

ABSTRACT
C-terminal Binding Proteins (CtBP) 1 and 2 are oncogenic transcriptional co-regulators overexpressed in many cancer types, with their expression level correlating to worse prognostic outcomes and aggressive tumor features. CtBP negatively regulates the expression of many tumor suppressor genes, while coactivating genes that promote proliferation, epithelial-mesenchymal transition, and cancer stem cell self-renewal activity. In light of this evidence, the development of novel inhibitors that mitigate CtBP function may provide clinically actionable therapeutic tools. This review article focuses on the progress made in understanding CtBP structure, role in tumor progression, and discovery and development of CtBP inhibitors that target CtBP's dehydrogenase activity and other functions, with a focus on the theory and rationale behind the designs of current inhibitors. We provide insight into the future development and use of rational combination therapy that may further augment the efficacy of CtBP inhibitors, specifically addressing metastasis and cancer stem cell populations within tumors.
KEYWORDS: C-terminal Binding Protein, CtBP, transcriptional co-regulator, corepressor, dehydrogenase, EMT, HIPP, MTOB, oncogene
Abbreviations
- BAX
Bcl-2-associated X protein
- BIK
Bcl-2-interacting killer
- CDH1
E-cadherin
- CtBP
C-terminal Binding Protein
- EMT
epithelial to mesenchymal transition
- E1A
Adenovirus early region 1A
- HDAC
histone deacetylase
- HIPP
Hydroxyimino-3-phenylpropanoic acid
- HMT
histone methyltransferase
- KLF
Krüppel-like factor
- MTOB
2-keto-4-methylthio-2-oxo butyric acid
- NADH
nicotanamide adenine dinucleotide hydride
- PTEN
Phosphatase and tensin homolog
- SUMO
Small Ubiquitin-like Modifier
- TIAM1
T-lymphoma invasion and metastasis-inducing protein 1
Introduction
C-terminal Binding Proteins (CtBP1/2) are evolutionarily conserved metazoan transcriptional co-regulators, with the founding paralog CtBP1 initially identified as a phospho-protein that interacted with the C-terminus of the adenovirus E1A protein.1-3 Extensive genetic studies in model organisms demonstrate that CtBP is required for embryonic development and also adult lifespan. Homozygous loss of'mCtBP2 results in developmental defects and embryonic lethality, while mCtBP1 homozygous loss yields runted offspring with limited lifespan.4,5 In Drosophila, reduction of maternal dCtBP activity caused severely disrupted embryonic segmentation patterning.6-8 Conversely, in Caenorhabditis elegans (C. elegans), inactivation of CtBP either by depletion or mutation resulted paradoxically in extended life span.9,10 In addition to its role in developmental biology, transcriptional co-regulation by CtBP has been found to play a role in several diseases, including cancer.11-14 CtBP promotes a neoplastic phenotype by suppressing tumor suppressor gene expression and by facilitating upregulation of oncogenic factors.15 The aim of this review is to revisit the existing literature relevant to CtBP structure as it correlates to its oncogenic functions, and then to provide prospective on a small but growing body of work characterizing the development of antineoplastic CtBP-targeted pharmacological inhibitors.
CtBP: Domain structure and post translational modifications
In vertebrates, CtBP family proteins are encoded by 2 paralogous genes, CtBP1 and CtBP2. These proteins display redundant functional and structural similarities, although each of them also has many distinct roles.5,16,17 CtBP1 has 2 splice isoforms, CtBP1-L (previously known as CtBP1) and the shorter form CtBP1-S, which splices out exon 1 that encodes the N-terminal 15 amino acids, and localizes predominantly to the cytoplasm.18,19 CtBP1-S is also referred to as CtBP1/BARS (Brefeldin A-ADP Ribosylated Substrate), which has been reported to play a critical role in regulation of lipid storage and golgi membrane fission.20,21 On the other hand, CtBP2 has 3 splice isoforms, namely CtBP2-L, CtBP2-S, and RIBEYE, which is expressed from an alternative promoter and contains an extended N-terminus. RIBEYE is a major structural component of synaptic ribbons that are important for the precise and accurate transmission of sensory signals.19,22
CtBP comprises 3 functional domains (Fig. 1): an N-terminal transcription factor-binding domain, a central dehydrogenase domain, and a C-terminal extension that is further involved in interactions with transcription factors and additionally acts to stabilize substrate binding.
Figure 1.

(a) Domains of CtBP1- Role, functions and post-translational modification sites. R, E and H signify the catalytic triad within the CtBP dehydrogenase domain (b) Graphical representation of CtBP mediated co-repression, where CtBP nucleates a complex that bridges chromatin modifiers (including histone deacteylases [HDAC] and histone methyltransferases [HMT]) to specific DNA sites recognized by a DNA-interacting protein (DNA-IP) that encodes a PxDLS domain.
N-terminus
The transcription factor-binding domain in the N-terminus of CtBP is responsible for CtBP's association with the majority of its known transcription factor and co-repressor partners.15,23,24 This occurs through CtBP recognition of a consensus PxDLS (Pro-x-Asp-Leu-Ser; x is commonly a hydrophobic amino acid) peptide motif in its partner proteins. Studies across different species depict the importance of the PxDLS motif for CtBP binding.25 For example, histone deacetylases such as HDAC4, HDAC5 and HDAC7 contain a PxDLS motif through which CtBP proteins can interact to promote gene silencing.26 Similarly, DNA-binding mammalian Kruppel-like factors (KLF) such as KLF3, KLF8, and KLF12 bind to CtBP via their PVDLS motif.27 Although most interacting partners of CtBP harbor the PxDLS binding motif, it is worth noting that some of them do not contain this sequence, but are still able to interact efficiently either directly, or indirectly through co-association with other components of the corepressor complex. The end result is recruitment of several different complexes that partake in CtBP mediated transcriptional regulation.28,29
Dehydrogenase domain
The dehydrogenase domain of CtBP contains an NADH or nucleotide-binding domain (NBD), a catalytic domain (CD), and an RRT motif that binds to certain transcription factors. The NBD in CtBP was found to share homology with other NADH-dependent D-2-hydroxyacid dehydrogenases (D2-HDH).17 Specifically, CtBP contains a sequence of amino acids that resembles the classic ‘Rossmann Fold’ topology (GXGXXG (17x) D) found in bacterial dehydrogenases that aides in nucleotide binding, and also contains a triad of amino acids (CtBP1/2: His315/321-Glu295/301-Arg266/272; the catalytic triad) that are essential to its substrate-binding and catalytic activities. In the presence of NADH, CtBP undergoes a conformational change that brings the substrate and NBD into close proximity, forming a “closed state,” which is a critical step in formation of CtBP oligomers.30
Oligomerization of CtBP is generally considered to be central to its role as a transcriptional corepressor and has been studied extensively in human and animal models alike.31-33 Increased concentrations of NADH intensify oligomer formation, consequently influencing its ability to associate with transcription factors. Mutations in the NBD diminished CtBP's oligomerization ability and reduced its ability to interact with transcription factors, which further highlights the importance of the NBD in CtBP-mediated transcriptional regulation.34,35 In addition to the mutations in the NBD, the Cadigan and Leiter groups recognized a cluster of homologous mutations outside the NBD in both dCtBP and hCtBP1 which diminished the ability of the protein to oligomerize, importantly reducing transcriptional activity.31,33
In conjunction with the NBD, the CD encodes an enzymatic function that catalyzes reduction of α-keto acids (substrate) to α-hydroxy acids in the presence of NADH (cofactor). Kumar et al, proposed that the catalytic triad acts in unison to facilitate the above conversion, wherein CtBP1 His315 acts as an acid or base catalyst and Glu295 helps in lowering the pKa of this histidine by stabilizing the protonated state. Arg266 facilitates the reaction by polarizing the 2-oxo functional group of the substrate for catalysis.30 Thus, CtBP holds a rare distinction as both a transcriptional co-regulator and active dehydrogenase. Furthermore, the dehydrogenase domain also contains an RRT binding-motif that is functionally similar to the PxDLS interaction domain, specifically recognizing the RRTGXPPXL sequence, featured in some transcription factors, such as RIZ, ZNF217, and others.18,25
Though CtBP has the ability to catalyze substrate reduction, it remains unclear whether this activity is of any relevance for transcriptional coregulation. Although there have been several attempts to dissect the importance of catalysis in CtBP's coregulatory activity, the answers till now have been puzzling at best. For example, Kumar et al. noted that a cluster of mutations in the catalytic domain diminished the protein's ability to associate with its interacting partners.30 In an attempt to unravel the importance of each CtBP domain, Kuppuswamy et al mutated key amino acids that were responsible either for PxDLS binding, nucleotide binding, or catalytic activity. The results from their reporter assay showed that the mutations in each of these domains interfered with CtBP's ability to interact with E1A.36 In contrast, other reports implied that mutations in the catalytic domain did not alter CtBP's activity. For instance, Grooteclaes et al used a combination of pull-down and reporter assays to show that a mutation in the catalytic site (CtBP1-H315Q) had limited impact on CtBP's transcriptional activity.37 Likewise, Madison et al using reporter and pulldown assays suggested that mutations in the NBD and CD did not affect interaction with E1A, whereas mutations in the PxDLS binding domain diminished E1A interaction.35
Importantly, the dehydrogenase and biologic activity of CtBP can be regulated by signaling events in the cell, as Ser158 within the dehydrogenase domain of CtBP1 can be phosphorylated by p21-activated kinase (Pak1; activated by heregulin growth factor (HRG)),38 facilitating CtBP1 localization to the cytoplasm, and thereby downregulating its transcriptional activity. Additionally, Pak1 preferentially phosphorylates CtBP1 bound to NADH, completely blocking its dehydrogenase activity. In addition, AMPK and Akt have also been shown to negatively regulate CtBP1 through phosphorylation at Ser158 and Thr176 respectively, consequently leading to proteasomal degradation.39,40
C-terminus
The C-terminal end of CtBP1/2 (∼90 residues) was initially presumed to be less crucial for its functional activity. This was likely a result of early in vitro studies that were performed without CtBP's C-terminus. However, later studies elucidated that the C-terminus is unstructured due to its proline/glycine-rich sequence, which contributes to conformational disorder and could explain the inability to crystallize full-length CtBP.41 Nevertheless, the C-terminus promotes the formation of CtBP tetramers, as evidenced from chromatographic and X-ray scattering experiments.35,41 Further studies elucidated that the C-terminus encodes domains responsible for PDZ interaction (CtBP1 only) and interaction with the ARF tumor suppressor.35,41-44 Below, we discuss the interactions and modifications of the C-terminus of CtBP in further detail.
Phosphorylation
HIPK2 (Homeodomain Interacting Protein Kinase 2), a serine/threonine kinase known to be activated by checkpoint kinase ATM under genotoxic stress, phosphorylates CtBP1 and CtBP2 at Ser422 and Ser428 respectively, leading to CtBP degradation and induction of cell death.45 An analogous role is played by c-Jun NH2-terminal kinase (JNK1) that phosphorylates CtBP1 at Ser422 in a p53-independent manner.46 On a different note, there are other reports that have indicated that HIPK2 regulates JNK1 activation and apoptosis in cancer cells, by participating in the TGF-β signaling pathway.47 Taken together, there is a strong possibility that HIPK2 plays both direct and indirect roles (via JNK1) in regulating CtBP and CtBP-mediated apoptosis and likely other cellular functions.
SUMOylation
In addition to the phosphorylation site, Lin et al reported the presence of a SUMOylation motif (427-VKPE-430) in the C-terminus of CtBP1, which is SUMOylated by PIAS1 and PIASxβ E3 ligases that regulate both CtBP1 localization and transcriptional co-regulation activities.43,48 Mutation of Lys428 to Arg, which would block SUMOylation, resulted in re-localization of CtBP1 from nucleus to cytoplasm and loss of the protein's transcriptional activity. A similar observation was made by Riefler et al, who described that the SUMOylation of CtBP1 was found to be inhibited by the PDZ domain of nNOS thereby resulting in enhanced cytoplasmic retention.42 Additionally, CtBP1s association with other transcriptional factors via the PxDLS domain was not affected upon mutation of Lys428. Interestingly, this site is not present in CtBP2.43
ARF interaction
An additional level of CtBP regulation is afforded through a negative regulatory interaction of the p14/p19 Alternative Reading Frame (ARF) tumor suppressor with the CtBP C-terminus.44 ARF encodes a tumor suppressor that interacts with CtBP and leads to its proteasome-dependent degradation. The potential functional importance of the ARF/CtBP axis was highlighted by the finding that CtBP1/2 was overexpressed in 65% of colorectal adenocarcinoma specimens, specifically those where ARF was absent, whereas those tumors with high ARF levels showed absent CtBP1/2 expression.44 In addition, depletion of ARF or overexpression of CtBP2 in a p53-null human colon cancer cell line led to the down regulation of PTEN expression, and activation of PI3K signaling and migration. Conversely, exogenous ARF was able to suppress migration of tumor cells by antagonizing CtBP and also sensitize these cells to stress induced apoptosis. Subsequent studies have shown that CtBP repression of Bik and other BH3 only proteins such as Bim and Bmf, may play a critical role in ARF induced p53-independent apoptosis.44,49,50
In summary, the domains of CtBP are universally conserved and carry similar functional activities, namely catalysis and oligomerization, across different species.24 Despite clear understanding about the functions of each domain individually, several major questions remain concerning the specific dependence of CtBP's transcriptional activity on its domains. It still remains to be deciphered whether oligomerization of protein and catalysis are part of a concerted mechanism or whether oligomerization of CtBP alone is essential for coregulation. Hence, a thorough study to disentangle the role of catalysis and oligomerization in CtBP's activity in different systems is highly desirable.
Role of CtBP in cancer
Transforming activity and tumor expression
The notion that CtBP could play a role in tumorigenesis first originated from the studies that involved the human adenovirus type 5 E1A gene.3 E1A can immortalize rodent cells through sequences encoded in its N-terminal exon-1 region, and cooperatively transforms rodent cells when coexpressed with an activated H-Ras allele.51,52 Exon-2 sequences of E1A actually negatively modulate E1A/Ras transformation, and CtBP1 interacts with a PLDLS sequence within E1A exon-2. E1A proteins that are deficient in CtBP interaction either due to mutations in the PLDLS domain, or its deletion, exhibited a hyper-transforming activity in rodent cells, leading to speculation that CtBP1 could possibly be acting as a tumor suppressor. However, it was later found that CtBP1 along with Evi-1, a leukemia oncogene, was able to mediate in vitro transformation of Rat1 cells, suggesting a possible role for CtBP1 in leukemogenesis.53 The early seemingly contradictory findings about CtBP's oncogenic role led to many further studies that have broadened our understanding about CtBP's likely oncogenic role in cancer (reviewed below), and important roles in other diseases as well.11 Indeed, recent data from our laboratory conclusively demonstrate that CtBP2 is a cellular transforming oncogene, cooperating with SV40 viral oncoproteins to oncogenically transform primary mouse and human cells with an efficiency similar to activated H-Ras.54
Highlighting the potential importance of CtBP in human cancer, CtBP1 and 2 overexpression occurs across the spectrum of solid human tumors, including breast, ovarian, prostate, colon, and gastric cancer, and is correlated with worse survival in many cases.55-59 Indeed, analysis of TCGA data demonstrates high frequency recurrent genomic amplification of CtBP1 and 2 in high grade serous ovarian cancer as well as uterine cancer (Fig. 2). To better understand the possible mechanisms by which CtBP contributes to oncogenesis, we review the literature that has demonstrated how CtBP contributes to many of the “hallmarks of cancer”60 such as abnormal cell survival, migration/invasion/metastasis, and disordered cellular metabolism, as well as key oncogenic biologic processes/pathways that include epithelial to mesenchymal transition, (EMT), activation of the Wnt pathway, and promotion of the cancer stem cell phenotype.
Figure 2.
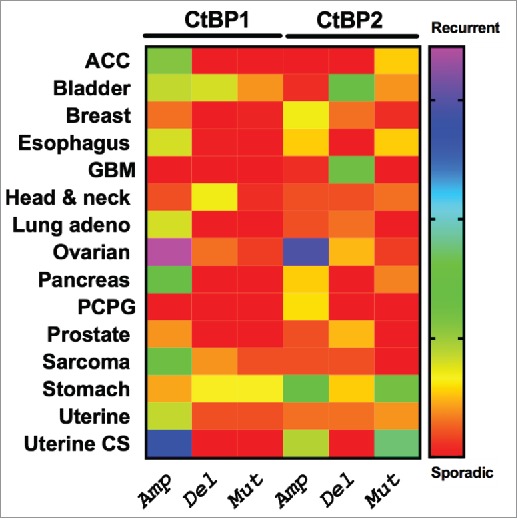
Patterns of genetic alteration of CtBP1 and CtBP2 in human solid tumors. The Cancer Genome Atlas (TCGA) Project data were extracted using cBioportal (http://www.cbioportal.org/public-portal/), and show copy number alterations or mutations in TCGA tumor collections from 15 different cancer types.
Cancer biology of CtBP
Apoptosis
Global expression profiling performed by Frisch and colleagues on mouse embryonic fibroblasts (MEFs) null for Ctbp1 and Ctbp2 expression showed upregulation of many genes regulating programmed cell death such as PERP, p21, Noxa, Bax, etc.37 In light of this seminal finding, multiple reports suggested that cells that evade apoptosis have increased activity of CtBP,58,61 and evading apoptosis or programmed cell death, is one of the key attributes of cancer cells, which also positively influences chemo-resistance. Following on the MEF study, we identified CtBP as an anti-apoptotic protein that negatively regulates genes responsible for cell death in human cancer cells as well. For instance, several pro-apoptotic proteins that belong to Bcl-2 family such as Bik, were upregulated when expression of CtBP was knocked-down using siRNA in human colon cancer cells, and in another instance, depletion of CtBP using siRNA has been found to induce cleavage of caspase-3, a signal that initiates programmed cell death.44,49
EMT
EMT is a critical phenomenon in cancer, during which cells lose their epithelial status and acquire mesenchymal traits.62,63 E-cadherin (CDH1), a molecule involved in intercellular adhesion and maintenance of epithelial status of cells, is downregulated during EMT, along with other markers of epithelial differentiation, such as keratin. As a direct consequence of lowered CDH1, cells tend to disassociate and disseminate to neighboring tissues from the primary tumor site. Indeed, E-cadherin and keratin-8, were both upregulated in CtBP1/2 knockout MEFs, as compared with fibroblasts with wildtype CtBP expression. Analogous to MEFs, cancer cells display an inverse correlation between gene expression of CtBP and CDH1, creating a cogent argument that CtBP could be involved in regulation of EMT.56,64 CtBP represses CDH1 via recruitment to the CDH1 promoter region through CtBP's interaction with the DNA interacting protein ZEB1. Repression is due to CtBP oligomerization and recruitment of chromatin re-modeling enzymes, such as HDACs, HMTs and other proteins, to form a “super-complex” that induces repressive chromatin marks, which eventually is responsible for CDH1 downregulation.15
CtBP-dependent induction of EMT appears regulated and not constitutive, as ERK2, but not ERK1 was found to induce EMT and CDH1 downregulation by phosphorylation of MCRIP1 (MAPK regulated corepressor interacting protein 1), a protein involved in inhibiting the interaction of the CtBP super-complex with ZEB1.65 In the absence of ERK2 activity, MCRIP1 binds to CtBP and inhibits its interaction with ZEB1, thereby preventing downregulation of CDH1.66 Functional crosstalk between CtBP and ERK2, as may occur during MAPK activation during oncogenic transformation may therefore be a key regulator of CtBP-mediated EMT.
Metastasis
Metastasis is one of the direct consequences of EMT, which relates to dissemination of cancer cells to neighboring tissues from the primary tumor site. It is a complex process that involves multiple signaling molecules, including loss of CDH1 expression that facilitates this movement. Several genes that were previously found to be responsible for migration of cancer cells have been found to be direct targets of CtBP. In addition, CtBP's expression and activity has been found to be upregulated in metastatic cancer types.61,67 For example, TIAM1 (T-lymphoma invasion and metastasis-inducing protein 1) and PTEN (Phosphatase and tensin homolog) are 2 genes that play regulatory roles in tumor migration and invasion.68,69 Recently, we have shown that TIAM1 and CtBP2 exhibit a strong positive correlation in their expression levels.70 RNAi knockdown of CtBP2 in colorectal cancer cells downregulated TIAM1, whereas ectopic expression of CtBP2 upregulated TIAM1 expression, indicating that TIAM1 is a direct transcriptional activation target of CtBP. As anticipated, when CtBP2 expression was depleted, the rate of migration of cancer cells was reduced significantly. Likewise, overexpression of CtBP2 in cancer cells activated TIAM1 expression and repressed PTEN expression, thereby activating PI3K/AKT signaling, and increasing the rate of migration.50 Importantly, CtBP-dependent migration was impeded if TIAM1 was depleted by siRNA, demonstrating that TIAM1 is downstream of CtBP and a critical mediator of CtBP-induced cell migration.
Aerobic glycolysis
Another essential characteristic of many cancers are their dependence on glycolytic metabolism.60 This altered metabolism in cancer cells, commonly termed the “Warburg effect,” influences levels of NAD+/NADH, which in turn, directly influences CtBP mediated co-regulation.55 For instance, increasing the ratio of NAD+/NADH in breast cancer cells evicts CtBP from the BRCA1 promoter, elevates histone acetylation and increases BRCA1 transcriptional levels.71 Likewise, hypoxia decreases this ratio, influencing CtBP mediated co-regulation and promoting enhanced migration.63
On the other hand, the NAD+/NADH ratio could be decreased by increasing caloric intake. According to a report by Moiola et al, a high-fat diet (HFD) fed to mice with xenografted orthotopic prostate tumors (PC3 cells), induced hormonal changes such as decreased levels of testosterone, increased levels of cholesterol, increased body weight and also, decreased the intra-tumoral NAD+/NADH ratio, which is responsible for the hyperactivity of CtBP. When control diet (CD) was fed to mice with wild-type CtBP1 (PC3-pGIPZ) or depleted levels of CtBP1 (PC3-shCtBP1), the tumor growth was almost similar. However, when mice were on HFD, there was significant reduction of tumor size in PC3-shCtBP1 mice group. This indicates that despite the presence of low NAD+/NADH ratio in HFD, depleted activity of CtBP1 due to lower levels of the protein negatively influenced tumorigenesis. Additionally, whole-genome expression array performed on tumors with depleted CtBP1 showed that the levels of CDH1 increased, with simultaneous decrease in cyclin D1 expression. These results indicate that CtBP has a significant role to play in cancer progression related to metabolic syndromes.72,73
Wnt signaling
Wnt signaling plays a vital role in developmental and cancer biology. CtBP intersects with Wnt signaling in multiple and complex ways as revealed by work in both model organisms and human cancer cells and tumors. The key effector of Wnt signaling, β-catenin, is degraded by ubiquitination and proteolysis as a result of interaction with the APC complex, which includes scaffolding proteins and kinases such as APC (Adenomatous Polyposis Coli), GSK-3β (glycogen synthase kinase-3b), and Axin-1/2. Oncogenic APC or β-catenin mutations cause β-catenin localization to the nucleus, where it interacts with TCF4/LEF transcription factors causing multiple downstream effects including upregulation of c-Myc, cyclin D1, and several other genes that are associated with tumor initiation and growth, metastasis, and cancer stemness.74
dCtBP plays a context-dependent role in Drosophila Wnt signaling, where it activates as well as represses Wnt target genes, though for unknown reasons.32 In human cancer cells, CtBP associates with the APC destruction complex to sequester levels of β-catenin.75,76 Thus, CtBP contributes toward inhibition of β-catenin's interaction with TCF/LEF and possibly paradoxically acts as a tumor suppressor.77,78 However, APC is often mutated in sporadic colon cancer, yielding an APC protein product that is truncated at the C-terminus. These products undergo oligomerization, resulting in its impaired ability to degrade β-catenin, resulting in β-catenin nuclear localization and interaction with TCF/LEF complex. In this mutant APC setting, CtBP promotes oligomerization of truncated APC by binding to 15 amino acid-repeats,76 facilitating release of β-catenin into the nucleus and activation of downstream oncogenic β-catenin transcriptional targets, such as cyclin D1 (Fig. 3). Additionally, CtBP may directly coactivate TCF4/LEF at key target promoters (c-Myc, LGR5) to promote cancer stem cell self-renewal,79 indicating that CtBP may activate Wnt signaling in cancer cells at multiple nodes- both by interacting with APC and by interaction with TCF4 directly. In further direct support of a role for CtBP in Wnt signaling, our recent work suggests that Ctbp2 haplo-insufficiency rescues polyposis induced by mutation of Apc in the min mouse model of the human disease Familial Adenomatous Polyposis.54
Figure 3.
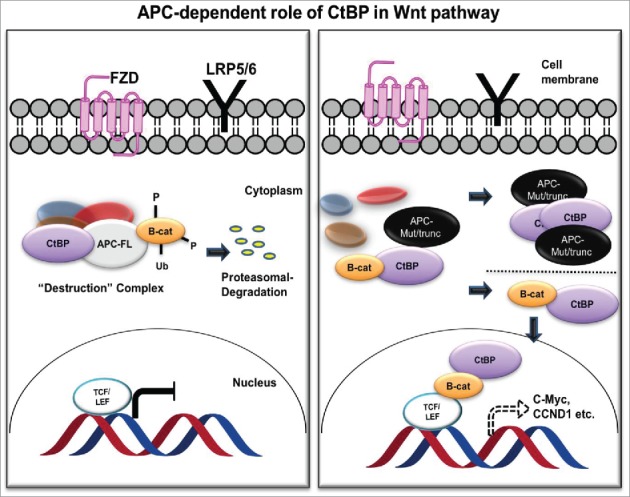
APC-dependent role of CtBP in the Wnt/ β-catenin pathway. In the presence of normal APC (APC-FL; left), CtBP participates in the destruction complex to sequester β-catenin (B-cat) and target it for proteasomal degradation. With mutant APC (APC-Mut/trunc; right), CtBP facilitates oligomerization and cytoplasmic sequestration of APC, promoting β-catenin release to the nucleus to induce downstream TCF/LEF mediated cell signaling, in which CtBP also plays a direct role as a coactivator at TCF target genes.75,76
Cancer Stem Cells
Taking into account the role of CtBP in the Wnt/ β-catenin pathway alongside other factors, it can be expected that CtBP may play a role in the formation of CSCs. In fact, genome-wide profiling by ChIP-seq in breast cancer cells showed CtBP repression of genes that are generally known to be repressed to maintain a stem cell-like phenotype.58 For example, an increased ratio of CD44/CD24 expression corresponds to the enhanced ability of breast cancer cells to form tumors80 and when the levels of CtBP in MCF-7 and MDA-MB-231 cells were depleted using siRNA, the ratio of CD44/CD24 decreased while the ratio increased when CtBP was overexpressed.58 In addition to these breast cancer data demonstrating CtBP regulation of CSC phenotypic markers, CtBP also supports colon CSC self-renewal as noted above, via its interaction with TCF4/LEF.79
CtBP Inhibitors
Based on the totality of human, mouse, and cellular data suggesting that hyperactivity of CtBP mediates aggressive and metastatic neoplastic behavior, 15 we predict that pharmacological inhibition of CtBP could be beneficial in cancer treatment. Recently, several approaches for targeting CtBP have been proposed. These targeting agents, most commonly small molecules, act by inhibiting the enzymatic activity, interfering with dimerization or, inhibiting the interaction of CtBP with other co-regulators (see Table 1). In the following section, we discuss the mechanism of action and biologic effects of each of these inhibitor types in detail.
Table 1.
Name and the chemical structures of CtBP inhibitors.
| Name of the compound | Structure of compound | Mode of action |
|---|---|---|
| MTOB |  |
Substrate at low concentrations, but dehydrogenase inhibitor at high concentrations.86,88 |
| 2-Keto-4-(methylthio)butyric acid | ||
| HIPP derivatives |  |
Dehydrogenase inhibitors.82,89 |
| (2-Hydroxyimino-3-phenyl-propionic acid) | ||
| Cyclic Peptide CP61 |  |
Inhibitor of homo/ hetero-dimerization of CtBP1 and CtBP2.92 |
| (cyclo-SGWTVVRMY) | ||
| NSC95397 |  |
Inhibitor of CtBP interaction with partners such as E1A that contain PxDLS sequences.93 |
Dehydrogenase inhibitors
The structural resemblance of CtBP with 2-hydroxyacid dehydrogenases led to an initial hypothesis that CtBP could also be mimicking their enzymatic activity, wherein NADH is converted to NAD+, in the presence of a substrate.30 A detailed chemical reaction is shown in Fig. 4. Indeed, upon testing recombinant protein for enzymatic activity, CtBP was able to catalyze the conversion of pyruvate (α-keto acid-substrate), to lactate (α-hydroxyacid) in the presence of NADH (cofactor), in a dose-dependent manner.30 In addition, mutations in the catalytic domain of CtBP inhibited the reaction. Further analysis with a library of potential substrates comprised of α-keto acids, showed that CtBP1 was able to reduce many of these substrates at varying efficiencies, but through a “compulsory-order mechanism,” wherein NADH binds first followed by substrate. Among the substrates that were tested, 2-keto-4-methylthiobutyrate (KMTB or MTOB, Table 1), which is reduced to 4-Methylthio-hydroxybutyric acid (MTHB), displayed the highest substrate conversion efficiency (∼80X higher than pyruvate). To achieve such high enzyme efficiency, either a high Kcat (rapid turnover or rate of conversion of substrate) or a small Km (high affinity of enzyme toward substrate) are required.81 Of note for future development of substrate competitive inhibitors based on the MTOB scaffold, MTOB exhibits substrate inhibitor activity in CtBP dehydrogenase reactions when in excess over NADH concentration.82
Figure 4.
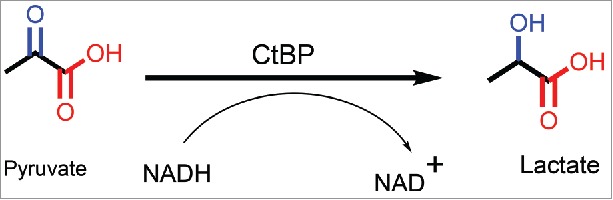
CtBP reduces pyruvate in the presence of NADH to form lactate and NAD+.
MTOB is the penultimate compound in the methionine-salvage pathway, and is converted to methionine in presence of transaminase.83 At low concentrations, MTOB was found to significantly decrease ornithine decarboxylase (ODC) activity, an enzyme upregulated in tumors and found to be a direct transcriptional target of the c-Myc oncogene.84,85 At concentrations above 1mM, there was also a significant amount of cell death in different types of cancer cells, though the mechanism of cell death was unclear.
Based on its properties of substrate inhibition in vitro, we explored the possibility whether MTOB could functionally inhibit CtBP. Cell viability, ChIP, real-time quantitative PCR (RT-QPCR), and Western blot analyses showed that MTOB treatment of colorectal cancer cells modulated BIK and other BH3 gene expression to induce apoptosis by eviction of at least CtBP2 from the BIK promoter.86 As a demonstration of on-target specificity of MTOB action, overexpression of CtBP rescued MTOB cytotoxicity. Additionally, MTOB was also found to be highly effective in vivo, reducing tumor size and increasing survival of immune compromised mice harbouring colon cancer peritoneal xenografts. This was the first description of biologically successful CtBP targeting using a pharmacological agent to inhibit its transcriptional regulation.
Our findings in colon cancer cells were extended to breast cancer cells, as Gardner and colleagues demonstrated in 2 different breast cancer cell lines that changes in mRNA levels of a 30 gene panel were similar between MTOB treatment and CtBP siRNA knockdown.58 These authors further demonstrated that this was due to the ability of MTOB to evict CtBP from the occupied promoter regions, thereby influencing cellular processes such as EMT and breast cancer CSC activity. Taking these results into consideration, MTOB was tested for its influence on the CSC phenotype and specifically spheroid forming ability (the ability of certain cells to form spherical colonies and be propagated as such in suspension culture- a measure of CSC self-renewal capability). As expected, MTOB robustly inhibited spheroid formation and self-renewal abilities in CSCs in a dose-dependent manner in colorectal cancer cell lines with EC50s around 250 μM. Mechanistically, MTOB action was traced to interruption of the binding of CtBP to TCF4/LEF, a known regulator of colon cancer CSCs.79
Despite many existing theories,58,87 how MTOB affects CtBP's activity still remains unclear. Although, it could be hypothesized that MTOB acts as a substrate at a lower concentration, at increasing concentration it assumes the role of allosteric inhibitor and locks the conformation of CtBP, thereby blocking recruitment of other co-repressors and downstream functions. It can also be speculated that excess MTOB affects the methionine-salvage pathway, but whether that is of any relevance to CtBP related pathways is yet to be discovered. Additionally, further studies could be focused on understanding the role of the product of MTOB hydrogenation, MTHB, in MTOB action. Of note, preliminary studies testing MTHB effects on cell viability in colon cancer cells revealed no significant effect (Straza and Grossman, unpublished observation).
With the above results that demonstrate that MTOB could possibly be the first CtBP-specific substrate / inhibitor, we investigated the structural detail of CtBP/ MTOB interaction using X-ray crystallography. From the MTOB/NAD+/CtBP1/2 crystal structures (Fig. 5), we found that despite binding in slightly different conformations, CtBP1 and CtBP2 both bind MTOB in their active site clefts within their substrate-binding domains, and MTOB interacts with catalytically conserved residues, such as Arg 266/272 and His315/321, of the catalytic triad.88 These residues form a scaffold between the coenzyme- and substrate-binding domains and position MTOB adjacent to the nictonamide ring of NADH. CtBP family proteins differ from other D2-dehydrogenases in that they contain a unique tryptophan residue within their active site (Trp 318/324 for CtBP1/2). MTOB is positioned in this active cleft in an orientation such that its thioether is within van der Waals distance from Trp 318/324 and is able to interact with the indolyl ring of this amino acid. This information led to rational designs of second generation inhibitors, which were developed to increase the strength of interaction between protein and inhibitors, simultaneously improving therapeutic efficacy.82,89
Figure 5.

Structure of CtBP/substrate/coenzyme complex. (a) CtBP1 dehydrogenase domain structure (monomer) with domains indicated. Conformations of MTOB (light or dark blue) when interacting with (b) CtBP1 and (c) CtBP2 in presence of NAD+. (Reprinted with permission from Hilbert et al.88 Copyright 2014 John Wiley & Sons, Inc.)
Previous studies have indicated that the CtBP 318/324 tryptophan residue may function as a “keystone,” integral for dimerization, dehydrogenase function, and higher oligomerization of the protein.35 We therefore sought to enhance the binding capacity of the MTOB substrate by replacing the thiol group with a benzene ring that would allow for pi-pi interactions with the catalytic tryptophan. The α-keto acid with this aromatic substitution is phenylpyruvic acid. Testing of this molecule showed a mild increase in inhibition capacity in an enzyme assay that measures the turnover of NADH to NAD+ in the presence of CtBP and MTOB substrate.82 It should be noted that CtBP is known to have a greater than 100-fold affinity for NADH over NAD+, allowing for unidirectional measurement within enzymatic assays. However, phenylpyruvate was not shown to be cytotoxic to cancer cell lines. It was then hypothesized that we could further increase inhibition potential by rendering phenylpyruvate non-reducible. A series of analogs with non-reducible ketone bioisosteres were designed, synthesized, and tested for CtBP inhibition.89 From these analogs based on phenylpyruvate, 2-hydroxyimino-3-phenylpropionic acid (HIPP, Table 1), was identified as a potent CtBP inhibitor. Crystallographic studies of CtBP1 showed that the phenyl-group interaction with W318 in both phenylpyruvate and HIPP increased favorable interactions 2–3-fold over MTOB (Fig. 6).82 Interestingly, the oxime of HIPP does not interact with NAD+, but rather binds in a flipped, noncanonical conformation. This surprising conformation serves to allow HIPP to fully occupy the active site.
Figure 6.

Ball and stick model representing interaction of CtBP1 with substrate MTOB, and substrate competitive inhibitors phenyl pyruvate (PPy) and HIPP (in stereo). (A) The hydrogen bond network of MTOB as reported in Hilbert et al82 (B) Phenylpyruvate (orange) possesses a similar hydrogen bond network to MTOB (orange dashes). (C) The non-canonical phenylpyruvate conformation (green) has a distinct hydrogen bond network (green dashes). Orientation of the carboxylate toward R266 of CtBP1 maximizes hydrogen bond potential as well as coulombic interactions (yellow dashes) with R97. (D) HIPP (cyan) forms a similar hydrogen bonding network (cyan dashes) to the non-canonical phenylpyruvate conformation, with the exception of losing the interaction with the nicotinamide ribose. (Reprinted with permission from Hilbert et al.85 Copyright 2015 American Chemical Society).
Notably, introduction of the oxime reduced the IC50 for HIPP to 240 nM, substantially improved over the IC50 of phenylpyruvate (IC50 = 135 μM). We further determined the inhibition constant of HIPP (Ki = 928 nM), showing that it more closely modeled non-competitive inhibition versus competitive inhibition (R2 = 0.7556 vs. R2 = 0.4995). This contrast to the crystal structure data, which indicates that MTOB and HIPP occupy the same binding site, can be explained by allosteric interactions. The current hypothesis suggests that HIPP-mediated CtBP inhibition may function by creating an abortive ternary complex, similar to the D2-dehydrogenase family member D-2-hydroxy-4-methyvalerate dehydrogenase.90 The hypothesis follows that upon NADH binding to CtBP, there is a conformational change in CtBP which likely causes domain closure around NAD+ once it undergoes its redox reaction with substrate. This closure is predicted to hinder the quick release of NAD+ and allow inhibitor binding to sequester the CtBP complex from participating in further reactions.82 Hence, HIPP is a substrate competitive inhibitor. Additionally, CtBPs are also unique among D2-dehydrogenases in that they have a large catalytic site that permits binding of water molecules throughout the cavity. With HIPP, 2 of the 4 water molecules within the active site are displaced, and a third is altered. It may be possible to further develop inhibitors that take advantage of this open cavity and displace all the water molecules to increase favorable binding interactions.
HIPP was tested in several cancer cell systems and found to cause cytotoxicity at millimolar concentrations (HCT116, p53-/- IC50 = 4.12 mM).89 By chromatin immunoprecipitation analysis (ChIP), it was shown that HIPP promotes this cytotoxicity, in part, by alleviating CtBP-mediated repression of the Bik promoter (S. Paliwal and S. R. Grossman, unpublished). Despite these encouraging results, an ideal therapeutic should be effective at doses several orders of magnitude lower. To this end, we took the structure of HIPP and created a series of substitutions along the benzene ring to assess for greater therapeutic potential. Two compounds using a chlorine functional group at the meta- and para- positions on the phenyl ring of HIPP (referred to as 3-Chloro HIPP and 4-Chloro HIPP, respectively) had an enzymatic IC50 that were improved over HIPP (0.17 vs 0.18 vs. 0.24 µM) and were found to be significantly more cytotoxic (cellular EC50: 0.85 mM and 1.74 mM vs 4.12 mM). To assess whether this increase in cytotoxicity may be due to CtBP inhibition, we performed BIK promoter/reporter experiments in HCT116, p53−/− cell lines. After 24 hours with 2 mM drug concentration, both 3-Cl HIPP and 4-Cl HIPP showed significantly higher BIK expression than negative controls, MTOB or HIPP at 4 mM.89 This suggests that the increased cytotoxicity of chlorinated HIPP derivatives is due, at least in part, to CtBP inhibition. Interestingly, HIPP also limited polyp formation in the Apc min mouse model, suggesting it can attain therapeutically relevant concentrations in vivo that may be an order of magnitude lower than concentrations required for activity in cell culture.54,89 Ongoing work is addressing the possible mechanisms for enhanced activity of CtBP inhibitors in the in vivo vs. in vitro settings.
Targeting dimerization
Macromolecular structures such as cyclic peptides have also been used to inhibit CtBP's activity, by inhibiting its ability to dimerize. The success of these molecules is based on their resistance to proteolytic degradation and large surface areas that can more effectively intervene in protein-protein interactions.91
Using this approach, Tavassoli and coworkers identified a cyclic peptide, CP6192 (Table 1) using a library screening method known as SICLOPPS (Split-Intein Circular Ligation of Peptides and Proteins). CP61 (cyclo-SGWTVVRMY) is a cyclic nona-peptide that is cyclized using NH2-terminal serine and COOH-terminal tyrosine, thus forming a loop-like structure. In vitro analysis showed that CP61 was able to inhibit dimerization of CtBP1, with an IC50 value of 19 ± 4 μM. In cellulo analysis also reflected the fact that CP61 was able to inhibit heterodimer formation between CtBP1 and CtBP2, and thereby restricted the ability of CtBP1 to colocalize into the nucleus. Mechanistic analysis of mode of inhibition by CP61 showed that the macrocyclic peptide selectively inhibited interaction between CtBP1 and NADH, most likely by an allosteric mechanism and not by directly binding in the NADH binding pocket.
In a previous report by the same group, CtBP promoted cell survival through maintenance of mitotic fidelity in breast cancer cells, with siRNA knock-down of CtBP causing lowered mitotic indices and increased fraction of micronuclei.16 Treating MCF-7 cells with increasing doses of CP61 coupled to the TAT peptide (an arginine and lysine rich peptide that enhances cellular permeability of chimerized or conjugated peptides and proteins), reflected the results seen with CtBP knockdown. Specifically, CP61 treatment resulted in increased number of micronuclei and increased the percentage of aberrant mitosis. In addition, the CP61-TAT inhibitor was able to induce a 3-fold decrease in colony formation as compared with DMSO control in MCF-7 breast cancer cells.
Targeting CtBP's interaction with its partners
As described in the foregoing sections, CtBP's transcriptional regulation is primarily due to its interaction with other transcriptional regulators, via a conserved PxDLS domain present in the binding partners. Blevins et al have reported inhibition of PxDLS mediated interaction of CtBP with other partners using a small molecule inhibitor.93 In this approach, a small molecule inhibitor NSC95397 (Table 1) was identified from the LOPAC (Library of Pharmacologically Active Compounds) library of 1280 compounds using an α-screen assay that measured the efficiency of a small molecule to inhibit interaction between CtBP1 and E1A (αScreen IC50 = 2.9 μM). Initial comparisons between NSC95397 and the previously published inhibitor of CtBP1, MTOB, using an “NADH disappearance assay” 82 showed that it was a weaker CtBP inhibitor than MTOB, but was able to inhibit interaction between CtBP1 and E1A proteins better than MTOB, as demonstrated using a fluorescence polarization assay. Additionally, NSC95397 was able to disrupt the CtBP1-mediated transcriptional complex, and de-repress E-cadherin expression. However, like MTOB, NSC85397 was a weak substrate for CtBP1. The authors speculate that NSC95397, being larger than MTOB, induces CtBP conformational change and locking CtBP in a conformation that prevents its interaction with transcription partners.
Interestingly, NSC95397 has been previously identified in multiple other reports not related to inhibition of CtBP, which includes inhibition of Mitogen-activated protein kinase phosphatase (MKP)-1/3, S100A4/myosin-IIA interaction, and inhibition of S100A4-mediated depolymerization of myosin-IIA filaments etc, which raises several questions about its specificity as a CtBP inhibitor.94-97
Future perspectives
Mounting experimental and clinical evidence suggests that CtBP's role in cancer is significant and warrants serious consideration as a therapeutic target. Given that CtBP's transcriptional activity contributes to neoplastic progression, pharmacological inhibition of CtBP, while simultaneously inhibiting other cancer therapeutic targets in combination, could lead to synergistically effective, low toxicity, therapies. Recently, several approaches have been explored, wherein small molecules that target other co-repressors, pathways or metabolic conditions were used in cells with altered CtBP expression or CtBP-inhibition.50,57,86 For instance, Berroilhet et al showed that RNAi mediated downregulation of CtBP2 expression and its activity in mucinous ovarian adenocarcinoma (MCAS), increased sensitivity toward HDAC inhibitors such as belinostat, and vorinostat, while cells with wild-type expression of CtBP2 remained unaffected.67 This suggests that the targeting of CtBP sensitizes cancer cells toward other established treatments.
Currently, inhibitors that target CtBP are still in early developmental stages and specificity has not been adequately tested. For the clinical application of CtBP-based inhibitors, additional effort will be required to design inhibitors that have limited-to-no off-target activity with pre-clinical efficacy alone, or in combination. Whether CtBP therapeutics will be of more benefit in tumor types and individual patients with higher CtBP expression, as in high-grade glioma, colon, breast, prostate and ovarian cancers, is an open question.67,98,99 Nonetheless, CtBP has generated tremendous interest in terms of being a co-repressor that targets multiple tumor suppressors, as well as an activator of key oncogenes, and as such, holds great promise as a wholly novel target for future cancer therapeutic development.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgement
We would like to thank Dr. Joseph Landry, Department of Human and Molecular Genetics, Virginia Commonwealth University School of Medicine, for helpful suggestions.
References
- 1.Boyd JM, Subramanian T, Schaeper U, La Regina M, Bayley S, Chinnadurai G. A region in the C-terminus of adenovirus 2/5 E1a protein is required for association with a cellular phosphoprotein and important for the negative modulation of T24-ras mediated transformation, tumorigenesis and metastasis. EMBO J 1993; 12:469-78; PMID:8440238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaeper U, Subramanian T, Lim L, Boyd JM, Chinnadurai G. Interaction between a cellular protein that binds to the C-terminal region of adenovirus E1A (CtBP) and a novel cellular protein is disrupted by E1A through a conserved PLDLS motif. J Biol Chem 1998; 273:8549-52; PMID:9535825; https://doi.org/ 10.1074/jbc.273.15.8549 [DOI] [PubMed] [Google Scholar]
- 3.Chinnadurai G, Chinnadurai G. CtBP family proteins: Unique transcriptional regulators in the nucleus with diverse cytosolic functions. In: Madame Curie Bioscience Database. Austin (TX): Landes Bioscience; 2000–2013; https://doi.org/ 10.1007/978-0-387-39973-7_1 [DOI] [Google Scholar]
- 4.Hildebrand JD, Soriano P. Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol Cell Biol 2002; 22:5296-307; PMID:12101226; https://doi.org/ 10.1128/MCB.22.15.5296-5307.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chinnadurai G. CtBP family proteins In: Chinnadurai G, ed. GtBP Family Proteins. New York, NY: Springer New York; 2007:1-17; https://doi.org/ 10.1007/978-0-387-39973-7_1 [DOI] [Google Scholar]
- 6.Nibu Y, Zhang H, Bajor E, Barolo S, Small S, Levine M. dCtBP mediates transcriptional repression by knirps, kruppel and snail in the drosophila embryo. EMBO J 1998; 17:7009-7020; PMID:9843507; https://doi.org/ 10.1093/emboj/17.23.7009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nibu Y, Zhang H, Levine M. Interaction of short-range repressors with drosophila CtBP in the embryo. Science 1998; 280:101-4; PMID:9525852; https://doi.org/ 10.1126/science.280.5360.101 [DOI] [PubMed] [Google Scholar]
- 8.Poortinga G, Watanabe M, Parkhurst SM. Drosophila CtBP: A hairy-interacting protein required for embryonic segmentation and hairy-mediated transcriptional repression. EMBO J 1998; 17:2067-78; PMID:9524128; https://doi.org/ 10.1093/emboj/17.7.2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reid A, Yucel D, Wood M, Llamosas E, Kant S, Crossley M, Nicholas H. The transcriptional repressor CTBP-1 functions in the nervous system of caenorhabditis elegans to regulate lifespan. Exp Gerontol 2014; 60:153-65; PMID:25456848; https://doi.org/ 10.1016/j.exger.2014.09.022 [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Whetstine JR, Ghosh S, Hanover JA, Gali RR, Grosu P, Shi Y. The conserved NAD(H)-dependent corepressor CTBP-1 regulates caenorhabditis elegans life span. Proc Nat Acad Sci 2009; 106:1496-501; PMID:19164523; https://doi.org/ 10.1073/pnas.0802674106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stankiewicz TR, Gray JJ, Winter AN, Linseman DA. C-terminal binding proteins: Central players in development and disease. Biomol Concepts 2014; 5:489-511; PMID:25429601; https://doi.org/ 10.1515/bmc-2014-0027 [DOI] [PubMed] [Google Scholar]
- 12.Garriga-Canut M, Schoenike B, Qazi R, Bergendahl K, Daley TJ, Pfender RM, Morrison JF, Ockuly J, Stafstrom C, Sutula T, Roopra A. 2-deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci 2006; 9:1382-87; PMID:17041593; https://doi.org/ 10.1038/nn1791 [DOI] [PubMed] [Google Scholar]
- 13.Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell 2011; 145:584-95; PMID:21565615; https://doi.org/ 10.1016/j.cell.2011.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huard CC, Tremblay CS, Helsper K, Delisle MC, Schindler D, Levesque G, Carreau M. Fanconi anemia proteins interact with CtBP1 and modulate the expression of the wnt antagonist dickkopf-1. Blood 2013; 121:1729-39; PMID:23303816; https://doi.org/ 10.1182/blood-2012-02-408997 [DOI] [PubMed] [Google Scholar]
- 15.Chinnadurai G. The transcriptional corepressor CtBP: A foe of multiple tumor suppressors. Cancer Res 2009; 69:731-4; PMID:19155295; https://doi.org/ 10.1158/0008-5472.CAN-08-3349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergman LM, Morris L, Darley M, Mirnezami AH, Gunatilake SC, Blaydes JP. Role of the unique N-terminal domain of CtBP2 in determining the subcellular localisation of CtBP family proteins. BMC Cell Biol 2006; 7:35; PMID:16999872; https://doi.org/ 10.1186/1471-2121-7-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lundblad JR. Structural determinants of CtBP function In: Chinnadurai G, ed. GtBP Family Proteins. New York, NY: Springer New York; 2007:83-92; https://doi.org/ 10.1007/978-0-387-39973-7_1 [DOI] [Google Scholar]
- 18.Kuppuswamy M, Vijayalingam S, Zhao LJ, Zhou Y, Subramanian T, Ryerse J, Chinnadurai G. Role of the PLDLS-binding cleft region of CtBP1 in recruitment of core and auxiliary components of the corepressor complex. Mol Cell Biol 2008; 28:269-81; PMID:17967884; https://doi.org/ 10.1128/MCB.01077-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verger A, Quinlan KG, Crofts LA, Spano S, Corda D, Kable EP, Braet F, Crossley M. Mechanisms directing the nuclear localization of the CtBP family proteins. Mol Cell Biol 2006; 26:4882-94; PMID:16782877; https://doi.org/ 10.1128/MCB.02402-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corda D, Colanzi A, Luini A. The multiple activities of CtBP/BARS proteins: The golgi view. Trends Cell Biol 2006; 16:167-73; PMID:16483777; https://doi.org/ 10.1016/j.tcb.2006.01.007 [DOI] [PubMed] [Google Scholar]
- 21.Bartz R, Seemann J, Zehmer JK, Serrero G, Chapman KD, Anderson RG, Liu P. Evidence that mono-ADP-ribosylation of CtBP1/BARS regulates lipid storage. Mol Biol Cell 2007; 18:3015-25; PMID:17538025; https://doi.org/ 10.1091/mbc.E06-09-0869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmitz F, Konigstorfer A, Sudhof TC. RIBEYE, a component of synaptic ribbons: A protein's journey through evolution provides insight into synaptic ribbon function. Neuron 2000; 28:857-72; PMID:11163272; https://doi.org/ 10.1016/S0896-6273(00)00159-8 [DOI] [PubMed] [Google Scholar]
- 23.Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, Shi Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003; 422:735-8; PMID:12700765; https://doi.org/ 10.1038/nature01550 [DOI] [PubMed] [Google Scholar]
- 24.Chinnadurai G. CtBP family proteins: More than transcriptional corepressors. Bioessays 2003; 25:9-12; PMID:12508276; https://doi.org/ 10.1002/bies.10212 [DOI] [PubMed] [Google Scholar]
- 25.Quinlan KG, Verger A, Kwok A, Lee SH, Perdomo J, Nardini M, Bolognesi M, Crossley M. Role of the C-terminal binding protein PXDLS motif binding cleft in protein interactions and transcriptional repression. Mol Cell Biol 2006; 26:8202-13; PMID:16940173; https://doi.org/ 10.1128/MCB.00445-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang CL, McKinsey TA, Lu J, Olson EN. Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J Biol Chem 2001; 276:35-9; PMID:11022042; https://doi.org/ 10.1074/jbc.M007364200 [DOI] [PubMed] [Google Scholar]
- 27.Vliet Jv, Turner J, Crossley M. Human krüppel-like factor 8: A CACCC-box binding protein that associates with CtBP and represses transcription. Nucleic Acids Res 2000; 28:1955-62; PMID:10756197; https://doi.org/ 10.1093/nar/28.9.1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 2005; 19:857-64; PMID:16140033; https://doi.org/ 10.1016/j.molcel.2005.08.027 [DOI] [PubMed] [Google Scholar]
- 29.Ray SK, Li HJ, Metzger E, Schüle R, Leiter AB. CtBP and associated LSD1 are required for transcriptional activation by NeuroD1 in gastrointestinal endocrine cells. Mol Cell Biol 2014; 34:2308-17; PMID:24732800; https://doi.org/ 10.1128/MCB.01600-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar V, Carlson JE, Ohgi KA, Edwards TA, Rose DW, Escalante CR, Rosenfeld MG, Aggarwal AK. Transcription corepressor CtBP is an NAD(+)-regulated dehydrogenase. Mol Cell 2002; 10:857-869; PMID:12419229; https://doi.org/ 10.1016/S1097-2765(02)00650-0 [DOI] [PubMed] [Google Scholar]
- 31.Bhambhani C, Chang JL, Akey DL, Cadigan KM. The oligomeric state of CtBP determines its role as a transcriptional co-activator and co-repressor of wingless targets. EMBO J 2011; 30:2031-43; PMID:21468031; https://doi.org/ 10.1038/emboj.2011.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang M, Li J, Blauwkamp T, Bhambhani C, Campbell N, Cadigan KM. C-terminal-binding protein directly activates and represses wnt transcriptional targets in drosophila. EMBO J 2006; 25:2735-45; PMID:16710294; https://doi.org/ 10.1038/sj.emboj.7601153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ray SK, Li HJ, Leiter AB. Oligomeric form of C-terminal-binding protein coactivates NeuroD1-mediated transcription. FEBS Lett 2017; 591:205-12; PMID:27880001; https://doi.org/ 10.1002/1873-3468.12501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science 2002; 295:1895-7; PMID:11847309; https://doi.org/ 10.1126/science.1069300 [DOI] [PubMed] [Google Scholar]
- 35.Madison DL, Wirz JA, Siess D, Lundblad JR. Nicotinamide adenine dinucleotide-induced multimerization of the co-repressor CtBP1 relies on a switching tryptophan. J Biol Chem 2013; 288:27836-48; PMID:23940047; https://doi.org/ 10.1074/jbc.M113.493569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuppuswamy M, Vijayalingam S, Zhao L, Zhou Y, Subramanian T, Ryerse J, Chinnadurai G. Role of the PLDLS-binding cleft region of CtBP1 in recruitment of core and auxiliary components of the corepressor complex. Mol Cell Biol 2008; 28:269-81; PMID:17967884; https://doi.org/ 10.1128/MCB.01077-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grooteclaes M, Deveraux Q, Hildebrand J, Zhang Q, Goodman RH, Frisch SM. C-terminal-binding protein corepresses epithelial and proapoptotic gene expression programs. Proc Natl Acad Sci U S A 2003; 100:4568-73; PMID:12676992; https://doi.org/ 10.1073/pnas.0830998100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnes CJ, Vadlamudi RK, Mishra SK, Jacobson RH, Li F, Kumar R. Functional inactivation of a transcriptional corepressor by a signaling kinase. Nat Struct Mol Biol 2003; 10:622-8 PMID:12872159; https://doi.org/ 10.1038/nsb957 [DOI] [PubMed] [Google Scholar]
- 39.Kim J, Choi S, Kang B, Lee S, Park HS, Kang G, Bang JY, Cho E, Youn H. AMP-activated protein kinase phosphorylates CtBP1 and down-regulates its activity. Biochem Biophys Res Commun 2013; 431:8-13; PMID:23291169; https://doi.org/ 10.1016/j.bbrc.2012.12.117 [DOI] [PubMed] [Google Scholar]
- 40.Merrill JC, Kagey MH, Melhuish TA, Powers SE, Zerlanko BJ, Wotton D. Inhibition of CtBP1 activity by akt-mediated phosphorylation. J Mol Biol 2010; 398:657-71; PMID:20361981; https://doi.org/ 10.1016/j.jmb.2010.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nardini M, Svergun D, Konarev PV, Spano S, Fasano M, Bracco C, Pesce A, Donadini A, Cericola C, Secundo F, Luini A, Corda D, Bolognesi M. The C-terminal domain of the transcriptional corepressor CtBP is intrinsically unstructured. Protein Sci 2006; 15:1042-1050; PMID:16597837; https://doi.org/ 10.1110/ps.062115406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riefler GM, Firestein BL. Binding of neuronal nitric-oxide synthase (nNOS) to carboxyl-terminal-binding protein (CtBP) changes the localization of CtBP from the nucleus to the cytosol: a novel function for targeting by the PDZ domain of nNOS. J Biol Chem 2001; 276:48262-8; PMID:11590170; https://doi.org/ 10.1074/jbc.M106503200 [DOI] [PubMed] [Google Scholar]
- 43.Lin X, Sun B, Liang M, Liang YY, Gast A, Hildebrand J, Brunicardi FC, Melchior F, Feng XH. Opposed regulation of corepressor CtBP by SUMOylation and PDZ binding. Mol Cell 2003; 11:1389-96; PMID:12769861; https://doi.org/ 10.1016/S1097-2765(03)00175-8 [DOI] [PubMed] [Google Scholar]
- 44.Paliwal S, Pande S, Kovi RC, Sharpless NE, Bardeesy N, Grossman SR. Targeting of C-terminal binding protein (CtBP) by ARF results in p53-independent apoptosis. Mol Cell Biol 2006; 26:2360-72; PMID:16508011; https://doi.org/ 10.1128/MCB.26.6.2360-2372.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Q, Yoshimatsu Y, Hildebrand J, Frisch SM, Goodman RH. Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell 2003; 115:177-86; PMID:14567915; https://doi.org/ 10.1016/S0092-8674(03)00802-X [DOI] [PubMed] [Google Scholar]
- 46.Wang S, Iordanov M, Zhang Q. C-jun NH2-terminal kinase promotes apoptosis by down-regulating the transcriptional co-repressor CtBP. J Biol Chem 2006; 281:34810-15; PMID:16984892; https://doi.org/ 10.1074/jbc.M607484200 [DOI] [PubMed] [Google Scholar]
- 47.Hofmann TG, Stollberg N, Schmitz ML, Will H. HIPK2 regulates transforming growth factor-ß-induced c-jun NH2-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res 2003; 63:8271-77; PMID:14678985 [PubMed] [Google Scholar]
- 48.Yang SH, Galanis A, Witty J, Sharrocks AD. An extended consensus motif enhances the specificity of substrate modification by SUMO. EMBO J 2006; 25:5083-93; PMID:17036045; https://doi.org/ 10.1038/sj.emboj.7601383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kovi RC, Paliwal S, Pande S, Grossman SR. An ARF/CtBP2 complex regulates BH3-only gene expression and p53-independent apoptosis. Cell Death Differ 2010; 17:513-21; PMID:19798104; https://doi.org/ 10.1038/cdd.2009.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paliwal S, Kovi RC, Nath B, Chen YW, Lewis BC, Grossman SR. The alternative reading frame tumor suppressor antagonizes hypoxia-induced cancer cell migration via interaction with the COOH-terminal binding protein corepressor. Cancer Res 2007; 67:932229; PMID:17909040; https://doi.org/ 10.1158/0008-5472.CAN-07-1743 [DOI] [PubMed] [Google Scholar]
- 51.Chinnadurai G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol Cell 2002; 9:213-24; PMID:11864595; https://doi.org/ 10.1016/S1097-2765(02)00443-4 [DOI] [PubMed] [Google Scholar]
- 52.Lin HJ, Eviner V, Prendergast GC, White E. Activated H-ras rescues E1A-induced apoptosis and cooperates with E1A to overcome p53-dependent growth arrest. Mol Cell Biol 1995; 15:4536-44; PMID:7623844; https://doi.org/ 10.1128/MCB.15.8.4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palmer S, Brouillet J, Kilbey A, Fulton R, Walker M, Crossley M, Bartholomew C. Evi-1 transforming and repressor activities are mediated by CtBP co-repressor proteins. J Biol Chem 2001; 276:25834-40; PMID:11328817; https://doi.org/ 10.1074/jbc.M102343200 [DOI] [PubMed] [Google Scholar]
- 54.Sumner ET, Chawla A, Cororaton A, Koblinski J, Kovi R, Love I, Szomju B, Korwar S, Ellis K, Grossman S. Transforming activity and therapeutic targeting of C-terminal binding protein 2 in apc mutated neoplasia. Oncogene 2017; in press; PMID:28414304; https://doi.org/ 10.1038/onc.2017.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Byun JS, Gardner K. C-terminal binding protein: A molecular link between metabolic imbalance and epigenetic regulation in breast cancer. Int J Cell Biol 2013; 2013:647975; PMID:23762064; https://doi.org/ 10.1155/2013/647975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng X, Song T, Dou C, Jia Y, Liu Q. CtBP2 is an independent prognostic marker that promotes GLI1 induced epithelial-mesenchymal transition in hepatocellular carcinoma. Oncotarget 2015; 6:3752-69; PMID:25686837; https://doi.org/ 10.18632/oncotarget.2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Birts CN, Harding R, Soosaipillai G, Halder T, Azim-Araghi A, Darley M, Cutress RI, Bateman AC, Blaydes JP. Expression of CtBP family protein isoforms in breast cancer and their role in chemoresistance. Biol Cell 2010; 103:1-19; PMID:20964627; https://doi.org/ 10.1042/BC20100067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Di LJ, Byun JS, Wong MM, Wakano C, Taylor T, Bilke S, Baek S, Hunter K, Yang H, Lee M, Zvosec C, Khramtsova G, Cheng F, Perou CM, Miller CR, Raab R, Olopade OI, Gardner K. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat Commun 2013; 4:1449; PMID:23385593; https://doi.org/ 10.1038/ncomms2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jin W, Scotto KW, Hait WN, Yang J. Involvement of CtBP1 in the transcriptional activation of the MDR1 gene in human multidrug resistant cancer cells. Biochem Pharmacol 2007; 74:851-9; PMID:17662696; https://doi.org/ 10.1016/j.bcp.2007.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanahan D, Weinberg R. Hallmarks of cancer: The next generation. Cell 2011; 144:646-74; PMID:21376230; https://doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 61.Wang R, Asangani IA, Chakravarthi BV, Ateeq B, Lonigro RJ, Cao Q, Mani RS, Camacho DF, McGregor N, Schumann TE, Jing X, Menawat R, Tomlins SA, Zheng H, Otte AP, Mehra R, Siddiqui J, Dhanasekaran SM, Nyati MK, Pienta KJ, Palanisamy N, Kunju LP, Rubin MA, Chinnaiyan AM, Varambally S. Role of transcriptional corepressor CtBP1 in prostate cancer progression. Neoplasia 2012; 14:905-14; PMID:23097625; https://doi.org/ 10.1593/neo.121192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119:1420-28; PMID:19487818; https://doi.org/ 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Q, Wang SY, Nottke AC, Rocheleau JV, Piston DW, Goodman RH. Redox sensor CtBP mediates hypoxia-induced tumor cell migration. Proc Natl Acad Sci U S A 2006; 103:9029-33; PMID:16740659; https://doi.org/ 10.1073/pnas.0603269103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang XL, Huang CX, Zhang J, Inoue A, Zeng SE, Xiao SJ. CtBP1 is involved in epithelial-mesenchymal transition and is a potential therapeutic target for hepatocellular carcinoma. Oncol Rep 2013; 30:809-14; PMID:23756565; https://doi.org/ 10.3892/or.2013.2537 [DOI] [PubMed] [Google Scholar]
- 65.Shin S, Dimitri CA, Yoon S, Dowdle W, Blenis J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell 2010; 38:114-127; PMID:20385094; https://doi.org/ 10.1016/j.molcel.2010.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ichikawa K, Kubota Y, Nakamura T, Weng J, Tomida T, Saito H, Takekawa M. MCRIP1, an ERK substrate, mediates ERK-induced gene silencing during epithelial-mesenchymal transition by regulating the co-repressor CtBP. Mol Cell 2015; 58:35-46; PMID:25728771; https://doi.org/ 10.1016/j.molcel.2015.01.023 [DOI] [PubMed] [Google Scholar]
- 67.Barroilhet L, Yang J, Hasselblatt K, Paranal RM, Ng SK, Rauh-Hain JA, Welch WR, Bradner JE, Berkowitz RS, Ng SW. C-terminal binding protein-2 regulates response of epithelial ovarian cancer cells to histone deacetylase inhibitors. Oncogene 2013; 32:3896-903; PMID:22945647; https://doi.org/ 10.1038/onc.2012.380 [DOI] [PubMed] [Google Scholar]
- 68.Minard ME, Kim LS, Price JE, Gallick GE. The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast Cancer Res Treat 2004; 84:21-32; PMID:14999151; https://doi.org/ 10.1023/B:BREA.0000018421.31632.e6 [DOI] [PubMed] [Google Scholar]
- 69.Yamada KM, Araki M. Tumor suppressor PTEN: Modulator of cell signaling, growth, migration and apoptosis. J Cell Sci 2001; 114:2375-82; PMID:11559746 [DOI] [PubMed] [Google Scholar]
- 70.Paliwal S, Ho N, Parker D, Grossman SR. CtBP2 promotes human cancer cell migration by transcriptional activation of Tiam1. Genes Cancer 2012; 3:481-90; PMID:23264848; https://doi.org/ 10.1177/1947601912463695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Di L, Fernandez AG, De Siervi A, Longo DL, Gardner K. Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol 2010; 17:1406-13; PMID:21102443; https://doi.org/ 10.1038/nsmb.1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moiola CP, De Luca P, Zalazar F, Cotignola J, Rodriguez-Segui SA, Gardner K, Meiss R, Vallecorsa P, Pignataro O, Mazza O, Vazquez ES, De Siervi A. Prostate tumor growth is impaired by CtBP1 depletion in high-fat diet-fed mice. Clin Cancer Res 2014; 20:4086-95; PMID:24842953; https://doi.org/ 10.1158/1078-0432.CCR-14-0322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Luca P, Dalton GN, Scalise GD, Moiola CP, Porretti J, Massillo C, Kordon E, Gardner K, Zalazar F, Flumian C, et al.. CtBP1 associates metabolic syndrome and breast carcinogenesis targeting multiple miRNAs. Oncotarget 2016; 7(14):18798-811; PMID:26933806; https://doi.org/ 10.18632/oncotarget.7711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 2013; 13:11-26; PMID:23258168; https://doi.org/ 10.1038/nrc3419 [DOI] [PubMed] [Google Scholar]
- 75.Hamada F, Bienz M. The APC tumor suppressor binds to C-terminal binding protein to divert nuclear beta-catenin from TCF. Dev Cell 2004; 7:677-85; PMID:15525529; https://doi.org/ 10.1016/j.devcel.2004.08.022 [DOI] [PubMed] [Google Scholar]
- 76.Schneikert J, Brauburger K, Behrens J. APC mutations in colorectal tumours from FAP patients are selected for CtBP-mediated oligomerization of truncated APC. Human Mol Genet 2011; 20:3554-64; PMID:21665989; https://doi.org/ 10.1093/hmg/ddr273 [DOI] [PubMed] [Google Scholar]
- 77.Choi SH, Estaras C, Moresco JJ, Yates JR, Jones KA 3rd.. Alpha-catenin interacts with APC to regulate beta-catenin proteolysis and transcriptional repression of wnt target genes. Genes Dev 2013; 27:2473-88; PMID:24240237; https://doi.org/ 10.1101/gad.229062.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sierra J, Yoshida T, Joazeiro CA, Jones KA. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at wnt target genes. Genes Dev 2006; 20:586-600; PMID:16510874; https://doi.org/ 10.1101/gad.1385806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Patel J, Baranwal S, Love IM, Patel NJ, Grossman SR, Patel BB. Inhibition of C-terminal binding protein attenuates transcription factor 4 signaling to selectively target colon cancer stem cells. Cell Cycle 2014; 13:3506-18; PMID:25483087; https://doi.org/ 10.4161/15384101.2014.958407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003; 100:3983-88; PMID:12629218; https://doi.org/ 10.1073/pnas.0530291100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Achouri Y, Noël G, Van Schaftingen E. 2-keto-4-methylthiobutyrate, an intermediate in the methionine salvage pathway, is a good substrate for CtBP1. Biochem Biophys Res Commun 2007; 352:903-6; PMID:17157814; https://doi.org/ 10.1016/j.bbrc.2006.11.111 [DOI] [PubMed] [Google Scholar]
- 82.Hilbert BJ, Morris BL, Ellis KC, Paulsen JL, Schiffer CA, Grossman SR, Royer WE Jr.. Structure-guided design of a high affinity inhibitor to human CtBP. ACS Chem Biol 2015; 10:1118-1127; PMID:25636004; https://doi.org/ 10.1021/cb500820b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sauter M, Moffatt B, Saechao MC, Hell R, Wirtz M. Methionine salvage and S-adenosylmethionine: Essential links between sulfur, ethylene and polyamine biosynthesis. Biochem J 2013; 451:145-54; PMID:23535167; https://doi.org/ 10.1042/BJ20121744 [DOI] [PubMed] [Google Scholar]
- 84.Tang B, Kadariya Y, Murphy ME, Kruger WD. The methionine salvage pathway compound 4-methylthio-2-oxobutanate causes apoptosis independent of down-regulation of ornithine decarboxylase. Biochem Pharmacol 2006; 72:806-15; PMID:16870157; https://doi.org/ 10.1016/j.bcp.2006.06.018 [DOI] [PubMed] [Google Scholar]
- 85.Bello-Fernandez C, Packham G, Cleveland JL. The ornithine decarboxylase gene is a transcriptional target of c-myc. Proc Nat Acad Sci 1993; 90:7804-08; PMID:8356088; https://doi.org/ 10.1073/pnas.90.16.7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Straza MW, Paliwal S, Koi RC, Rajeshkumar B, Trenh P, Parker D, Whalen GF, Lyle S, Schiffer CA, Grossman SR. Therapeutic targeting of C-terminal binding protein in human cancer. Cell Cycle 2010; 9:3740-50; PMID:20930544; https://doi.org/ 10.4161/cc.9.18.12936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li M, Riddle S, Zhang H, D'Alessandro A, Flockton A, Serkova NJ, Hansen KC, Moldvan R, McKeon BA, Frid M, et al.. Metabolic reprogramming regulates the proliferative and inflammatory phenotype of adventitial fibroblasts in pulmonary hypertension through the transcriptional corepressor C-terminal binding protein-1. Circulation 2016; 134:1105-21; PMID:27562971; https://doi.org/ 10.1161/CIRCULATIONAHA.116.023171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hilbert BJ, Grossman SR, Schiffer CA, Royer WE Jr.. Crystal structures of human CtBP in complex with substrate MTOB reveal active site features useful for inhibitor design. FEBS Lett 2014; 588:1743-48; PMID:24657618; https://doi.org/ 10.1016/j.febslet.2014.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Korwar S, Morris BL, Parikh HI, Coover RA, Doughty TW, Love IM, Hilbert BJ, Royer WE Jr, Kellogg GE, Grossman SR, Ellis KC. Design, synthesis, and biological evaluation of substrate-competitive inhibitors of C-terminal binding protein (CtBP). Bioorg Med Chem 2016; 24:2707-15; PMID:27156192; https://doi.org/ 10.1016/j.bmc.2016.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alvarez JA, Gelpí JL, Johnsen K, Bernard N, Delcour J, Clarke AR, Holbrook JJ, Cortés A. D-2-hydroxy-4-methylvalerate dehydrogenase from lactobacillus delbrueckii subsp. bulgaricus? I. kinetic mechanism and pH dependence of kinetic parameters, coenzyme binding and substrate inhibition. Eur J Biochem 1997; 244:203-12; PMID:9063465; https://doi.org/ 10.1111/j.1432-1033.1997.00203.x [DOI] [PubMed] [Google Scholar]
- 91.Gao M, Cheng K, Yin H. Targeting protein-protein interfaces using macrocyclic peptides. Biopolymers 2015; 104:310-6; PMID:25664609; https://doi.org/ 10.1002/bip.22625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Birts CN, Nijjar SK, Mardle CA, Hoakwie F, Duriez PJ, Blaydes JP, Tavassoli A. A cyclic peptide inhibitor of C-terminal binding protein dimerization links metabolism with mitotic fidelity in breast cancer cells. Chem Sci 2013; 4:3046-57; https://doi.org/ 10.1039/c3sc50481f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blevins MA, Kouznetsova J, Krueger AB, King R, Griner LM, Hu X, Southall N, Marugan JJ, Zhang Q, Ferrer M, Zhao R. Small molecule, NSC95397, inhibits the CtBP1-protein partner interaction and CtBP1-mediated transcriptional repression. J Biomol Screen 2015; 20:663-672; PMID:25477201; https://doi.org/ 10.1177/1087057114561400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang Y, Yang WS, Yu T, Yi Y, Park JG, Jeong D, Kim JH, Oh JS, Yoon K, Kim J, Cho JY. Novel anti-inflammatory function of NSC95397 by the suppression of multiple kinases. Biochem Pharmacol 2014; 88:201-15; PMID:24468133; https://doi.org/ 10.1016/j.bcp.2014.01.022 [DOI] [PubMed] [Google Scholar]
- 95.Dulyaninova NG, Hite KM, Zencheck WD, Scudiero DA, Almo SC, Shoemaker RH, Bresnick AR. Cysteine 81 is critical for the interaction of S100A4 and myosin-IIA. Biochemistry 2011; 50:7218-27; PMID:21749055; https://doi.org/ 10.1021/bi200853y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Larsson DE, Wickstrom M, Hassan S, Oberg K, Granberg D. The cytotoxic agents NSC-95397, brefeldin A, bortezomib and sanguinarine induce apoptosis in neuroendocrine tumors in vitro. Anticancer Res 2010; 30:149-56; PMID:20150630 [PubMed] [Google Scholar]
- 97.Vogt A, McDonald PR, Tamewitz A, Sikorski RP, Wipf P, Skoko JJ, Lazo JS 3rd.. A cell-active inhibitor of mitogen-activated protein kinase phosphatases restores paclitaxel-induced apoptosis in dexamethasone-protected cancer cells. Mol Cancer Ther 2008; 7:330-40; PMID:18245669; https://doi.org/ 10.1158/1535-7163.MCT-07-2165 [DOI] [PubMed] [Google Scholar]
- 98.Zhang C, Gao C, Xu Y, Zhang Z. CtBP2 could promote prostate cancer cell proliferation through c-myc signaling. Gene 2014; 546:73-9; PMID:24835310; https://doi.org/ 10.1016/j.gene.2014.05.032 [DOI] [PubMed] [Google Scholar]
- 99.Liu B, Di G. C-terminal binding protein is involved in promoting to the carcinogenesis of human glioma. Mol Neurobiol 2016; PMID:27699603; https://doi.org/ 10.1007/s12035-016-0159-x [DOI] [PubMed] [Google Scholar]