

Abstract
The COP9 signalosome removes Nedd8 modifications from the Cullin subunits of ubiquitin ligase complexes, reducing their activity. Here, we show that mutations in the Drosophila COP9 signalosome subunit 1b (CSN1b) gene increase the activity of ubiquitin ligases that contain Cullin 1. Analysis of CSN1b mutant phenotypes revealed a requirement for the COP9 signalosome to prevent ectopic expression of Epidermal growth factor receptor (EGFR) target genes. It does so by protecting Capicua, a transcriptional repressor of EGFR target genes, from EGFR pathway-dependent ubiquitylation by a Cullin 1/SKP1-related A/Archipelago E3 ligase and subsequent proteasomal degradation. The CSN1b subunit also maintains basal Capicua levels by protecting it from a separate mechanism of degradation that is independent of EGFR signaling. As a suppressor of tumor growth and metastasis, Capicua may be an important target of the COP9 signalosome in cancer.
KEY WORDS: Capicua, COP9 signalosome, Drosophila melanogaster, EGFR, Nedd8, Ubiquitin ligase, Wing disc
Summary: Mutations in the COP9 signalosome component CSN1b promote degradation of the tumor suppressor Capicua, by both a cullin-mediated EGFR-dependent mechanism and by an alternative route.
INTRODUCTION
Regulation of protein stability is an important feature of many signaling pathways. Crucial pathway components can be constitutively degraded in the absence of the signal, or signaling can induce the removal of inhibitors of pathway function. Protein stability is often regulated by post-translational modifications that affect protein recognition by ubiquitin ligases. Polyubiquitylation targets these proteins for degradation by the 26S proteasome. Compared with reversible post-translational modifications, such as phosphorylation, the regulation of signaling pathways by protein degradation is long lasting and irreversible (Hunter, 2007; Lim et al., 2013).
Ubiquitin ligases themselves are also regulated by post-translational modifications. One such modification is addition of the small ubiquitin-like protein Nedd8 to Cullin proteins, the scaffolding subunits of Cullin-RING E3 ubiquitin ligase complexes (CRLs) (Merlet et al., 2009). Neddylation is required for the activity of these complexes, as it induces a conformational change that allows the RING domain of Rbx1 (Roc1a in Drosophila) more flexibility to catalyze ubiquitin transfer (Boh et al., 2011; Saha and Deshaies, 2008). However, deneddylation of CRLs in the absence of substrate is thought to prevent their auto-ubiquitylation and to allow adaptor subunit exchange (Pierce et al., 2013; Saha and Deshaies, 2008; Wu et al., 2005; Zhou et al., 2003). The COP9 signalosome (CSN), which catalyzes deneddylation, is an essential factor in this modification cycle (Lyapina et al., 2001). CSN is a nine-subunit complex (Rozen et al., 2015) that is evolutionarily related to the lid of the 26S proteasome; its enzymatic activity resides in the CSN5 subunit (Cope et al., 2002; Pick and Bramasole, 2014). The CSN may also inhibit Cullin-based ubiquitin ligases non-catalytically, by directly binding to them and occluding sites for interaction with the substrate and ubiquitin-conjugating E2 enzyme (Enchev et al., 2012). However, recent structural and kinetic analyses show that deneddylated CRLs rapidly dissociate from the CSN (Cavadini et al., 2016; Mosadeghi et al., 2016). Other reported functions of the CSN and its associated proteins include phosphorylation, direct transcriptional regulation and de-ubiquitylation (Chamovitz, 2009; Wei and Deng, 2003). These activities make the CSN a major regulator of diverse cellular processes that include the cell cycle, the DNA damage response, gene expression and transduction of signals that depend on CRL activity, such as Hedgehog (Hh) and Wingless (Wg) (Wu et al., 2011; Schutz et al., 2012).
Epidermal growth factor receptor (EGFR) signaling controls cell growth, differentiation and survival in numerous contexts, and its misregulation contributes to many forms of cancer (Appert-Collin et al., 2015). Ligand binding to the EGFR leads to the activation of a kinase cascade, which culminates in phosphorylation of transcription factors by Mitogen-activated protein kinase (MAPK) (McKay and Morrison, 2007). MAPK phosphorylation activates transcriptional activators of EGFR target genes, including ETS family members, such as Pointed (Pnt) in Drosophila, and inactivates repressors, including the ETS protein Yan/TEL and the HMG-box protein Capicua (Cic) (Jimenez et al., 2012; O'Neill et al., 1994). Inactivation of Cic by EGFR signaling is essential to specify wing vein formation, which is prefigured by expression of argos (aos) in the wing imaginal disc (Roch et al., 2002). In the eye disc, EGFR signaling has a dual role, inactivating Cic to promote growth and activating Pnt to induce photoreceptor differentiation (Tseng et al., 2007; Yang and Baker, 2003). The human Cic homolog CIC acts as a suppressor of tumorigenesis and metastasis, consistent with a conserved function in growth regulation (Bettegowda et al., 2011; Dissanayake et al., 2011; Okimoto et al., 2016). In addition, CIC interactions with the corepressor Ataxin-1 may underlie the neurodegeneration defects in spinocerebellar ataxia (Lam et al., 2006; Lasagna-Reeves et al., 2015).
The mechanism by which receptor tyrosine kinase (RTK) signaling inactivates Cic is not fully understood. Activated MAPK phosphorylates Cic after binding to its conserved C2 domain; phosphorylation of potential target sites in human CIC interferes with binding of its nuclear localization signal to importin-α (Astigarraga et al., 2007; Dissanayake et al., 2011; Futran et al., 2015). Consistent with this model, EGFR signaling results in cytoplasmic localization of Cic (Astigarraga et al., 2007), accompanied in some tissues by its degradation (Roch et al., 2002). It has been proposed that the primary event in Cic downregulation by the embryonic RTK Torso is its exclusion from the nucleus, as degradation occurs primarily in the cytoplasm (Grimm et al., 2012). However, inactivation of the repressor activity of Cic appears to precede its relocalization, at least in the early embryo (Lim et al., 2013). It is not known whether cytoplasmic localization or protein degradation is essential to neutralize Cic activity in response to RTK signaling.
We identified mutations in CSN1b, which encodes a subunit of the COP9 signalosome, based on phenotypes that reflected reduced levels of Cullin 1 (Cul1) substrates in the Hh and Wg signaling pathways (Janody et al., 2004). These data suggest that failure of deneddylation leads to increased Cul1 activity (Wu et al., 2011). In the wing disc, cells mutant for CSN1b or other CSN subunits misexpressed EGFR target genes. We found that the CSN restricts EGFR target gene expression by protecting Cic from ubiquitylation and proteasomal degradation. Although Cic degradation in CSN4 or CSN5 mutant cells requires its interaction with MAPK, CSN1b also protects Cic from another mechanism of degradation that is independent of MAPK, thus controlling its level of expression in the absence of RTK signaling. These results provide evidence for functional heterogeneity among the CSN subunits.
RESULTS
CSN1b mutations affect eye and wing development
In a genetic mosaic screen for genes required for normal photoreceptor differentiation (Janody et al., 2004), we recovered two non-complementing lethal alleles, T12 and T942, that had the same mutant phenotype. Clones of cells homozygous for either mutation disrupted normal eye patterning and did not survive in the adult eye. In the third larval instar eye imaginal disc, homozygous mutant clones showed reduced levels of the pan-neuronal marker Elav, which labels photoreceptors forming posterior to the morphogenetic furrow (Fig. 1B,C). Genetic mapping followed by sequencing of candidate genes in the mapped region identified likely null mutations in the CSN1b gene for both alleles. T12 had a 122 bp deletion after V64, leading to a frameshift and early stop codon, whereas T942 had a nonsense mutation at K366 (Fig. 1A). CSN1b encodes a subunit of the COP9 signalosome (CSN), a complex that removes the ubiquitin-like peptide Nedd8 from Cullins, which can form functional ubiquitin ligase complexes only when neddylated (Cope and Deshaies, 2003; Lyapina et al., 2001; Wei and Deng, 2003).
Fig. 1.

CSN1b mutations affect multiple signaling pathways. (A) Diagram of the CSN1b gene, indicating the positions of the intron, the T12 deletion and the T942 nonsense mutation. (B-F) Third instar eye discs (B,C) and third instar wing discs (D-F) containing CSN1bT12 (B,D) or CSN1bT942 (C,E,F) clones marked by the absence of GFP (D′″, green in B-F) on a wild-type (B) or Minute (C-F) background. Anterior is to the left and dorsal up in this and all subsequent figures. (B) Staining is with anti-Elav (B′, blue in B) and anti-β-galactosidase reflecting dpp-lacZ (red in B). (C) Staining is with anti-Elav (C′, red in C) and anti-Ato (blue in C). In CSN1b clones, Elav expression is reduced and delayed, dpp-lacZ is strongly reduced, but Ato is unaffected. (D) Staining is with anti-β-galactosidase reflecting dpp-lacZ (D′, red in D) and anti-Ptc (D″, blue in D). (E) Staining is with anti-β-galactosidase reflecting Dll-lacZ (E′, magenta in E). (F) Staining is with anti-β-galactosidase reflecting wg-lacZ (F′, magenta in F). dpp-lacZ and Dll-lacZ are strongly reduced, wg-lacZ at the wing margin is expanded (although expression at the wing hinge, which is Notch-independent, is unaffected) and Ptc is unaffected in CSN1b clones. This is consistent with a requirement for CSN1b to activate low-threshold targets of Hh and Wg signaling and to inhibit Notch signaling. n≥10 discs for all stainings shown in this and subsequent figures.
Several conserved signaling pathways were affected in CSN1b mutant clones. A reporter for decapentaplegic (dpp) expression, dpp-lacZ, was lost from the morphogenetic furrow in the eye disc, indicating a likely defect in Hh signaling (Fig. 1B). Expression of dpp-lacZ at the anterior-posterior compartment boundary of the wing disc was also greatly diminished, although patched (ptc), a target gene that requires high levels of Hh signaling, was still expressed (Fig. 1D). Other CSN subunits were previously reported to have similar effects on the expression of dpp in the wing disc and of Elav in the eye disc (Suh et al., 2002; Wu et al., 2011), supporting the conclusion that the CSN1b phenotype is attributable to a loss of CSN function. CSN1b may also affect the Wg signaling pathway in the wing disc. Expression of Distalless-lacZ (Dll-lacZ), a target gene reporter activated by low levels of Wg signaling, was reduced in CSN1b mutant clones in wing discs despite increased levels of the ligand Wg at the wing margin, where it is regulated by Notch signaling, a pathway known to be affected by CSN subunits (Mummery-Widmer et al., 2009) (Fig. 1E,F).
The effect of loss of CSN function on Hh signaling was attributed to increased neddylation of the Cul1-Slimb ubiquitin ligase, which enhances its ability to cleave the transcription factor Cubitus interruptus (Ci) and convert it from an activator of dpp expression into a repressor (Wu et al., 2011). Enhanced ubiquitylation of the Wg pathway transcription factor β-catenin/Armadillo (Arm) by the Cul1-Slimb complex, leading to its degradation (Jiang and Struhl, 1998), could likewise explain the reduced expression of Dll in CSN1b mutant clones. These results suggest that in vivo, deneddylation by the CSN reduces the activity of CRL complexes.
The COP9 signalosome restricts the expression of EGFR target genes
EGFR signaling is crucial for Drosophila wing vein development. In the late third instar larval wing imaginal disc, the EGFR pathway activates target genes such as argos (aos) and kekkon (kek) in cells that will differentiate into wing veins, forming a stereotypical striped pattern (Roch et al., 2002) (Fig. 2A). In CSN1b clones, we observed ectopic expression of aos-lacZ, a phenotype that had not previously been described for CSN subunits (Fig. 2B,C). This ectopic aos expression was not simply attributable to the presence of two copies of the aos-lacZ transgene within the mutant clone, as clones homozygous for aos-lacZ expressed aos only within its normal domain (Fig. S1A). To determine whether aos misregulation in CSN1b clones was attributable to the loss of CSN function, we examined aos-lacZ expression in clones mutant for other CSN subunits. Cells mutant for either CSN4 or CSN5, the catalytic subunit, also showed ectopic expression of aos-lacZ (Fig. 2D,E) and kek-lacZ (Fig. S1B). Accordingly, adult wings containing clones mutant for any of the three subunits developed extra wing veins (Fig. 2G-J; Fig. S1D). Cultured S2 cells stably expressing EGFR (D2F cells) produce Aos in response to treatment with the ligand Spitz (Spi) (Schweitzer et al., 1995). Knocking down CSN1b or CSN5 by RNA interference (RNAi) in these cells increased both basal and Spi-induced Aos levels (Fig. 2K) and transcription of the EGFR target gene pnt-P1 (Fig. S1C), confirming a requirement for CSN subunits to restrict EGFR target gene expression in several contexts.
Fig. 2.
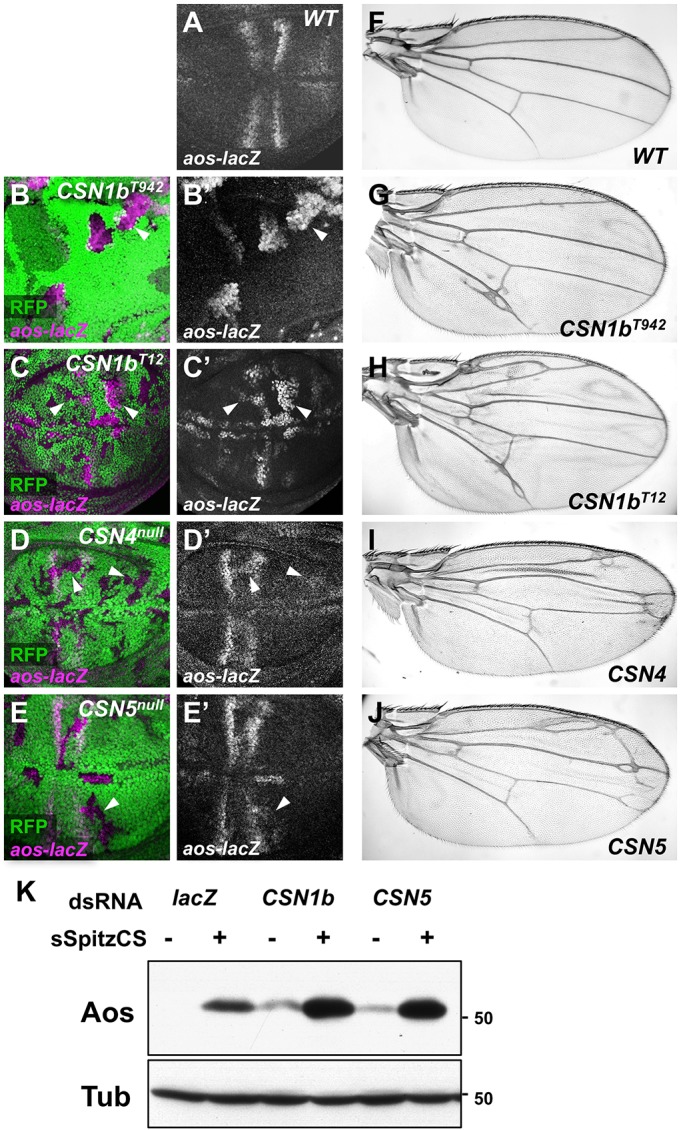
CSN subunits are required for EGFR signaling. (A-E) Wing discs stained with anti-β-galactosidase reflecting aos-lacZ (A,B′-E′, magenta in B-E). (A) Wild type (WT). (B-E) Clones homozygous for CSN1bT942 (B), CSN1bT12 (C), CSN4null (D) or CSN5null (E) are marked by the absence of RFP (green in B-E). aos is misexpressed in clones mutant for all three CSN subunits. Note that the aos-lacZ transgene is on the same chromosome arm as CSN1b, and is therefore not present in the wild-type twin spots in (B,C). Arrowheads indicate representative clones. (F-J) Adult wings that are wild type (F) or contain clones mutant for CSN1bT942 (G), CSN1bT12 (H), CSN4null (I) or CSN5null (J). Loss of CSN subunits results in extra wing veins. (K) Lysates of D2F cells treated (+) or not treated (−) with a purified soluble form of the EGFR ligand Spitz (Miura et al., 2006) and with dsRNA targeting lacZ, CSN1b or CSN5 as indicated, blotted with antibodies to Aos and β-tubulin. Knocking down CSN subunits increases both basal and Spitz-induced Aos levels. n≥3 for all western blots shown in this and subsequent figures.
The COP9 signalosome promotes Capicua accumulation
Upon binding of the ligand Spi to the EGFR, the GTP-bound form of Ras activates the protein kinase Raf, initiating a phosphorylation cascade. This cascade culminates with the phosphorylation of MAPK, which can then translocate into the nucleus and phosphorylate specific transcription factors to regulate target gene expression (see Fig. 6C) (McKay and Morrison, 2007). To determine the level at which EGFR signaling is affected by loss of CSN function, we examined MAPK phosphorylation using a phospho-specific antibody. No ectopic phospho-MAPK was detected in CSN1b or CSN4 clones (Fig. S2A,B). Additionally, knocking down CSN1b or CSN5 in D2F cells using double stranded RNA (dsRNA) did not increase MAPK phosphorylation (Fig. S2C). The increase in target gene expression despite unchanged MAPK phosphorylation suggests that COP9 exerts its effects on one or more transcription factors downstream of MAPK.
Fig. 6.

CSN subunits protect Cic from EGFR-dependent and -independent modes of degradation. (A,B) Wing discs expressing UAS-CicΔC2-HA (HA stained in A′,B′, blue in A,B) in the dorsal domain using ap-GAL4, with clones mutant for CSN5 (A) or CSN1bT12 DroncI29 (B) marked by the absence of RFP (A′″,B′″, red in A,B). Anti-β-galactosidase staining reflects aos-lacZ (A″,B″, green in A,B). CicΔC2 is stable throughout the dorsal domain and can repress aos-lacZ in CSN5 clones, but is degraded and fails to repress aos-lacZ in CSN1b clones. Arrows point to representative clones. (C) Diagram showing a model for the effects of CSN subunits on Cic. The CSN promotes Cic stabilization by deneddylating Cul1 and reducing the ability of a Cul1-SkpA-Ago complex to ubiquitinate Cic. CSN1b also protects Cic from a MAPK-independent mode of degradation.
Phosphorylation by MAPK can change the stability or activity of transcription factors. In the wing disc, MAPK promotes aos expression by binding to and phosphorylating the transcriptional repressor Cic, inhibiting its function and triggering its degradation (Fig. 3A) (Astigarraga et al., 2007; Futran et al., 2015; Roch et al., 2002). This makes Cic a good candidate to mediate the effect of the CSN on EGFR signaling. Indeed, we found that Cic protein levels were reduced in CSN1b, CSN4 and CSN5 clones, in regions of the wing pouch in which the EGFR pathway is not strongly active (Fig. 3B-D). Quantification of Cic levels in the mutant clones relative to adjacent wild-type tissue showed that Cic staining was reduced by ∼20% in CSN-depleted cells (Fig. 3H). However, measuring Cic levels in heterozygous cic/+ tissue, in which the level of background staining observed in homozygous cic mutant clones could be subtracted, revealed that the reduction in CSN clones is likely to be closer to 50% (Fig. S3). We examined whether this reduction had functional consequences. The CUASC-lacZ reporter for Cic repression, in which four Cic binding sites are introduced into UAS-lacZ (Ajuria et al., 2011), is expressed only in prospective veins when driven throughout the wing pouch in a wild-type background (Fig. 3E), but showed increased expression in cells mutant for CSN4 (Fig. 3F). Cic has also been shown to regulate EGFR-mediated growth in the eye disc by repressing cyclin E (cycE) expression (Tseng et al., 2007). CSN5 clones had higher levels of CycE than wild-type tissue, suggesting that the CSN regulates Cic levels in this tissue also (Fig. 3G). Notably, CycE protein is a target of Cul1 degradation (Ou et al., 2002), so its increased transcription must be robust enough to counteract the reduction in protein stability caused by increased Cul1 activity in CSN5 clones. By contrast, the CSN was not essential for EGFR-induced photoreceptor differentiation (Fig. 1B,C), which is mediated by activation of the Pnt-P2 transcription factor rather than relief of Cic repression (Yang and Baker, 2003). Overall, these results suggest that the CSN promotes Cic stabilization.
Fig. 3.

CSN subunits stabilize Cic. (A-G) Wing discs (A-F) and an eye disc (G) that are wild type (A,E) or contain clones mutant for CSN1bT942 (B), CSN4null (C,F) or CSN5null (D,G), marked by the absence of RFP (green in B-D,F) or GFP (green in G). Discs are stained with antibodies to Cic (A′-D′,E, green in A, magenta in B-D), β-galactosidase reflecting aos-lacZ (magenta in A) or CUASC-lacZ driven by C5-GAL4 (E′,F′, magenta in F) or CycE (G′, magenta in G). Cic levels are reduced, whereas CUASC-lacZ and CycE, targets of Cic repression, are increased in the absence of CSN subunits. Representative clones are marked by arrows. (H) Quantification of Cic levels in wild-type, CSN1b, CSN4, CSN5 or Cul1 clones compared with adjacent wild-type tissue. Box and whiskers plot shows median bounded by minimum, first quartile, third quartile and maximum. WT, n=27 clones in nine wing discs; CSN1b, n=73 clones in eight discs; CSN4, n=29 clones in 14 discs; CSN5, n=43 clones in 15 discs; cul1, n=78 clones in 15 discs; ***P<0.005 and ****P<0.0001 by one-way ANOVA.
Cul1-mediated ubiquitylation of Cic targets it for proteasomal degradation
The Cic protein is known to be destabilized by receptor tyrosine kinase signaling in the wing disc and embryo (Grimm et al., 2012; Roch et al., 2002), but the mechanism of its degradation has not been determined. Cullins, the scaffold subunits of CRLs, are substrates for deneddylation by the CSN. We therefore hypothesized that upon phosphorylation by MAPK, Cic is ubiquitinated by a CRL and targeted for proteasomal degradation. To test this, we determined whether Cic could be stabilized by blocking the proteasome or Cullins. In S2 cells, knocking down CSN1b using dsRNA reduced the levels of transfected HA-Cic driven from a UAS promoter, and Cic levels were restored by the proteasome inhibitor MG132 (Fig. 4A) (Lee and Goldberg, 1998). This indicates that loss of CSN induces Cic degradation in a proteasome-dependent manner. In the presence of MG132, an HA-tagged form of Cic could be co-immunoprecipitated with Myc-tagged ubiquitin (Fig. 4B), suggesting that ubiquitylation targets Cic for proteasomal degradation. In vivo, wing discs treated with MLN4924, which inhibits the Nedd8 activating enzyme and thus prevents neddylation of all Cullins (Soucy et al., 2009), or with Suramin, which inhibits CRLs by preventing their interactions with E2 ubiquitin conjugating enzymes (Wu et al., 2016), showed increased Cic levels in the regions where it is normally degraded in response to EGFR signaling (Fig. 4C,D; Fig. S4A).
Fig. 4.

Cic is targeted for degradation by a Cul1-Ago ubiquitin ligase. (A) Lysates from D2F cells co-transfected with Actin-GAL4, UAS-HA-Cic, UAS-GFP and dsRNA targeting luciferase or CSN1b, and treated with the indicated concentrations of MG132. Western blots with anti-HA, anti-Arm, anti-β-tubulin and anti-GFP are shown. The proteasome inhibitor MG132 stabilizes Arm and restores Cic stability in the absence of CSN1b. (B) Lysates of cells treated with MG132 and transfected with ubiquitin-Myc, with or without Cic-HA, were immunoprecipitated using anti-HA beads. Lysates and immunoprecipitates are blotted for anti-HA and anti-Myc. Immunoprecipitated Cic is ubiquitinated in these conditions. (C,D) Wing discs expressing cic-GFP from the endogenous promoter and treated with DMSO (C) or 50 µM MLN4924 in DMSO (D) and stained with anti-Ci (C,D) and anti-GFP (C′,D′). The neddylation inhibitor MLN4924 stabilizes both Ci in the anterior and Cic in the wing vein primordia (arrowheads). (E) Cul1EX clones are marked by the absence of RFP (green) and stained for Cic (E′, blue in E) and CUASC-lacZ driven by C5-GAL4 (E″, red in E). (F) ago1 clones are marked by the absence of RFP (F″, green in F) and stained for Cic (F′, magenta in F). Arrows indicate representative clones. (G) ago1 clones are marked by the absence of RFP (green) in a disc in which RasV12 is expressed in the dorsal domain (above the yellow arrowheads), causing destabilization of Cic (G′, magenta in G) except within the ago clones. White arrows indicate clones in which Cic is stabilized.
Examination of mutations and RNAi lines targeting the six Drosophila Cullins identified Cul1 as the most likely regulator of Cic stability. Cul1EX clones in EGFR-responsive regions of the wing disc failed to degrade Cic (Fig. 3H; Fig. 4E; Fig. S4B), leading to loss of some wing veins in the adult (Fig. S4G). Consistent with this, cells lacking SKP1-related A (SkpA), the subunit that links Cul1 to substrate-binding F-box proteins (Bocca et al., 2001), also showed accumulation of Cic (Fig. S4C). To identify the F-box subunit that binds Cic, we screened RNAi lines by expressing them in the dorsal domain of the wing disc under the control of apterous (ap)-GAL4. Only archipelago (ago) (Moberg et al., 2001) RNAi increased Cic-GFP levels in the wing vein primordia (Fig. S4D-F). ago1 mutant clones in EGFR-responsive regions also failed to degrade Cic (Fig. 4F). When Cic phosphorylation and degradation was induced throughout the dorsal wing disc by expressing activated Ras with ap-GAL4, ago mutant clones still accumulated Cic (Fig. 4G). Ago/Fbw7 recognizes the phosphodegron pTPPXS (Welcker and Clurman, 2008), a sequence that is present at position 910-914 of the Cic-PA isoform. Together, these data indicate that the SCFAgo complex is responsible for ubiquitinating Cic and targeting it for proteasomal degradation.
CSN1b protects Cic from a MAPK-independent mechanism of degradation
If the ubiquitylation and proteasomal degradation of Cic in the absence of CSN are triggered by MAPK-mediated phosphorylation, they should require EGFR signaling. Indeed, the reduction in Cic protein and ectopic expression of aos are most consistent in the central region of the wing disc between the two endogenous aos stripes, where EGFR pathway activity is likely to be highest. We took two approaches to block both EGFR signaling and CSN activity. First, we found that the ectopic induction of aos-lacZ caused by CSN6 RNAi expression in the dorsal wing disc (Fig. 5A,B) was completely blocked by coexpression of mapk RNAi (Fig. 5C,D). Our second epistasis experiment used mutations in Ras85D, an essential EGFR pathway component (Diaz-Benjumea and Hafen, 1994). Like single Ras85D mutant clones (Fig. 5F) but opposite to single CSN5 mutant clones (Fig. 5E), Ras85D CSN5 double mutant clones showed decreased aos-lacZ expression and strong Cic accumulation (Fig. 5G). These results indicate that reduced Cic stability and ectopic aos expression in the absence of CSN function require input from the EGFR signaling pathway. Of note, Cic levels increased in Ras85D and Ras85D CSN5 double mutant clones even outside the prospective vein stripes, indicating that low level EGFR signaling throughout the wing pouch reduces Cic levels sufficiently to allow cell growth. These low levels of EGFR activity make Cic stability dependent on the CSN.
Fig. 5.

EGFR signaling is required for Cic degradation in the absence of CSN subunits other than CSN1b. (A-D) Wing discs in which aos-lacZ is stained with X-gal. ap-GAL4 drives no RNAi construct (A), CSN6 RNAi (B), mapk RNAi (C) or CSN6 RNAi and mapk RNAi (D). Increased aos expression in the absence of CSN6 requires MAPK. (E-G) Wing discs with clones mutant for CSN5 (E), Ras85D (F) or CSN5 and Ras85D (G), marked by the absence of RFP (green). Anti-β-galactosidase reflecting aos-lacZ (E′,F′,G′, red in E-G) and anti-Cic (E″,F″,G″, blue in E-G) are shown. Yellow arrows point to representative clones. Double mutant clones show reduced aos-lacZ and increased Cic, like Ras85D clones and opposite to CSN5 clones. (H) Wing disc with CSN1bT12 DroncI29 clones marked by the absence of RFP (green), expressing mapk RNAi in the dorsal compartment under the control of ap-GAL4, stained for anti-β-galactosidase reflecting aos-lacZ (H′, red in H) and Cic (H″, blue in H). mapk RNAi blocks aos-lacZ expression and Cic degradation in wild-type cells, but not in CSN1b clones (arrow).
The requirement for EGFR pathway activity upstream of aos expression in cells lacking CSN4 or CSN5 function could indicate that only Cic that has been phosphorylated by MAPK requires the CSN for its stability. To test this, we used a form of Cic that lacks the C2 domain necessary for interaction with MAPK (Andreu et al., 2012; Astigarraga et al., 2007). CicΔC2 fully blocked both endogenous aos expression and ectopic aos expression within CSN5 mutant clones, and its stability was unaffected by loss of CSN5 (Fig. 6A). Preventing phosphorylation of Cic by MAPK thus renders it independent of the deneddylation function of the CSN. By contrast, in CSN1b clones CicΔC2 was degraded and did not prevent misexpression of aos (Fig. 6B; Fig. S5A,B). Consistent with this result, endogenous Cic could be degraded and aos expressed in CSN1b clones even when EGFR-induced Cic degradation was blocked by mapk RNAi (Fig. 5H). Both experiments suggest that CSN1b has a function separate from CSN5 that allows it to protect Cic from a MAPK-independent mechanism of degradation. We found that CicΔC2 was also ubiquitinated in S2 cells, providing additional evidence for MAPK-independent degradation of Cic (Fig. S5C). In addition, Myc-tagged CSN1b co-immunoprecipitated with both Cic and CicΔC2 (Fig. S5D), suggesting that direct binding could contribute to its ability to protect Cic from degradation.
DISCUSSION
Our analysis of the CSN1b subunit of the COP9 signalosome has uncovered both a new function for the entire complex, in regulating EGFR signaling through Cullin-mediated control of Cic stability, and an independent protective effect of CSN1b that sets the level of the Cic repressor in the absence of a signal. These findings emphasize the complex effects of the CSN as a modulator of protein degradation.
Regulation of Capicua degradation
MAPK phosphorylation of Cic has been shown to relocalize it to the cytoplasm, and in the wing disc and embryo, this results in its degradation (Astigarraga et al., 2007; Dissanayake et al., 2011; Grimm et al., 2012). CIC degradation has been assigned a crucial role in the metastasis of lung and gastric adenocarcinomas (Okimoto et al., 2016). However, the mechanism by which Cic is targeted for degradation was previously unknown. Our data showing that Cul1, SkpA and the F-box protein Ago negatively regulate Cic levels, and that inhibition of CRLs or the proteasome stabilizes Cic, argue that MAPK phosphorylation targets Cic for ubiquitylation by the SCFAgo complex and subsequent proteasomal degradation (Fig. 6C). The ubiquitin modification itself may be sufficient to block Cic activity even before its degradation; the moderate reduction in Cic protein we observed in CSN mutant cells is similar to the reduction in cic heterozygous cells, which is not sufficient for ectopic aos expression (Fores et al., 2015). These findings suggest that drugs that target CRLs, such as MLN4924 or suramin, might be useful in treating cancers for which CIC is a suppressor of growth or metastasis (Bettegowda et al., 2011; Choi et al., 2015; Dissanayake et al., 2011; Nawrocki et al., 2012; Okimoto et al., 2016; Wu et al., 2016). Conversely, drugs that antagonize the CSN might help reduce the accumulation of toxic Cic-Ataxin complexes in spinocerebellar ataxia (Lam et al., 2006; Lasagna-Reeves et al., 2015).
Our data extend previous observations reporting distinct modes of Cic regulation. In ovarian follicle cells, EGFR activation leads to cytoplasmic localization of phosphorylated Cic, but not to its degradation (Astigarraga et al., 2007). We found that the subcellular distribution of Cic and expression of its target genes were unaffected in CSN1b or CSN5 mutant follicle cells (data not shown), suggesting that Cic is not ubiquitinated, and therefore does not require CSN function, in this tissue. Conversely, our data show that Cic stability can be regulated independently of MAPK and of the C2 domain that is required for MAPK binding (Astigarraga et al., 2007; Futran et al., 2015), through a mechanism that is specifically antagonized by CSN1b. The competition between CSN1b and this alternative mode of degradation would establish the basal level of Cic in the absence of RTK activity. The other highly conserved and essential domains of Cic, the high-mobility group (HMG)-box DNA-binding domain and the C1 motif (Astigarraga et al., 2007), might influence its stability in unstimulated cells. The kinase Minibrain (Mnb) was recently found to phosphorylate Cic on its N-terminus, inhibiting its repressive activity (Yang et al., 2016). Mnb and its adaptor Wings apart do not affect Cic stability, but this finding hints that other mechanisms for Cic regulation may remain to be discovered.
Deneddylase-independent functions for CSN1b
Our analysis confirms that CSN1b, which had not previously been studied in vivo, shares functions with CSN4, CSN5 and CSN8 in eye development (Cope et al., 2002; Suh et al., 2002), Hedgehog signaling (Wu et al., 2011) and Notch signaling (Mummery-Widmer et al., 2009). However, we find that CSN1b differs from other CSN subunits in its requirement to stabilize unphosphorylated Cic. CSN1b mutant clones are also smaller and rounder in shape than CSN4 or CSN5 clones (Figs 2, 3, 6), and are difficult to recover without inhibiting cell death by removing Dronc, suggesting that this subunit may have additional CSN-independent functions. Various CSN subcomplexes and free subunits have been proposed to function independently of the CSN (Dubiel et al., 2015); for instance, CSN2 and CSN7 can act as transcriptional regulators (Dressel et al., 1999; Singer et al., 2014). Flies mutant for CSN4, CSN5 and CSN8 show different phenotypes in vivo, although this could reflect differential stability of their maternally provided gene products (Oren-Giladi et al., 2008; Oron et al., 2002, 2007). The CSN5 subunit contains the active site for deneddylation and is the last to assemble into the CSN (Lingaraju et al., 2014). Although the other subunits are thought to have primarily structural roles, CSN1, CSN2 and CSN4 also form contacts with subunits and substrates of CRLs (Lee et al., 2013; Lingaraju et al., 2014). During Drosophila oogenesis, Bag of marbles has been reported to bind to and sequester CSN4, altering the function of the CSN (Pan et al., 2014).
A mass spectrometry study in S2R+ cells did not detect CSN1b in a complex with the other CSN subunits (Guruharsha et al., 2011), although this result is difficult to reconcile with the central position of CSN1 in the human and fungal signalosomes (Beckmann et al., 2015; Lingaraju et al., 2014). The C-terminal Proteasome-COP9 complex-Initiation factor 3 (PCI) domain of CSN1 is sufficient for its incorporation into the CSN, whereas the N-terminal domain has been assigned other functions (Wang et al., 2002), including transcriptional repression (Tsuge et al., 2001), regulation of JNK activity (Li et al., 2007; Tsuge et al., 2011) and interactions with the calcium-binding protein TSA1 (Li et al., 2011) and inositol 1,3,4-trisphosphate 5/6-kinase (Sun et al., 2002). The C-terminal tail of human CSN1 also directly interacts with IκB (Lee et al., 2013). We likewise detected a physical interaction between CSN1b and Cic, consistent with a previous mass spectrometry study (Yang et al., 2016), suggesting that direct binding may underlie the effect of CSN1b on Cic stability. Interestingly, Drosophila has a second CSN1 subunit, CSN1a, which lacks the PCI domain and is primarily expressed in the testis (Flybase), and human CSN1 has multiple alternatively spliced isoforms and can be phosphorylated (Fang et al., 2008; Fuzesi-Levi et al., 2014). Differently spliced or modified forms of CSN1 could influence CSN deneddylase activity or mediate functions of CSN1 independent of the rest of the complex. Our CSN1b mutations will be useful tools for further analysis of the relationship of this subunit to the COP9 signalosome.
MATERIALS AND METHODS
Drosophila genetics
The T12 and T942 mutations were identified in the screen described by Janody et al. (2004). The lethality of the two mutations was mapped by recombination with P(w+) elements, using the FRT80 site on the chromosome as an additional marker, and was localized 0.06 cM distal to P{EPgy2}CG18135EY07333. Homozygous CSN1b clones were generated by crossing CSN1b, FRT80/TM6B or aos-lacZ, CSN1b, FRT80/TM6B or DroncI29, aos-lacZ, CSN1b, FRT80/TM6B or UAS-CicΔC2HA; DroncI29, aos-lacZ, CSN1b, FRT80/SM6-TM6B or UAS-mapk RNAi; DroncI29, aos-lacZ, CSN1b, FRT80/SM6-TM6B males to ey-FLP; Ubi-GFP, FRT80 or ey-FLP; M(3)67C, Ubi-GFP, FRT80 or hs-FLP; M(3)67C, Ubi-GFP, FRT80/TM6B or Ubx-FLP; His2AvRFP, FRT80 or tsh-GAL4, UAS-FLP; His2AvRFP, FRT80/SM6-TM6B or Ubx-FLP; ap-GAL4/CyO; His2AvRFP, FRT80/TM6c females. CSN4 clones were generated by crossing FRT42, CSN4null; aos-lacZ/SM6-TM6B males to Ubx-FLP; FRT42, Ubi-RFPNLS or ey-FLP; FRT42, Ubi-RFPNLS or Ubx-FLP; FRT42, P(y+) females. CSN5 clones were generated by crossing aos-lacZ, FRT82, CSN5null/TM6B or aos-lacZ, FRT82, Ras85DΔC40B, CSN5null/TM6B or UAS-CicΔC2HA; FRT82, CSN5null/TM6B males to Ubx-FLP; FRT82, Ubi-RFPNLS or UbxFLP; ap-GAL4/CyO; FRT82, Ubi-RFPNLS/TM6c females. Cul1 clones were generated by crossing FRT42, Cul1EX; C5-GAL4, CUASC-lacZ/SM6-TM6B males to Ubx-FLP; FRT42, Ubi-RFPNLS females. ago clones were generated by crossing ago1, FRT80/TM6B or UAS-rasV12; ago1, FRT80/SM6-TM6B males to Ubx-FLP; His2AvRFP, FRT80 or tsh-GAL4, UAS-FLP; His2AvRFP, FRT80/SM6-TM6B or Ubx-FLP; ap-GAL4/CyO; His2AvRFP, FRT80/TM6c females. SkpA clones were generated by crossing SkpA7D9, FRT19A/FM6, P(Tb, RFP) females (Legent et al., 2012) to hs-FLP, FRT19, Ubi-RFPNLS males. cic clones were generated by crossing FRT82, cicQ474X/TM6B males to hs-FLP; FRT82, Ubi-GFP females and heat shocking for 1 h at 37°C in first and second instar. aos-lacZ, dpp-lacZ, Dll-lacZ and wg-lacZ reporters are described in Flybase. The F-box RNAi screen was performed by crossing UAS-RNAi males to ap-GAL4, cic-GFP; UAS-dcr2/SM6-TM6B females and examining Cic-GFP expression in third instar wing discs. UAS-RNAi lines were obtained from the Bloomington Drosophila Stock Center and the Vienna Drosophila Resource Center. Cic-GFP is a C-terminally GFP- and FLAG-tagged form of Cic expressed from its endogenous promoter, made by recombineering a BAC transgene (http://flybase.org/reports/FBrf0220060.html).
Immunohistochemistry and western blotting
Eye and wing discs were stained as described by Hazelett et al. (1998). Antibodies used were rat anti-Elav [1:100; Developmental Studies Hybridoma Bank (DSHB), 7E8A10], chicken anti-GFP (1:300; Aves, GFP-1020), mouse anti-β-galactosidase (1:10; DSHB, 40-1a), rabbit anti-Ato (Jarman et al., 1995), mouse anti-Ptc (1:10; DSHB, Apa1), rat anti-Ci (Motzny and Holmgren, 1995), guinea pig anti-Cic (Tseng et al., 2007), mouse anti-CycE (1:1; Richardson et al., 1995), rat anti-HA (1:100; Roche, 3F4) and mouse anti-dpERK (1:100; Sigma, M8159; signal amplified using the TSA Cyanine 5 System from PerkinElmer). Fluorescent secondary antibodies were from Jackson ImmunoResearch (1:200; guinea pig Cy3, 706-165-148; rabbit Cy3, 711-165-152; mouse Cy3, 715-165-151; rat Cy3, 712-165-153; mouse Cy5, 715-175-151; guinea pig DyLight 649, 706-496-148; rat Cy5, 712-175-153; rabbit Cy5, 711-175-152) or Life Technologies (1:1000; mouse Alexa 488, A-21202; rabbit Alexa 488, A21206; chicken Alexa 488, A11039; rat Alexa 488, A-21208; guinea pig Alexa 488, A-11073), and images were obtained using a Leica SP5 confocal microscope. Quantification of Cic signal intensity was performed using ImageJ by measuring average signal intensity in RFP-negative clones outside the prospective wing veins, compared with adjacent RFP-positive regions of the same size. In discs containing cic mutant clones, the intensity in these homozygous clones was subtracted as background. To block CRLs, wing discs were dissected in PBS and incubated in Schneider's complete media supplemented with 50 µM MLN4924 or 500 µm Suramin for 5 h at room temperature, before fixing and staining. To make whole fly protein extracts, 20 individuals were lysed in 100 µl RIPA buffer supplemented with EDTA-free cOmplete protease inhibitor (Roche), 5 mM NaF and 1 mM Na3VO4. The lysate was then cleared by centrifugation and an equal volume of 2× Laemmli buffer was added. For western blots, the samples were boiled for 5 min at 95°C and loaded on 8% SDS-PAGE gels. The gels were transferred onto nitrocellulose membranes (Bio-Rad), which were blocked for 30 min in TBST [20 mM Tris (pH 7.6), 137 mM NaCl, 0.2% Tween-20] supplemented with 5% low-fat milk, before incubation with primary antibodies overnight at 4°C in TBST with 5% milk. Blots were washed with TBST for 30 min and incubated with horseradish peroxidase-conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch; rabbit HRP, 711-035-152; rat HRP, 712-035-153; mouse HRP, 715-035-151; chicken HRP, 703-035-155) for 1 h in TBST plus 5% milk. Blots were developed with enhanced chemiluminescence (Thermo SuperSignal WestPico). Primary antibodies used were as follows: rat anti-HA (1:1000; Roche, 3F4), mouse anti-Arm (1:100; DSHB, N2 7A1), mouse anti-β-tubulin (1:10,000; Sigma, T4026), mouse anti-Argos (1:50, DSHB; 85/2/16), chicken anti-GFP (1:10,000; Aves, GFP-1020), mouse anti-Myc (1:1000; Cell Signaling, 9B11), mouse anti-phospho-MAPK (1:1000; Sigma, M8159) and rabbit anti-MAPK (1:20,000; Sigma, M5670).
Molecular biology
The pUAST-CicHA and pUAST-CicΔC2HA plasmids were a gift from Gerardo Jimenez (Astigarraga et al., 2007). pUAST-Ub-Myc was a gift from Hyung Don Ryoo (NYU School of Medicine, NY, USA). To make the Myc-CSN1b construct, the CSN1b coding region was amplified by PCR from wild-type adult genomic DNA using the following primers: 5′-TTGGGAATTCCAAAAGCAGAAGCTGATCTCCGAGGAGGACCTGATGCCCGTGCTGCCGAT-3′ and 5′-ATCCTCTAGACTAGATTCGGGCGAGTTGAGCAG-3′. The amplicon was cloned into pUASTattB as a EcoRI-XbaI fragment.
Cell culture and immunoprecipitation
S2 cells and D2F cells were maintained in Schneider's medium supplemented with 10% fetal calf serum. D2F cells (Schweitzer et al., 1995) were provided by Erika Bach (NYU School of Medicine, NY, USA), and induction of EGFR by Cu2SO4 treatment was confirmed by western blotting. These cells were additionally treated with 150 µg/ml G418. dsRNAs were generated using the MEGAScript T7 and T3 kit (Ambion) as described by Worby et al. (2001). We chose dsRNA templates using the DRSC database (www.flyrani.org) for the genes CSN1b (DRSC11108) and CSN5 (DRSC22381) and amplified them from wild-type fly genomic DNA. For luciferase dsRNA, we used the primers FWD 5′-TCGTCACATCTCATCTACCTCCC-3′ and REV 5′-ATGGAACAACTTTACCGACCGC-3′ and amplified our dsRNA template from the TOP-Flash plasmid. Total RNA was extracted using Trizol (Invitrogen). RT-PCR was performed on 1 µg of total RNA using the Invitrogen SuperScript First-Strand Kit. Primer sequences are available on request.
To look at endogenous protein and mRNA levels, 106 cells/well were treated with 15 µg dsRNA. When assessing exogenous protein levels, 106 cells/well were transfected with 0.9 µg total plasmid DNA and 0.9 µg dsRNA using Effectene (Qiagen). Four days later, cells were harvested and lysed in ice-cold RIPA buffer supplemented with EDTA-free cOmplete protease inhibitor (Roche), 5 mM NaF and 1 mM Na3VO4. Laemmli buffer was added. To activate the EGFR pathway, EGFR production was induced on the previous day for 3 h with 60 µM CuSO4 before adding purified sSpiCS to the medium overnight at a concentration empirically determined to induce Aos expression (Miura et al., 2006).
For immunoprecipitation, the transfection was scaled up to 6 cm plates, and cells were harvested and lysed in ice-cold lysis buffer [75 mM NaCl, 50 mM Tris-HCl pH 7.5, 1 mM EDTA, 0.2% NP-40, 5 mM NaF, 1 mM Na3VO4, EDTA-free cOmplete protease inhibitor (Roche)]. Lysates were incubated for 1 h with anti-HA affinity matrix (Roche 3F10). Washes were performed using 75 mM NaCl, 10 mM Tris-HCl pH 7.5, 1 mM EDTA, 5 mM NaF, 1 mM Na3VO4, EDTA-free cOmplete protease inhibitor (Roche) and precipitates were eluted with 2× Laemmli buffer.
Acknowledgements
We thank Erika Bach, Daniel Chamovitz, Cheng-Ting Chien, Iswar Hariharan, Andrew Jarman, Gerardo Jimenez, Qiuling Li, Ken Moberg, Hyung Don Ryoo, the Bloomington Drosophila Stock Center, the Vienna Drosophila Resource Center and the Developmental Studies Hybridoma Bank for fly stocks and reagents. We are grateful to Hui Hua Liu, Juhee Pae and Tony Huang for technical assistance and advice. The manuscript was improved by the critical comments of Jessica Douthit, Cheuk Hei Ho, Carolyn Morrison, Nicholas Stavropoulos and Josefa Steinhauer.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: A.S., K.L., J.E.T.; Validation: A.S., D.H., J.E.T.; Investigation: A.S., D.H., K.L., J.E.T.; Writing - original draft: A.S., J.E.T.; Writing - review & editing: D.H., K.L.; Visualization: A.S., J.E.T.; Supervision: J.E.T.; Project administration: J.E.T.; Funding acquisition: J.E.T.
Funding
This work was supported by the National Institutes of Health (EY13777 to J.E.T.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.148767.supplemental
References
- Ajuria L., Nieva C., Winkler C., Kuo D., Samper N., Andreu M. J., Helman A., Gonzalez-Crespo S., Paroush Z., Courey A. J. et al. (2011). Capicua DNA-binding sites are general response elements for RTK signaling in Drosophila. Development 138, 915-924. 10.1242/dev.057729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu M. J., Ajuria L., Samper N., Gonzalez-Perez E., Campuzano S., Gonzalez-Crespo S. and Jimenez G. (2012). EGFR-dependent downregulation of Capicua and the establishment of Drosophila dorsoventral polarity. Fly 6, 234-239. 10.4161/fly.21160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appert-Collin A., Hubert P., Cremel G. and Bennasroune A. (2015). Role of ErbB receptors in cancer cell migration and invasion. Front. Pharmacol. 6, 283 10.3389/fphar.2015.00283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astigarraga S., Grossman R., Diaz-Delfin J., Caelles C., Paroush Z. and Jimenez G. (2007). A MAPK docking site is critical for downregulation of Capicua by Torso and EGFR RTK signaling. EMBO J. 26, 668-677. 10.1038/sj.emboj.7601532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann E. A., Kohler A. M., Meister C., Christmann M., Draht O. W., Rakebrandt N., Valerius O. and Braus G. H. (2015). Integration of the catalytic subunit activates deneddylase activity in vivo as final step in fungal COP9 signalosome assembly. Mol. Microbiol. 97, 110-124. 10.1111/mmi.13017 [DOI] [PubMed] [Google Scholar]
- Bettegowda C., Agrawal N., Jiao Y., Sausen M., Wood L. D., Hruban R. H., Rodriguez F. J., Cahill D. P., McLendon R., Riggins G. et al. (2011). Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 333, 1453-1455. 10.1126/science.1210557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocca S. N., Muzzopappa M., Silberstein S. and Wappner P. (2001). Occurrence of a putative SCF ubiquitin ligase complex in Drosophila. Biochem. Biophys. Res. Commun. 286, 357-364. 10.1006/bbrc.2001.5394 [DOI] [PubMed] [Google Scholar]
- Boh B. K., Smith P. G. and Hagen T. (2011). Neddylation-induced conformational control regulates cullin RING ligase activity in vivo. J. Mol. Biol. 409, 136-145. 10.1016/j.jmb.2011.03.023 [DOI] [PubMed] [Google Scholar]
- Cavadini S., Fischer E. S., Bunker R. D., Potenza A., Lingaraju G. M., Goldie K. N., Mohamed W. I., Faty M., Petzold G., Beckwith R. E. et al. (2016). Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 531, 598-603. 10.1038/nature17416 [DOI] [PubMed] [Google Scholar]
- Chamovitz D. A. (2009). Revisiting the COP9 signalosome as a transcriptional regulator. EMBO Rep. 10, 352-358. 10.1038/embor.2009.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi N., Park J., Lee J.-S., Yoe J., Park G. Y., Kim E., Jeon H., Cho Y. M., Roh T.-Y. and Lee Y. (2015). miR-93/miR-106b/miR-375-CIC-CRABP1: a novel regulatory axis in prostate cancer progression. Oncotarget 6, 23533-23547. 10.18632/oncotarget.4372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope G. A. and Deshaies R. J. (2003). COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell 114, 663-671. 10.1016/S0092-8674(03)00722-0 [DOI] [PubMed] [Google Scholar]
- Cope G. A., Suh G. S., Aravind L., Schwarz S. E., Zipursky S. L., Koonin E. V. and Deshaies R. J. (2002). Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 298, 608-611. 10.1126/science.1075901 [DOI] [PubMed] [Google Scholar]
- Diaz-Benjumea F. J. and Hafen E. (1994). The sevenless signalling cassette mediates Drosophila EGF receptor function during epidermal development. Development 120, 569-578. [DOI] [PubMed] [Google Scholar]
- Dissanayake K., Toth R., Blakey J., Olsson O., Campbell D. G., Prescott A. R. and MacKintosh C. (2011). ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor Capicua. Biochem. J. 433, 515-525. 10.1042/BJ20101562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressel U., Thormeyer D., Altincicek B., Paululat A., Eggert M., Schneider S., Tenbaum S. P., Renkawitz R. and Baniahmad A. (1999). Alien, a highly conserved protein with characteristics of a corepressor for members of the nuclear hormone receptor superfamily. Mol. Cell. Biol. 19, 3383-3394. 10.1128/MCB.19.5.3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiel D., Rockel B., Naumann M. and Dubiel W. (2015). Diversity of COP9 signalosome structures and functional consequences. FEBS Lett. 589, 2507-2513. 10.1016/j.febslet.2015.06.007 [DOI] [PubMed] [Google Scholar]
- Enchev R. I., Scott D. C., da Fonseca P. C., Schreiber A., Monda J. K., Schulman B. A., Peter M. and Morris E. P. (2012). Structural basis for a reciprocal regulation between SCF and CSN. Cell Rep. 2, 616-627. 10.1016/j.celrep.2012.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L., Wang X., Yamoah K., Chen P.-L., Pan Z.-Q. and Huang L. (2008). Characterization of the human COP9 signalosome complex using affinity purification and mass spectrometry. J. Proteome Res. 7, 4914-4925. 10.1021/pr800574c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fores M., Ajuria L., Samper N., Astigarraga S., Nieva C., Grossman R., Gonzalez-Crespo S., Paroush Z. and Jimenez G. (2015). Origins of context-dependent gene repression by capicua. PLoS Genet. 11, e1004902 10.1371/journal.pgen.1004902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futran A. S., Kyin S., Shvartsman S. Y. and Link A. J. (2015). Mapping the binding interface of ERK and transcriptional repressor Capicua using photocrosslinking. Proc. Natl. Acad. Sci. USA 112, 8590-8595. 10.1073/pnas.1501373112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuzesi-Levi M. G., Ben-Nissan G., Bianchi E., Zhou H., Deery M. J., Lilley K. S., Levin Y. and Sharon M. (2014). Dynamic regulation of the COP9 signalosome in response to DNA damage. Mol. Cell. Biol. 34, 1066-1076. 10.1128/MCB.01598-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm O., Sanchez Zini V., Kim Y., Casanova J., Shvartsman S. Y. and Wieschaus E. (2012). Torso RTK controls Capicua degradation by changing its subcellular localization. Development 139, 3962-3968. 10.1242/dev.084327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruharsha K. G., Rual J.-F., Zhai B., Mintseris J., Vaidya P., Vaidya N., Beekman C., Wong C., Rhee D. Y., Cenaj O. et al. (2011). A protein complex network of Drosophila melanogaster. Cell 147, 690-703. 10.1016/j.cell.2011.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelett D. J., Bourouis M., Walldorf U. and Treisman J. E. (1998). decapentaplegic and wingless are regulated by eyes absent and eyegone and interact to direct the pattern of retinal differentiation in the eye disc. Development 125, 3741-3751. [DOI] [PubMed] [Google Scholar]
- Hunter T. (2007). The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730-738. 10.1016/j.molcel.2007.11.019 [DOI] [PubMed] [Google Scholar]
- Janody F., Lee J. D., Jahren N., Hazelett D. J., Benlali A., Miura G. I., Draskovic I. and Treisman J. E. (2004). A mosaic genetic screen reveals distinct roles for trithorax and Polycomb group genes in Drosophila eye development. Genetics 166, 187-200. 10.1534/genetics.166.1.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarman A. P., Sun Y., Jan L. Y. and Jan Y. N. (1995). Role of the proneural gene, atonal, in formation of Drosophila chordotonal organs and photoreceptors. Development 121, 2019-2030. [DOI] [PubMed] [Google Scholar]
- Jiang J. and Struhl G. (1998). Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature 391, 493-496. 10.1038/35154 [DOI] [PubMed] [Google Scholar]
- Jimenez G., Shvartsman S. Y. and Paroush Z. (2012). The Capicua repressor--a general sensor of RTK signaling in development and disease. J. Cell Sci. 125, 1383-1391. 10.1242/jcs.092965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam Y. C., Bowman A. B., Jafar-Nejad P., Lim J., Richman R., Fryer J. D., Hyun E. D., Duvick L. A., Orr H. T., Botas J. et al. (2006). ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 127, 1335-1347. 10.1016/j.cell.2006.11.038 [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves C. A., Rousseaux M. W., Guerrero-Munoz M. J., Park J., Jafar-Nejad P., Richman R., Lu N., Sengupta U., Litvinchuk A., Orr H. T. et al. (2015). A native interactor scaffolds and stabilizes toxic ATAXIN-1 oligomers in SCA1. Elife 4 10.7554/eLife.07558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. H. and Goldberg A. L. (1998). Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397-403. 10.1016/S0962-8924(98)01346-4 [DOI] [PubMed] [Google Scholar]
- Lee J.-H., Yi L., Li J., Schweitzer K., Borgmann M., Naumann M. and Wu H. (2013). Crystal structure and versatile functional roles of the COP9 signalosome subunit 1. Proc. Natl. Acad. Sci. USA 110, 11845-11850. 10.1073/pnas.1302418110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legent K., Steinhauer J., Richard M. and Treisman J. E. (2012). A screen for X-linked mutations affecting Drosophila photoreceptor differentiation identifies Casein kinase 1 alpha as an essential negative regulator of Wingless signaling. Genetics 190, 601-616. 10.1534/genetics.111.133827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.-Y., Chai B.-X., Zhang W., Liu Y.-Q., Ammori J. B. and Mulholland M. W. (2007). Ankyrin repeat and SOCS box containing protein 4 (Asb-4) interacts with GPS1 (CSN1) and inhibits c-Jun NH2-terminal kinase activity. Cell. Signal. 19, 1185-1192. 10.1016/j.cellsig.2006.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Zang B., Liu C., Lu L., Wei N., Cao K., Deng X. W. and Wang X. (2011). TSA1 interacts with CSN1/CSN and may be functionally involved in Arabidopsis seedling development in darkness. J. Genet. Genomics 38, 539-546. 10.1016/j.jgg.2011.08.007 [DOI] [PubMed] [Google Scholar]
- Lim B., Samper N., Lu H., Rushlow C., Jimenez G. and Shvartsman S. Y. (2013). Kinetics of gene derepression by ERK signaling. Proc. Natl. Acad. Sci. USA 110, 10330-10335. 10.1073/pnas.1303635110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingaraju G. M., Bunker R. D., Cavadini S., Hess D., Hassiepen U., Renatus M., Fischer E. S. and Thoma N. H. (2014). Crystal structure of the human COP9 signalosome. Nature 512, 161-165. 10.1038/nature13566 [DOI] [PubMed] [Google Scholar]
- Lyapina S., Cope G., Shevchenko A., Serino G., Tsuge T., Zhou C., Wolf D. A., Wei N., Shevchenko A. and Deshaies R. J. (2001). Promotion of NEDD8-CUL1 conjugate cleavage by COP9 signalosome. Science 292, 1382-1385. 10.1126/science.1059780 [DOI] [PubMed] [Google Scholar]
- McKay M. M. and Morrison D. K. (2007). Integrating signals from RTKs to ERK/MAPK. Oncogene 26, 3113-3121. 10.1038/sj.onc.1210394 [DOI] [PubMed] [Google Scholar]
- Merlet J., Burger J., Gomes J.-E. and Pintard L. (2009). Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell. Mol. Life Sci. 66, 1924-1938. 10.1007/s00018-009-8712-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura G. I., Buglino J., Alvarado D., Lemmon M. A., Resh M. D. and Treisman J. E. (2006). Palmitoylation of the EGFR ligand Spitz by Rasp increases Spitz activity by restricting its diffusion. Dev. Cell 10, 167-176. 10.1016/j.devcel.2005.11.017 [DOI] [PubMed] [Google Scholar]
- Moberg K. H., Bell D. W., Wahrer D. C., Haber D. A. and Hariharan I. K. (2001). Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature 413, 311-316. 10.1038/35095068 [DOI] [PubMed] [Google Scholar]
- Mosadeghi R., Reichermeier K. M., Winkler M., Schreiber A., Reitsma J. M., Zhang Y., Stengel F., Cao J., Kim M., Sweredoski M. J. et al. (2016). Structural and kinetic analysis of the COP9-Signalosome activation and the cullin-RING ubiquitin ligase deneddylation cycle. Elife 5 10.7554/eLife.12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzny C. K. and Holmgren R. (1995). The Drosophila Cubitus interruptus protein and its role in the Wingless and Hedgehog signal transduction pathways. Mech. Dev. 52, 137-150. 10.1016/0925-4773(95)00397-J [DOI] [PubMed] [Google Scholar]
- Mummery-Widmer J. L., Yamazaki M., Stoeger T., Novatchkova M., Bhalerao S., Chen D., Dietzl G., Dickson B. J. and Knoblich J. A. (2009). Genome-wide analysis of Notch signalling in Drosophila by transgenic RNAi. Nature 458, 987-992. 10.1038/nature07936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki S. T., Griffin P., Kelly K. R. and Carew J. S. (2012). MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin. Investig. Drugs 21, 1563-1573. 10.1517/13543784.2012.707192 [DOI] [PubMed] [Google Scholar]
- Okimoto R. A., Breitenbuecher F., Olivas V. R., Wu W., Gini B., Hofree M., Asthana S., Hrustanovic G., Flanagan J., Tulpule A. et al. (2016). Inactivation of Capicua drives cancer metastasis. Nat. Genet. 49, 87-96. 10.1038/ng.3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill E. M., Rebay I., Tjian R. and Rubin G. M. (1994). The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell 78, 137-147. 10.1016/0092-8674(94)90580-0 [DOI] [PubMed] [Google Scholar]
- Oren-Giladi P., Krieger O., Edgar B. A., Chamovitz D. A. and Segal D. (2008). Cop9 signalosome subunit 8 (CSN8) is essential for Drosophila development. Genes Cells 13, 221-231. 10.1111/j.1365-2443.2008.01164.x [DOI] [PubMed] [Google Scholar]
- Oron E., Mannervik M., Rencus S., Harari-Steinberg O., Neuman-Silberberg S., Segal D. and Chamovitz D. A. (2002). COP9 signalosome subunits 4 and 5 regulate multiple pleiotropic pathways in Drosophila melanogaster. Development 129, 4399-4409. [DOI] [PubMed] [Google Scholar]
- Oron E., Tuller T., Li L., Rozovsky N., Yekutieli D., Rencus-Lazar S., Segal D., Chor B., Edgar B. A. and Chamovitz D. A. (2007). Genomic analysis of COP9 signalosome function in Drosophila melanogaster reveals a role in temporal regulation of gene expression. Mol. Syst. Biol. 3, 108 10.1038/msb4100150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou C.-Y., Lin Y. F., Chen Y. J. and Chien C. T. (2002). Distinct protein degradation mechanisms mediated by Cul1 and Cul3 controlling Ci stability in Drosophila eye development. Genes Dev. 16, 2403-2414. 10.1101/gad.1011402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L., Wang S., Lu T., Weng C., Song X., Park J. K., Sun J., Yang Z. H., Yu J., Tang H. et al. (2014). Protein competition switches the function of COP9 from self-renewal to differentiation. Nature 514, 233-236. 10.1038/nature13562 [DOI] [PubMed] [Google Scholar]
- Pick E. and Bramasole L. (2014). Moonlighting and pleiotropy within two regulators of the degradation machinery: the proteasome lid and the CSN. Biochem. Soc. Trans. 42, 1786-1791. 10.1042/BST20140227 [DOI] [PubMed] [Google Scholar]
- Pierce N. W., Lee J. E., Liu X., Sweredoski M. J., Graham R. L., Larimore E. A., Rome M., Zheng N., Clurman B. E., Hess S. et al. (2013). Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell 153, 206-215. 10.1016/j.cell.2013.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson H., O'Keefe L. V., Marty T. and Saint R. (1995). Ectopic cyclin E expression induces premature entry into S phase and disrupts pattern formation in the Drosophila eye imaginal disc. Development 121, 3371-3379. [DOI] [PubMed] [Google Scholar]
- Roch F., Jimenez G. and Casanova J. (2002). EGFR signalling inhibits Capicua-dependent repression during specification of Drosophila wing veins. Development 129, 993-1002. [DOI] [PubMed] [Google Scholar]
- Rozen S., Fuzesi-Levi M. G., Ben-Nissan G., Mizrachi L., Gabashvili A., Levin Y., Ben-Dor S., Eisenstein M. and Sharon M. (2015). CSNAP is a stoichiometric subunit of the COP9 signalosome. Cell Rep. 13, 585-598. 10.1016/j.celrep.2015.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A. and Deshaies R. J. (2008). Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol. Cell 32, 21-31. 10.1016/j.molcel.2008.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz A. K., Hennes T., Jumpertz S., Fuchs S. and Bernhagen J. (2012). Role of CSN5/JAB1 in Wnt/beta-catenin activation in colorectal cancer cells. FEBS Lett. 586, 1645-1651. 10.1016/j.febslet.2012.04.037 [DOI] [PubMed] [Google Scholar]
- Schweitzer R., Shaharabany M., Seger R. and Shilo B. Z. (1995). Secreted Spitz triggers the DER signaling pathway and is a limiting component in embryonic ventral ectoderm determination. Genes Dev. 9, 1518-1529. 10.1101/gad.9.12.1518 [DOI] [PubMed] [Google Scholar]
- Singer R., Atar S., Atias O., Oron E., Segal D., Hirsch J. A., Tuller T., Orian A. and Chamovitz D. A. (2014). Drosophila COP9 signalosome subunit 7 interacts with multiple genomic loci to regulate development. Nucleic Acids Res. 42, 9761-9770. 10.1093/nar/gku723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy T. A., Smith P. G., Milhollen M. A., Berger A. J., Gavin J. M., Adhikari S., Brownell J. E., Burke K. E., Cardin D. P., Critchley S. et al. (2009). An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732-736. 10.1038/nature07884 [DOI] [PubMed] [Google Scholar]
- Suh G. S., Poeck B., Chouard T., Oron E., Segal D., Chamovitz D. A. and Zipursky S. L. (2002). Drosophila JAB1/CSN5 acts in photoreceptor cells to induce glial cells. Neuron 33, 35-46. 10.1016/S0896-6273(01)00576-1 [DOI] [PubMed] [Google Scholar]
- Sun Y., Wilson M. P. and Majerus P. W. (2002). Inositol 1,3,4-trisphosphate 5/6-kinase associates with the COP9 signalosome by binding to CSN1. J. Biol. Chem. 277, 45759-45764. 10.1074/jbc.M208709200 [DOI] [PubMed] [Google Scholar]
- Tseng A. S., Tapon N., Kanda H., Cigizoglu S., Edelmann L., Pellock B., White K. and Hariharan I. K. (2007). Capicua regulates cell proliferation downstream of the receptor tyrosine kinase/ras signaling pathway. Curr. Biol. 17, 728-733. 10.1016/j.cub.2007.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuge T., Matsui M. and Wei N. (2001). The subunit 1 of the COP9 signalosome suppresses gene expression through its N-terminal domain and incorporates into the complex through the PCI domain. J. Mol. Biol. 305, 1-9. 10.1006/jmbi.2000.4288 [DOI] [PubMed] [Google Scholar]
- Tsuge T., Menon S., Tong Y. and Wei N. (2011). CSN1 inhibits c-Jun phosphorylation and down-regulates ectopic expression of JNK1. Protein Cell 2, 423-432. 10.1007/s13238-011-1043-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Kang D., Feng S., Serino G., Schwechheimer C. and Wei N. (2002). CSN1 N-terminal-dependent activity is required for Arabidopsis development but not for Rub1/Nedd8 deconjugation of cullins: a structure-function study of CSN1 subunit of COP9 signalosome. Mol. Biol. Cell 13, 646-655. 10.1091/mbc.01-08-0427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei N. and Deng X. W. (2003). The COP9 signalosome. Annu. Rev. Cell Dev. Biol. 19, 261-286. 10.1146/annurev.cellbio.19.111301.112449 [DOI] [PubMed] [Google Scholar]
- Welcker M. and Clurman B. E. (2008). FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8, 83-93. 10.1038/nrc2290 [DOI] [PubMed] [Google Scholar]
- Worby C. A., Simonson-Leff N. and Dixon J. E. (2001). RNA interference of gene expression (RNAi) in cultured Drosophila cells. Sci. STKE 2001, pl1 10.1126/stke.2001.95.pl1 [DOI] [PubMed] [Google Scholar]
- Wu J. T., Lin H. C., Hu Y. C. and Chien C. T. (2005). Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nat. Cell Biol. 7, 1014-1020. 10.1038/ncb1301 [DOI] [PubMed] [Google Scholar]
- Wu J.-T., Lin W.-H., Chen W.-Y., Huang Y.-C., Tang C.-Y., Ho M. S., Pi H. and Chien C.-T. (2011). CSN-mediated deneddylation differentially modulates Ci(155) proteolysis to promote Hedgehog signalling responses. Nat. Commun. 2, 182 10.1038/ncomms1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Chong R. A., Yu Q., Bai J., Spratt D. E., Ching K., Lee C., Miao H., Tappin I., Hurwitz J. et al. (2016). Suramin inhibits cullin-RING E3 ubiquitin ligases. Proc. Natl. Acad. Sci. USA 113, E2011-E2018. 10.1073/pnas.1601089113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. and Baker N. E. (2003). Cell cycle withdrawal, progression, and cell survival regulation by EGFR and its effectors in the differentiating Drosophila eye. Dev. Cell 4, 359-369. 10.1016/S1534-5807(03)00059-5 [DOI] [PubMed] [Google Scholar]
- Yang L., Paul S., Trieu K. G., Dent L. G., Froldi F., Fores M., Webster K., Siegfried K. R., Kondo S., Harvey K. et al. (2016). Minibrain and Wings apart control organ growth and tissue patterning through down-regulation of Capicua. Proc. Natl. Acad. Sci. USA 113, 10583-10588. 10.1073/pnas.1609417113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C., Wee S., Rhee E., Naumann M., Dubiel W. and Wolf D. A. (2003). Fission yeast COP9/signalosome suppresses cullin activity through recruitment of the deubiquitylating enzyme Ubp12p. Mol. Cell 11, 927-938. 10.1016/S1097-2765(03)00136-9 [DOI] [PubMed] [Google Scholar]