

Abstract
The concept of innate immunity has been expanded to recognize environmental pathogens other than microbial components. However, whether and how the innate immunity is initiated by epithelium in response to environmental physical challenges such as low humidity and high osmolarity in an autoimmune disease, dry eye, is still largely unknown. Using two experimental dry eye models, primary human corneal epithelial cultures exposed to hyperosmolarity and mouse ocular surface facing desiccating stress, we uncovered novel innate immunity pathway by ocular surface epithelium, where oxidized mitochondrial DNA induces imbalanced activation of NLRP3/NLRP6 inflammasomes via stimulation of caspase-8 and BRCC36 in response to environmental stress. Activated NLRP3 with suppressed NLRP6 stimulates caspase-1 activation that leads to IL-1β and IL-18 maturation and secretion. NLRP3-independent caspase-8 noncanonically activates caspase-1 via reciprocal regulation of NLRP3/NLRP6-mediated inflammasomes. Reactive oxygen species-induced mitochondrial DNA oxidative damage and BRCC36 deubiquitinating activity provide a missing link and mechanism by which innate immunity responds to environmental stress via caspase-8-involved NLRP3/NLRP6 inflammasomes.
Keywords: Innate immunity, environmental stress, NLR proteins, Inflammasome activation, caspase activity, mitochondrial DNA
1. Introduction
Innate immunity is the first defense against pathogen associated molecular patterns (PAMPs), a process mediated by germ line-encoded pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), nucleotide-binding oligomerization domain receptors (NLRs) and others. Increasing evidence suggests that the concept of innate immunity has been largely changed. Traditionally, the essential role of innate immunity is recognition of invading microorganisms such as bacteria, virus, and fungus. Recent breakthroughs reveal that the innate immunity has been expanded to recognize and respond to environmental PAMPs other than conserved microbial components [1, 2]. Furthermore, the advances in innate immunity have also changed the view on epithelial cells. In addition to a long-recognized property of their physical barrier function, epithelial cells are known to play important roles in initiation and regulation of immune responses [3–5].
Traditionally, innate immunity cells are white blood cells or leukocytes including basophils, dendritic cells, eosinophils, Langerhans cells, mast cells, monocytes and macrophages, neutrophils and NK cells [6, 7]. However, epithelial cells are now recognized to participate in innate immune responses, and serve as a bridge linking the innate immunity to adaptive immunity [3]. Moutsopoulos and his colleagues have done pioneer works at early years and revealed that epithelial cells can play important role in the initiation of autoimmune responses, such as in Sjogren’s syndrome [8–10]. We have recently uncovered a novel pollen/TLR4 innate immunity pathway where a plant product, short ragweed pollen triggers allergic inflammation via TLR4-dependent innate immunity by mucosal epithelium, which produces pro-allergic cytokine TSLP that initiates TSLPR/OX40L/OX40/Th2 signaling pathways [11]. This breakthrough demonstrated that in addition to recognize microbial components, mucosal epithelium also respond to plant products like pollen proteins through TLR4-depedent innate immune response.
However, whether and how the innate immunity is initiated by epithelial cells in response to environmental stress is still largely unknown. Corneal and conjunctival epithelium, facing environmental changes all the time, is the first line of eye defense against infectious and non-infectious pathogens. For example, multiple environmental factors are potential risks of developing human ocular surface diseases [12]. Dry eye can be the result of autoimmune processes and one of the most prevalent eye diseases, suffered by millions of people with bothersome irritation and possibly decrease quality of life. The environmental and occupational risk factors, including the exposure to low humidity, high wind velocity, air conditioning and extended viewing video display terminals, could increase tear evaporation, decrease tear volume, lead tear hyperosmolarity, and cause desiccating stress [13–15]. The environmental desiccating stress has been well known to cause ocular surface epithelial disease with inflammatory disorder, such as eye irritation, conjunctival desquamation, disrupted cell membranes and intercellular connections, as well as blunting and loss of microplicae, which often lead to keratoconjunctivitis sicca [16, 17].
NLRs are highly conserved cytosolic PRRs and play key roles in regulation of innate immune response. They are intracellular sensors of PAMPs that enter the cell via phagocytosis or pores, and also for damage associated molecular patterns that are associated with cell stress [18, 19]. NLRs-mediated inflammasome is a recent advance in innate immunity-initiated inflammation. Through adaptor apoptosis-associated speck-like protein containing a CARD (ASC), NLR recruits pro-caspase-1 to form the multiprotein complex known as inflammasome [12, 20], which activates one or more caspases, leading to the processing and secretion of pro-inflammatory cytokines, interleukin (IL) 1β and IL-18 [21, 22]. We have previously observed that desiccating stress induces IL-1β maturation and release, which plays a pivotal pathogenic role in dry eye of both human patients and experimental mouse model [23–26].
However, the role and mechanism by which NLRs-mediated innate immunity triggers inflammation in response to environmental challenges such as humidity and osmolality in dry eye disease, remain to be elucidated. In this study, we uncovered an innate immunity pathway that mitochondrial DNA (mtDNA) oxidation induces reciprocal regulation of NLRP3/NLRP6 inflammasomes in ocular epithelium under environmental stress using two models, a culture model of primary human corneal epithelial cells (HCECs) exposed to hyperosmolarity and a murine model exposed to desiccating environment.
2. Materials and Methods
2.1. Primary cultures of HCECs and in vitro hyperosmotic stress model
Fresh human corneoscleral tissues (<72 hours after death) not suitable for clinical use, with donors aged 18 to 65 years, were obtained from the Lions Eye Bank of Texas (Houston, TX). Primary HCECs were cultured in 12-well plates using explants from corneal limbal rims in a supplemented hormonal epidermal medium (SHEM) containing 5% FBS using our previous methods [27]. Confluent cultures in 14–18 days were switched to an equal volume (0.5 ml/well) of serum-free medium (SHEM without FBS) for 24 hours, and treated for 4–24 hours in medium with iso- and hyper- osmolarity (312, 400, 450 and 500 mOsM), which was achieved by adding 0, 44, 69 or 94 mM sodium chloride (NaCl). The osmolarity of the culture media was measured by a vapour pressure osmometer in the Body Fluid Chemistry Clinical Laboratory of the Methodist Hospital (Houston, TX). The cells treated for 4 hours were lysed in RLT buffer from Qiagen RNeasy Plus Mini kit for total RNA extraction. The cells treated for 24 hours were lysed in RIPA buffer for cellular proteins. The supernatants of the conditioned media were stored at −80 °C before use.
2.2. Experimental murine model exposed to environmental desiccating stress (DS)
This animal research protocol was approved by the Institutional Animal Care and Use Committee (IACUC), Center for Comparative Medicine, Baylor College of Medicine. All animals used in this study were maintained in specific pathogen-free conditions in microisolator cages and were treated in accordance with the Guide for the Care and Use of Laboratory Animals by the National Institute of Health.
For a mouse model under DS, C57BL/6 female mice (Jackson Laboratory, Bar Harbor, ME) at 8–10 weeks were fed in an environmentally controlled room with humidity ≤30% and placed in customized cages with two sides constructed of wire to allow exposure to air drafts created by fans. Tear secretion was pharmacologically inhibited by subcutaneous injection of scopolamine hydrobromide (0.5 mg/0.2 ml; Sigma-Aldrich, St. Louis) four times a day for 5 consecutive days as previously described [28, 29]. Control mice were age- and gender-matched, and maintained in a normal environment at 50–75% relative humidity. Whole eye balls, corneal epithelium and conjunctival tissues were collected for different assays.
2.3. RNA extraction, reverse transcription and quantitative real time PCR (RT-qPCR)
Total RNA was extracted with a RNeasy Plus Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions, quantified with a spectrophotometer (NanoDrop ND-1000; Thermo Fisher Scientific, Waltham, MA), and stored at −80°C before use. RT-qPCR was performed as previously described [30, 31]. In brief, the first strand cDNA was synthesized by RT from 1.0μg of total RNA using Ready-To-Go You-Prime First-Strand Beads (GE Healthcare, Piscataway, NJ), and qPCR was performed in a Mx3005P QPCR System (Stratagene, La Jolla, CA) with 20 μl reaction volume containing 5μl of cDNA, 1μl gene expression assay and 10μl TaqMan gene expression master mix. TaqMan gene expression assays (Thermo Fisher Scientific) used for this study were: mGAPDH (Mm99999915_g1), mNLRP3 (Mm00840904_m1), mNLRP6 (Mm00460229_m1), mASC (Mm00445747_ml), mCaspase-1 (Mm00438023_ml), hGAPDH (Hs02758991_g1), hNLRP3 (Hs00918082_m1), hNLRP6 (Hs00373246_m1), hPYCARD (Hs01547324_ml). The thermocycler parameters were 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. A non-template control was included to evaluate DNA contamination. The results were analyzed by the comparative threshold cycle (Ct) method and normalized by GAPDH as an internal control.
2.4. Immunofluorescent and immunohistochemical Staining
HCEC cultures on 8-chamber slides and mouse eye sections were fixed with freshly prepared 2% paraformaldehyde at 4°C for 10 minutes. Cell cultures were permeabilized with 0.2% Triton X-100 in PBS at room temperature for 10 min. Indirect immunofluorescent and immunohistochemical staining was performed using our previous methods [32, 33]. Primary rabbit polyclonal antibodies against human and mouse NLRP3 (NBP1-77080), caspase-1 (NB100-56565), caspase 8 (NB100-56116, Novus Biologicals, Littleton, CO), NLRP6 (PA5-21022), IL-1β (P420B), BRCC36 (PA5-20426, Thermo Scientific, Rochford, IL), ASC (N-15, SC-22514R), IL-18 (SC-6179, Santa Cruz Biotechnology, Dallas, TX), aconitase-2 (ab129105), 8-OHdG (ab62623, Abcam, Cambridge, MA) were used. Alexa-Fluor 488 conjugated secondary antibodies (R&D Systems) were applied, and propidium iodide (PI) or 4′, 6-diamidino-2-phenylindole (DAPI) was used for nuclear counterstaining. Secondary antibodies were applied alone were as a negative control and compared to isotype goat IgG. The staining will be photographed with Zeiss laser scanning confocal microscope (LSCM510META, Thornwood, NY).
2.5. Western blot analysis
Western blotting was performed as previously described [34]. In brief, Equal amounts of protein measured by a BCA protein assay kit, were mixed with 6×SDS reducing sample buffer and boiled for 5 min before loading. The proteins (50μg/lane) were separated on an SDS polyacrylamide gel and transferred electronically to PVDF membranes. The membranes were blocked with 5% nonfat milk in TTBS (50 mM Tris [pH 7.5], 0.9% NaCl, and 0.1% Tween-20) for 1 h at room temperature and incubated with primary antibodies against human and mouse NLRP3, ASC, NLRP6, caspase-1 (NB100-56565), caspase-8 (NB100-56527), IL-1β, IL-18, BRCC36 or β-actin overnight at 4°C. After three washes with TTBS for 10min each, the membranes were incubated with HRP conjugated goat anti-mouse IgG (1:2000) or goat anti-rabbit IgG (1:2000) for 1h at room temperature. The signals were detected with a chemiluminescence reagent (ECL; GE Healthcare), and the images were acquired by an imaging station (model 4000R; Eastman Kodak, Rochester, NY).
2.6. Enzyme-linked immunosorbent assay (ELISA)
Double-sandwich ELISA for human IL-1β and IL-18 was performed according to the manufacturer’s protocol and our previous publication [34]. Absorbance was read at a wavelength of 450 nm by Tecan Infinite M200 Multimode Microplate Reader (Tecan US, Inc. Morrisville, NC).
2.7. Caspase-1 and Caspase-8 Activity Assay
Caspase-1 and Caspase-8 activity fluorometric assay kits were used according to manufactory’s protocol. In bief, living cells (1×106) or cell lysates (50 μg) were resuspended in 50 μl of chilled Cell Lysis Buffer and incubated on ice for 10 mins. After adding 50 μl of 2x Reaction Buffer (containing 10 mM DTT) to each sample, 5 μl of 1 mM substrate, YVAD-AFC or IETD-AFC, was added respectively for caspase-1 or caspapse-8 activity assay (50mM final concentration), and incubated at 37°C for 1 hour. Read samples in Tecan Infinite M200 Multimode Microplate Reader (Tecan US, Inc. Morrisville, NC) with a 400-nm excitation filter and 505-nm emission filter. Fold-increase in Caspase-1 activity can be determined by comparing the results of induced samples with the levels of the untreated cells or normal mouse tissue lysate. Cell images of caspase-8 activity were taken using a fluorescent microscope.
2.8. Measurement of cellular production of reactive oxygen species (ROS)
Cellular ROS production was measured using 2′, 7′-dichlorofluorescein diacetate (DCFDA) assay kit. DCFDA, a cell-permeable fluorogenic dye, is deacetylated by cellular esterases to a non-fluorescent compound, it could be oxidized by ROS into highly fluorescent 2′,7′-dichlorofluorescein (DCF) which measures hydroxyl, peroxyl and other ROS activity within the cell. HCECs were grown on the 96-well plates or 8-chamber slides. When confluent, HCECs were incubated with 25 μM DCFDA at 37°C for 45 min, and then exposed to hyperosmotic medium (450 mOsM) alone or in the presence of 10 μM of L-carnitine or N-Acetyl-L-cysteine (NAC) for different time periods (5–120 min). Cell fluorescence in 96-well plates was measured at 488 nm excitation and 525 nm emission using Tecan Infinite M200 Microplate Reader (Tecan US, Inc. Morrisville, NC). Relative changes of DCF fluorescence were expressed as fold increase over the control cells at isosmolar condition.
2.9. Anti-oxidative and anti-inflammatory effects of L-carnitine, NAC and 8-OHdG
To explore the direct link of oxidative damage and oxidized mtDNA with imbalanced activation of NLRP3/NLRP6 inflammasomes and inflammation caused by environmental stress, antioxidants L-carnitine and NAC (10μM each), as well as competitive inhibitor 8-OHdG (8-hydroxy-2-deoxyguanosine 5–50μM) were included in the hyperosmotic culture model 1 hour prior, with dG used as negative control.
2.10. Statistical analysis
Student’s t-test was used to compare differences between two groups. One-way ANOVA test was used to make comparisons among three or more groups, followed by Dunnett’s post-hoc test. P <0.05 was considered statistically significant.
3. Results
3.1. Environmental hyperosmolarity activates NLRP3 inflammasome while suppresses NLRP6 production in primary HCECs
The ocular surface epithelium has been well known as the first line of defense. To explore the innate immune responses of epithelium to environmental stress, a hyperosmolarity model representing the consequence of low humidity, high wind velocity and over-evaporation, was created using in vitro primary HCEC cultures exposed to the media with increasing osmolarity (400, 450, and 500 mOsM) from normal isomolar medium (312 mOsM). The results showed that mRNA expression and protein production of NLRP3 and its adaptor protein ASC were significantly increased by the epithelial cells exposed to hyperosmotic medium (400–500 mOsM), and peaked at 450 and/or 500 mOsM, as evaluated by RT-qPCR (Fig. 1A) and Western-blot analysis (Fig. 1B).
Fig. 1. Environmental hyperosmolarity activates NLRP3 inflammasome while suppresses NLRP6 production in primary HCECs.
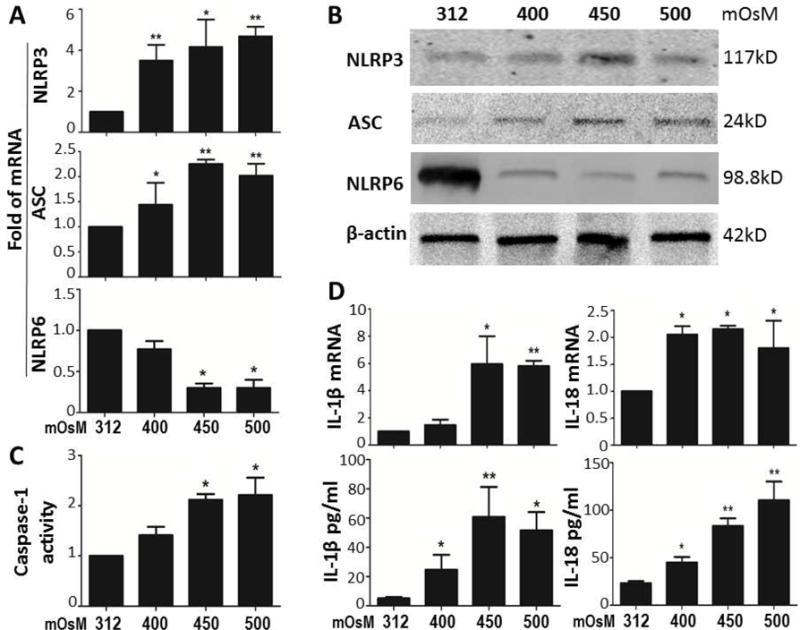
(A) mRNA expressions of NLRP3, ASC and NLRP6 by HCECs in hyperosmotic medium (400, 450 and 500mOsM) with isomolar 312mOsM as control, evaluated by RT-qPCR. (B) The protein levels of NLRP3, ASC and NLRP6 in HCECs exposed to iso- and hyper-osmotic medium, evaluated by Western blotting with β-actin as internal control. (C) Caspase-1 activity assay in HCECs with iso- and hyper-osmotic medium. (D) mRNA expression and protein secretion of IL-1β and IL-18 in HCECs exposed to iso- and hyper-osmotic medium, evaluated by RT-qPCR and ELISA. Data shown are representative of three or more independent experiments (mean ± SD). *P < 0.05, **P < 0.01, vs. controls.
NLRP6 is a newly identified NLR protein that has been shown to participate in inflammasome signaling and to play critical roles in defense against inflammation [19, 35]. But the involvement of NLRP6 in epithelial innate immunity is largely unknown. Interestingly, we observed the significantly suppressed mRNA expression (Fig. 1A) and protein production (Fig. 1B) of NLRP6, a reciprocal response in contrast of NLRP3, under the hyperosmotic stress.
Caspase-1 is an important component for assembling NLR-mediated inflammasomes, which actively participate in processing proinflammatory cytokines, IL-1β and IL-18, in inflammation pathogenesis [36]. The imbalance of activated NLRP3 and suppressed NLRP6 leads the significant activation of caspase-1 in corneal epithelial cells exposed to hyperosmotic stress, as evaluated by Caspase-1 activity assay (Fig. 1C), which resulted in maturation of pro IL-1β and IL-18 into the cleaved active forms that released to culture medium, as evaluated by ELISA (Fig. 1D). Interestingly, IL-1β and IL-18 transcripts measured by RT-qPCR were also stimulated (Fig. 1D). The increase of mRNA expression of NLRP3 and IL-1β are recognized to be important for NLRP3 priming before activation [37]. Immunofluorescent double staining vividly showed that NLRP3/ASC inflammasome was activated in HCECs exposed to hyperosmotic environment (Fig. 4B).
Fig. 4. Hyperosmolarity-activated caspse-8 reciprocally regulates NLRP3/NLRP6 mediated inflammasomes in HCECs.
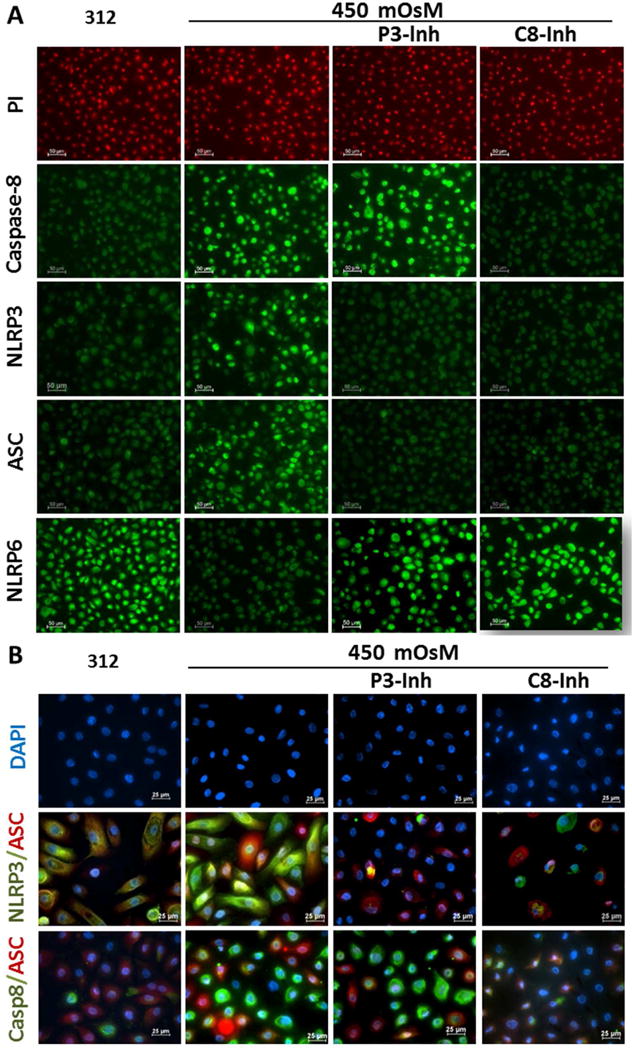
(A) Immunofluorescent staining results showed the increased number of caspase-8, NLRP3 or ASC positive cells with decreased number of NLRP6 positive cells in HCECs at 450mOsM, which was completely reversed by 20μM C8-Inh, but partly by P3-Inh. (B) Double Immunofluorescent staining showed that caspase-8 involved in NLRP3/ASC/caspase-8 inflammasomes in HCECs exposed to 450mOsM medium, which was completely reversed by 20μM C8-Inh, but partly by P3-Inh. Data shown are representative of three or more independent experiments.
3.2. Inhibition of NLRP3 or caspase-1 activity reverses the reciprocal regulation of NLRP3 and NLRP6, and blocked caspase-1-dependent secretion of IL-1β and IL-18
To functionally confirm that the reciprocal regulation of NLRP3 and NLRP6 mediated inflammasomes promotes inflammation, a NLRP3 inhibitor, glybenclamide (30μM) was prior added into the hyperosmotic culture model. As shown in Fig. 2 A and B, glybenclamide effectively attenuated the stimulated expression of NLRP3 and ASC, and reversed or restored the suppressed NLRP6 production, at both transcription and translation levels in HCECs exposed to 450mOsM hyperosmotic medium. Caspase-1 activity was also reduced to near normal levels by glybenclamide in HCECs under hyperosmotic stress (Fig. 2C), which resulted in the suppressed levels of active forms of IL-1β an IL-18 and their secretion as evaluated by Western blotting (Fig. 2D) and ELISA (Fig. 2E). Inhibition by siRNA interference further confirmed the role of NLRP3 (see Supplementary Material and Fig. S1).
Fig. 2. Inhibition of NLRP3 or caspase-1 activity reverses the reciprocal regulation of NLRP3 and NLRP6, and blocked caspase-1-dependent secretion of IL-1β and IL-18.
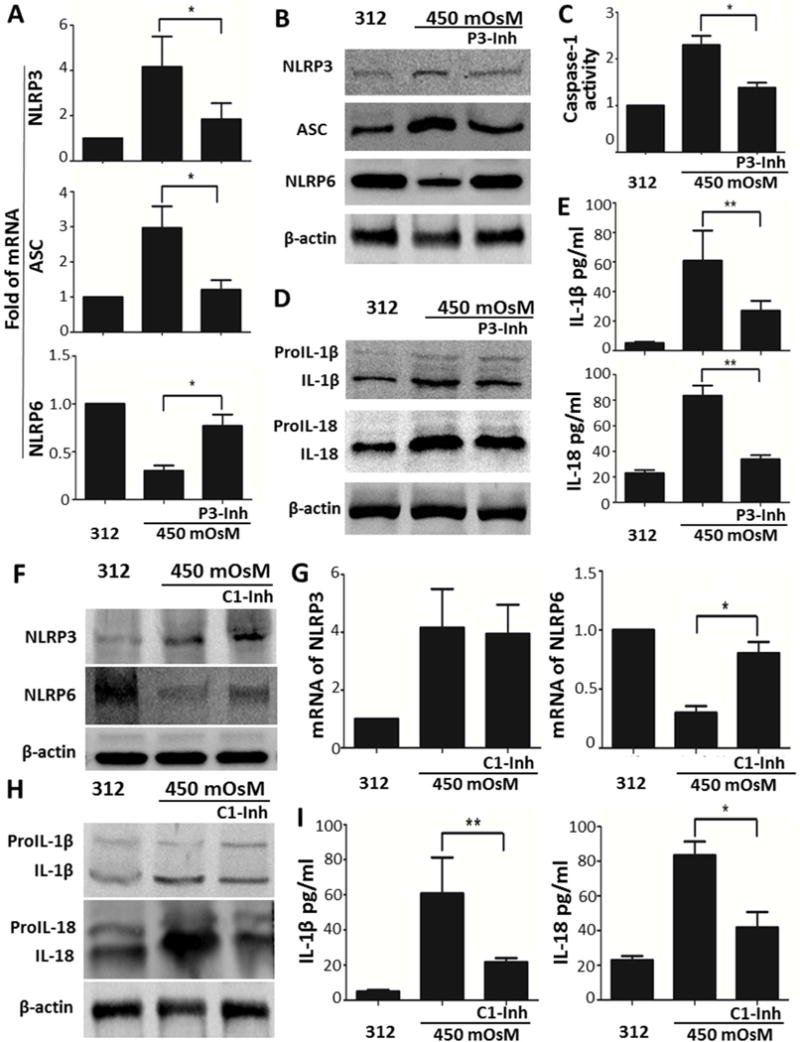
(A and B) mRNA and protein levels of NLRP3, ASC and NLRP6 in HCECs at 450mOsM without or with addition of glybenclamide (30μM), a NLRP3 inhibitor (P3-Inh), compared with isomolar controls at 312 mOsM. (C) Caspase-1 activity assay in HCECs at 450mOsM without or with P3-Inh. (D and E) Cleavage and secretion of proIL-1β and proIL-18 in HCECs at 450mOsM without or with P3-Inh. (F and G) Protein and mRNA levels of NLRP3 and NLRP6 in HCECs at 450mOsM without or with 20μM of caspase-1 inhibitor z-YVAD fmk (C1-Inh). (H and I) Cleavage and secretion of proIL-1β and proIL-18 in HCECs at 450mOsM without or with C1-Inh. Data shown are representative of three or more independent experiments (mean ± SD). *P < 0.05, **P < 0.01, vs. controls.
The roles of caspase-1-dependent NLRP3 inflammasome in processing proIL-1β and proIL-18 maturation and in suppressing NLRP6 production were further investigated by prior addition of 20 μM of caspase-1 inhibitor z-YVAD fmk in HCECs exposed to hyperosmotic environment. Serving as the downstream signaling molecule of NLRP3, caspase-1 inhibitor had no effect on the NLPR3 upregulation and activation, but it significantly restored the NRLP6 production from downregulation induced by hyperosmolarity stress (Fig. 2, F and G). The finding may indicate that caspase-1-dependent inflammasome could suppress the production and activity of NLRP6. As predicted, caspase-1 inhibitor significantly blocked the maturation and secretion of IL-1β and IL-18 stimulated by environmental stress (Fig. 2, H and I), implying that caspase-1-dependent inflammasome exerts the inflammatory effects via activation and secretion of proIL-1β and proIL-18.
3.3. NLRP3-independent caspase-8 signaling uniquely regulates NLRP3/NLRP6 mediated inflammation in HCECs exposed to hyperosmotic environment
Caspase-8 has been recently found to regulate IL-1β expression and maturation via NLR-mediated inflammasome pathway in addition to its traditional role in regulating cell death [38, 39]. In the hyperosmotic culture model, caspase-8 production and activity were significantly upregulated with peak levels induced by 450mOsM (Fig. 3A). However, different from caspase-1, caspase-8 activity appears NLRP3-independent. Pre-incubation of NLRP3 inhibitor glybenclamide or caspase-1 inhibitor z-YVAD fmk did not suppress caspase-8 production and activity, which was only blocked by z-IETD fmk, a caspase-8 inhibitor, as evaluated by caspase-8 activity fluorescent assay (Fig. 3B).
Fig. 3. NLRP3-independent caspase-8 signaling uniquely regulates NLRP3/NLRP6 mediated inflammation in HCECs exposed to hyperosmotic environment.
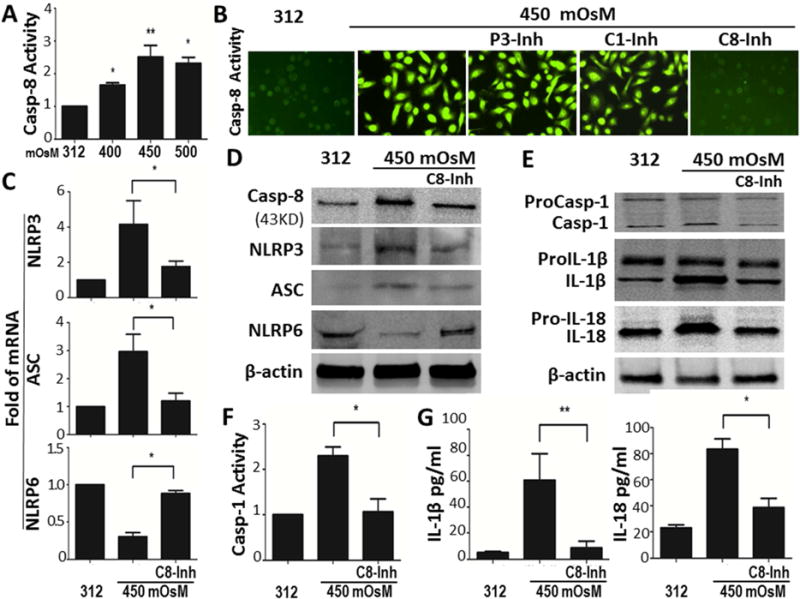
(A) Caspase-8 (Casp-8) activity assay in HCECs with iso- and hyper-osmotic medium. (B) Capase-8 fluorescent activity in HCECs at 450mOsM without or with addition of P3-Inh, C1-Inh or 20μM of caspase-8 inhibitor z-IETD fmk (C8-Inh), compared with isomolar controls at 312 mOsM. (C and D) mRNA and protein levels of NLRP3, ASC and NLRP6 in HCECs at 450mOsM without or with addition of C8-Inh, compared with isomolar controls at 312 mOsM. (E–G) Caspase-1 (Casp-1) activity and processing of IL-1β and IL-18 in HCECs at 450mOsM without or with C8-Inh, compared with isomolar controls at 312 mOsM. Data shown are representative of three or more independent experiments (mean ± SD). *P < 0.05, **P < 0.01, vs. controls.
Interestingly, caspase-8 was observed to promote activation of NLRP3 inflammasome in HCECs exposed to hyperosmotic stress. Prior incubation with 20μM of caspase-8 inhibitor z-IETD fmk largely blocked hyperosmolarity-induced NLRP3/ASC upregulation and NLRP6 downregulation in HCECs (Fig. 3, C and D). The role of caspase-8 in processing proIL-1β and proIL-18 was further revealed by addition of caspase-8 inhibitor in HCECs under hyperosmotic stress. Caspase-8 inhibitor at 20μM completely blocked the caspase-1 activation (Fig. 3F), thus largely prevent maturation and release of IL-1β and IL-18 as measured by ELISA (Fig. 3G). Overall, these results indicate that environmental hyperosmolarity uniquely induces upregulation and activation of caspase-8 in HCECs, which appears to be NLRP3-independent but it stimulates activation of NLRP3 inflammasome, and promotes maturation of proIL-1β and proIL-18, whereas it inhibits NLRP6 production.
Immunofluorescent staining (Fig. 4A) further showed that caspase-8 intensity and positive cells were significantly increased by 450mOsM, while suppressed by caspase-8 inhibitor, but not by inhibitors of NLRP3 and caspase-1. Caspase-8 inhibitor further suppressed NLRP3/ASC inflammasome activation while restored NLRP6 production in HCECs under hyperosmotic stress. Immunofluorescent double staining of NLRP3/ASC and caspase-8/ASC with DAPI nuclear counterstaining vividly validated the formation and activation of caspase-8-mediated NLRP3/ASC inflammasomes in HCECs, which was stimulated by 450mOsM while suppressed by caspase-8 inhibitor (Fig. 4B).
3.4. Oxidized mitochondrial DNA induces the caspase-8-mediated imbalance of NLRP3/NLRP6 activation via BRCC36 deubiquitinating activity
Mitochondrial generated ROS and oxidative stress have been recognized to activate NLRP3 inflammasome [40, 41]. To explore the mechanism by which environmental hyperosmotic stress triggers inflammation in HCECs via NLR-mediated innate immune response, we investigated whether the environmental stress stimulates ROS overproduction and potential oxidative damage to the epithelial cells.
DCFDA assay was used to detect all forms of ROS generated during cell metabolism, including superoxide ion (O2−), hydroxy radicle (HO•) and hydrogen peroxide (H2O2). We observed that hyperosmotic stress stimulated intracellular ROS production. As shown in Fig. 5A, a time-course study (5–120 min) of DCF fluorescence intensities suggests that ROS generation increased in a time-dependent manner in HCECs exposure to 450 mOsM, whereas ROS levels of cells in isomolar condition remain at relatively stable low levels. Pretreatment of antioxidants N-Acetyl-L-cysteine (NAC) or L-carnitine at 10mM significantly suppressed ROS overproduction, indicating hyperosmolarity induces ROS overproduction (Fig. 5A).
Fig. 5. Oxidized mitochondrial DNA induces the caspase-8-mediated imbalance of NLRP3/NLRP6 activation via BRCC36 deubiquitinating activity.
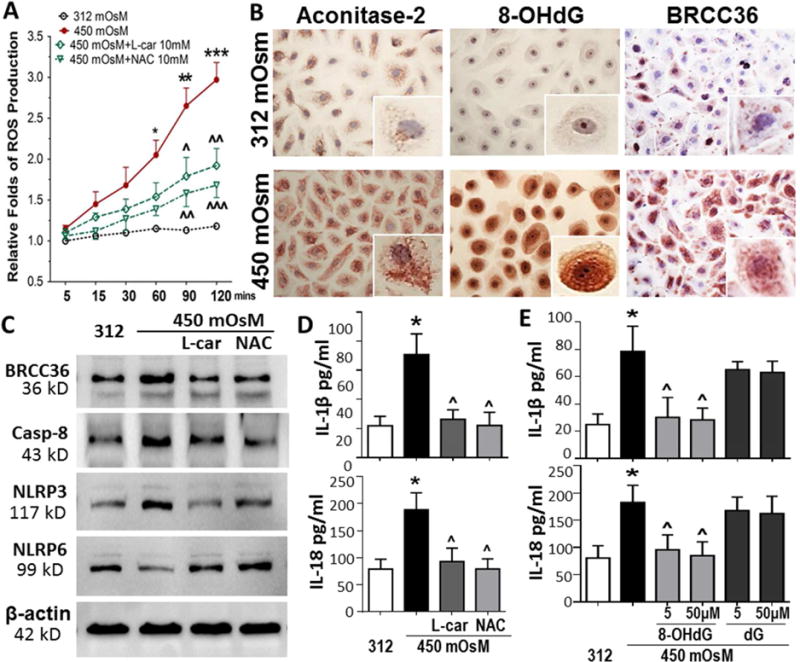
(A) Hyperosomtic stress induced the ROS overproduction in a time-dependent manner in HCECs, which was significantly reduced by antioxidants, 10 mM of N-Acetyl-L-cysteine (NAC) or L-carnitine. (B) Immunohisctochemical staining showed stimulated aconitase-2, 8-OHdG and BRCC36, markers indicating mitochondrial DNA oxidative damage. (C) Western blotting showed that hyperosmolarity-stimulated BRCC36/Caspase-8 and imbalanced NLRP3/NLRP6 were reversed by antioxidants, 10 mM of NAC or L-carnitine. (D) ELISA showed that hyperosmolarity-activated IL-1β and IL-18 were reversed by antioxidants, 10 mM of NAC or L-carnitine. (E) Exogenous 8-OHdG (blocking oxidized mitochondrial DNA) at 5–50 μM significantly suppressed hyperosmolarity-stimulated maturation and secretion of IL-1β and IL-18 with dG as negative control. Data shown are representative of three or more independent experiments (mean ± SD). *P < 0.05, **P < 0.01, ***P < 0.001, vs. controls; ΛP < 0.05, ΛΛP <0.01, ΛΛΛP < 0.001, vs. stimulated levels by 450mOsM.
ROS overproduction causes cell oxidative damage. To examine mtDNA damage, the levels of two major biomarkers, aconitase-2 and 8-OHdG, as well as BRCA1/BRCA2-containing complex subunit 36 (BRCC36), a DNA damage-associated deubiquitinase were evaluated. Aconitase-2, an enzyme participating in the tricarboxylic acid cycle, acts as a biosensor for oxidative stress and mtDNA oxidative damage [42, 43]. 8-OHdG is one of the predominant forms of free radical-induced oxidative lesions in mtDNA [44, 45]. As shown in Fig. 5B, immunohistochemical staining demonstrates that expressions of these two biomarkers increased dramatically in HCECs exposed to hyperosmotic medium for 24 hours when compared their very low levels under normal isomolar condition. The positively stained cells of aconitase-2 and 8-OHdG increased 5.5 to 12.1 fold respectively. The single cell images clearly showed the punctate staining of aconitase 2 and 8-OHdG in cytoplasm, representing a structural pattern of mitochondria, while 8-OHdG staining was homogenous pattern in nuclear. Interestingly, we observed the stimulated production of deubiquitinase BRCC36 in cytoplasm of cells exposed to 450mOsM (Fig. 5B). BRCC36 staining also displayed a punctate mitochondrial pattern in cytoplasm. BRCC36 is known to participate in DNA damage response in the BRCA1-A complex recruited by the chromatin at double-strand break sites, as well as in the cytosolic BRISC complex. Recently BRCC36 is also known as a critical regulator of NLRP3 deubiquitination and inflammasome activation [46, 47]. Thus we hypothesize that mtDNA oxidative damage may stimulate BRCC36 activity to priming and activate NLRP3 inflammasome, an innate immune response to environmental stress via ROS-mediated oxidative injury.
Western blot analysis provides further evidence for the hypothesis. We observed that the hyperosmolarity stimulated caspase-8 activity, which was suppressed by antioxidants, 10mM of L-carnitine or NAC. Interestingly, these antioxidants also significantly inhibited the stimulated production of BRCC36 and NLRP3, but attenuated or restored the suppressed production of NLRP6 in cells exposed to 450 mOsM medium (Fig. 5C). L-carnitine and NAC further suppressed maturation and secretion of IL-1β and IL-18 by the cells in 450mOsM, measured by ELISA (Fig. 5D). Furthermore, the exogenous 8-OHdG or dG was applied into the cultures of stress model to confirm the role of oxidized mtDNA in hyperosmolarity-induced inflammation. We observed that 8-OHdG at 5–50 μM significantly suppressed hyperosmolarity-stimulated maturation and secretion of IL-1β and IL-18 while dG had no effect (Fig. 5E).
3.5. Imbalanced activation of NLRP3/NLRP6 inflammasomes triggers inflammation in ocular surface of experimental mice under desiccating stress
To explore innate immune responses to environmental stress in vivo, a mouse model facing environmental DS was used. These mice developed ocular surface disorders in 5 days after exposure to low humidity and high wind velocity. The eye displayed redness, reduced tear volume and positive corneal fluorescent staining, the manifestations similar to human dry eye disease caused by environmental desiccating stress, which suggests a DS-induced dry eye murine model as previous reported [28].
Interestingly, we observed NLRs-mediated innate immune responses by ocular surface epithelium of DS mice. The immunofluorescent staining clearly showed the increased production of NLRP3 with decreased NLRP6 in corneal and conjunctival epithelia (Fig. 6A). The imbalanced mRNA expression by RT-qPCR (Fig. 6B) and protein productions by Western blotting (Fig. 6C) were also observed in corneal and conjunctival epithelia of DS mice, which displayed the stimulated NLRP3 and ASC while a suppressed NLRP6, compared with untreated control mice. Pro-caspase-1 was significant activated into cleaved form in corneal and conjunctival epithelia of DS mice, evaluated by Western blotting, staining and activity assay (Fig. 6, C–E). Interestingly, the corneal and conjunctival epithelia of DS mice disclosed a significantly higher activity of caspase-8 (Fig. 6, D and E) and BRCC3, an analogous of human BRCC36 (Fig. 6F), than untreated normal mice. As the consequence, IL-1β and IL-18 were significantly activated and secreted by ocular surface epithelium of DS mice as shown by immunofluorescent staining (Fig. 6F). These unique findings in vivo mouse ocular surface under desiccating environment are similar to that in vitro culture model exposed to hyperosmotic stress, which strongly suggest a potential mechanism by which environmental physical challenge causes inflammatory response of ocular surface via oxidative stress-mediated imbalanced activation of NLRP3/NLRP6 inflammasomes, an innate immunity pathway.
Fig. 6. Imbalanced activation of NLRP3/NLRP6 inflammasomes triggers inflammation in ocular surface of experimental mice under desiccating stress.
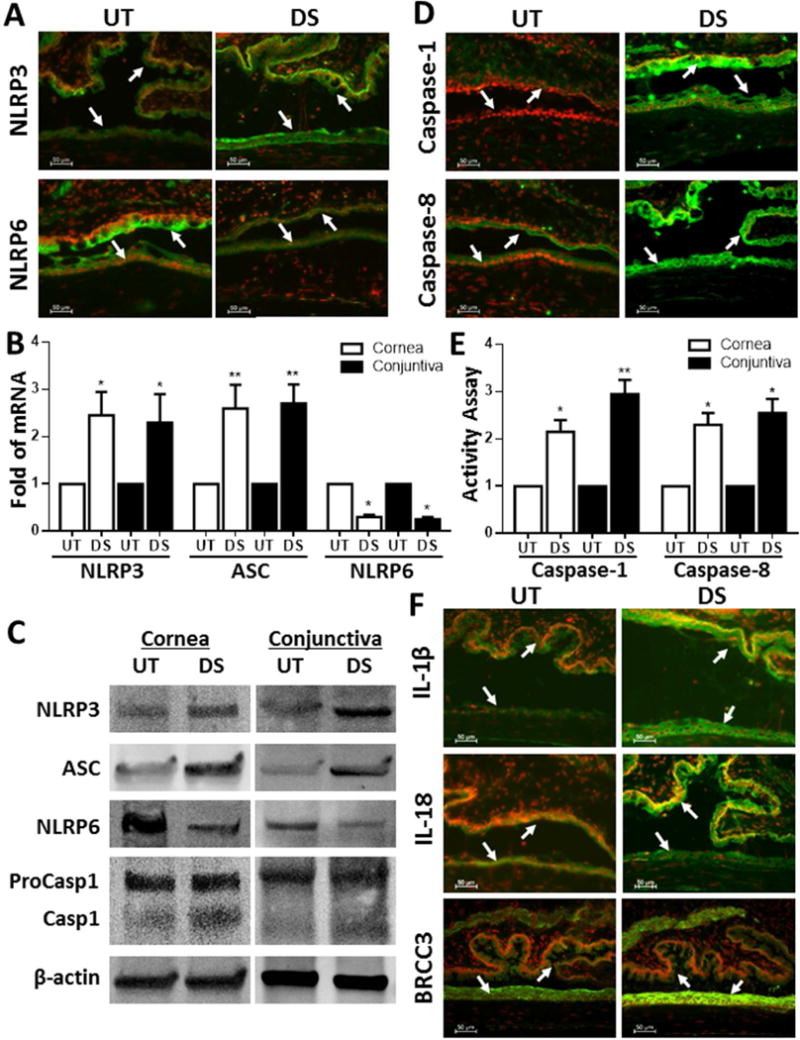
(A and B) Immunofluorescent staining and RT-qPCR showed stimulated immunoreactivity and/or mRNA of NLRP3 and ASC with decreased NLRP6 in corneal and conjunctival epithelia of desiccating (DS) mice with untreated (UT) mice as controls. (C) Western blotting showed the imbalanced activation between NLRP3/ASC and NLRP6 as well as caspase-1 activation in corneal and conjunctival epithelia of DS mice compared with UT mice. (D and E) Activity of caspase-1 and caspase-8 was significantly increased in corneal and conjunctival epithelia of DS mice, evaluated by immunofluorescent staining and activity assay. (F) Immunofluorescent staining showed enhanced production of BRCC3, IL-1β and IL-18 in ocular surface epithelia of DS mice. Data shown are representative of three or more independent experiments (mean ± SD). *P < 0.05, **P < 0.01, vs. controls.
4. Discussion
Ocular surface has great advantages for studying innate immunity because it encounters all types of environmental challenges all the time. Ocular surface faces not only a variety of microorganisms and many non-microbial pathogens including pollen allergen, but also different environmental challenges from changes of physical conditions. To explore whether and how the innate immunity responds to environmental stress by epithelium, we created two ocular surface models mimicking the consequence of physical environmental challenges such as high temperature, low humidity, and rapid wind velocity, which cause tear over-evaporation and hyperosmolarity, as well as desiccating condition. One is primary human corneal epithelial cultures exposed to hyperosmotic medium, and another is a murine model under desiccating stress, mimicking human dry eye disease due to multiple factors from environmental stress [12].
In this study, we uncovered that environmental hyperosmotic stress initiated the innate immune responses of HCECs by a reciprocal regulation of NLRs-mediated inflammasomes, the activated inflammatory NLRP3 and suppressed anti-inflammatory NLRP6, which results in caspase-1 activation to process maturation and secretion of IL-1β and IL-18 from their inactivated proforms, thus causing ocular surface inflammation. Interestingly, we revealed a unique role of stimulated caspase-8, which is NLRP3-independent, but mediated the reciprocal regulation of NLRP3/NLRP6 inflammasomes. We further identified that the ROS overproduction and mtDNA oxidative damage with upregulated deubiquitinase BRCC36, a potential molecular mechanism by which ocular surface epithelium initiates innate immune response to the environmental stress. Finally, our animal model shows interesting data in mouse ocular surface under desiccating stress, that almost validate major findings from in vitro cell culture model.
In previous studies, we have demonstrated that IL-1β is a key proinflammatory cytokine produced by ocular surface epithelium in response to desiccating environment and hyperosmolar stress [23, 25, 26]. However, it is largely unknown whether and how innate immunity plays a role in ocular surface inflammation under environmental stress. NLRs, a member of key PRR family, play a vital role in recognizing PAMP and initiating the innate immune response by forming multi-protein complexes termed as inflammasomes, which consist of one or two NLR proteins, adapter molecule ASC and pro-caspase-1. On inflammasome platforms, pro-caspase-1 is catalyzed into activated caspase-1, which processes proIL-1β and proIL-18 into activated mature form of IL-1β and IL-18 for secretion from cells. NLRP3 inflammasome is the most versatile and important one, which involved in many inflammatory diseases in human [48–51].
In view of the crucial role of inflammasome in initiating innate immune response and IL-1β in desiccating stress-induced inflammation, we hypothesize that environmental challenges may initiate NLRs-mediated inflammasomes to promote inflammation. Our findings support our hypothesis by showing that NLRP3-ASC-caspase-1 inflammsome was significantly activated in both models, in vitro hyperosmolarity cultures and desiccating environment-induced DS mice. Inhibition of NLRP3 inflammasome activation significantly suppressed maturation and secretion of IL-1β and IL-18 in HCECs, indicating the stimulatory effects of NLRP3 inflammasome on the processing of proinflammatory cytokines, IL-1β and IL-18.
Besides classical inflammasomes that promote inflammatory response, certain NLRs-mediated inflammasomes show anti-inflammatory functions. NLRP6 is just such a novel one, serving as a negative regulator of inflammatory signaling. NLRP6 was found to inhibit activation of mitogen-activated protein kinase (MAPK) and canonical NF-κB pathway. Infected NLRP6-deficient mice had increased numbers of monocytes and neutrophils in circulation, and contributed to increased susceptibility. Infected NLRP6-deficient cells produced increased levels of NF-κB- and MAPK-dependent cytokines and chemokines [19, 35]. However, the role of NLRP6 in response to environmental stress, especially by mucosal epithelium, has not been investigated yet. The present study revealed that NLRP6 was significantly downregulated in both the hyperosmolarity culture model and desiccating-challenged animal, and inhibitors of NLRP3 and caspase-1 largely restored NLRP6 production, suggesting that an imbalanced activation of NLRP3/NLRP6 mediates the innate immune responses to environmental stress by ocular epithelium.
Caspase-8, initially discovered as an initiator protease in apoptosis, is now known to have an apparently confounding opposing effect in cell survival and inflammation. The activated NLRP3/ASC or NLRC4/ASC inflammasomes were found to recruit caspase-8 to drive proIL-1β processing in murine dendritic cells and macrophages [39, 52, 53]. Our previous study also found that caspase-8 plays a crucial role in the maturation of IL-1β in retinal ischemic reperfusion injury via innate immune pathways [38]. However, the role of caspase-8 mediated innate response by epithelium is largely unknown. Using environment challenged ocular surface, the present study demonstrated that caspase-8 was significantly activated in corneal and conjunctival epithelial cells in our hyperosmotic cultures and desiccant stressed mice. Interestingly, caspase-8 activation was not NLRP3-dependent because NLRP3 inhibitor did not block the caspase-8 activity. However, blockage of caspase-8 activation by its inhibitor z-IETD fmk also largely suppressed activity of NLRP3 and caspase-1, and resulted in decreased processing of proIL-1β and proIL-18 in HCECs exposed to hyperosmotic stress.
Moreover, inhibition of caspase-8 or NLRP3 activity reversed or restored the expression and production of NLRP6 from downregulated levels in HCECs exposed hyperosmotic medium, suggesting that caspase-8 promotes inflammatory response not only by activating NLRP3 inflammsome, but also via suppressing production and activity of NLRP6 that possesses anti-inflammatory functions. These findings reveal an uniquely noncanonical IL-1β processing via caspase-8 activation, in addition to canonical molecular platforms via caspase-1 activation, demonstrating that caspase-8 activation contributes to the imbalanced regulation of NLRP3/NLRP6-mediated inflammasomes in ocular surface epithelium under environmental stress.
Furthermore, we explored a potential signaling pathway where environmental stress triggeres inflammation with an imbalanced activation of NLRP3/NLRP6 inflammasomes via ROS-induced mtDNA oxidative damage and BRCC36 deubiquitinase activation. ROS and redoxin have been recognized to mediate the NLRP3 activation [40, 54, 55], and BRCC36 deubiquitinating activity has been found to be essential in NLRP3 priming and activation [47]. However, direct link between environmental stress and mucosal epithelium in innate immune responses is largely unknown. The present study for the first time demonstrates that ROS-oxidized mtDNA, evidenced by increase immuno-reactivity of aconitase-2 and OHdG, induces reciprocal regulation of NLRP3/NLRP6 inflammasomes by BRCC36 activation in ocular epithelium exposed to environmental stress. Anti-oxidants L-carnitine and NAC significantly suppress ROS overproduction and BRCC36 production, block caspase-8 activation, and largely reverse or restore the imbalance of activated NLRP3 and suppressed NLRP6. The process and secretion of mature IL-1β and IL-18 were also suppressed by L-carnitine and NAC.
8-OHdG was then used as a competitive agent to block the oxidized mitochondrial 8-OHdG DNA in this culture model of hyperosmotic stress, with dG as a negative control. Interestingly, 8-OHdG, but not dG, competitively inhibited the maturation and secretion of IL-1β and IL-18. The findings provide strong evidence that oxidized mtDNA may be a direct signaling pathway through which environmental stress initiates NLR-mediated innate immune responses that lead to ocular surface inflammation.
5. Conclusion
This study for the first time uncovers a novel innate immunity pathway by mucosal epithelium, where the oxidized mtDNA induces the imbalanced activation of NLRP3/NLRP6 inflammasomes via stimulation of caspase-8 and BRCC36 in response to environmental stress in dry eye using our culture and animal models. In addition to traditional NLRP3 activation, the suppressed production of anti-inflammatory NLRP6 contributes to inflammatory response. NLRP3-independent caspase-8 activation has been identified as a unique pro-inflammatory signaling in addition to canonical way for caspase-1 activation. Oxidized mtDNA and BRCC36 deubiquitinase have been revealed to be a direct link and mechanism by which the innate immune responses are initiated. Our findings provide new insight into the innate immune responses of ocular epithelium to environmental stress, and suggest potential therapeutic targets in preventing and treating eyes from environmental stress.
Supplementary Material
Highlights.
Using two experimental dry eye models, primary human corneal epithelial cultures exposed to hyperosmolarity and mouse ocular surface facing desiccating stress, we uncovered a novel innate immunity pathway by ocular surface epithelium, where oxidized mitochondrial DNA induces imbalanced activation of NLRP3/NLRP6 inflammasomes leading to caspase-1 activation and IL-1β/IL-18 secretion via NLRP3-independent caspase-8 and BRCC36 deubiquitinating activities in response to environmental stress and created a new intervention strategy to treat dry eye disease.
Acknowledgments
We thank Dr. Marshall Bowes Hamill for his kind support and the Lions Eye Bank of Texas for providing human corneoscleral tissues.
This work was supported by National Institutes of Health, National Eye Institute grants R01 EY023598 (DQL), EY011915 (SCP) and Core Grant for Vision Research EY002520, Natural Science Foundations of China 81570830, an unrestricted grant from Research to Prevent Blindness, the Oshman Foundation and the William Stamps Farish Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
D. L., Y. L., and W.C. designed research; W.C., X.H., X.C., F.B., X. Y., L.Z., X.W., D.C., and R.D. performed research; C.D. and S.P. contributed new reagents/analytic tools; W.C., X.H., D. L., and Y. L. analyzed data; W.C. and D. L. wrote the paper. All authors contributed to the revision of the final manuscript.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
- 1.Bauer S, Muller T, Hamm S. Pattern recognition by Toll-like receptors. Adv Exp Med Biol. 2009;653:15–34. doi: 10.1007/978-1-4419-0901-5_2. [DOI] [PubMed] [Google Scholar]
- 2.Fukata M, Vamadevan AS, Abreu MT. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders. Semin Immunol. 2009;21:242–253. doi: 10.1016/j.smim.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: at the interface of innate and adaptive immune responses. J Allergy Clin Immunol. 2007;120:1279–1284. doi: 10.1016/j.jaci.2007.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bulek K, Swaidani S, Aronica M, Li X. Epithelium: the interplay between innate and Th2 immunity. Immunol Cell Biol. 2010 doi: 10.1038/icb.2009.113. [DOI] [PubMed] [Google Scholar]
- 5.Cario E. Heads up! How the intestinal epithelium safeguards mucosal barrier immunity through the inflammasome and beyond. Curr Opin Gastroenterol. 2010;26:583–590. doi: 10.1097/MOG.0b013e32833d4b88. [DOI] [PubMed] [Google Scholar]
- 6.Fritz JH, Le BL, Magalhaes JG, Philpott DJ. Innate immune recognition at the epithelial barrier drives adaptive immunity: APCs take the back seat. Trends Immunol. 2008;29:41–49. doi: 10.1016/j.it.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Kumar S, Dwivedi PD, Das M, Tripathi A. Macrophages in food allergy: An enigma. Mol Immunol. 2013;56:612–618. doi: 10.1016/j.molimm.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Yannopoulos DI, Roncin S, Lamour A, Pennec YL, Moutsopoulos HM, Youinou P. Conjunctival epithelial cells from patients with Sjogren’s syndrome inappropriately express major histocompatibility complex molecules, La(SSB) antigen, and heat-shock proteins. J Clin Immunol. 1992;12:259–265. doi: 10.1007/BF00918149. [DOI] [PubMed] [Google Scholar]
- 9.Moutsopoulos HM. Sjogren’s syndrome: autoimmune epithelitis. Clin Immunol Immunopathol. 1994;72:162–165. doi: 10.1006/clin.1994.1123. [DOI] [PubMed] [Google Scholar]
- 10.Kapsogeorgou EK, Moutsopoulos HM, Manoussakis MN. Functional expression of a costimulatory B7.2 (CD86) protein on human salivary gland epithelial cells that interacts with the CD28 receptor, but has reduced binding to CTLA4. J Immunol. 2001;166:3107–3113. doi: 10.4049/jimmunol.166.5.3107. [DOI] [PubMed] [Google Scholar]
- 11.Li DQ, Zhang L, Pflugfelder SC, De Paiva CS, Zhang X, Zhao G, et al. Short ragweed pollen triggers allergic inflammation through Toll-like receptor 4-dependent thymic stromal lymphopoietin/OX40 ligand/OX40 signaling pathways. J Allergy Clin Immunol. 2011;128:1318–1325 e1312. doi: 10.1016/j.jaci.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The definition and classification of dry eye disease: report of the Definition and Classification Subcommittee of the International Dry Eye WorkShop (2007) Ocul Surf. 2007;5:75–92. doi: 10.1016/s1542-0124(12)70081-2. [DOI] [PubMed] [Google Scholar]
- 13.Pflugfelder SC, Stern ME. Mucosal environmental sensors in the pathogenesis of dry eye. Expert Rev Clin Immunol. 2014;10:1137–1140. doi: 10.1586/1744666X.2014.944163. [DOI] [PubMed] [Google Scholar]
- 14.Alex A, Edwards A, Hays JD, Kerkstra M, Shih A, de Paiva CS, et al. Factors Predicting the Ocular Surface Response to Desiccating Environmental Stress. Invest Ophthalmol Vis Sci. 2013 doi: 10.1167/iovs.12-11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsubota K, Nakamori K. Dry eyes and video display terminals. N Eng J Med. 1993;328:584–584. doi: 10.1056/NEJM199302253280817. [DOI] [PubMed] [Google Scholar]
- 16.Begley CG, Chalmers RL, Abetz L, Venkataraman K, Mertzanis P, Caffery BA, et al. The relationship between habitual patient-reported symptoms and clinical signs among patients with dry eye of varying severity. Invest Ophthalmol Vis Sci. 2003;44:4753–4761. doi: 10.1167/iovs.03-0270. [DOI] [PubMed] [Google Scholar]
- 17.Niederkorn JY, Stern ME, Pflugfelder SC, de Paiva CS, Corrales RM, Gao J, et al. Desiccating stress induces T cell-mediated Sjogren’s Syndrome-like lacrimal keratoconjunctivitis. J Immunol. 2006;176:3950–3957. doi: 10.4049/jimmunol.176.7.3950. [DOI] [PubMed] [Google Scholar]
- 18.Kersse K, Bertrand MJ, Lamkanfi M, Vandenabeele P. NOD-like receptors and the innate immune system: coping with danger, damage and death. Cytokine Growth Factor Rev. 2011;22:257–276. doi: 10.1016/j.cytogfr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Lupfer C, Kanneganti TD. Unsolved Mysteries in NLR Biology. Front Immunol. 2013;4:285. doi: 10.3389/fimmu.2013.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 21.Guarda G, So A. Regulation of inflammasome activity. Immunology. 2010;130:329–336. doi: 10.1111/j.1365-2567.2010.03283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol. 2012;13:333–332. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solomon A, Dursun D, Liu Z, Xie Y, Macri A, Pflugfelder SC. Pro- and anti-inflammatory forms of interleukin-1 in the tear fluid and conjunctiva of patients with dry-eye disease, Invest Ophthalmol. Vis Sci. 2001;42:2283–2292. [PubMed] [Google Scholar]
- 24.Li DQ, Luo L, Chen Z, Kim HS, Song XJ, Pflugfelder SC. JNK and ERK MAP kinases mediate induction of IL-1beta, TNF-alpha and IL-8 following hyperosmolar stress in human limbal epithelial cells. Exp Eye Res. 2006;82:588–596. doi: 10.1016/j.exer.2005.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo L, Li DQ, Corrales RM, Pflugfelder SC. Hyperosmolar saline is a proinflammatory stress on the mouse ocular surface. Eye Contact Lens. 2005;31:186–193. doi: 10.1097/01.icl.0000162759.79740.46. [DOI] [PubMed] [Google Scholar]
- 26.Narayanan S, Corrales RM, Farley W, McDermott AM, Pflugfelder SC. Interleukin-1 receptor-1-deficient mice show attenuated production of ocular surface inflammatory cytokines in experimental dry eye. Cornea. 2008;27:811–817. doi: 10.1097/ICO.0b013e31816bf46c. [DOI] [PubMed] [Google Scholar]
- 27.Kim HS, S Jun X, de Paiva CS, Chen Z, Pflugfelder SC, Li D-Q. Phenotypic characterization of human corneal epithelial cells expanded ex vivo from limbal explant and single cell cultures. Exp Eye Res. 2004;79:41–49. doi: 10.1016/j.exer.2004.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dursun D, Wang M, Monroy D, Li DQ, Lokeshwar BL, Stern M, et al. Experimentally induced dry eye produces ocular surface inflammation and epithelial disease. Adv Exp Med Biol. 2002;506:647–655. doi: 10.1007/978-1-4615-0717-8_91. [DOI] [PubMed] [Google Scholar]
- 29.Pflugfelder SC, Farley W, Luo L, Chen LZ, de Paiva CS, Olmos LC, et al. Matrix metalloproteinase-9 knockout confers resistance to corneal epithelial barrier disruption in experimental dry eye. Am J Pathol. 2005;166:61–71. doi: 10.1016/S0002-9440(10)62232-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li DQ, Zhou N, Zhang L, Ma P, Pflugfelder SC. Suppressive effects of azithromycin on zymosan-induced production of proinflammatory mediators by human corneal epithelial cells, Invest Ophthalmol. Vis Sci. 2010;51:5623–5629. doi: 10.1167/iovs.09-4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hua X, Yuan X, Li Z, Coursey TG, Pflugfelder SC, Li DQ. A Novel Innate Response of Human Corneal Epithelium to Heat-killed Candida albicans by Producing Peptidoglycan Recognition Proteins. PLoS One. 2015;10:e0128039. doi: 10.1371/journal.pone.0128039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li DQ, Tseng SC. Three patterns of cytokine expression potentially involved in epithelial-fibroblast interactions of human ocular surface. J Cell Physiol. 1995;163:61–79. doi: 10.1002/jcp.1041630108. [DOI] [PubMed] [Google Scholar]
- 33.Chen Z, de Paiva CS, Luo L, Kretzer FL, Pflugfelder SC, Li DQ. Characterization of putative stem cell phenotype in human limbal epithelia. Stem Cells. 2004;22:355–366. doi: 10.1634/stemcells.22-3-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li DQ, Luo L, Chen Z, Kim HS, Song XJ, Pflugfelder SC. JNK and ERK MAP kinases mediate induction of IL-1beta, TNF-alpha and IL-8 following hyperosmolar stress in human limbal epithelial cells. Exp Eye Res. 2006;82:588–596. doi: 10.1016/j.exer.2005.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature. 2012;488:389–393. doi: 10.1038/nature11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu HB, Finlay BB. The caspase-1 inflammasome: a pilot of innate immune responses, Cell Host Microbe 4. 2008:198–208. doi: 10.1016/j.chom.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci. 2014;1319:82–95. doi: 10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chi W, Li F, Chen H, Wang Y, Zhu Y, Yang X, et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc Natl Acad Sci U S A. 2014;111:11181–11186. doi: 10.1073/pnas.1402819111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, Kaiser WJ, et al. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J Biol Chem. 2015;290:20167–20184. doi: 10.1074/jbc.M115.652321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubartelli A. Redox control of NLRP3 inflammasome activation in health and disease. J Leukoc Biol. 2012;92:951–958. doi: 10.1189/jlb.0512265. [DOI] [PubMed] [Google Scholar]
- 41.Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40:616–619. doi: 10.1002/eji.200940168. [DOI] [PubMed] [Google Scholar]
- 42.Kim SJ, Cheresh P, Williams D, Cheng Y, Ridge K, Schumacker PT, et al. Mitochondria-targeted Ogg1 and aconitase-2 prevent oxidant-induced mitochondrial DNA damage in alveolar epithelial cells. J Biol Chem. 2014;289:6165–6176. doi: 10.1074/jbc.M113.515130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen XJ, Wang X, Kaufman BA, Butow RA. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science. 2005;307:714–717. doi: 10.1126/science.1106391. [DOI] [PubMed] [Google Scholar]
- 44.Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009;27:120–139. doi: 10.1080/10590500902885684. [DOI] [PubMed] [Google Scholar]
- 45.Pilger A, Rudiger HW. 8-Hydroxy-2′-deoxyguanosine as a marker of oxidative DNA damage related to occupational and environmental exposures. Int Arch Occup Environ Health. 2006;80:1–15. doi: 10.1007/s00420-006-0106-7. [DOI] [PubMed] [Google Scholar]
- 46.Ng HM, Wei L, Lan L, Huen MS. The K63-Deubiquitylating Enzyme BRCC36 Limits DNA Break Processing and Repair. J Biol Chem. 2016;291:16197–16207. doi: 10.1074/jbc.M116.731927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49:331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 48.Cassel SL, Sutterwala FS. Sterile inflammatory responses mediated by the NLRP3 inflammasome. Eur J Immunol. 2010;40:607–611. doi: 10.1002/eji.200940207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease, Trends. Immunol. 2011;32:373–379. doi: 10.1016/j.it.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol. 2011;166:1–15. doi: 10.1111/j.1365-2249.2011.04440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salvesen GS, Walsh CM. Functions of caspase 8: the identified and the mysterious. Semin Immunol. 2014;26:246–252. doi: 10.1016/j.smim.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao Y, Sui X, Ren H. From procaspase-8 to caspase-8: revisiting structural functions of caspase-8. J Cell Physiol. 2010;225:316–320. doi: 10.1002/jcp.22276. [DOI] [PubMed] [Google Scholar]
- 54.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 55.Zheng Q, Ren Y, Reinach PS, She Y, Xiao B, Hua S, et al. Reactive oxygen species activated NLRP3 inflammasomes prime environment-induced murine dry eye. Exp Eye Res. 2014;125:1–8. doi: 10.1016/j.exer.2014.05.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.