

Abstract
Advances in public health in the last century have seen a sharp increase in human life expectancy. With these changes have come increased incidence of age-related pathologies and health burdens in the elderly. Patient age is the biggest risk factor for multiple chronic conditions that often occur simultaneously within one individual. An alternative to disease centric therapeutic approaches is that of ‘geroscience’, which aims to define molecular mechanisms that link age to overall disease risk. One such mechanism is deregulation of CREB-regulated transcriptional coactivators, CRTCs. Initially identified for their role in modulating CREB transcription, the last five years has seen an expansion in knowledge of new cellular regulators and roles of CRTCs beyond CREB. CRTCs have been shown to modulate organismal aging in C. elegans and to impact age-related diseases in humans. Here, we discuss CRTC deregulation as a new driver of aging, and integrating link between age and disease risk.
Keywords: AGING, CREB-REGULATED TRANSCRIPTIONAL COACTIVATORS, METABOLIC DISEASE, CANCER, NEURODEGENERATION
Public Health and Medical advances in the last century have yielded significant increases in human life expectancy. In 1955, worldwide average life expectancy was 46, yet, by 2015 this number had risen by nearly 20 years to 65 (United Nations World Population Prospects 2015). As developing countries continue to get access to better health care this trend is set to continue, such that by 2100, at least half of the human population worldwide can expect to live to 83 years of age (United Nations World Population Prospects 2015, Figure 1). The resulting shift in our population demographics means that by 2050, there will be 1.5 billion people over the age of 65 (United Nations World Population Prospects 2015, Figure 1). Although this change is something to be lauded, our success in increasing our survival has come at a cost, as for the majority of us, these added years are not healthy. Age is the biggest risk factor for the majority of chronic diseases, and worse still, many of these conditions often occur simultaneously in the same individual - in the US over half of people older than 65 have two or more chronic conditions. This pandemic of co-morbidities lessens the impact disease-centric medical approaches have on years spent disease free; cure one disease completely and the patient is still left with the remaining ailments. An emerging alternative approach is that of ‘geroscience’ [1]. Rather than focussing on proximal mechanisms of individual age-related diseases in isolation, geroscience focuses on the commonality between all of them, namely aging itself, and cellular components that might link patient age to overall disease risk.
Figure 1. The downsides of increased life expectancy: an aged population suffering from multiple chronic diseases and increased economic burden.
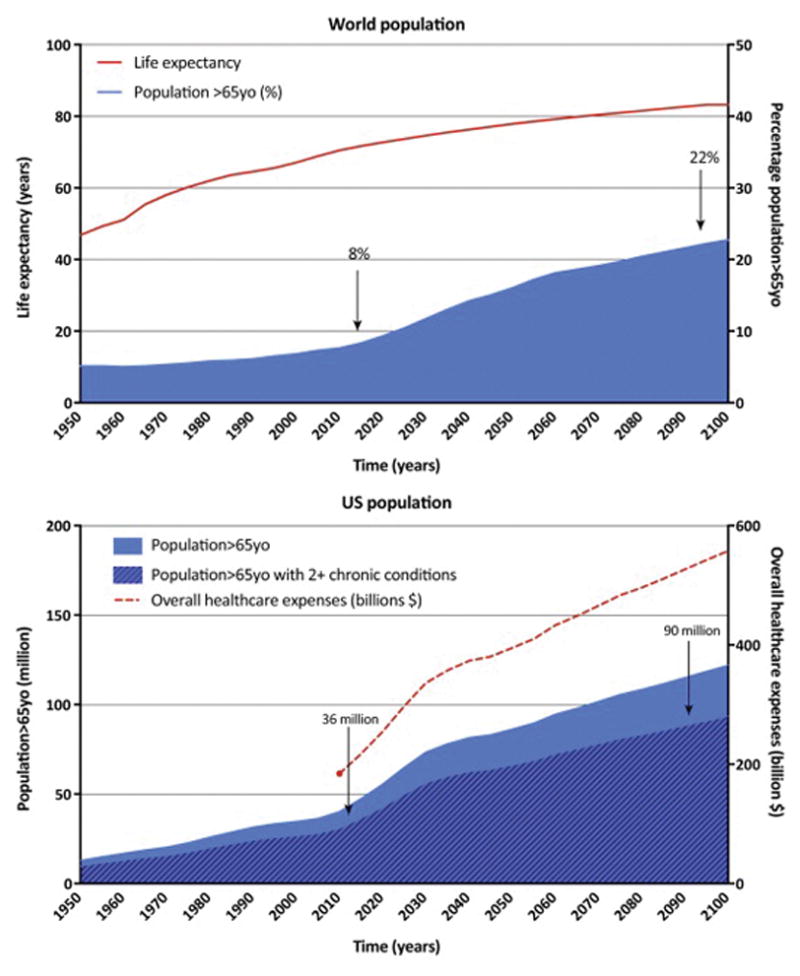
A) Increase in lifespan expectancy in the world from 1950 to 2015 and projected increase from 2015 to 2050. This increase is followed by an increase in the percentage of population aged >65, representing 8% and is predicted to represent up to 22% of the world population by 2050. B) Proportional increase in people >65 year old suffering from 2 or more chronic diseases as population ages. This comorbidity will cause a subsequent increase in the overall health care expenses (data extrapolated from expenses measured in 2015). (Sources: United Nations World Population Prospects 2015, AHRQ Medical Expenditure Panel Survey 2005, Center for Medicare and Medicaid Services 2012 Edition)
The last two decades have seen the discovery of genetic, environmental and pharmacological perturbations that slow ageing and decrease age-related pathology of model organisms. Although many of these interventions seem to function through non-overlapping mechanisms, one commonality is that lifespan extension is often coupled to reductions in energy requiring processes and the rate at which energy stores are depleted. Decreasing food intake during dietary restriction (DR) increases longevity in species ranging from yeast to primates [2], and has protective effects against multiple age-onset conditions. Conserved mechanisms that integrate upstream energy sensing pathways with targeted downstream transcriptional targets might therefore be ideal targets to specifically recapitulate the physiological benefits of DR for therapeutic purposes.
Recently, work in the nematode worm C. elegans has highlighted a family of newly discovered co-factors, cAMP response element binding protein (CREB)-regulated transcriptional co-activators (CRTCs), as novel modulators of aging that link energy sensing to transcriptional regulation of longevity [3,4]. Although the role of CRTCs in longevity to date has only been shown in C. elegans, the last five years have seen the discovery of novel cellular roles for CRTCs and links between CRTC dysfunction and multiple human age-related pathologies. Discovery and canonical regulation of CRTCs has been outlined extensively previously [5]. Here we will briefly outline new cellular roles (outputs) for CRTCs along with known CRTC regulators (inputs), before focusing on mechanistic links between CRTC regulation/function and multiple age-onset pathologies and finally, whether CRTCs might be targeted to promote healthy aging in mammals
Outputs Beyond CREB: Emerging Roles for CRTCs
Initially named transducers of regulated CREB (TORCs) or mucoepidermoid carcinoma translocated protein (MECTs), CRTC family members were first identified as co-activators of the transcription factor CREB, due to their ability to induce CREB target gene expression in the absence of a cAMP stimulus [6–9]. However, recent work has identified expanding roles for CRTCs outside the confines of their name, showing CRTCs act both as regulators of transcription factors beyond CREB, and of processes other than transcription. Three isoforms of CRTC (CRTC1-3) have been described in mammals, with CRTC2 and 3 showing 32% homology with CRTC1 [7]. In humans, CRTC2 and CRTC3 are ubiquitously expressed, whereas CRTC1 is mainly expressed in the brain, with little expression in other tissues [6,10–12]. CRTC2 and CRTC3 are both expressed in the immune tissue, in B and T lymphocytes [6] as well as in lungs [12]. CRTC2 is highly expressed in the liver [13] and muscles [12], whereas CRTC3 is predominantly found in white adipose tissue (WAT) and brown adipose tissue (BAT) [14]. CRTCs do not directly bind DNA, but rather act as cofactors for bZIP transcription factors. CRTCs contain a highly conserved N-terminal coil-coil domain required for CREB binding (amino acids 1–56) [6], an invariant sequence matching protein kinase A (PKA) phosphorylation consensus sequence (RKXS), a serine/glutamine rich domain and negatively charged C-terminus domain sufficient for transcriptional activation (last 200 amino acids) [7] (Figure 2). The N-terminal CREB-binding domain of CRTCs binds to the bZIP domain of CREB [15] and facilitates recruitment of the transcriptional apparatus [5–7]. However, recently the role of CRTCs as critical transcriptional coactivators has expanded beyond CREB, with CRTCs both directly binding and co-activating additional bZIP transcription factors, including AP-1 and activating transcription factor (ATF) 6 [16,17], and competing with ATF3 and farnesoid X receptor (FXR) for occupancy over promoters [18,19].
Figure 2. Structure and post translational modifications of CRTC2 protein.
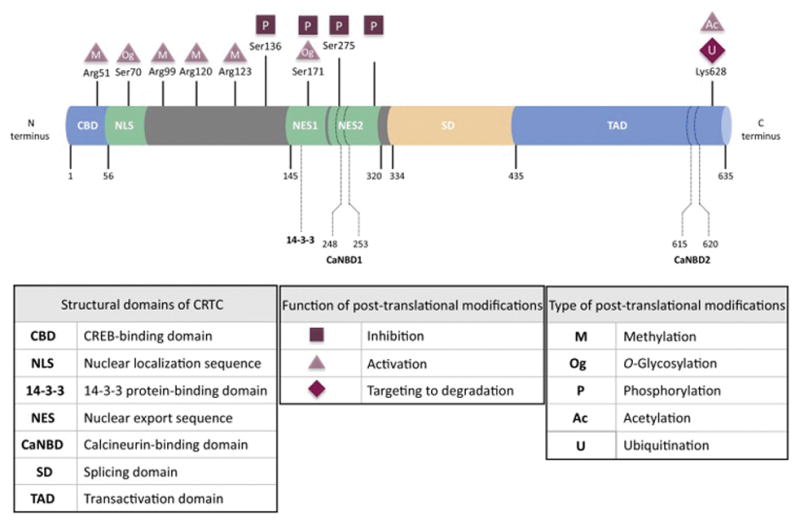
Representation of CRTC2 structural domains location and function as well as the main post translational modifications sites, type and effect on the protein activity.
In addition to expanding roles in transcriptional regulation, CRTCs have now been shown to have functions beyond transcription and the nucleus. Both ER>Golgi trafficking regulation and RNA splicing have emerged as new cellular roles of CRTCs (Box 2) [20,21]. CRTC2 contains a proline-rich region at residues 334–435, which display similarities to regions found in splicing factors (Figure 2). This domain is required for CRTC-mediated alternative splicing of transcripts with CRE containing promoters [20]. However, CRTCs do not contain known definable RNA-binding domains, such as an RNA recognition motif, arginine-serine domain or a K homology domain [20]. Therefore more work is needed to define how CRTCs modulate RNA splicing and affect spliceosome function, along with whether CRTCs impact known roles for RNA splicing in DR longevity [22]. In addition to splicing, CRTCs play a key role in Coat protein complex II (COPII)-mediated vesicle trafficking from the ER to the golgi, via direct binding and competitive inhibition of Sec31. The mTORC1 pathway, a critical regulator of cellular growth and organismal longevity, promotes lipogenesis via directly phosphorylating CRTC2 and de-repressing Sec31, thereby promoting cleavage and activation of the master lipogenesis regulator SREBP1 [21]. These new roles for CRTCs expand their function to new biological processes beyond transcriptional regulation of gluconeogenesis. Coupled with the effect of CRTCs on aging in C. elegans, CRTCs may therefore represent new cellular links between aging, metabolic homeostasis and disease risk.
Box 2. CRTCs and RNA Splicing.
Through a screen for CRTC interacting partners in mammalian cells, NONO (p54nrb), a protein implicated in transcription and RNA processing, has been demonstrated to interact with CRTC2 upon cAMP signaling to regulate the tethering of the CREB/CRTC complex with RNA polymerase II on cAMP responsive promoters [110]. Given the role of NONO in pre-mRNA splicing, it was further discovered that CRTCs affect pre-mRNA splice site selection. These distinct effects of CRTC on alternative splicing and transcription are independent of each other, yet both require functional CRE containing promoters. The recruitment of CRTCs to the promoter by CREB is required to promote alternative splicing of transcripts, in a cell type specific manner. However, this does not affect transcriptional activity of the target, suggesting mechanistically separable functions of the CRTCs. These two activities are mediated by two distinct domains on CRTC, a trans-activation domain (residues 435–635) and a proline rich domain (residues 334–434), which when mutated abrogate CRTCs’ effects on alternative splicing (Figure 2). Although CRTCs lack any conserved canonical RNA binding domain, they might serve as a scaffold for other proteins to form a complex in order to bind and process pre-mRNA transcripts [20]. Taken together, these observations demonstrate the ability of CRTC to couple extracellular signals to gene expression through modulation of both transcriptional activity and alternative splicing.
Inputs: Post-translational Regulators of CRTCs and Aging
CRTCs are extensively post-translationally regulated, with many CRTC regulators having known roles in the modulation of aging (Table 1). CRTCs are negatively regulated through phosphorylation by AMP-activated protein kinase (AMPK) family kinases, including salt-inducible kinase 1, 2 and 3 (SIK1/2/3) [13,23,24], AMPK [9,13] and microtubule affinity regulating kinase 2 (MARK2) [25]. Phosphorylation by these kinases facilitates 14-3-3 protein binding and retention of CRTCs in the cytoplasm (Figure 3). Conversely, CRTCs are activated via dephosphorylation by the serine/threonine phosphatase calcineurin, and putatively the suppressor of MEK null 1 and 2 (SMEK1/2), both of which modulate aging in C. elegans [3,26]. Dephosphorylation of CRTCs induces nuclear translocation and consequent activation of CREB targets (Figure 3) [8,27]. How CRTCs shuttle in out of the nucleus has not been extensively studied. However, XPO1 (also known as exportin1 or CRM1) has been shown to actively export CRTCs from the nucleus. XPO1 is a member of the importin beta superfamily of nuclear transport receptors which recognizes leucine-rich nuclear export signal sequences and its inhibition induces the nuclear sequestering of CRTCs [8]. AMPK and calcineurin have antagonistic effects on longevity in C. elegans and inhibition of the sole worm CRTC family member, ‘CRTC-1’ is critical for these effects. AMPK is a key energy sensor in all eukaryotes and a mediator of DR longevity [28]. Expression of a constitutively active AMPK increases lifespan in C. elegans, while also promoting phosphorylation and inhibition of CRTC-1. Conversely, inhibition of calcineurin increases lifespan in the worm, and reduces CRTC-1 dephosphorylation. Blocking CRTC-1 phosphorylation at two conserved serine residues renders it constitutively nuclear and active, and suppresses both AMPK- and calcineurin-mediated longevity. Supporting a role for CRTCs inhibition in aging, inactivation of CRTC-1 via RNAi is sufficient to extend lifespan in a CREB dependent manner [3].
Table 1.
Post translational modifications of CRTC2.
| CRTC2’s post translational modifications | |||
|---|---|---|---|
| Enzyme | Type of modification | Site | References |
| Activators of CRTC2 | |||
| Calcineurin | Dephosphorylation | Ser275 | [25] |
| SMEK1/2 | Dephosphorylation | Ser171 | [14] |
| OGT | O-glycosylation | Ser70, Ser171 | [32] |
| p300 | Acetylation | Lys628 | [29] |
| CBP | Acetylation | Lys628 | [29] |
| PRMT6 | Methylation | Arg51, Arg99, Arg120, Arg123 | [33] |
| Inhibitors of CRTC2 | |||
| AMPK | Phosphorylation | Ser171, Ser275 | [89] |
| SIK1 | Phosphorylation | Ser307 | [24] |
| SIK2/3 | Phosphorylation | Ser171, Ser275 | [23,89] |
| MARK2 | Phosphorylation | Ser275 | [25,89] |
| mTOR | Phosphorylation | Ser136 | [21] |
| SIRT1 | Deacetylation | Lys628 | [29] |
| COP1 | Ubiquitination | Lys628 | [29,31] |
Post translational modifications of CRTC2, representative of the other CRTC isoforms. Table lists the enzymes responsible for modification as well as the type, amino acid location and effect (activating/inhibiting).
Figure 3. Regulation of CRTC2 nuclear localization.
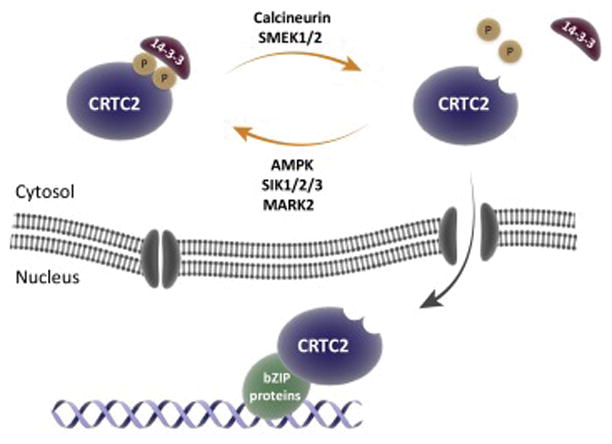
CRTC2 can be phosphorylated by multiple LKB1 regulated kinases (AMPK, SIK1/2/3, MARK2). The phosphorylated form of CRTC2 is bound by 14-3-3 proteins and sequestered in the cytoplasm where is it inactive. Dephosphorylation of CRTC1 by phosphatases such as calcineurin releases 14-3-3 binding and induces CRTC2 nuclear translation. In the nucleus, CRTC2 binds to bZIP transcription factors to promote transcription.
Besides the classical phosphorylation sites in CRTC2 (Ser171 and Ser275) [9,25], growing evidence points to new residues and domains that regulate the activity of this protein and may be linked to aging (Figure 2). Gluconeogenesis during fasting is activated by glucagon stimulation of CRTC2 association with CREB binding protein (CBP)/p300 [29]. After prolonged fasting, CRTC2 is downregulated via the nutrient-sensing deacetylase sirtuin 1 (SIRT1), which is a critical modulator of longevity in model systems from yeast to mice [30]. In liver, SIRT1 deacetylates CRTC2 at Lys628, allowing its ubiquitination by the E3 ligase constitutive photomorphogenic protein (COP1), and targeting CRTC2 for proteasome degradation [29,31]. However, although activating SIRT1 is known to promote longevity, the role of CRTC-1 inhibition in SIRT1 mediated longevity remains untested.
In addition to phosphorylation, acetylation and ubiquitination, CRTCs are also regulated via O-glycosylation. O-glycosyl transferase (OGT) O-glycosylates serines 70 and 171 in CRTC2, blocking phosphorylation and enriching nuclear CRTC2 to drive hepatic gluconeogenesis [32]. In addition, arginine methylation of CRTC2 plays an important role in the transcriptional control of hepatic glucose metabolism. Indeed, the protein arginine methyltransferase 6 (PRMT6) can promote fasting-induced transcriptional activation of the gluconeogenic program through CRTC2 [33]. As yet, little is known about how changes to these newly identified post-translational modifications impacts CRTC function with age. However, as newly discovered inputs and outputs of CRTCs have been identified, their ties to multiple age-related pathologies have increased (Figure 4). Below we will focus on how deregulation of CRTCs inputs and outputs are causally associated with three of the greatest public health burdens in old age - namely metabolic disease, cancer and neurodegeneration.
Figure 4. CRTCs dysregulation is linked to disease in multiple organs.
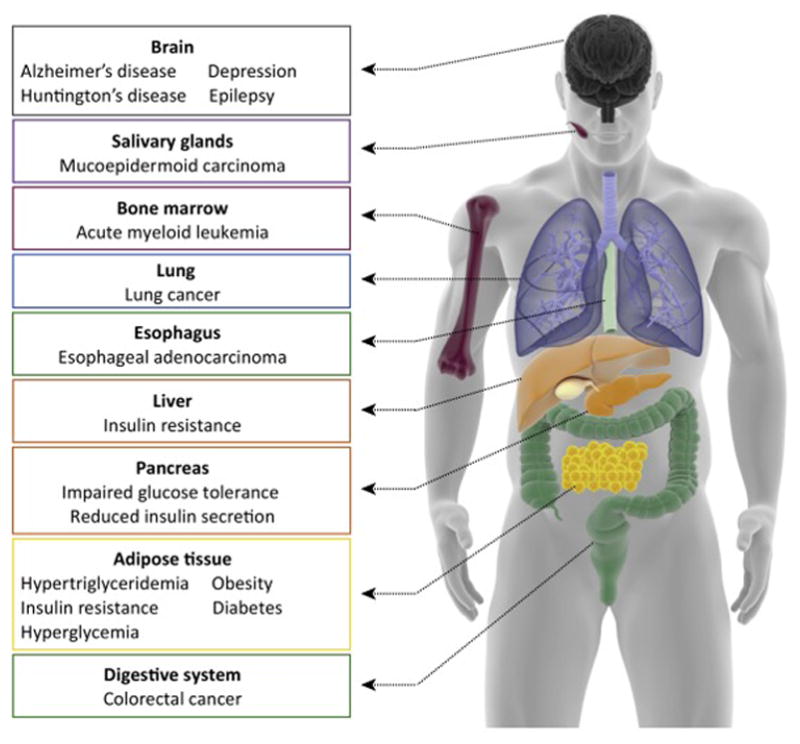
Human body scheme with organs in which the activity of one CRTC family member has been linked to physiological dysregulation or disease.
CRTCs and Metabolic Disorders
Homeostatic regulation of metabolism is critical for maintaining health throughout life. Metabolic disorders such as obesity not only impact the quality of life but also reduce life expectancy and expedite the onset of multiple age related diseases beyond type II diabetes, including cancer and neurodegeneration [34]. As such, obesity can be viewed as an accelerated aging phenotype, adding the pandemic of obesity as a contributor to the increase in age onset diseases [35]. CRTC family proteins are implicated in a variety of metabolic disorders in humans (Figure 4). Polymorphisms of CRTC1 and CRTC3 are associated with total cholesterol level in plasma, as well as high BMI, obesity and hypertriglyceridemia in Mexican-Americans, Chinese populations and psychiatric patients [14,36,37]. In addition, CRTC3 is found in circulation, partly as a result of adipose tissue secretion in childhood obesity [38], suggesting CRTC3 might have effects in other tissues beyond adipose and act as a “physiological messenger” during obesity. Most recently, polymorphisms in the CRTC1 loci show a significance genome-wide association with body fat percentage. This association is sex specific, being stronger in women compared to men [39]. Furthermore, in the same study, two variants present in the intron 1 of CRTC1 showed histone marks characteristic of active transcription and Pol2 binding [39]. Epigenetic deregulation of CRTC genes may therefore contribute to the development of metabolic disorders, which is a key area to be explored in the future.
Lipid Metabolism
In murine models, CRTC family members have also been causally linked to obesity and metabolic disease. Homozygous mice mutant in Crtc1 develop obesity, while CRTC2 contributes to insulin resistance and lipogenesis, and deregulation of both CRTC1 and 2 enhances gluconeogenesis and leads to high blood glucose. Lastly, CRTC3 promotes obesity through disruption of catecholamine signaling [10,14,21,40]. CRTC3 is highly expressed in white adipose tissue (WAT) and less in brown adipose tissue (BAT) [14]. CRTC3 mutant mice have smaller adipocytes in WAT and 50% lower adipose mass compared to wild-type animals. Under HFD (high fat diet) conditions, CRTC3 mutants are protected from the development of obesity, insulin resistance, and hepatic steatosis. This suggests CRTC3 may promote obesity by attenuating catecholamine signaling. One potential mechanism for this may be upregulation of the Regulator of G protein signaling (RGS2), which in turns disrupts lipolysis and fatty acid oxidation [14]. Recently, CRTC2 has also been implicated in lipid metabolism via a novel non-transcriptional function.
CRTC2 controls hepatic lipid metabolism by regulation of sterol regulatory element-binding protein 1 (SREBP1). SREBP1 is a transcription factor synthesized as an inactive precursor bound to the endoplasmic reticulum. In response to insulin signaling, SREBP1 is transported to the Golgi through COPII-mediated vesicle trafficking, and then shuttled to the nucleus to induce the expression of genes involved in cholesterol and fatty acid synthesis [41]. CRTC2 modulates COPII-dependent SREBP1 activation, via an inhibitory association with Sec31, a subunit of COPII complex at Trp143. Phosphorylation of CRTC2 by mTOR dissociates the CRTC2/Sec31 interaction and releases Sec31, promoting its binding to another subunit COPII complex Sec23. This process allows increased SREBP1 activation and promotes lipogenesis [21]. Overall, these data expand metabolic regulation by CRTC family proteins beyond their well-defined role in gluconeogenesis to lipid metabolism, and suggest these CRTCs deregulations might be a central link in the loss of metabolic homeostasis with age.
Autoimmune diseases
Obesity promotes insulin resistance in part by activation of the innate immune system. In this regard, obesity causes an M2-to-M1 (alternatively activated M2 macrophages to classically activated M1 macrophages) shift in adipose tissue that leads to insulin resistance [42]. CRTC3 controls the interconversion from M1 to regulatory macrophages (M2b) [43], and alternatively activated M2 macrophages can promote insulin sensitivity [44]. Increases in cAMP signaling promote M2 macrophage polarization, and CRTC2 and CRTC3 are required for this induction [45]. In particular, this induction is not directly via CRTC/CREB regulation of M2 marker genes, since these genes do not have conserved CREB binding sites on their promoters [45]. In contrast to the effects of CRTCs on HFD induced obesity described above, these data show CRTC2/3 promote M2 polarization in macrophages to protect adipose tissue from insulin resistance during obesity. However, the positive or negative role of CRTCs in immune response remains unclear, as CRTC2 also promotes autoimmune disease by stimulating T helper (Th)17 differentiation [46]. Th17 cells promote clearance of pathogens, but they are also implicated in autoimmune diseases such as multiple sclerosis and rheumatoid arthritis, and CRTC2 promotes Th17 differentiation from CD4 T cells, stimulating the expression of cytokines IL-17A and IL-17F, and increasing immune response [46]. More work is therefore required to elucidate the roles of CRTCs in autoimmune diseases and inflammation, in addition to how CRTCs function in macrophages links to other disorders such as cancer (see cancer section).
Glucose Homeostasis
Acute regulation of metabolic flexibility is particularly critical during fasting, and short term or intermittent fasting has been shown in multiple species to restore metabolic homeostasis and positively impact lifespan [47]. CRTCs were also identified as key regulators of the metabolic shifts during fasting [13]. In response to fasting, SIK proteins (SIK1-3) are phosphorylated, reducing CRTC2 and CRTC3 phosphorylation and triggering gluconeogenic gene expression [13,23,24,31,48,49]. Nuclear translocation of CRTC2 enhances the transcription of CREB-dependent gluconeogenic genes including phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6P) [13,31]. PEPCK and G6P are also targets of glucocorticoid receptor (GR), and CRTC2 functions as a coactivator enhancing transcriptional activity of GR [50]. Animals with constitutively active CRTC2 in liver are unable to reduce blood glucose concentrations compared to control animals following insulin administration [51]. Supporting this, SIK2 knockout mice have increased blood glucose and serum insulin levels, as well as increased population of larger fat cells, phenotypes that all depend upon CRTC2 [52]. In these mutant animals, the CRTC2/CREB complex leads to increased expression of ATF3, and subsequent downregulation of the glucose transporter GLUT4 and adiponectin in white adipose tissue, affecting whole-body glucose metabolism [52]. Thus, constitutive activation of the CRTC2 pathway in liver is sufficient to promote insulin resistance. Conversely, CRTC2 knockout mice show reduced gluconeogenic gene expression (G6Pase and PEPCK), lowered hepatic glucose production during fasting, and reduced circulating glucose, insulin, triglycerides and cholesterol concentrations [40,51]. Along with transcriptional regulation of gluconeogenic gene expression, CRTCs also modulate gene expression by epigenetics changes; CRTC2 modulates gluconeogenic gene expression during fasting and diabetes by promoting histone H3 acetylation at Lys 9 (H3K9Ac) [53]. Recruitment of the lysine acetyltransferase 2B (KAT2B) and WD repeat–containing protein 5 (WDR5) by CRTC2 is required for this acetylation [53].
Along with its effects on hepatic glucose homeostasis, CRTC2 has been implicated in the regulation of pancreatic metabolism [25,54–56]. CRTC2 has a protective role in pancreas, promoting insulin secretion and cell survival [55]. CRTC2 contributes to β-cell proliferation by inducing the expression of the antiapoptotic Bcl-2 gene [57]. In addition, CRTC2 promotes insulin secretion by stimulating MAF bZIP transcription factor A (MAFA) expression, which is a β-cell-restricted factor [54]. In vivo, mice with β-cell-specific knockout of CRTC2 secrete less insulin in response to oral glucose gavage, suggesting that CRTC2 is required for β-cell function [54].
Although multiple groups have shown that activated CRTC proteins translocate to nucleus to regulate glucose metabolism, Han et al. showed that this shift is not always required [33]. The protein arginine methyltransferase PRMT6 binds to CRTC2 and enhances CRTC2-dependent CRE gene targets expression to induce gluconeogenic program in the liver. However, the methylation of conserved CRTC2 arginine residues by PRMT6 does not affect its subcellular localization [33]. These results suggest that CRTC proteins can impact metabolic signaling from different subcellular compartments, and that their functions are not restricted to nucleus. Clarifying the importance of CRTCs subcellular localization and their conserved domains in metabolism should therefore reveal how changes to CRTC function with age impacts metabolic disease risk.
Neuronal regulation of metabolism
In Drosophila, CRTC family proteins link metabolism to stress response. Expression of the Drosophila crtc is increased in response to fasting [58]. crtc mutant flies are sensitive to fasting and oxidative stress (paraquat), and have lower stored glycogen and lipids. Expression of Crtc in neurons rescues the starvation- and paraquat-sensitive phenotypes, as well as lipid levels, indicating Crtc regulates stress response and lipid storage cell nonautonomously from the nervous system [58]. Furthermore, overexpression of Crtc in sNPF (short NeuroPeptide F) neurons of wild type flies further enhances starvation resistance [59]. Crtc maintains metabolic homeostasis in response to stress and inflammation from neurons to gut, regulating the expression of short neuropeptide F (sNPF), an ortholog of mammalian neuropeptide Y. Crtc together with CREB binds to the sNPF promoter to regulate its expression, and the sNPF receptor in the gut mediates effects of the neuronal Crtc/sNPF pathway [59]. In support of a key role in central regulation of metabolism for this pathway, salt-inducible kinase (SIK) in flies regulates body lipids, glycogen and fasting resistance from neurons through Crtc [60].
In addition to peripheral regulation, e.g. by liver, adipose, muscle and pancreas, the central nervous system critically maintains systemic metabolic homeostasis during aging [61]. Supporting the data from Drosophila, CRTCs in the brain can also impact organismal metabolism in mammals. By immunolocalization, mammalian CRTC2 is detected both in nuclei and cytosol of brain tissues. CRTC2 nuclear expression is present in hippocampus, trigeminal and hypoglossal [62], and its cytoplasmic expression has been detected specifically in lateral hypothalamic area of dissected tissues [62], but also in hypothalamic cell cultures [62,63]. Therefore CRTC2 likely has cell-type specific functions in brain that depend upon its subcellular localization. Hypothalamic CRTC2 phosphorylation reflects nutrient state, being dephosphorylated in fed mice, phosphorylated after fasting and dephosphorylated after glucose [62]. Coupled with this, CRTC2 translocates into hypothalamic cell nuclei during exposure to high glucose [63]. CREB expression is also increased after glucose treatment [63]. In addition, expression of CREB target genes, such as insulin receptor substrate 2 (Irs2) and thyrotropin releasing hormone (TRH), require activation of CRTC2, via inhibition of AMPK in hypothalamic cells [62,63]. These data demonstrate that the AMPK/CRTC2/CREB axis is conserved in the mammalian central nervous system.
Work in C. elegans suggests regulation of the neuronal AMPK/CRTC/CREB axis underlies the effects of CRTCs on systemic longevity and metabolism. Lifespan extension via ubiquitous activation of AMPK is associated with a remodelling of mitochondrial metabolism. Blocking phosphorylation of CRTC-1 by AMPK only in neurons is sufficient both to suppress AMPK longevity and reverse effects on metabolism. Furthermore, neuronal CRTC-1 activation alone can drive fragmentation of mitochondrial networks in muscle. Neuronal CRTC-1 modulates systemic longevity and metabolism via octopamine, a monoamine neurotransmitter, and mutations in octopamine synthesis enzymes derepress the effect of neuronal CRTC-1 on AMPK and calcineurin mediated longevity [4]. These data establish a CRTC-1 cell nonautonomous role in communicating perception of energy status in neurons to systemic regulation of metabolism and lifespan in C. elegans and examining whether these effects are conserved in mammals now becomes a priority [3,4]. Initial data suggest that the effect of neural regulation of metabolism by CRTCs on longevity may be conserved in mammals [64]. Mice lacking the transient receptor potential cation channel subfamily V member 1 (TRPV1), a pain receptor, are long-lived putatively because of inactivation of the CRTC1/CREB pathway in sensory neurons [64]. In TRPV1 neurons, CRTC1 induces the release of neuropeptide calcitonin gene-related peptide (CGRP), which in turn antagonizes insulin release from pancreatic β cells [64]. Given that CRTC2 regulates pancreatic β cell function by induction of β-cell-restricted transcription factor, MAFA, promoting insulin gene expression and secretion [54], it would be interesting to determine if CRTC1 and CRTC2 might indirectly interact from different tissues to regulate systemic metabolism.
Taken together, although CRTCs were first discovered due to their role in modulating CREB-mediated in gluconeogenesis in fasting, as new metabolic roles for CRTCs are discovered, CRTC family proteins are emerging as key links between organismal metabolism and aging. CRTCs may therefore provide novel therapeutic avenues for maintaining metabolic homeostasis in old age.
CRTCs and Cancer Proliferation
Another critical disease for which age is a potent risk factor is cancer (Figure 4), and CRTCs have been implicated in carcinogenesis in several conditions, including lung cancer, colorectal cancer, acute myeloid leukemia, mucoepidermoid carcinoma, and esophageal adenocarcinoma [3,4,65–73]. Mechanistic links between CRTCs and cancer are only just beginning to emerge but often involve hypophosphorylation of CRTC1 and upregulation of target gene expression. Liver kinase B1, LKB1, is a serine/threonine kinase that acts as a critical direct activator of AMPK and the 12 AMPK-related kinases [74]. LKB1 is downregulated in a subset of human esophageal cancer cell lines and patient samples with lung cancer and cervical carcinomas [66]. Since multiple AMPK family kinases (i.e. SIK2) can directly inhibit CRTCs, LKB1 loss therefore indirectly leads to CRTC1 hypophosphorylation and activation, and unregulated expression of oncogenic CRTC1 targets, such as glycosylphosphatidylinositol (GPI)-anchored metastasis-associated protein (LYPD3), cyclooxygenase-2 (COX-2), and the long coding RNA LINC00473 [65,67,68,75]. Dysregulation of LKB1/SIK2/CRTC1 pathway may therefore contribute to cancer progression.
Mucoepidermoid carcinoma (MEC) is the most common malignant form of salivary gland tumor. MECs are induced by recurrent chromosomal translocations that fuse the segment encoding the N-terminal CREB-binding domain (CBD) of CRTC1 with substantial portions of the coding region of another transcriptional coactivator, mastermind-like (MAML)2 [76]. This specific chromosomal translocation joins exon 1 of CRTC1 to exons 2–5 of MAML2, resulting in expression of a CRTC1-MAML2 fusion. This CRTC1-MAML2 fusion controls expression of genes that contribute to tumor growth [6]. One such target is an autocrine signal governed by Amphiregulin (AREG)-epidermal growth factor receptor (EGFR) [69]. Secreted AREG ligand then activates EGFR signaling in an autocrine manner to induce cell growth and survival of MEC cells [69]. An additional molecular mechanism linking CRTC1-MAML2 to tumorigenesis is via aberrant activation of CREB. CRTC1-MAML2 fusion is constitutively nuclear, and binds to CBP/p300, recruiting CREB, and enhancing CREB’s oncogenic activity [71]. In addition, more than 800 human genes have been identified as CRTC1-MAML2 targets, involved in cellular movement, development, death and survival, growth and proliferation, cell-to-cell signaling and interaction, and metabolism [69,70], indicating that CRTC1-MAML2 might be a promising therapeutic target.
Taken together, these data establish CRTC proteins as potential targets for treatments of cancer [77]. Indeed, this idea is supported by data on endogenous regulation of CRTC1 by microRNA-22. miR-22 has been suggested to have an anti-tumor role in acute myeloid leukemia (AML) [78]. During leukemia, miR-22 is downregulated via transcriptional regulators TET1/GFI1/EZH2/SIN3A, which increase histone H3K27 trimethylation (me3) at the miR-22 promoter. Repression of miR-22 de-represses CRTC1, and upregulates oncogenic CREB targets (CDK6, HOXA7, BMI1, FASN and HMGA1) [78]. However, the role of CRTCs as a friend or foe of cancer remains controversial. Besides putative oncogenic roles of CRTCs during cancer development, recent data also suggest that CRTC2 can act as a lymphoma tumor suppressor gene. CRTC2 plays an important role in maintaining genome integrity by promoting transcription of DNA mismatch repair (MMR) genes EXO1, MSH6, PMS1 and POLD2. CRTC2 knockout HeLa cells have reduced MMR activity, which in turn leads to a 25-fold higher spontaneous mutation rate [79]. Furthermore, CRTC2 expression is reduced in lymphoma cell lines and patient samples. This reduction results from low levels of acetylated histone H3 at the CRTC2 promoter, leading to increased mutation frequency as well as microsatellite instability [79]. These data suggest that deregulation of CRTCs and their targets may have causal links to multiple forms of cancer, yet the directionality of these links may well be context specific. Therefore if and how CRTCs might be targeted for cancer therapeutics will depend upon the specific malignancy in question.
CRTCs Function in Learning, Memory and Neurodegenerative Diseases
CREB has an evolutionarily conserved fundamental role in regulating memory formation and memory consolidation [80–82]. The cAMP-CREB pathway controls memory for example by enhancing synaptic transmission or increasing neuronal excitability [80]. Modulating CREB levels or its binding partner the histone acetyltransferase CBP (CREB binding protein) by overexpression or knockout, results in either enhanced memory formation or memory deficits respectively, in mammalian models [80]. Even though CREB phosphorylation at Ser133 is an important activating post translational modification, it is not always sufficient to induce CREB-dependent gene transcription [83], suggesting additional mechanisms regulating CREB function. CRTC1 has therefore been mainly studied in the context of memory formation and has been characterized for its role in neurodegenerative diseases [84]. Intriguingly, in brain tissue, activation of CRTC1 seems beneficial, in contrast to the detrimental effects of CRTCs hyperactivation discussed earlier in metabolic disease and cancer.
Crtc1 knockout mice do not display any brain developmental defects [10], however these mouse models have been informative in highlighting the importance of CRTC1 in neuronal function [85,86]. Crtc1 knockout mice display a treatment-resistant depression-like phenotype [86] as well as symptoms associated with mood disorders such as impulsive aggressiveness, social withdrawal and decreased sexual motivation. These phenotypes are likely caused by decreased levels of dopaminergic and serotoninergic activity in the mice pre-frontal cortex, and reduced expression of CREB susceptibility genes involved in neuroplasticity [85].
Synaptic activity plays a major role in regulating CRTC1 function in neurons. Synaptic stimuli induced by AMPA (α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate) receptor, NMDA (N-methyl-D-aspartic acid) receptor, LVGCC (L-type voltage-gated calcium channels) calcium influx or GABA (gamma-aminobutyric acid) receptor activation are required to trigger CRTC1 nuclear import and transcription dependent plasticity effects. Indeed, CRTC1 localizes to dendrites and spines in electrically silenced hippocampal neurons and translocates to the nucleus in a calcium and calcineurin dependent manner upon stimulation [87]. Post-translational modifications and subsequent CRTC1 subcellular localization changes integrate this signal. Activation of synaptic NMDA or AMPA glutamate receptors, or LVGCC induces a local increase in calcium concentration at the synapse (as opposed to a general rise in intracellular calcium), which triggers the dynein-mediated retrograde transport of CRTC1 along microtubules from the synapse to the nucleus [88]. The activity-dependent nuclear shuttling of CRTC1 in neurons occurs within minutes and conversely, once the stimulus stops, CRTC1 returns to the cytoplasm in 30 minutes [89]. In hippocampal neurons, basal constitutive phosphorylation of CRTC1 is controlled by SIK2/AMPK and MARK2 enzymes at Serine 151 and Serine 245, whereas the stimulus triggered dephosphorylation is mediated by the phosphatase calcineurin [89]. Dephosphorylation of 3 serine residues (Ser64, Ser151, Ser145) on CRTC1 are necessary and sufficient for dissociation from 14-3-3 protein and nuclear accumulation [88]. Activity stimulated dephosphorylation of CRTC1 at Serine 151 can induce transcriptional activation of CRTC1 in mice hippocampus [90]. Nuclear CRTC1 then drives CRE-dependent (cAMP response element) transcription even in the absence of CREB phosphorylation on Serine 133 [89]. However CREB is required for CRTC1 to bind to CRE sites in the promoters of Immediate Early Genes (IEG), in an activity dependent manner. Long term potentiation (LTP) is an activity dependent long lasting increase in synaptic strength. LTP is a form of synaptic plasticity, which requires gene transcription and protein synthesis, and is considered an electrophysiological model for the basic mechanisms involved in learning and memory formation [91,92]. CRTC1 in neurons senses the coincidence of calcium and cAMP signals and induces a CREB-dependent transcriptional response, leading to enhanced synaptic transmission (through for example the expression of Brain Derived Neurotrophic Factor BDNF) [11,93,94]. CRTC1 is involved in the maintenance of late phase LTP in mice hippocampal slices [11] and induction of late phase LTP triggers a robust nuclear accumulation of CRTC1 in neurons. Consistently, nuclear localization of CRTC1 facilitates late phase LTP induction in mouse hippocampal slices, whereas expression of a dominant negative CRTC1 suppresses the maintenance of late phase LTP [93,95]. Similarly, increasing CRTC1 or CREB function in excitatory dentate granule cells of the dentate gyrus region of the dorsal hippocampus of mice is sufficient to enhance memory stabilization as well as memory reactivation without compromising memory quality in a fear conditioning assay, suggesting a region specific role of CRTC1 in controlling memory [96]. More recent work has shown that in mouse hippocampus, CRTC1 induces chromatin changes following associative learning to promote sustained target gene transcription and enhance memory strength [97].
In Drosophila, Crtc nuclear activity is required specifically in the mushroom body of the fly for memory formation induced in appetitive long term memory (fLTM: a single training preceded by a period of fasting). The fasting period downregulates the insulin signaling pathway in Drosophila, which normally phosphorylates Crtc via SIK2 leading to CRTC degradation [58]. Nuclear translocation of CRTC after fasting can be mimicked by the expression of a constitutively active form of CRTC, which is necessary and sufficient to form fLTM in the mushroom body. Supporting this mechanism, Drosophila chico mutants that display reduced insulin signaling mimic fLTM through the activation of CRTC in the mushroom body [98]. Interestingly, the Drosophila chico mutation, which extends lifespan via downregulation of the insulin signaling pathway [99], is shown to activate CRTC to promote beneficial effects [58,98]. These observations contrast with the requirement of CRTC-1 inhibition in AMPK mediated longevity in C. elegans [3], suggesting that CRTCs might display opposite roles in the regulation of lifespan or metabolism, and in neuronal functions and memory. Lastly, in addition to being involved in the formation of memory in Drosophila, the CRTC/CREB complex is also involved in early maintenance of Long Term Memory (LTM), targeting genes required for memory maintenance and extinction in mushroom bodies [100].
Alzheimer’s Disease
Data in mammals and Drosophila therefore highlight the importance of CRTC in neuronal plasticity and memory formation. Not surprisingly therefore, CRTC deregulation has been linked to the pathogenesis of age onset diseases associated with memory defects such as Alzheimer’s Disease (AD) and Huntington’s Disease (HD). In the APPsw/ind mice model of AD (transgenic mice expressing mutant human APP695 isoform harboring the FAD-linked Swedish K670N/M671L and Indiana V717F mutations in the neurons), accumulation of the toxic amyloid beta peptide in neurons reduces calcineurin activity by disrupting calcium influx through L-type calcium channels. Calcineurin dependent CRTC1 dephosphorylation and activation is thus impaired, leading to the disruption of CRTC1 dependent gene expression program. On a functional level, the APPsw/ind mice display a hippocampal-dependent memory deficit in the Morris Water Maze spatial memory task, which correlates with a specific reduction in the expression of CRTC1 dependent genes normally upregulated during training [101]. Furthermore, over expression of CRTC1 by AAV (Adeno-Associated Virus) delivery in mice hippocampus improves learning and memory of the AD mice in the Morris Water Maze test without affecting wild type animals performance [90]. Human data support the hypothesis that CRTC1 function is altered in Alzheimer’s Disease patients, and might therefore represent a therapeutic target. Studies reveal a reduction of both total and phosphorylated CRTC1, as well as decreased expression of CRTC1 targets in human hippocampal samples of AD pathological cases (classified according to Braak stages) [90,102]. In addition, DNA methylation at the CRTC1 promoter is significantly lower in hippocampus samples of AD patients, and methylation levels inversely correlate with phospho-tau and beta-amyloid pathology, suggesting a link between CRTC1 activity and neurodegeneration pathophysiology.
Huntington’s Disease
Huntington’s Disease (HD) is caused by an abnormal autosomal dominant glutamine repeat expansion of 3 nucleotides (CAG) in the huntingtin (Htt) protein leading to cognitive, psychiatric, motor dysfunctions and loss of autonomy during the final stages of the disease [103]. Similar to Alzheimer’s disease, CRTC1 expression levels are decreased in vivo in postmortem striatal tissue from HD patients, in the striatum of various transgenic mice models (NLS-N171-82Q, R6/2 and HdhQ111 HD) and in vitro in STHdhQ111 cells expressing mutant Htt [104]. Mutant huntingtin interacts with SIRT1 and disrupts SIRT1-mediated activation of CRTC1 [105]. Under normal conditions, SIRT1 promotes deacetylation of CRTC1 (lysines 13,20) and activates CRTC1 by promoting its dephosphorylation and interaction with CREB, which leads to activation of neuronal target genes such as bdnf. Interestingly this activating effect of SIRT1 on CRTC1 in neurons is in contrast with the effect of SIRT1 on CRTC2 in the liver. Indeed, fasting induces SIRT1-mediated deacetylation of CRTC2 and its subsequent ubiquitination and degradation by the proteasome [29,31]. These opposite regulations of SIRT1 on different CRTC isoforms might indicate tissue specific modulations and physiological roles for the CRTCs. In a mouse model of HD (R6/2), SIRT1 overexpression is neuroprotective and CRTC1 and CREB are at least partially required for SIRT1 mediated neuroprotection in primary cortical neurons. The requirement for CRTC1 in SIRT1 mediated neuroprotection in vivo in a HD mouse model remains to be explored [105].
CRTC plays an important role in the expression of an additional key metabolic regulator, the transcriptional co-regulator PGC-1α (peroxisome proliferator-activated receptor gamma coactivator 1α) [12]. PGC-1α is a master regulator of mitochondrial biogenesis, energy homeostasis, adaptive thermogenesis and glucose metabolism whose expression is impaired in HD [106,107]. Chaturvedi et al therefore asked whether PGC-1α might mediate beneficial effects of CRTC1 in HD models. CRTC1 overexpression in wild type or huntingtin-expressing striatal cells (Htt cells) increased PGC-1α promoter activity, expression of downstream targets (NRF-1, Tfam, CytC), as well as mitochondrial DNA content, mitochondrial activity and mitochondrial membrane potential [104]. Consistent with this result, CRTC1 knockdown enhances mitochondrial dysfunction caused by a mitochondrial toxin (3-NP, inhibits Complex II of ETC) in Htt cells, and this effect can be partially rescued by PGC-1α overexpression. Finally, specific knock down of CRTC1 in the striatum in NLS-N171-82Q HD transgenic mice induces neurodegeneration and cell death [104].
Given the results of these studies, CRTCs represent a potential therapeutic target for various age related diseases ranging from metabolic dysfunctions to neurological disorders. However, recent work highlights the complex and sometimes opposing roles of CRTCs in aging versus specific diseases. It is therefore necessary to better elucidate the mechanism of CRTCs activity, and how their dysregulation affects physiology and pathogenesis in aging individuals.
Concluding Remarks
As recent work has conclusively shown, CREB-regulated transcriptional coactivators play broader cellular roles than initially thought (Box 1,3). Being neither regulated by CREB nor exclusively transcriptional coactivators, perhaps the time has come for a rethink of their - with hindsight - rather myopic name. Although ‘cyclic AMP regulated transcriptional coactivator’ has been suggested, until more work is done on these understudied proteins a name that adequately emcompasses their multiple roles and binding partners (Figure 5, Key Figure) may be beyond reach. As discussed above, despite sequence similarity, different CRTC family members have differing functions and roles in both physiology and disease (Table 2). This is in part due to their largely non-overlapping tissue expression, but may also reflect the how recent many of the newly described roles for these proteins are - full characterization of all three proteins is still lacking. The role of CRTCs in organismal aging itself has only been described in C. elegans, yet as outlined here, deregulation of mammalian CRTCs is linked to multiple age-onset pathologies. CRTCs can be both protective and destructive depending on the disease in question. Therefore more work is needed to determine how different CRTCs become deregulated with age across different tissues (see outstanding questions). However, what is increasingly clear is that age-onset deregulation of CRTC family members underlies the pathophysiology of age onset disorders beyond high blood glucose and Type II diabetes. CRTCs are therefore molecular examples geroscience targets - linking age to a multitude of diseases of the elderly.
Box 1. CRTCs and Stem cells regulation.
CRTC has recently been described as an inducer of intestinal stem cells proliferation (Figure 5, Key Figure). In Drosophila, dietary L-glutamate acts on the metabotropic receptor mGluR to influence cytoplasmic Ca2+ oscillations via phospholipase C and IP3Rc, leading to an overall increase in cytoplasmic calcium. High cytoplasmic calcium induces nuclear translocation of CRTC through activation of its upstream phosphatase calcineurin [108]. CRTC therefore integrates signals from the diet to induce intestinal stem cell activity in Drosophila. These discoveries are the first steps in the study of stem cells regulation by CRTCs, and suggest CRTCs may have a role in other type of stem cells. For instance, CRTC1/2/3 have been identified in human testis samples [109], suggesting a role in the development of germ stem cells.
Box 3. CRTCs in circadian rhythm.
Circadian rhythm is a tightly regulated internal biological clock, which modulates various physiological processes and undergoes changes with age [111–114]. In particular, dysregulation of circadian rhythm is associated with alterations in metabolism and development of cancers [115–117]. The master circadian pacemaker is located in the suprachiasmatic nuclei (SCN) in the hypothalamus, and responds to light stimulus through retinal photoreceptors. CRTC1 displays both a rhythmic expression pattern in SCN and a strong nuclear accumulation following light stimulus [118]. Indeed, upon light exposure, intraneuronal calcium levels rise, leading to both phosphorylation of CREB and nuclear import and activation of CRTC1. The resulting CRTC-CREB complex induces the expression of the major circadian rhythm regulator Per, as well as the kinase Sik1. SIK1 serves as a negative feedback mechanism by phosphorylating CRTC1 and deactivating it to suppress further effects of light on the clock [119]. In addition to modulating circadian clock in response to light, a more recent study in Drosophila suggests a light-independent role of CRTC in sustaining circadian behaviours, by acting as a transcriptional co-activator of another essential circadian gene, timeless [120]. These findings thus highlight a novel role for CRTCs in regulating another major physiological process that become deregulated with age, the circadian clock (Figure 5, Key Figure).
Figure 5, Key Figure.
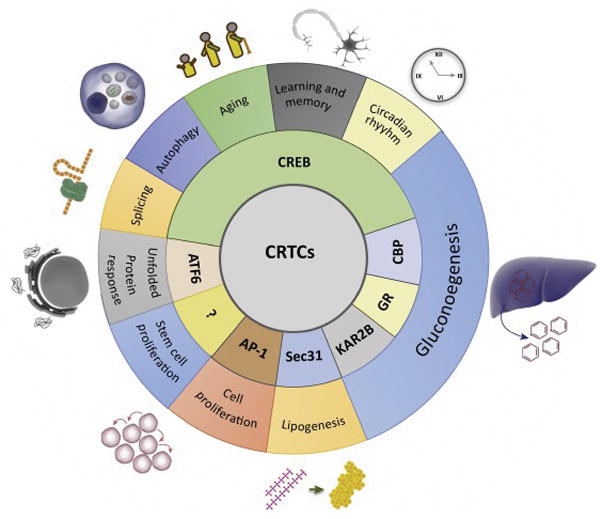
Physiological and cellular processes modulated by CRTCs.
Inner circle lists the proteins known to interact directly with at least one CRTC family member. Outer circle represents the physiological and cellular processes specifically controlled by the interaction between CRTC and its interacting partner. CRTC can either interact with a unique partner to modulate a unique pathway (eg: CRTC-Sec31 influencing lipogenesis), or multiple pathways (eg: CRTC-CREB acting on splicing, autophagy, etc.), or can interact with multiple proteins to act on the same physiological process (eg: CRTC-CREB or CRTC-CBP modulating gluconeogenesis).
Table 2.
Expression and physiological roles of CRTCs’ isoforms.
| Organism | Isoform | Tissue specific expression | Physiological process regulated | References |
|---|---|---|---|---|
 Mouse |
CRTC1 | Brain | Energy balance, fertility | [10] |
| (Hippocampus) Memory formation | [97] | |||
| (Suprachiasmatic nuclei) Circadian rhythm | [118,119] | |||
| CRTC2 | White Adipose Tissue | Insulin Resistance | [52] | |
| Lung | Unknown | [12] | ||
| Liver | Regulation of lipogenesis | [21] | ||
| Gluconeogenesis | [13,29,31] | |||
| Muscle | Mitochondrial biogenesis | [12] | ||
| Immune System | Macrophage M1 to M2 interconversion (promotes insulin sensitivity) | [45] | ||
| T helper (Th)17 cells differentiation | [46] | |||
| Brain | Hypothalamic glucose sensing | [62,63] | ||
| Pancreas | (Beta cells) Insulin secretion | [25,54,55] | ||
| CRTC3 | Lung | Unknown | [12] | |
| White Adipose Tissue | Attenuates response to catecholamine signals | [14] | ||
| Brown Adipose Tissue | Unknown | [14] | ||
| Immune System | Macrophage M1 to M2 interconversion | [71] | ||
 Drosophila |
Crtc | Neurons | Central regulation of metabolism | [58,59,60] |
| (mushroom body) Memory formation | [98,100] | |||
| Circadian rhythm | [120] | |||
| Intestine | Stem cells proliferation | [108] | ||
 C. elegans |
crtc-1 | Neurons | Lifespan Central regulation of metabolism | [3,4] |
| Intestine | Unknown | [3] |
List of CRTCs’ isoforms in mouse, Drosophila and C. elegans, their specific expression and physiological roles.
Outstanding Questions Box.
How do CRTCs become deregulated with age?
Which cellular role of CRTCs contributes to their effect on organismal aging
Is the role of CRTCs in AMPK longevity conserved?
Can we target CRTCs for neuroprotective benefits?
Do CRTCs have antagonistic effects on aging and disease in different tissues?
Can CRTC family members cell nonautonomously co-regulate each other across tissues to regulate metabolism and aging?
Trends Box.
Novel cellular regulators and targets of the CREB-regulated transcriptional co-activator (CRCT) family have recently been identified.
In C. elegans CRCTs have been shown to modulate aging.
Recently CRTC dysfunction has been associated with age-related human disease.
CRTCs could provide a target for healthy human aging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kennedy BK, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159:709–713. doi: 10.1016/j.cell.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mair W, Dillin A. Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem. 2008;77:727–754. doi: 10.1146/annurev.biochem.77.061206.171059. [DOI] [PubMed] [Google Scholar]
- 3.Mair W, et al. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature. 2011;470:404–408. doi: 10.1038/nature09706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burkewitz K, et al. Neuronal CRTC-1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell. 2015;160:842–855. doi: 10.1016/j.cell.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conkright MD, et al. TORCs: transducers of regulated CREB activity. Mol Cell. 2003;12:413–423. doi: 10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Iourgenko V, et al. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci U S A. 2003;100:12147–12152. doi: 10.1073/pnas.1932773100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bittinger MA, et al. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol. 2004;14:2156–2161. doi: 10.1016/j.cub.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Screaton RA, et al. The CREB Coactivator TORC2 Functions as a Calcium- and cAMP-Sensitive Coincidence Detector. Cell. 2004;119:61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 10.Altarejos JY, et al. The Creb1 coactivator Crtc1 is required for energy balance and fertility. Nat Med. 2008;14:1112–1117. doi: 10.1038/nm.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovács KA, et al. TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104:4700–4705. doi: 10.1073/pnas.0607524104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Z, et al. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc Natl Acad Sci U S A. 2006;103:14379–14384. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koo SH, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 14.Song Y, et al. CRTC3 links catecholamine signalling to energy balance. Nature. 2010;468:933–939. doi: 10.1038/nature09564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo Q, et al. Mechanism of CREB recognition and coactivation by the CREB-regulated transcriptional coactivator CRTC2. Proc Natl Acad Sci U S A. 2012;109:20865–20870. doi: 10.1073/pnas.1219028109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, et al. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460:534–537. doi: 10.1038/nature08111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canettieri G, et al. The coactivator CRTC1 promotes cell proliferation and transformation via AP-1. Proc Natl Acad Sci U S A. 2009;106:1445–1450. doi: 10.1073/pnas.0808749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seok S, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516:108–111. doi: 10.1038/nature13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai WW, et al. ATF3 mediates inhibitory effects of ethanol on hepatic gluconeogenesis. Proc Natl Acad Sci U S A. 2015;112:2699–2704. doi: 10.1073/pnas.1424641112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amelio AL, et al. Bipartite functions of the CREB co-activators selectively direct alternative splicing or transcriptional activation. EMBO J. 2009;28:2733–2747. doi: 10.1038/emboj.2009.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han J, et al. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature. 2015;524:243–246. doi: 10.1038/nature14557. [DOI] [PubMed] [Google Scholar]
- 22.Heintz C, et al. Splicing factor 1 modulates dietary restriction and TORC1 pathway longevity in C. elegans. Nature. 2016 doi: 10.1038/nature20789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katoh Y, et al. Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J. 2006;273:2730–2748. doi: 10.1111/j.1742-4658.2006.05291.x. [DOI] [PubMed] [Google Scholar]
- 24.Uebi T, et al. Phosphorylation of the CREB-specific coactivator TORC2 at Ser307 regulates its intracellular localization in COS-7 cells and in the mouse liver. American Journal of Physiology - Endocrinology and Metabolism. 2010;299:E413–E425. doi: 10.1152/ajpendo.00525.2009. [DOI] [PubMed] [Google Scholar]
- 25.Jansson D, et al. Glucose controls CREB activity in islet cells via regulated phosphorylation of TORC2. Proc Natl Acad Sci U S A. 2008;105:10161–10166. doi: 10.1073/pnas.0800796105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolff S, et al. SMK-1, an essential regulator of DAF-16-mediated longevity. Cell. 2006;124:1039–1053. doi: 10.1016/j.cell.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 27.Yoon YS, et al. Suppressor of MEK null (SMEK)/protein phosphatase 4 catalytic subunit (PP4C) is a key regulator of hepatic gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107:17704–17709. doi: 10.1073/pnas.1012665107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burkewitz K, et al. AMPK at the nexus of energetics and aging. Cell Metab. 2014;20:10–25. doi: 10.1016/j.cmet.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giblin W, et al. Sirtuins: guardians of mammalian healthspan. Trends Genet. 2014;30:271–286. doi: 10.1016/j.tig.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dentin R, et al. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–369. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- 32.Dentin R, et al. Hepatic glucose sensing via the CREB coactivator CRTC2. Science. 2008;319:1402–1405. doi: 10.1126/science.1151363. [DOI] [PubMed] [Google Scholar]
- 33.Han H-S, et al. Arginine methylation of CRTC2 is critical in the transcriptional control of hepatic glucose metabolism. Sci Signal. 2014;7:ra19. doi: 10.1126/scisignal.2004479. [DOI] [PubMed] [Google Scholar]
- 34.Jura M, Kozak LP. Obesity and related consequences to ageing. Age. 2016;38:23. doi: 10.1007/s11357-016-9884-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kramer CK, et al. Are metabolically healthy overweight and obesity benign conditions?: A systematic review and meta-analysis. Ann Intern Med. 2013;159:758–769. doi: 10.7326/0003-4819-159-11-201312030-00008. [DOI] [PubMed] [Google Scholar]
- 36.Choong E, et al. Influence of CRTC1 polymorphisms on body mass index and fat mass in psychiatric patients and the general adult population. JAMA Psychiatry. 2013;70:1011–1019. doi: 10.1001/jamapsychiatry.2013.187. [DOI] [PubMed] [Google Scholar]
- 37.Ou Z, et al. CRTC3 polymorphisms were associated with the plasma level of total cholesterol and the risks of overweight and hypertriglyceridemia in a Chinese Han population. Mol Biol Rep. 2014;41:125–130. doi: 10.1007/s11033-013-2844-4. [DOI] [PubMed] [Google Scholar]
- 38.Prats-Puig A, et al. Soluble CRTC3: A Newly Identified Protein Released by Adipose Tissue That Is Associated with Childhood Obesity. Clin Chem. 2016;62:476–484. doi: 10.1373/clinchem.2015.249136. [DOI] [PubMed] [Google Scholar]
- 39.Lu Y, et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat Commun. 2016;7:10495. doi: 10.1038/ncomms10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, et al. Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc Natl Acad Sci U S A. 2010;107:3087–3092. doi: 10.1073/pnas.0914897107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldstein JL, et al. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 42.Dalmas E, et al. Defining macrophage phenotype and function in adipose tissue. Trends Immunol. 2011;32:307–314. doi: 10.1016/j.it.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 43.Clark K, et al. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc Natl Acad Sci U S A. 2012;109:16986–16991. doi: 10.1073/pnas.1215450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luan B, et al. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U S A. 2015;112:15642–15647. doi: 10.1073/pnas.1519644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hernandez JB, et al. The CREB/CRTC2 pathway modulates autoimmune disease by promoting Th17 differentiation. Nat Commun. 2015;6:7216. doi: 10.1038/ncomms8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014;19:181–192. doi: 10.1016/j.cmet.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel K, et al. The LKB1-salt-inducible kinase pathway functions as a key gluconeogenic suppressor in the liver. Nat Commun. 2014;5:4535. doi: 10.1038/ncomms5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henriksson E, et al. SIK2 regulates CRTCs, HDAC4 and glucose uptake in adipocytes. J Cell Sci. 2015;128:472–486. doi: 10.1242/jcs.153932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hill MJ, et al. CRTC2 Is a Coactivator of GR and Couples GR and CREB in the Regulation of Hepatic Gluconeogenesis. Mol Endocrinol. 2016;30:104–117. doi: 10.1210/me.2015-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hogan MF, et al. Hepatic Insulin Resistance Following Chronic Activation of the CREB Coactivator CRTC2. J Biol Chem. 2015;290:25997–26006. doi: 10.1074/jbc.M115.679266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park J, et al. SIK2 is critical in the regulation of lipid homeostasis and adipogenesis in vivo. Diabetes. 2014;63:3659–3673. doi: 10.2337/db13-1423. [DOI] [PubMed] [Google Scholar]
- 53.Ravnskjaer K, et al. Glucagon regulates gluconeogenesis through KAT2B- and WDR5-mediated epigenetic effects. J Clin Invest. 2013;123:4318–4328. doi: 10.1172/JCI69035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blanchet E, et al. Feedback inhibition of CREB signaling promotes beta cell dysfunction in insulin resistance. Cell Rep. 2015;10:1149–1157. doi: 10.1016/j.celrep.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eberhard CE, et al. CRTC2 is required for β-cell function and proliferation. Endocrinology. 2013;154:2308–2317. doi: 10.1210/en.2012-2088. [DOI] [PubMed] [Google Scholar]
- 56.Kim SJ, et al. Glucose-dependent insulinotropic polypeptide-mediated up-regulation of beta-cell antiapoptotic Bcl-2 gene expression is coordinated by cyclic AMP (cAMP) response element binding protein (CREB) and cAMP-responsive CREB coactivator 2. Mol Cell Biol. 2008;28:1644–1656. doi: 10.1128/MCB.00325-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim SJ, et al. Glucose-dependent insulinotropic polypeptide-mediated up-regulation of β-cell antiapoptotic Bcl-2 gene expression is coordinated by cyclic AMP (cAMP) response element binding protein (CREB) and cAMP-responsive CREB coactivator 2. Mol Cell Biol. 2008;28:1644–1656. doi: 10.1128/MCB.00325-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang B, et al. The insulin-regulated CREB coactivator TORC promotes stress resistance in Drosophila. Cell Metab. 2008;7:434–444. doi: 10.1016/j.cmet.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen R, et al. Neuronal energy-sensing pathway promotes energy balance by modulating disease tolerance. Proc Natl Acad Sci U S A. 2016;113:E3307–14. doi: 10.1073/pnas.1606106113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choi S, et al. Drosophila salt-inducible kinase (SIK) regulates starvation resistance through cAMP-response element-binding protein (CREB)-regulated transcription coactivator (CRTC) J Biol Chem. 2011;286:2658–2664. doi: 10.1074/jbc.C110.119222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weir HJ, Mair WB. SnapShot: Neuronal Regulation of Aging. Cell. 2016;166:784–784. e1. doi: 10.1016/j.cell.2016.07.022. [DOI] [PubMed] [Google Scholar]
- 62.Lerner RG, et al. A role for the CREB co-activator CRTC2 in the hypothalamic mechanisms linking glucose sensing with gene regulation. EMBO Rep. 2009;10:1175–1181. doi: 10.1038/embor.2009.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Breit A, et al. Glucose Enhances Basal or Melanocortin-Induced cAMP-Response Element Activity in Hypothalamic Cells. Mol Endocrinol. 2016;30:748–762. doi: 10.1210/me.2016-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Riera CE, et al. TRPV1 pain receptors regulate longevity and metabolism by neuropeptide signaling. Cell. 2014;157:1023–1036. doi: 10.1016/j.cell.2014.03.051. [DOI] [PubMed] [Google Scholar]
- 65.Gu Y, et al. Altered LKB1/CREB-regulated transcription co-activator (CRTC) signaling axis promotes esophageal cancer cell migration and invasion. Oncogene. 2012;31:469–479. doi: 10.1038/onc.2011.247. [DOI] [PubMed] [Google Scholar]
- 66.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feng Y, et al. The CRTC1-NEDD9 signaling axis mediates lung cancer progression caused by LKB1 loss. Cancer Res. 2012;72:6502–6511. doi: 10.1158/0008-5472.CAN-12-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Z, et al. cAMP/CREB-regulated LINC00473 marks LKB1-inactivated lung cancer and mediates tumor growth. J Clin Invest. 2016;126:2267–2279. doi: 10.1172/JCI85250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Z, et al. Aberrantly activated AREG-EGFR signaling is required for the growth and survival of CRTC1-MAML2 fusion-positive mucoepidermoid carcinoma cells. Oncogene. 2014;33:3869–3877. doi: 10.1038/onc.2013.348. [DOI] [PubMed] [Google Scholar]
- 70.Chen J, et al. Gene expression profiling analysis of CRTC1-MAML2 fusion oncogene-induced transcriptional program in human mucoepidermoid carcinoma cells. BMC Cancer. 2015;15:803. doi: 10.1186/s12885-015-1827-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Clark MD, et al. Molecular Basis for the Mechanism of Constitutive CBP/p300 Coactivator Recruitment by CRTC1-MAML2 and Its Implications in cAMP Signaling. Biochemistry. 2015;54:5439–5446. doi: 10.1021/acs.biochem.5b00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Levine DM, et al. A genome-wide association study identifies new susceptibility loci for esophageal adenocarcinoma and Barrett’s esophagus. Nat Genet. 2013;45:1487–1493. doi: 10.1038/ng.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He Y, et al. Identification and validation of PROM1 and CRTC2 mutations in lung cancer patients. Mol Cancer. 2014;13:19. doi: 10.1186/1476-4598-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lizcano JM, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cao C, et al. Role of LKB1-CRTC1 on glycosylated COX-2 and response to COX-2 inhibition in lung cancer. J Natl Cancer Inst. 2015;107:358. doi: 10.1093/jnci/dju358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tonon G, et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet. 2003;33:208–213. doi: 10.1038/ng1083. [DOI] [PubMed] [Google Scholar]
- 77.Liang Y, et al. CREB-regulated transcription coactivator 1 enhances CREB-dependent gene expression in spinal cord to maintain the bone cancer pain in mice. Mol Pain. 2016:12. doi: 10.1177/1744806916641679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang X, et al. miR-22 has a potent anti-tumour role with therapeutic potential in acute myeloid leukaemia. Nat Commun. 2016;7:11452. doi: 10.1038/ncomms11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fang M, et al. The CREB Coactivator CRTC2 Is a Lymphoma Tumor Suppressor that Preserves Genome Integrity through Transcription of DNA Mismatch Repair Genes. Cell Rep. 2015;11:1350–1357. doi: 10.1016/j.celrep.2015.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alberini CM, Chen DY. Memory enhancement: consolidation, reconsolidation and insulin-like growth factor 2. Trends Neurosci. 2012;35:274–283. doi: 10.1016/j.tins.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim R, et al. Molecular mechanisms for the destabilization and restabilization of reactivated spatial memory in the Morris water maze. Mol Brain. 2011;4:9. doi: 10.1186/1756-6606-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 84.Xue ZC, et al. CREB-regulated transcription coactivator 1: important roles in neurodegenerative disorders. Sheng Li Xue Bao. 2015;67:155–162. [PubMed] [Google Scholar]
- 85.Breuillaud L, et al. Deletion of CREB-regulated transcription coactivator 1 induces pathological aggression, depression-related behaviors, and neuroplasticity genes dysregulation in mice. Biol Psychiatry. 2012;72:528–536. doi: 10.1016/j.biopsych.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 86.Meylan EM, et al. The HDAC inhibitor SAHA improves depressive-like behavior of CRTC1-deficient mice: Possible relevance for treatment-resistant depression. Neuropharmacology. 2016;107:111–121. doi: 10.1016/j.neuropharm.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ch’ng TH, et al. Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell. 2012;150:207–221. doi: 10.1016/j.cell.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ch’ng TH, et al. Cell biological mechanisms of activity-dependent synapse to nucleus translocation of CRTC1 in neurons. Front Mol Neurosci. 2015;8:48. doi: 10.3389/fnmol.2015.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nonaka M, et al. Region-specific activation of CRTC1-CREB signaling mediates long-term fear memory. Neuron. 2014;84:92–106. doi: 10.1016/j.neuron.2014.08.049. [DOI] [PubMed] [Google Scholar]
- 90.Parra-Damas A, et al. Crtc1 activates a transcriptional program deregulated at early Alzheimer’s disease-related stages. J Neurosci. 2014;34:5776–5787. doi: 10.1523/JNEUROSCI.5288-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bliss TV, CGL A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 92.Martin SJ, et al. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 93.Zhou Y, et al. Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS One. 2006;1:e16. doi: 10.1371/journal.pone.0000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. 2004;22:123– 131. doi: 10.1080/08977190410001723308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu H, et al. Transducer of regulated CREB and late phase long-term synaptic potentiation. FEBS J. 2007;274:3218–3223. doi: 10.1111/j.1742-4658.2007.05891.x. [DOI] [PubMed] [Google Scholar]
- 96.Sekeres MJ, et al. Increasing CRTC1 function in the dentate gyrus during memory formation or reactivation increases memory strength without compromising memory quality. J Neurosci. 2012;32:17857–17868. doi: 10.1523/JNEUROSCI.1419-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Uchida S, et al. CRTC1 Nuclear Translocation Following Learning Modulates Memory Strength via Exchange of Chromatin Remodeling Complexes on the Fgf1 Gene. Cell Rep. 2017;18:352–366. doi: 10.1016/j.celrep.2016.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hirano Y, et al. Fasting launches CRTC to facilitate long-term memory formation in Drosophila. Science. 2013;339:443–446. doi: 10.1126/science.1227170. [DOI] [PubMed] [Google Scholar]
- 99.Clancy DJ, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 100.Hirano Y, et al. Shifting transcriptional machinery is required for long-term memory maintenance and modification in Drosophila mushroom bodies. Nat Commun. 2016;7:13471. doi: 10.1038/ncomms13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.España J, et al. β-Amyloid Disrupts Activity-Dependent Gene Transcription Required for Memory through the CREB Coactivator CRTC1. J Neurosci. 2010;30:9402–9410. doi: 10.1523/JNEUROSCI.2154-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mendioroz M, et al. CRTC1 gene is differentially methylated in the human hippocampus in Alzheimer’s disease. Alzheimers Res Ther. 2016;8:15. doi: 10.1186/s13195-016-0183-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huntington’s Disease Society of America. www.hdsa.org.
- 104.Chaturvedi RK, et al. Transducer of regulated CREB-binding proteins (TORCs) transcription and function is impaired in Huntington’s disease. Hum Mol Genet. 2012;21:3474–3488. doi: 10.1093/hmg/dds178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jeong H, et al. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med. 2011;18:159–165. doi: 10.1038/nm.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chaturvedi RK, et al. Impaired PGC-1a function in muscle in Huntington’s disease. Hum Mol Genet. 2009;18:3048–3065. doi: 10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cui L, et al. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 108.Deng H, et al. Signal integration by Ca(2+) regulates intestinal stem-cell activity. Nature. 2015;528:212–217. doi: 10.1038/nature16170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ham S, et al. Immunolocalisation of aromatase regulators liver kinase B1, phosphorylated AMP-activated protein kinase and cAMP response element-binding protein-regulated transcription co-activators in the human testis. Reprod Fertil Dev. 2016 doi: 10.1071/RD15390. [DOI] [PubMed] [Google Scholar]
- 110.Amelio AL, et al. A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc Natl Acad Sci U S A. 2007;104:20314–20319. doi: 10.1073/pnas.0707999105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cornelissen G, Otsuka K. Chronobiology of Aging: A Mini-Review. Gerontology. 2016 doi: 10.1159/000450945. [DOI] [PubMed] [Google Scholar]
- 112.Mattis J, Sehgal A. Circadian Rhythms, Sleep, and Disorders of Aging. Trends Endocrinol Metab. 2016;27:192–203. doi: 10.1016/j.tem.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen CY, et al. Effects of aging on circadian patterns of gene expression in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2016;113:206–211. doi: 10.1073/pnas.1508249112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nakamura TJ, et al. The suprachiasmatic nucleus: age-related decline in biological rhythms. J Physiol Sci. 2016;66:367–374. doi: 10.1007/s12576-016-0439-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Altman BJ. Cancer Clocks Out for Lunch: Disruption of Circadian Rhythm and Metabolic Oscillation in Cancer. Front Cell Dev Biol. 2016;4:62. doi: 10.3389/fcell.2016.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Blakeman V, et al. Circadian clocks and breast cancer. Breast Cancer Res. 2016;18:89. doi: 10.1186/s13058-016-0743-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Panda S. Circadian physiology of metabolism. Science. 2016;354:1008–1015. doi: 10.1126/science.aah4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sakamoto K, et al. Clock and light regulation of the CREB coactivator CRTC1 in the suprachiasmatic circadian clock. J Neurosci. 2013;33:9021–9027. doi: 10.1523/JNEUROSCI.4202-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jagannath A, et al. The CRTC1-SIK1 pathway regulates entrainment of the circadian clock. Cell. 2013;154:1100–1111. doi: 10.1016/j.cell.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim M, et al. CRTC Potentiates Light-independent timeless Transcription to Sustain Circadian Rhythms in Drosophila. Sci Rep. 2016;6:32113. doi: 10.1038/srep32113. [DOI] [PMC free article] [PubMed] [Google Scholar]