suggested that the naïve T-cell population is important since this subset may contain precursors for tumor-specific antigens that could contribute to tumor elimination (49). The question of whether naïve-derived T-cells displaying memory T-cell phenotypic characteristics have the capacity for long-term survival in vivo, particularly if they do not encounter their cognate antigen remains an important subject for investigation. Naïve to memory T-cell conversion is a very complex process that requires multiple encounters with antigen and occurs in secondary lymphoid organs that provide an environment for such conversion (50–53). Several studies reported effective in vitro T-cell priming for multiple viral and tumor antigens, however, their clinical efficacy remains to be demonstrated (54–62). In our studies, naïve-derived T-cells developed effector functions in vitro, but these were less potent than memory-derived T-cells. Whether such naïve-derived T-cells with a memory phenotype have long-term memory potential in vivo and whether in vivo CAR-stimulation by tumor cells can induce true memory conversion remains to be demonstrated.
Recently Sommermeyer et al. showed that when modified with a CD19.CAR, a combination of CD4+ naïve- and CD8+ TCM-derived T-cells showed the most potent anti-tumor function in vitro and in preclinical models and in combination had synergistic anti-tumor effects in an EBV-positive lymphoma xenograft model (Raji) (63). This combination is currently under clinical evaluation. A possible advantage of the TCM population is that it contains T-cells specific for endogenous viruses that may provide antigenic stimulation after infusion.
Gattinoni and colleagues described the existence of stem cell-like T-cells (TSCM) within the phenotypically naïve (CD45RA+CCR7+) T-cell population (31). The expression of CD95 distinguished TSCM (CD45RA+CCR7+CD95+) from Tnaive (CD45RA+CCR7+CD95−), and in a murine xenograft model only TSCM were able to serially transfer GVHD (41). The distinct functional and unique homeostatic properties of TSCM have also been demonstrated in a nonhuman primate model during the course of simian immunodeficiency virus infection. In this report, TSCM were described as the least-differentiated memory subset, functionally distinct from conventional memory cells and served as precursors of TCM (40). In a human study, Xu and colleagues suggested that the frequency of TSCM-like cells within the infused T-cell product correlated with in vivo expansion following adoptive transfer (38) while Biasco et al. showed that TSCM produced the greatest contribution to long-term reconstitution in gene therapy studies using T-cells (64). Hence, TSCM may have even greater potential for expansion and long-term persistence than TCM (41), but to date, the relative benefits of TCM- and TSCM-derived CAR T-cells in clinical trials have not been reported. Our data suggest that standard strategies for CAR T-cell manufacturing may preferentially expand naïve T-cells with some contribution from TCM but little contribution from TSCM, despite the fact that they expand exponentially in vitro when cultured alone or within PBMC. Further studies are required to evaluate the long-term fate of TCM and TSCM-derived cells, both in culture and in patients.
The ultimate T-cell subset composition will likely also be affected by the CAR structure and endodomains, but our system provides a way to test these effects on the final infusion product. We show that culture conditions profoundly affect the subset composition of expanded T-cells. IL-2 expansion produced lower total fold expansion than IL-7 and IL-15 and thus decreased the numbers of TSCM and TCM in the final product. Although not significant, IL-7 and IL-15 also produced higher frequencies of TCM– and TSCM–derived cells than IL-2. Further efforts to improve the relative frequency of TCM/ TSCM-derived cells over naïve-derived T-cells could be validated using our strategy. Other culture manipulations including culture vessels, media formulation and type of initial stimulus will likely also influence the degree to which each subset contributes to the final population. Indeed in our prior study, we found that TSCM-derived CMV-specific T-cells did not expand within the bulk population if they received stimulation with CMV peptides rather than CD3 and CD28 antibodies (24). A more direct way to obtain the desired T-cell subset is to separate it from the starting population prior to culture, but this increases the expense and complexity of manufacture (63). It would be instructive to know whether the manufacturing strategies used in various clinical trials promote the outgrowth of TCM and TSCM and useful to develop cell culture strategies that enhance their expansion within bulk cultures, without the requirement for separation strategies. We provide a strategy to evaluate the subsets of origin of T-cells grown under any conditions using PBMCs from patients or healthy donors. Our study supports the importance of IL-7 and IL-15 for the expansion of TCM and TSCM-derived effector T-cells, at least using our manufacturing strategy (38).
Although the different T-cell subsets were not differentially transduced by retroviral vectors, their dissimilar rates of proliferation led to changes in their relative frequencies in the final population, which was dominated by naïve-derived T-cells (76±19% of CD3+ T-cells). TCM-derived populations maintained their frequencies (30±12% of CD3+ T-cells) and TSCM-derived populations were maintained or slightly increased (mean of 1.5±0.3% of CD3+ T-cells), while TEM and TEMRA frequencies were reduced (mean of 4.7±1.6% and 2.8±1.3% of CD3+ T-cells, respectively). Although the majority of each subpopulation differentiated into cells with a TEM-like phenotype, their functionalities differed with respect to cytokine production and proliferation in response to CAR stimulation. Tnaive- and TEMRA-derived cells produced lower levels of cytokines and proliferated poorly in response to CAR stimulation compared to TCM and TSCM-derived cells. This may be of little import for TEMRA, since they comprise only a minor fraction of the final product, but a high frequency of poorly functional Tnaive-derived cells may be less effective after infusion. Our results confirm reports suggesting that naïve-derived T-cells possess poor effector functions (39). However, we cannot exclude the possibility that naïve-derived T-cells will acquire improved function after multiple stimulations in vivo via the CAR, although they responded relatively poorly to a single CAR stimulation in vitro. Our data confirms the recent report by Klebanoff and colleagues showing enhanced phenotypic and functional differentiation of naïve-derived T-cells that have been cultured in the presence of antigen-experienced memory T-cell (29). Naïve-derived T-cells cultured in bulk PBMC preparations show reduced re-expansion capacity in response to CAR stimulation, and hence may lack anti-tumor potential when transferred in vivo. Nevertheless, when we cultured naïve-derived T-cells separately, re-expansion to CAR stimulation was reduced compared to TSCM-, TCM- and TEM-derived cultures. Hence, if naïve T-cells are to be used for adoptive immunotherapy, we would propose culture in isolation to avoid the negative effects of culture with antigen-experienced memory T-cells. Further in vitro and in vivo studies over longer time periods are required to determine the long-term consequences of infusing a preponderance of Tnaive-derived cells, but without some form of differential gene-marking, this may be difficult.
Our strategy could also be used in cases in which short-lived effectors are desirable for safety reasons, for example to test a novel T-cell manipulation. Regardless, our strategy can be a powerful tool to evaluate the effects of manufacturing changes on the subset contribution to in vitro expanded T-cells. Certainly, a one-size fit all strategy for the generation of CAR-modified TEFF may not be expected because of tumor cell biology and patient characteristics, but a better understanding of the characteristics of the generated CAR-modified T-cell products will help identify the subsets with most clinical efficacy. To this end, our strategy provides a powerful tool to elucidate the characteristics of CAR T-cells polyclonally expanded from PBMC on CD3 and CD28 antibody-coated plates, and to reveal the functional properties of each expanded T-cell subset.
Supplementary Material
Acknowledgments
We thank Dr. Jason Millward (Institute of Medical Immunology, Charité University Medicine Berlin and Experimental and Clinical Research Center, a joint cooperation between the Charité Medical Faculty and the Max-Delbrück Center for Molecular Medicine) for statistical advice. This project was supported in part by the Cytometry and Cell Sorting Core at Baylor College of Medicine and the expert assistance of Joel M. Sederstrom and team. We thank Dr. Juan F. Vera (Center for Gene Therapy, Baylor College of Medicine, Houston, TX, US) for the generous provision of the SFG.IRES.mOrange construct, Tatiana Goltsova and Dr. Amos Gaikwad (Texas Children’s Hospital Flow Cytometry Core Laboratory) for technical flow cytometry assistance.
Funding: The study was generously supported in parts by the Deutsche Forschungsgemeinschaft (DFG-SFB-TR36-project A3), the German Federal Ministry of Education and Research (Berlin-Brandenburg Center for Regenerative Therapies grant), an Alex’s Lemonade Stand Foundation grant to CMR, NIH-NCI PO1 CA94234 and P50 CA126752, the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and The V-Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Disclosures
CMR is on the scientific advisory board of Cell Medica.
Author contributions
M.S.-H. led the project, designed the research, performed most of the experiments, analyzed and interpreted the data, and significantly contributed to writing of the manuscript. C.M.R designed the research, interpreted the data, and significantly contributed to writing of the manuscript. P.R. and H.-D.V. helped to design the research. B.O., T.S., H.T. helped to design the experiments, performed some of the experiments and helped to analyze data. M.M. and N.L. helped to design the experiments. S.S. and L.R. provided technical assistance. G.D. provided the CAR constructs and helped to design the experiments.
Bibliography
- 1.Rosenberg Sa, Restifo NP, Yang JC, Morgan Ra, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalos M, Levine BL, Porter DL, Katz S, Grupp Sa, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grupp Sa, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Dotti G. Brief report CD28 costimulation improves expansion and persistence of chimeric antigen receptor – modified T cells in lymphoma patients. 2011;121:1–5. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, Nagase F, Kurosawa Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149:960–968. doi: 10.1016/0006-291x(87)90502-x. [DOI] [PubMed] [Google Scholar]
- 7.Gross G, Eshhar Z. Endowing T cells with antibody specificity using chimeric T cell receptors. FASEB J. 1992;6:3370–8. doi: 10.1096/fasebj.6.15.1464371. [DOI] [PubMed] [Google Scholar]
- 8.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257:107–26. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milone CM, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp Sa, June CH. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong X, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Dotti G. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman Ha, Qian X, James SE, Raubitschek A, Forman SJ, Gopal AK, Pagel JM, Lindgren CG, Greenberg PD, Riddell SR, O. W. Press Adoptive immunotherapy for indolent non-hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–56. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman Sa, Maric I, Raffeld M, Nathan DAN, Lanier BJ, Morgan Ra, Rosenberg Sa. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, Hakim FT, Halverson DC, Fowler DH, Hardy NM, Mato AR, Hickstein DD, Gea-Banacloche JC, Pavletic SZ, Sportes C, Maric I, Feldman SA, Hansen BG, Wilder JS, Blacklock-Schuver B, Jena B, Bishop MR, Gress RE, Rosenberg Sa. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122:4129–4139. doi: 10.1182/blood-2013-08-519413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, Raubitschek A, Forman SJ, Greenberg PD, Riddell SR, O. W. Press CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood. 2012;119:3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, Chen K, Shin M, Wall DM, Hönemann D, Gambell P, Westerman Da, Haurat J, Westwood Ja, Scott AM, Kravets L, Dickinson M, Trapani Ja, Smyth MJ, Darcy PK, Kershaw MH, Prince HM. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21:2122–9. doi: 10.1038/mt.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cruz CRY, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, Diouf O, Liu E, Barrett AJ, Ito S, Shpall EJ, Krance RA, Kamble RT, Carrum G, Hosing CM, Gee AP, Mei Z, Grilley BJ, Heslop HE, Rooney CM, Brenner MK, Bollard CM, Dotti G. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122:2965–73. doi: 10.1182/blood-2013-06-506741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maus MV, Grupp Sa, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123:2625–35. doi: 10.1182/blood-2013-11-492231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257:107–26. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19:3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmueck-Henneresse M, Sharaf R, Vogt K, Weist BJD, Landwehr-Kenzel S, Fuehrer H, Jurisch A, Babel N, Rooney CM, Reinke P, Volk H-D. Peripheral Blood-Derived Virus-Specific Memory Stem T Cells Mature to Functional Effector Memory Subsets with Self-Renewal Potency. J Immunol. 2015 doi: 10.4049/jimmunol.1402090. [DOI] [PubMed] [Google Scholar]
- 25.Savoldo B, Goss Ja, Hammer MM, Zhang L, Lopez T, Gee AP, Lin Y-F, Quiros-Tejeira RE, Reinke P, Schubert S, Gottschalk S, Finegold MJ, Brenner MK, Rooney CM, Heslop HE. transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs) Blood. 2006;108:2942–9. doi: 10.1182/blood-2006-05-021782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bollard CM, Rooney CM, Heslop HE. T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nat Rev Clin Oncol. 2012;9:510–9. doi: 10.1038/nrclinonc.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brestrich G, Zwinger S, Fischer A, Schmück M, Röhmhild A, Hammer MH, Kurtz A, Uharek L, Knosalla C, Lehmkuhl H, Volk H-D, Reinke P. Adoptive T-cell therapy of a lung transplanted patient with severe CMV disease and resistance to antiviral therapy. Am J Transplant. 2009;9:1679–84. doi: 10.1111/j.1600-6143.2009.02672.x. [DOI] [PubMed] [Google Scholar]
- 28.Terakura S, Yamamoto TN, Gardner Ra, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012;119:72–82. doi: 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klebanoff CA, Scott CD, Leonardi AJ, Yamamoto TN, Cruz AC, Ouyang C, Ramaswamy M, Roychoudhuri R, Ji Y, Eil RL, Sukumar M, Crompton JG, Palmer DC, Borman ZA, Clever D, Thomas SK, Patel S, Yu Z, Muranski P, Liu H, Wang E, Marincola FM, Gros A, Gattinoni L, Rosenberg SA, Siegel RM, Restifo NP. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J Clin Invest. 2016;126:318–334. doi: 10.1172/JCI81217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 31.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmueck M, Fischer AM, Hammoud B, Brestrich G, Fuehrer H, Luu S-H, Mueller K, Babel N, Volk H-D, Reinke P. Preferential expansion of human virus-specific multifunctional central memory T cells by partial targeting of the IL-2 receptor signaling pathway: the key role of CD4+ T cells. J Immunol. 2012;188:5189–98. doi: 10.4049/jimmunol.1103763. [DOI] [PubMed] [Google Scholar]
- 34.Carrasco J, Godelaine D, Van Pel A, Boon T, Van Der Bruggen P. CD45RA on human CD8 T cells is sensitive to the time elapsed since the last antigenic stimulation. Blood. 2006;108:2897–2905. doi: 10.1182/blood-2005-11-007237. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood. 2011;117:1888–98. doi: 10.1182/blood-2010-10-310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Naranjo A, Brown CE, Bautista C, Wong CW, Chang W-C, Aguilar B, Ostberg JR, Riddell SR, Forman SJ, Jensen MC. Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T cells manufactured at clinical scale. J Immunother. 2012;35:689–701. doi: 10.1097/CJI.0b013e318270dec7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riddell SR, Sommermeyer D, Berger C, Liu LS, Balakrishnan A, Salter A, Hudecek M, Maloney DG, Turtle CJ. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J. 2014;20:141–4. doi: 10.1097/PPO.0000000000000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y, Zhang M, Ramos Ca, Durett A, Liu E, Dakhova O, Liu H, Creighton CJ, Gee AP, Heslop HE, Rooney CM, Savoldo B, Dotti G. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123:3750–9. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hinrichs CS, Borman Za, Gattinoni L, Yu Z, Burns WR, Huang J, Klebanoff Ca, Johnson La, Kerkar SP, Yang S, Muranski P, Palmer DC, Scott CD, Morgan Ra, Robbins PF, Rosenberg Sa, Restifo NP. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117:808–814. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K, Klatt NR, Brenchley JM, Vaccari M, Gostick E, Price Da, Waldmann Ta, Restifo NP, Franchini G, Roederer M. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest. 2013;123:594–9. doi: 10.1172/JCI66327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, Bondanza A, Bordignon C, Peccatori J, Ciceri F, Lupo-Stanghellini MT, Mavilio F, Mondino A, Bicciato S, Recchia A, Bonini C. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573–84. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
- 42.Lugli E, Gattinoni L, Roberto A, Mavilio D, Price Da, Restifo NP, Roederer M. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat Protoc. 2013;8:33–42. doi: 10.1038/nprot.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pulè Ma, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–41. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 44.Rossig C, Bollard CM, Nuchtern JG, Merchant DA, Brenner MK. Targeting of GD2-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int J Cancer. 2001;94:228–236. doi: 10.1002/ijc.1457. [DOI] [PubMed] [Google Scholar]
- 45.Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner MK. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. 2006;20:1819–28. doi: 10.1038/sj.leu.2404366. [DOI] [PubMed] [Google Scholar]
- 46.Sen G, Chakraborty M, Foon KA, Reisfeld RA, Bhattacharya-Chatterjee M. Preclinical evaluation in nonhuman primates of murine monoclonal anti- idiotype antibody that mimics the disialoganglioside GD2. Clin Cancer Res. 1997;3:1969–1976. [PubMed] [Google Scholar]
- 47.Hermans IF, Silk JD, Yang J, Palmowski MJ, Gileadi U, McCarthy C, Salio M, Ronchese F, Cerundolo V. The VITAL assay: A versatile fluorometric technique for assessing CTL- and NKT-mediated cytotoxicity against multiple targets in vitro and in vivo. J Immunol Methods. 2004;285:25–40. doi: 10.1016/j.jim.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 48.Hammoud B, Schmueck M, Fischer AM, Fuehrer H, Park S-J, Akyuez L, Schefold JC, Raftery MJ, Schönrich G, Kaufmann AM, Volk H-D, Reinke P. HCMV-specific T-cell therapy: do not forget supply of help. J Immunother. 2013;36:93–101. doi: 10.1097/CJI.0b013e31827b87cc. [DOI] [PubMed] [Google Scholar]
- 49.Wölfl M, Merker K, Morbach H, Van Gool SW, Eyrich M, Greenberg PD, Schlegel PG. Primed tumor-reactive multifunctional CD62L+ human CD8 + T cells for immunotherapy. Cancer Immunol Immunother. 2011;60:173–186. doi: 10.1007/s00262-010-0928-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grakoui A. The Immunological Synapse: A Molecular Machine Controlling T Cell Activation. Science (80-) 1999;285:221–227. [PubMed] [Google Scholar]
- 51.Bousso P, Robey E. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat Immunol. 2003;4:579–585. doi: 10.1038/ni928. [DOI] [PubMed] [Google Scholar]
- 52.Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science. 2002;296:1873–6. doi: 10.1126/science.1071065. [DOI] [PubMed] [Google Scholar]
- 53.Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–9. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 54.Berard F, Blanco P, Davoust J, Neidhart-Berard EM, Nouri-Shirazi M, Taquet N, Rimoldi D, Cerottini JC, Banchereau J, Palucka AK. Cross-priming of naive CD8 T cells against melanoma antigens using dendritic cells loaded with killed allogeneic melanoma cells. J Exp Med. 2000;192:1535–44. doi: 10.1084/jem.192.11.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moser JM, Sassano ER, Leistritz DC, Eatrides JM, Phogat S, Koff W, Drake DR. Optimization of a dendritic cell-based assay for the in vitro priming of naïve human CD4+ T cells. J Immunol Methods. 2010;353:8–19. doi: 10.1016/j.jim.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 56.Hanley PJ, Melenhorst JJ, Nikiforow S, Scheinberg P, Blaney JW, Demmler-Harrison G, Cruz CR, Lam S, Krance RA, Leung KS, Martinez CA, Liu H, Douek DC, Heslop HE, Rooney CM, Shpall EJ, Barrett AJ, Rodgers JR, Bollard CM. CMV-specific T cells generated from naïve T cells recognize atypical epitopes and may be protective in vivo. Sci Transl Med. 2015;7:285ra63. doi: 10.1126/scitranslmed.aaa2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ho WY, Nguyen HN, Wolfl M, Kuball J, Greenberg PD. In vitro methods for generating CD8+ T-cell clones for immunotherapy from the naive repertoire. J Immunol Methods. 2006;310:40–52. doi: 10.1016/j.jim.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 58.Wölfl M, Greenberg PD. Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nat Protoc. 2014;9:950–66. doi: 10.1038/nprot.2014.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schlienger K, Craighead N, Lee KP, Levine BL, June CH. Efficient priming of protein antigen-specific human CD4(+) T cells by monocyte-derived dendritic cells. Blood. 2000;96:3490–3498. [PubMed] [Google Scholar]
- 60.Santegoets SJAM, Schreurs MWJ, Masterson AJ, Liu YP, Goletz S, Baumeister H, Kueter EWM, Lougheed SM, Van Den Eertwegh AJM, Scheper RJ, Hooijberg E, De Gruijl TD. In vitro priming of tumor-specific cytotoxic T lymphocytes using allogeneic dendritic cells derived from the human MUTZ-3 cell line. Cancer Immunol Immunother. 2006;55:1480–1490. doi: 10.1007/s00262-006-0142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nguyen HH, Kim T, Song SY, Park S, Cho HH, Jung S-H, Ahn J-S, Kim H-J, Lee J-J, Kim H-O, Cho J-H, Yang D-H. Naïve CD8(+) T cell derived tumor-specific cytotoxic effectors as a potential remedy for overcoming TGF-β immunosuppression in the tumor microenvironment. Sci Rep. 2016;6:28208. doi: 10.1038/srep28208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tuettenberg A, Jonuleit H, Tüting T, Brück J, Knop J, Enk AH. Priming of T cells with Ad-transduced DC followed by expansion with peptide-pulsed DC significantly enhances the induction of tumor-specific CD8+ T cells: implications for an efficient vaccination strategy. Gene Ther. 2003;10:243–50. doi: 10.1038/sj.gt.3301880. [DOI] [PubMed] [Google Scholar]
- 63.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2015:1–9. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biasco L, Scala S, Basso Ricci L, Dionisio F, Baricordi C, Calabria A, Giannelli S, Cieri N, Barzaghi F, Pajno R, Al-Mousa H, Scarselli A, Cancrini C, Bordignon C, Roncarolo MG, Montini E, Bonini C, Aiuti A. In vivo tracking of T cells in humans unveils decade-long survival and activity of genetically modified T memory stem cells. Sci Transl Med. 2015;7:273ra13–273ra13. doi: 10.1126/scitranslmed.3010314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.