

SUMMARY
UPF1 is an RNA helicase that orchestrates nonsense-mediated decay and other RNA surveillance pathways. While UPF1 is best known for its basal cytoprotective role in degrading aberrant RNAs, UPF1 also degrades specific normally occurring mRNAs to regulate diverse cellular processes. Here, we describe a role for UPF1 in regulated protein decay, wherein UPF1 acts as an E3 ubiquitin ligase to repress human skeletal muscle differentiation. Suppressing UPF1 accelerates myogenesis, while ectopically increasing UPF1 levels slows myogenesis. UPF1 promotes the decay of MYOD protein, a transcription factor that is a master regulator of myogenesis, while leaving MYOD mRNA stability unaffected. UPF1 acts as an E3 ligase via its RING domain to promote MYOD protein ubiquitination and degradation. Our data characterize a regulatory role for UPF1 in myogenesis and demonstrate that UPF1 provides a mechanistic link between the RNA and protein decay machineries in human cells.
Keywords: UPF1, nonsense-mediated decay, myogenesis, RING domain, ubiquitin-proteasome
Graphical abstract
INTRODUCTION
Nonsense-mediated RNA decay (NMD) is a highly conserved post-transcriptional pathway that selectively degrades both abnormal and normally occurring RNA. NMD is best known for its role as an mRNA surveillance mechanism. In that capacity, NMD recognizes and degrades aberrant mRNAs containing premature termination codons that interrupt the normal reading frames of genes (Lykke-Andersen and Jensen, 2015; Popp and Maquat, 2013). Such aberrant mRNAs can arise from many sources, including genetic variation, somatic mutations, and errors introduced during RNA transcription or splicing. However, NMD is not just a cytoprotective pathway. In addition to suppressing aberrant RNAs, NMD also degrades many normally occurring mRNAs that contain sequence features that trigger NMD, including splice junctions downstream of stop codons, upstream open reading frames in the 5’ untranslated region (UTR), or a long 3’ UTR. Approximately 10-30% of normally occurring human mRNAs are predicted substrates for degradation by NMD (Hurt et al., 2013; Lewis et al., 2003; McIlwain et al., 2010).
By controlling the levels of many endogenous mRNAs, NMD can regulate diverse molecular processes in addition to functioning as a basal cytoprotective mechanism. For example, mRNAs encoding key effectors of the unfolded protein response are degraded by NMD (Karam et al., 2015), while endoplasmic reticulum stress in turn inhibits NMD (Wang et al., 2011). NMD also contributes to the regulation of the integrated stress response (Gardner, 2008; Martin and Gardner, 2014; Wang et al., 2011), apoptosis (Jia et al., 2015; Popp and Maquat, 2015), immune response (Gloggnitzer et al., 2014), response to viral infection (Balistreri et al., 2014; Ramage et al., 2015), and other core physiological processes. Ongoing attempts to identify endogenous NMD substrates will continue to reveal new regulatory roles for NMD.
The broad scope of NMD’s regulatory role is exemplified by recent evidence that NMD factors regulate cell differentiation and cell fate. Many lines of evidence implicate NMD in differentiation. NMD efficiency—the fraction of an NMD substrate that is recognized and degraded instead of escaping degradation—varies during development and differentiation. NMD substrates are differentially recognized during C. elegans development and NMD efficiency decreases during mammalian myogenesis and neurogenesis (Barberan-Soler et al., 2009; Bruno et al., 2011; Gong et al., 2009). Cellular requirements for NMD also vary during differentiation. Hematopoietic deletion of Upf2, encoding a core component of the NMD machinery, ablated hematopoietic stem and progenitor cells, but not more differentiated blood cells (Weischenfeldt et al., 2008). NMD is also required for licensing some cell types for differentiation. Deletion of Smg6, encoding a NMD factor with endonucleolytic activity, blocked differentiation of mouse embryonic stem cells without impairing their proliferation (T. Li et al., 2015). Similarly, conditional deletion of Upf2 prevented fetal liver cells from undergoing terminal differentiation (Thoren et al., 2010). Finally, NMD has been directly implicated in repressing neural differentiation. Overexpression or knockdown of Upf1, encoding an RNA helicase that is required for NMD, respectively inhibited or promoted murine neurogenesis. NMD influences neurogenesis in part by degrading the mRNA encoding SMAD7, which regulates neurogenesis via TGF-β signaling (Lou et al., 2014).
Muscle differentiation may represent a tractable and biomedically relevant system to study the role of NMD in cell differentiation for several reasons. First, a previous study reported that UPF1 levels and NMD efficiency decreased during skeletal myogenesis and that the mRNA encoding the myogenic marker MYOG was degraded by NMD (Gong et al., 2009). These data suggest that NMD factors might repress myogenesis, although that hypothesis has not been tested. Second, the regulatory factors controlling muscle differentiation are well-characterized, which may facilitate identifying direct mechanistic ties between NMD and master regulators of myogenesis. Third, we recently found that expression of the disease gene DUX4 in skeletal muscle—which causes facioscapulohumeral muscular dystrophy—results in UPF1 protein degradation and severe inhibition of NMD (Feng et al., 2015). Determining the normal role of NMD during myogenesis is therefore of direct biomedical relevance.
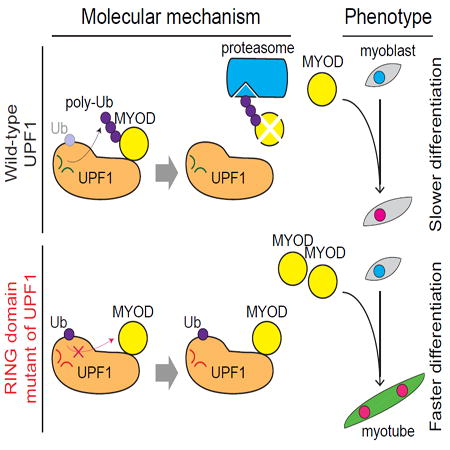
In this study, we describe a direct and mechanistic role for the central NMD factor UPF1 in repressing human muscle differentiation. UPF1 promotes proteasome-mediated degradation of MYOD protein, a master regulator of the myogenic process. UPF1 promotes MYOD proteolysis by acting as an E3 ubiquitin ligase via its RING domain while leaving MYOD mRNA stability unaffected. Mutating UPF1’s RING domain alleviates UPF1-dependent MYOD proteolysis and reverses UPF1’s repressive role in myogenesis.
RESULTS
UPF1 knockdown accelerates human myogenesis
To determine whether the reported decrease in UPF1 levels and NMD efficiency that occurs during myogenesis (Gong et al., 2009) plays a regulatory and causative role, rather than being a by-product of the myogenic process, we tested whether forced reductions in UPF1 levels altered the efficiency of myoblast differentiation. We transfected two genetically distinct myoblast cell lines generated from healthy human muscle (54-1 and MB135 cells) with two different siRNAs against UPF1, reducing UPF1 protein to 24.7% and 63.0% of levels in cells transfected with two different non-targeting siRNAs (Figure 1A-B, S1A-C). We induced differentiation two days post-transfection (day 0) by switching the confluent myoblasts from high-serum growth media to low-serum differentiation media, which promotes cell cycle stalling and the expression of myogenic markers.
Figure 1. UPF1 knockdown accelerates myoblast differentiation.
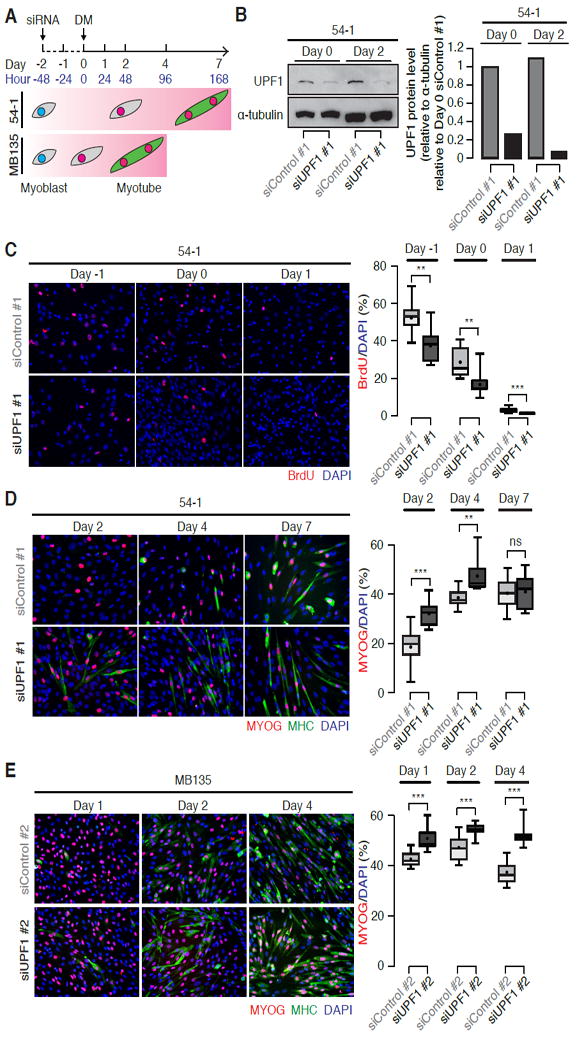
(A) Schematic of a time course of human myoblast differentiation following UPF1 knockdown (KD). Two genetically distinct human myoblast cell lines (54-1 and MB135 cells) were transfected with two different siRNAs against UPF1, as well as two different non-targeting siRNAs as controls (Day -2). When the transfected cells reached full confluency (Day 0), differentiation was induced by switching from high-serum growth media to low-serum differentiation media (DM). 54-1 and MB135 cells respectively differentiate less and more rapidly, and so were differentiated for seven and four days, respectively.
(B) Immunoblot for UPF1 protein from 54-1 cells following control or UPF1 KD immediately prior to (Day 0) or two days after (Day 2) induction of differentiation. Equal amounts of protein were loaded based on the BCA assay. α-tubulin, loading control. Bar plot, quantification of UPF1 protein levels relative to the loading control.
(C) Immunofluorescence labeling of 54-1 cells with an antibody against BrdU (red) at the indicated time points, prior to significant cell fusion. At each time point, cells were fixed after one hour of BrdU labeling. Box plot, percentage of BrdU+ nuclei measured over 10 fields. Whiskers, max and min over the fields. */**/***, p < 0.05/0.01/0.001.
(D-E) Immunofluorescence labeling of 54-1 or MB135 cells with antibodies against Myogenin (MYOG, red) and Myosin Heavy Chain (MHC, green) at the indicated time points. MB135 differentiate more rapidly than do 54-1 cells and so a shorter time course was used. Box plot, percentage of MYOG+ nuclei measured over 10 fields. Whiskers, max and min over the fields. */**/***, p < 0.05/0.01/0.001.
See also Figure S1.
UPF1 knockdown (KD) resulted in accelerated differentiation throughout the myogenic process in both 54-1 and MB135 myoblasts. We first analyzed cells from the 54-1 genetic background, finding that UPF1 KD promoted cell cycle exit even prior to the initiation of differentiation. One day after transfection with control versus UPF1-targeting siRNAs (day -1), 52.2% versus 37.8% of nuclei were BrdU+ (Figure 1C). We then quantified muscle differentiation by counting the fraction of nuclei expressing Myogenin (MYOG), a marker of myogenic commitment. Two days post-induction of differentiation (day 2), we observed 68.7% more MYOG+ nuclei in UPF1-KD cells compared to control cells, coincident with a 3.5-fold increase in MYOG mRNA (Figure 1D, S1D). This increase in MYOG occurred only after the induction of differentiation. UPF1-KD cells matured faster into myotubes marked by Myosin Heavy Chain (MHC) expression, with MHC staining readily visible at day 2 when essentially none was evident in control cells. The same phenotype of accelerated myogenesis occurred in MB135 myoblasts, which differentiate more rapidly than do the genetically distinct 54-1 myoblasts, following transfection with a distinct siRNA against UPF1 (Figure 1E, S1E).
We next confirmed that UPF1 KD drives accelerated myogenesis in the physiological setting of primary, rather than immortalized, cell differentiation. We repeated the above differentiation experiments with two genetically distinct cultures of primary human skeletal muscle cells. These primary MB135 and MB2401 myoblasts were obtained from muscle biopsies of two healthy individuals. (A different culture of primary MB135 cells was previously immortalized to generate the immortalized MB135 cell line used in this study.) Transduction with UPF1-targeting shRNAs resulted in efficient UPF1 KD and accelerated myogenesis (Figure S1F-H). We concluded that ectopic suppression of UPF1 levels promotes differentiation of both immortalized and primary human myoblasts.
UPF1 overexpression slows myogenesis
As UPF1 KD accelerated myogenesis, we hypothesized that UPF1 overexpression might conversely slow myogenesis. To test this hypothesis, we generated a clonal myoblast cell line in the MB135 genetic background that expressed doxycycline (Dox)-inducible FLAG-tagged UPF1 (Figure 2A). We induced UPF1 overexpression by adding Dox to the media 12 hours prior to induction of differentiation, resulting in robust 46- and 16-fold increases in UPF1 mRNA and protein levels (Figure 2B-C). Consistent with our hypothesis, UPF1 overexpression reversed both the cell cycle and myogenic phenotypes observed following UPF1 KD. UPF1-overexpressing cells contained 40.3% BrdU+ nuclei versus 24.7% for control (uninduced) cells at day 0, immediately prior to the induction of differentiation (Figure 2D). UPF1-overexpressing versus control cells similarly exhibited 41.6% versus 55.3% MYOG+ nuclei at day 2, two days post-differentiation (Figure 2E). Taken together, our UPF1 knockdown and overexpression experiments indicate that UPF1 represses myogenesis through an unknown mechanism.
Figure 2. UPF1 overexpression slows myoblast differentiation.
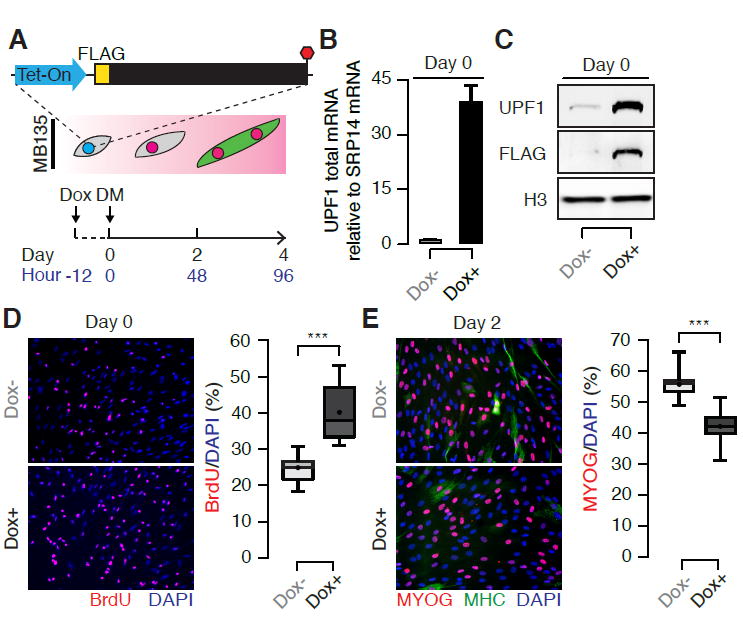
(A) Schematic illustrating a time course of human myoblast differentiation following the induction of UPF1 overexpression in MB135 myoblasts. Transgenic UPF1 was induced or not induced 12 hours prior to the induction of differentiation.
(B) Levels of UPF1 mRNA at Day 0, 12 hours after the addition of Dox to induce UPF1 expression. Error bars, standard deviation.
(C) Immunoblot for total and transgenic UPF1 protein at the same time point as in (B), measured using antibodies against UPF1 and FLAG. H3, loading control histone H3.
(D) Immunofluorescence labeling with an antibody against BrdU (red) at Day 0. Cells were fixed and labeled after incubation with BrdU-containing media for one hour. Box plot, percentage of BrdU+ nuclei measured over eight fields. Whiskers, max and min over the fields. */**/***, p < 0.05/0.01/0.001.
(E) Immunofluorescence labeling with antibodies against Myogenin (MYOG, red) and Myosin Heavy Chain (MHC, green) at Day 2. Box plot, percentage of MYOG+ nuclei measured over 10 fields. Whiskers, max and min over the fields. */**/***, p < 0.05/0.01/0.001.
UPF1 knockdown induces the MYOD transcriptional program
We next sought to determine the molecular mechanism by which UPF1 represses myogenesis. NMD represses diverse targets, including the gene encoding the marker of myogenic commitment MYOG (Gong et al., 2009). MYOG exhibits higher mRNA and protein levels two days after the induction of differentiation, but not before (Figure 1D-E, S1D-E). We therefore hypothesized that UPF1 might repress myogenesis by targeting the mRNA of a key earlier myogenic factor for degradation.
To identify myogenic regulatory factors that might connect UPF1 levels to the myogenic process, we characterized the transcriptomes of cells following UPF1 or control KD with RNA-seq two days post-differentiation. Coding genes that were up-regulated by ≥1.5-fold (365 genes) following UPF1 versus control KD were strongly enriched for myogenic processes in a Gene Ontology analysis, consistent with the cellular myogenic phenotype induced by UPF1 KD (Figure 3A-B). Subsequent analysis revealed that 6.6% of these up-regulated genes were known direct targets of the transcription factor MYOD, a master myogenic factor that is one of the earliest factors that initiates myogenesis and whose expression is sufficient to transdifferentiate fibroblasts into myoblasts (Tapscott et al., 1988). Furthermore, 13 of the up-regulated genes were MYOD-specific targets that are not promoted by other early myogenic transcription factors (Figure 3C). Global gene expression patterns following UPF1 KD therefore indicate that UPF1 KD activates the MYOD transcriptional cascade. Consistent with MYOD playing a central role in driving accelerated myogenesis following UPF1 KD, we observed a 2.7-fold increase in MYOD protein 24 hours after UPF1 KD, prior to the induction of differentiation (Figure 3D).
Figure 3. UPF1 knockdown promotes a myogenic gene expression program, including MYOD-specific targets.
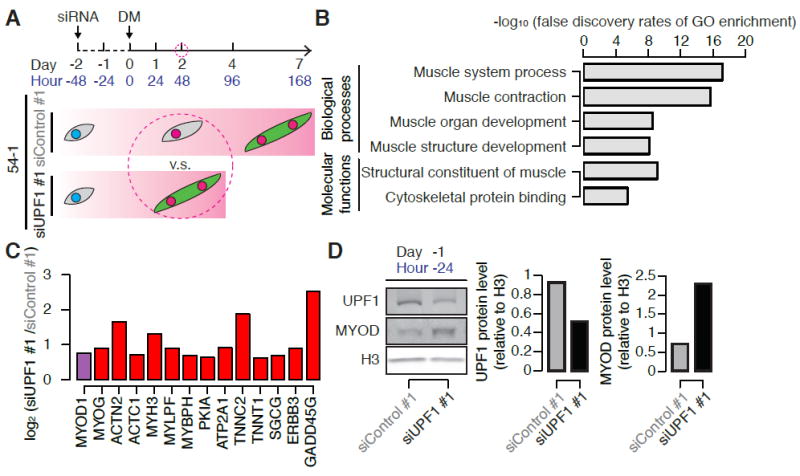
(A) Schematic illustrating when RNA was collected for RNA-seq (Day 2) during differentiation of 54-1 myoblasts following control or UPF1 KD.
(B) Gene Ontology (GO) enrichment analysis of genes that were up-regulated by ≥1.5-fold following UPF1 versus control KD at Day 2.
(C) Relative mRNA levels of genes that are specifically activated by MYOD and not other myogenic factors (red) (Conerly et al., 2016; Ishibashi et al., 2005), as well as MYOD mRNA itself (purple), following UPF1 versus control KD at Day 2. The illustrated genes exhibited increases in expression of ≥1.5-fold.
(D) Immunoblots for UPF1 and MYOD proteins one day following transfection with a control or UPF1-targeting siRNA (Day -1, or equivalently Hour -24), one day prior to the induction of differentiation. H3, loading control histone H3. Bar plots, quantification of UPF1 and MYOD relative to the loading control.
UPF1 knockdown induces MYOD protein, yet does not stabilize MYOD mRNA
Given UPF1’s central role in RNA surveillance, we hypothesized that UPF1 KD alleviated NMD-dependent degradation of the MYOD mRNA to promote increased MYOD protein levels. Two predictions follow from this hypothesis. First, MYOD mRNA levels should increase before or at the same time as do MYOD protein levels following UPF1 KD. Second, MYOD mRNA should be stabilized by UPF1 KD. To our surprise, neither prediction was correct. First, a time course revealed that MYOD protein up-regulation (which occurs between -30 and -27 hours) occurred 15 to 18 hours prior to MYOD mRNA up-regulation (which occurs between -24 and -12 hours) following UPF1 KD (red arrow, Figure 4A-C). Second, measurement of the MYOD mRNA half-life revealed that MYOD mRNA was not stabilized by UPF1 KD at day 0 despite clearly evident mRNA up-regulation (Figure 4D). We observed the same pattern, wherein MYOD protein up-regulation occurred 10 to 18 hours prior to MYOD mRNA up-regulation, following UFP1 KD in MB135 cells (Figure S2).
Figure 4. UPF1 knockdown induces MYOD protein in the absence of MYOD mRNA up-regulation.
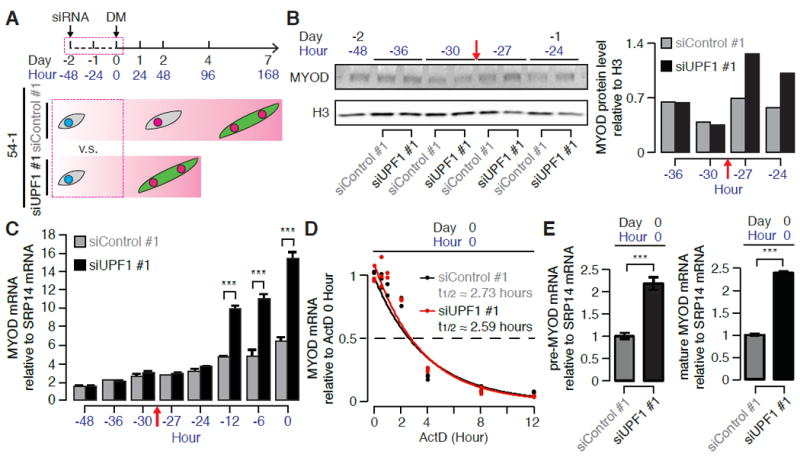
(A) Schematic illustrating time points for sample collection following UPF1 KD (Day -2 to 0, or equivalently Hour -48 to 0) in 54-1 myoblasts.
(B) Immunoblot for MYOD protein in the 24 hours immediately following transfection with a control or UPF1-targeting siRNA (Day -2 to Day -1, or equivalently Hour -48 to -24). H3, loading control histone H3. Bar plot, quantification of MYOD protein relative to the loading control. Red arrow indicates when MYOD protein levels detectably increased (between Hour -30 and -27).
(C) Relative levels of MYOD mRNA in the 48 hours immediately following transfection with a control or UPF1-targeting siRNA (Day -2 to Day 0, or equivalently Hour -48 to 0). Red arrow indicates when MYOD protein levels detectably increased (see B). */**/***, p < 0.05/0.01/0.001.
(D) Estimates of MYOD mRNA half-lives at Day 0 (Hour 0) in control- or UPF1-KD cells. MYOD mRNA levels were measured 0, 0.5, 1, 2, 4, 8 and 12 hours after the addition of actinomycin D (ActD, 2.5μg/mL) to inhibit transcription.
(E) Relative levels of MYOD pre-mRNA (left) and mature mRNA (right) at Day 0 (Hour 0) in control- or UPF1-KD cells, normalized to MYOD pre-mRNA or mature mRNA levels in control KD samples. Error bars, error propagation computed with the balanced repeated replication (BRR) method. */**/***, p < 0.05/0.01/0.001.
See also Figure S2.
Increased levels of MYOD mRNA following UPF1 KD are likely due to MYOD transcriptional up-regulation rather than alleviation of post-transcriptional repression. We observed similar increases in pre-mRNA and mature mRNA transcribed from MYOD following control versus UPF1 KD, consistent with MYOD transcriptional up-regulation (Figure 4E). Our data therefore indicates that UPF1 KD activates the MYOD transcriptional cascade, but does not support our original hypothesis that UPF1 represses myogenesis by regulating MYOD mRNA levels. In contrast, UPF1 KD induces MYOD protein while leaving its mRNA unaffected.
UPF1 regulates MYOD protein via the proteasome
We next sought to determine the molecular mechanism by which UPF1 KD resulted in up-regulation of MYOD protein. As we observed increased MYOD protein prior to up-regulation of MYOD mRNA following UPF1 KD, we hypothesized that UPF1 might repress MYOD protein by mediating MYOD proteolytic decay. To test this hypothesis, we returned to the inducible UPF1 overexpression system. Just as UPF1 KD induced MYOD protein, so did UPF1 overexpression result in decreased MYOD protein. MYOD protein levels decreased to 25.7% of their original levels within 12 hours of the addition of Dox to induce UPF1 overexpression (Figure 5A-B). UPF1-dependent suppression of MYOD protein was prevented when we treated cells with the proteasome inhibitor MG132 (Figure 5C). We concluded that UPF1 represses MYOD protein in a proteasome-dependent manner.
Figure 5. UPF1 promotes proteasome-dependent degradation of MYOD protein.
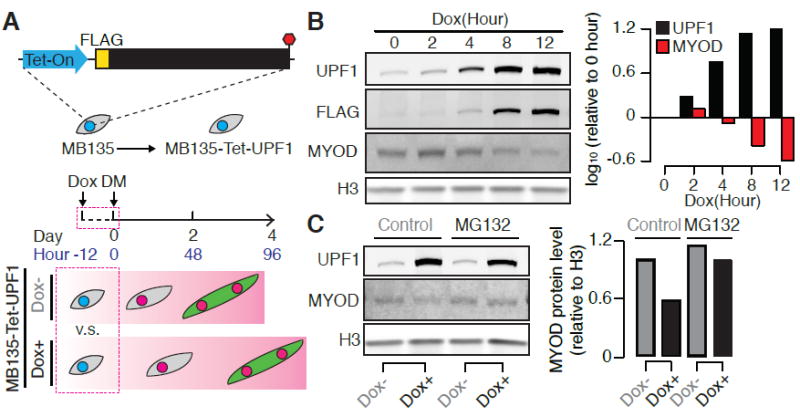
(A) Schematic illustrating a time course of differentiation following the induction of UPF1 overexpression in MB135 myoblasts. Samples were collected during the 12 hours immediately following Dox treatment (Hours -12 to 0).
(B) Immunoblot for total UPF1, FLAG-tagged transgenic UPF1, and MYOD proteins, measured during the 12 hours following Dox treatment. H3, loading control histone H3. Bar plot, quantification of total UPF1 and MYOD proteins relative to the loading control.
(C) Immunoblot for total UPF1 and MYOD proteins, measured 12 hours after Dox treatment, in cells treated with the proteasome inhibitor MG132 (10 μM, eight-hour treatment). H3, loading control histone H3. Bar plot, quantification of MYOD protein relative to the loading control.
UPF1 represses MYOD protein via its E3 ubiquitin ligase activity
How does UPF1 repress MYOD protein? As UPF1-mediated repression of MYOD protein is proteasome-dependent (Figure 5C), we hypothesized that UPF1 promoted MYOD protein decay via an unknown mechanism. While this mechanism could conceivably be indirect—for example, mediated by UPF1-dependent degradation of an mRNA encoding an E3 ubiquitin ligase that targets MYOD protein—three pieces of evidence led us to hypothesize a direct role for UPF1 in directing MYOD proteolysis. First, MYOD protein levels decreased concordantly with increased UPF1 expression, without the temporal lag expected of an indirect mechanism dependent upon mRNA stabilization of a tertiary factor (Figure 5B). Second, in addition to its RNA helicase domain, UPF1 possesses an N-terminal cysteine- and histidine-rich domain that is structurally similar to the RING domains frequently found in E3 ubiquitin ligases (Kadlec et al., 2006). Yeast Upf1 can self-ubiquitinate in vitro, suggesting that its RING-like domain functions as an E3 ligase, although it is unknown whether this E3 activity occurs in vivo or in human cells (Takahashi et al., 2008). Third, previous proteomics studies of the UPF1 interactome recovered components of the ubiquitin-proteasome system (Brannan et al., 2016; Flury et al., 2014). We therefore sought to test the hypothesis that human UPF1’s RING-like domain is responsible for repressing MYOD protein.
We first confirmed that the structure of human UPF1’s RING domain is consistent with the E3 ubiquitin ligase activity reported for yeast Upf1. UPF1’s RING domain resides near UPF1’s N-terminus and consists of two zinc fingers that were previously reported to provide a peripheral surface for UPF2 binding (Kadlec et al., 2006). We took advantage of a crystal structure of full-length UPF1 in complex with a C-terminal portion of UPF2 (Clerici et al., 2009) and compared the 2D and 3D structures of UPF1’s RING domain to the structures of known RING E3 ubiquitin ligases. This comparison revealed that the two zinc fingers that comprise UPF1’s RING domain are consistent with the canonical binding pocket formed by loops one and two of functional RING domains. This binding pocket of functional RING domains mediates interactions with E2 ubiquitin-conjugating proteins (Berndsen and Wolberger, 2014; Lorick et al., 1999; Metzger et al., 2014) (Figure 6A-C). We concluded that UPF1’s structure likely contains the E2-E3 binding pocket expected of a functional E3 ligase.
Figure 6. UPF1’s RING domain has E3 ubiquitin ligase activity.
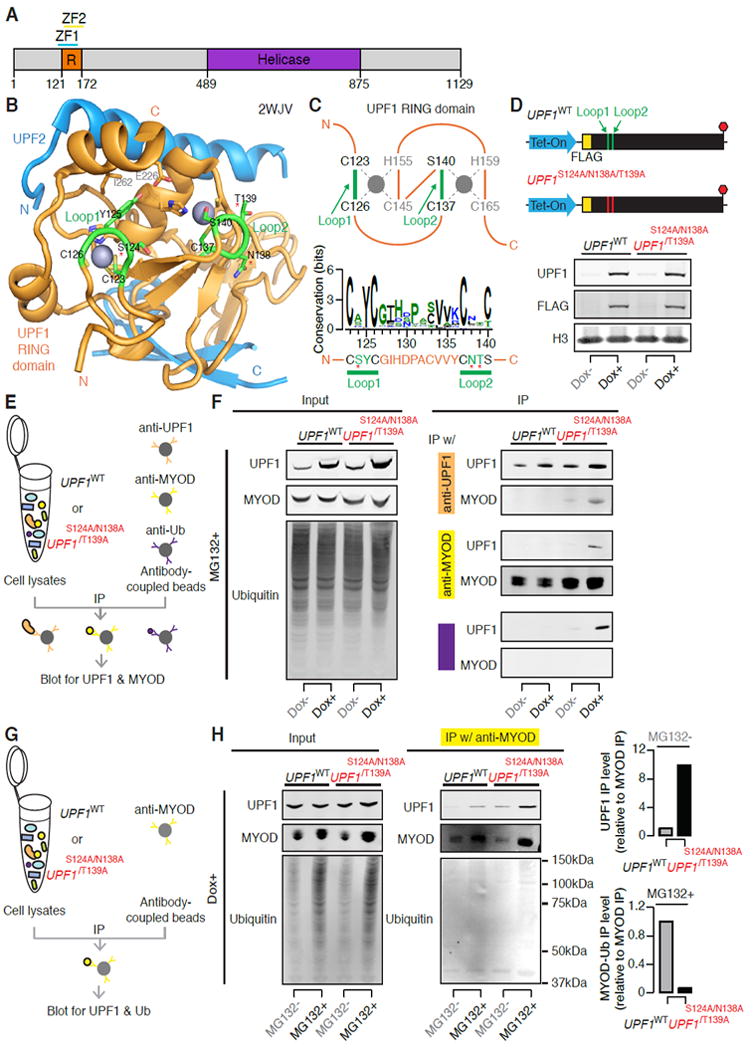
(A) Schematic of UPF1’s protein domain structure (Kadlec et al., 2006; UniProt Consortium, 2012). Orange, RING-like domain. Purple, RNA helicase domain.
(B) Protein structure of UPF1’s RING domain 1 (orange, residues 121-172 from PDB 2WJV; (Clerici et al., 2009)), illustrating two zinc fingers coordinating zinc ions (grey spheres), the canonical E2-RING E3 interaction pocket formed by Loop1 and Loop2 (green) which faces inside, and the UPF2 (cyan) binding surface on the periphery. Structure was visualized with PyMOL (Schrödinger, LLC, n.d.).
(C) Top, schematic diagram of UPF1’s RING domain 1 (orange, residues 121-172 from PDB 2WJV). The first zinc ion (grey circle) is held by a CCCH motif and the second zinc ion is held by a CSCH motif. Green, Loop1 and Loop2. Bottom, estimated amino acid sequence conservation of Loop1 and Loop2 (residues 123-140). Asterisks indicate the residues S124, N138, and T139 that we selected for mutagenesis.
(D) Top, schematic of construct to enable Dox-inducible expression of the mutant UPF1S124A/N138A/T139A. This construct was used to generate the clonal myoblast cell line MB135-Tet-UPF1S124A/N138A/T139A. Bottom, immunoblot for total and transgenic UPF1 protein from MB135-Tet-UPF1WT and MB135-Tet-UPF1S124A/N138A/T139A cells 12 hours following transgene induction with Dox. H3, loading control histone H3.
(E) Schematic of co-immunoprecipitation (co-IP) experiments with cell lysates from MB135-Tet-UPF1WT or MB135-Tet-UPF1S124A/N138A/T139A cells. Induced or uninduced cells were treated with MG132 for six hours to inhibit the proteasome. IP eluates from each pull-down were then probed with antibodies against UPF1 and MYOD. Ub, ubiquitin.
(F) Immunoblots of Input and IP eluates from (E). Left, Input total lysates were probed for UPF1, MYOD and Ubiquitin. Right, from top to bottom, IP eluates from the anti-UPF1, anti-MYOD, and anti-Ubiquitin pull-downs were probed for UPF1 and MYOD.
(G) As (E), but induced cells were treated or not treated with MG132 for six hours to inhibit the proteasome. IP eluates from the anti-MYOD pull-down were probed with antibodies against UPF1 and Ubiquitin.
(H) Immunoblots of Input and IP eluates from (G). Left, Input total lysates were probed for UPF1, MYOD and Ubiquitin. Right, IP eluates from the anti-MYOD pull-down were probed for UPF1, MYOD and Ubiquitin. Bar plots, quantification of UPF1 (top) or MYOD-Ub (bottom) in co-IP with MYOD relative to the level of MYOD IP.
See also Figure S3.
We sought to mutate UPF1’s RING domain in order to reduce its putative E3 activity while leaving its NMD activity intact. We constructed the mutant UPF1S124A/N138A/T139A by mutating three residues within loops one and two into alanine, with the goal of disrupting UPF1’s putative E2 binding pocket. These residues (S124/N138/T139) are proximal but not identical to the highly conserved cysteines and serine that are responsible for zinc ion coordination (C123 and C126 within loop one, and C137 and S140 within loop two). We did not mutate Y125 because it is responsible for holding the two α-helices on the RING domain periphery. We generated clonal MB135 myoblasts that inducibly expressed UPF1S124A/N138A/T139A, and compared these cells to our previously generated cells that inducibly expressed wild-type UPF1 (UPF1WT). The UPF1WT and UPF1S124A/N138A/T139A transgenes exhibited comparable UPF1 mRNA induction upon Dox treatment and produced similar levels of FLAG-tagged UPF1 protein (Figure 6D).
We first confirmed that mutating UPF1’s RING domain did not compromise its NMD activity. We measured levels of three endogenous NMD substrates in myoblasts overexpressing either wild-type or mutant UPF1. Overexpression of UPF1S124A/N138A/T139A did not substantially alter levels of these endogenous NMD substrates relative to overexpression of UPF1WT (Figure S3A). As we overexpressed mutant UPF1 in the context of endogenous (wild-type) UPF1 expression, these data demonstrate that UPF1S124A/N138A/T139A is not a dominant negative for NMD activity, but do not test whether UPF1S124A/N138A/T139A itself can function in NMD. We therefore specifically knocked down endogenous UPF1 with an shRNA targeting the 3’ UTR of UPF1 (which was not present in our transgenes), induced expression of UPF1WT or UPF1S124A/N138A/T139A, and measured levels of endogenous NMD substrates (Figure S3B, Figure S4). NMD substrate levels were not significantly higher in UPF1S124A/N138A/T139A-expressing versus UPF1WT-expressing cells following endogenous UPF1 KD. These data suggest that UPF1S124A/N138A/T139A is competent for NMD, and therefore is useful for our goal of deconvolving UPF1’s NMD and putative E3 ligase activities.
We next tested whether the S124A/N138A/T139A mutation had the intended effect of disrupting UPF1’s putative E2 ubiquitin conjugase binding pocket. The yeast E2 conjugase Ubc3 was previously reported to be a binding/activation partner for yeast Upf1’s E3 ligase activity (Takahashi et al., 2008). We therefore tested whether our RING domain mutation affected possible interactions between UPF1 and the E2 conjugase CDC34, the human ortholog of yeast Ubc3. Immunoprecipitation of FLAG-tagged UPF1 followed by immunoblotting against HA-tagged CDC34 revealed that UPF1 interacts with CDC34. The S124A/N138A/T139A mutation attenuates, but does not abolish, this interaction (Figure S3C). Suggestively, the E2 ubiquitin conjugase activity of CDC34 has been previously implicated in MYOD protein turnover (Song et al., 1998).
We sought to determine the specific mechanistic consequence of the S124A/N138A/T139A mutation for UPF1’s putative E3 ligase activity. The RING domains of many well-characterized E3 ligases are commonly involved in recruiting an E2 conjugase as well as transferring ubiquitin to the substrate, whereas other domains of the E3 ligases or co-factors govern substrate specificity (Berndsen and Wolberger, 2014; Deshaies and Joazeiro, 2009; Lorick et al., 1999; Lydeard et al., 2013; Skaar et al., 2013). As UPF1S124A/N138A/T139A exhibited reduced binding to the E2 conjugase CDC34 that was previously implicated in MYOD ubiquitination, we hypothesized that UPF1S124A/N138A/T139A would also exhibit reduced activity for ubiquitin transfer to the candidate substrate MYOD. This hypothesis generates several testable predictions. First, we expect UPF1 and MYOD to interact physically. Second, we expect UPF1S124A/N138A/T139A, but not UPF1WT, to remain charged with mono-ubiquitin upon proteasome inhibition. Third, we expect MYOD ubiquitination to decrease in UPF1S124A/N138A/T139A-expressing cells relative to UPF1WT-expresing cells. We systematically tested these hypotheses as follows.
We collected cell lysates from UPF1WT- or UPF1S124A/N138A/T139A-expressing cells following proteasome inhibition, immunoprecipitated UPF1, MYOD or ubiquitin, and probed for UPF1 and MYOD. UPF1S124A/N138A/T139A and MYOD robustly co-immunoprecipitated, while UPF1WT and MYOD interacted weakly. Immunoprecipitating ubiquitin revealed mono-ubiquitin associated with UPF1S124A/N138A/T139A but not UPF1WT (Figure 6E-F). We next compared MYOD ubiquitination in UPF1WT- and UPF1S124A/N138A/T139A-expressing cells. MYOD polyubiquitination was readily visible upon proteasome inhibition in UPF1WT-expressing cells, but not in UPF1S124A/N138A/T139A-expressing cells (Figure 6G-H). These data suggest that the RING domain mutation S124A/N138A/T139A reduces UPF1’s E3 activity by compromising the transfer of ubiquitin to substrates. The increased physical interaction between MYOD and UPF1S124A/N138A/T139A versus UPF1WT suggests, although does not prove, that release of MYOD from its association with UPF1 may be coupled to ubiquitin transfer. We concluded that UPF1 ubiquitinates MYOD in vivo.
UPF1 represses myogenesis via its RING domain
We next tested whether mutating UPF1’s RING domain reduced UPF1-mediated MYOD protein degradation and accompanying repression of myogenesis. We treated UPF1WT and UPF1S124A/N138A/T139A cells with Dox to induce transgenic UPF1 expression and measured MYOD protein stability following treatment with the translation inhibitor cycloheximide. Induction of UPF1WT resulted in rapid degradation of MYOD relative to uninduced cells, while induction of UPF1S124A/N138A/T139A did not. MYOD protein levels were reduced 6.5-fold in Dox-induced UPF1WT cells relative to uninduced UPF1WT cells six hours after cycloheximide treatment, but only reduced by 1.5-fold in induced relative to uninduced UPF1S124A/N138A/T139A cells (Figure 7A).
Figure 7. UPF1 RING domain mutations stabilize MYOD protein and promote myogenesis.
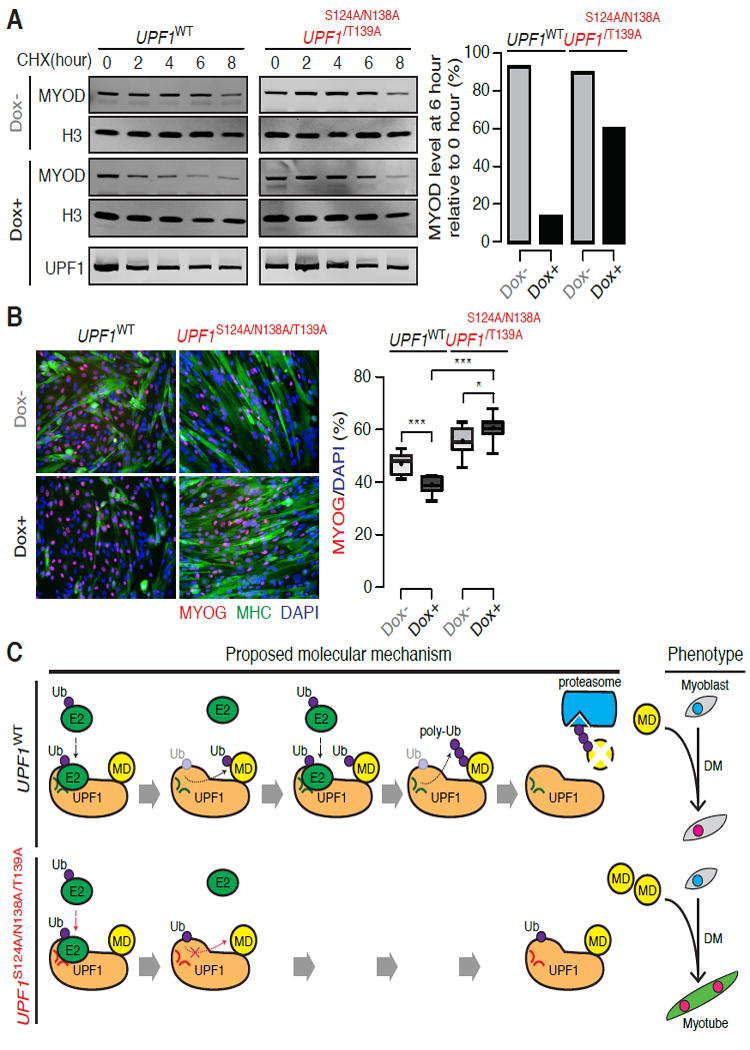
(A) Immunoblot for MYOD protein, measured throughout a time course following cycloheximide (CHX, 100 μg/mL) treatment to inhibit translation. Identical numbers of induced or uninduced MB135-Tet-UPF1WT or MB135-Tet-UPF1S124A/N138A/T139A cells were used. H3, loading control histone H3, which has a long half-life (Toyama et al., 2013). Lower blot, levels of induced UPF1 WT and UPF1S124A/N138A/T139A proteins.
(B) Immunofluorescence labeling of induced or uninduced MB135-Tet-UPF1WT or MB135-Tet-UPF1S124A/N138A/T139A cells with antibodies against Myogenin (MYOG, red) and Myosin Heavy Chain (MHC, green) at Day 2. Box plot, percentage of MYOG+ nuclei measured over 10 fields. Whiskers, max and min over the fields. */**/***, p < 0.05/0.01/0.001.
(C) Schematic of proposed interactions between UPF1 and MYOD (MD) during myogenesis. The E3 ligase activity of UPF1’s RING domain promotes proteasome-mediated degradation of MYOD protein in myoblasts. Mutating the E2-E3 binding pocket of UPF1 stabilizes MYOD protein, which in turn promotes myogenesis. DM, differentiation media. Green versus red loops within UPF1 indicate the wild-type versus mutated E2-E3 binding pockets. Black versus red dotted arrows indicate proposed RING domain activities for wild-type versus mutated UPF1, including recruiting an E2 conjugase and transferring ubiquitin (Ub) for the wild-type protein.
See also Figure S4.
Consistent with UPF1S124A/N138A/T139A’s impaired ability to ubiquitinate MYOD, expression of UPF1S124A/N138A/T139A reversed the anti-myogenic phenotype associated with UPF1WT expression. Induction of UPF1WT slowed myogenesis, while induction of UPF1S124A/N138A/T139A caused a modest but statistically significant increase in myogenesis. Cells expressing UPF1S124A/N138A/T139A versus UPF1WT exhibited a higher fraction of MYOG+ nuclei and a 2.5-fold increase in MYOG mRNA levels (Figure 7B, S4).
As the above experiments were conducted in the presence of endogenous (wild-type) UPF1, we speculated that the phenotypic consequences of UPF1S124A/N138A/T139A expression might be augmented when endogenous UPF1 was suppressed. We therefore measured the kinetics of differentiation of UPF1S124A/N138A/T139A- versus UPF1WT-expressing myoblasts in the context of endogenous UPF1 KD. Two days after the induction of differentiation, we observed a 10.4-fold increase in MYOG levels in cells expressing UPF1S124A/N138A/T139A versus UPF1WT in the context of endogenous UFP1 KD. In contrast, we observed a 2.5-fold increase in the context of unperturbed endogenous UFP1 expression (Figure S4). We concluded that UPF1’s RING domain is responsible for UPF1-mediated proteolysis of MYOD as well as UPF1’s repressive role in myogenesis (Figure 7C).
DISCUSSION
Recent studies provided clear evidence that UPF1 contributes to the regulation of cell differentiation and fate choice by selectively degrading mRNAs encoding key differentiation factors. Our data complement these previous reports by implicating UPF1 in regulating the kinetics of myogenesis. Surprisingly, however, UPF1 influences myogenesis by promoting protein, rather than mRNA, degradation. Given UPF1’s central role in nonsense-mediated decay and other RNA surveillance pathways, our findings therefore imply that UPF1 directly connects the RNA and protein decay machineries.
Given MYOD’s role as initiator of the myogenic cascade (Tapscott et al., 1988), our data strongly suggest that UPF1-dependent ubiquitination of MYOD explains how UPF1 represses myogenesis. Consistent with this hypothesis, mutations in UPF1’s RING domain that prevented UPF1-mediated MYOD ubiquitination also alleviated UPF1’s repressive role in myogenesis (Figure 7). However, we cannot rule out the possibility that UPF1 ubiquitinates factors in addition to MYOD which may contribute to UPF1’s repressive role in myogenesis.
Similarly, it is possible that UPF1 influences MYOD protein levels via mechanisms in addition to the ubiquitination characterized here. For example, UPF1 could potentially repress MYOD mRNA translation through an unknown mechanism. However, our data indicate that UPF1 primarily represses MYOD at the level of protein turnover rather than translation for two reasons. First, UPF1-dependent suppression of MYOD protein is proteasome-dependent (Figure 5C). Second, the three-fold increase in MYOD protein that occurs upon UPF1 KD (Figure 3D) is mimicked by expression of our UPF1S124A/N138A/T139A E3 ligase mutant, which results in an increase in MYOD protein stability of a similar magnitude (Figure 7A).
In many contexts, UPF1’s capacities to promote RNA as well as protein degradation may be closely linked rather than disjoint biochemical activities. For example, UPF1’s E3 activity may contribute to the degradation of abnormal and potentially deleterious peptides encoded by NMD substrates. Previous studies in yeast found that peptides encoded by reporter NMD substrates were degraded by the proteasome, and that Upf1 was important for this process (Kuroha et al., 2013; 2009; Verma et al., 2013). However, the method by which Upf1 promoted selective degradation of such peptides was not identified. We hypothesize that UPF1’s combined RNA helicase and E3 ligase activities might be directly responsible for recognizing NMD substrates as well as catalyzing the ubiquitination and degradation of the encoded peptides or proteins. However, further work is required to identify the spectrum of peptide products encoded by NMD substrates that are subjected to proteolytic decay, as well as test the hypothesis that UPF1’s E3 activity governs this process.
UPF1’s mechanistic role in both RNA and protein decay supports the conjecture that activation of translation-dependent mRNA quality control frequently results in peptide as well as mRNA degradation (Brandman and Hegde, 2016). Such direct connections between mRNA and protein quality control have been clearly elucidated for the ribosome quality-control complex (RQC). The RQC is a ribosome-associated quality control mechanism that degrades nascent peptides encoded by mRNAs lacking termination codons or containing rare codons, stem-loops, or other barriers to efficient translation (Brandman et al., 2012). These difficult-to-translate mRNA templates can induce ribosome stalling (Shao et al., 2013). The RQC may then recognize this stalled ribosome (Becker et al., 2011), split it into its constituent subunits (Shoemaker et al., 2010), expose the mRNA for endonucleolytic cleavage and/or exosome-mediated decay (Doma and Parker, 2006; van Hoof et al., 2002), and catalyze ubiquitination and degradation of the nascent peptide via the RING E3 ligase Listerin (Bengtson and Joazeiro, 2010). The canonical RQC components, including the E3 ligase Listerin, do not seem to be involved in degrading peptide products encoded by NMD substrates (Verma et al., 2013). However, deletion of Upf1 or the E3 ligase Ubr1 stabilized NMD substrate-encoded peptides in yeast, suggesting that Upf1 and/or Ubr1’s E3 ligase activities contribute to degradation of these aberrant peptides (Verma et al., 2013). It remains to be determined whether UPF1 and/or UBR1 promote degradation of NMD substrate-encoded peptides in human cells.
MYOD is probably not the only normally occurring protein that is targeted for regulated degradation in a UPF1-dependent manner. Just as UPF1 plays basal cytoprotective as well as regulatory roles in mRNA degradation, so may UPF1’s E3 activity result in degradation of abnormal peptides encoded by NMD substrates as well as normally occurring, well-formed proteins. Determining which specific features of MYOD mRNA and/or protein result in UPF1-dependent MYOD proteolysis may aid in the identification of other normally occurring proteins whose turnover is regulated by UPF1.
Finally, our results suggest that disease-associated perturbations of UPF1 may dysregulate protein quality control in addition to RNA surveillance. Impaired UPF1 function and/or NMD has been identified in genetic diseases, cancer, and viral infection. For example, the disease gene DUX4, which causes facioscapulohumeral muscular dystrophy, triggers UPF1 proteolysis (Feng et al., 2015); UPF1 is commonly subject to somatic mutations in pancreatic adenosquamous carcinoma (Liu et al., 2014) and inflammatory myofibroblastic tumors (Lu et al., 2016); the hepatitis C virus core protein interacts with a component of the exon-junction complex and disrupts NMD (Ramage et al., 2015). In each case, perturbations in UPF1 or associated NMD factors may inhibit normal protein decay as well as RNA decay, potentially implicating impaired protein quality control in these diseases.
STAR★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents and resources may be directed to and will be fulfilled by the corresponding author, Dr. Robert K. Bradley, at Fred Hutchinson Cancer Research Center (rbradley@fredhutch.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Three cell lines were used in this study: 54-1 (male), MB135 (female), and HEK293T (female, hypotriploid). Human myoblasts (54-1 and MB135 cells; (Krom et al., 2012; Snider et al., 2010)) were cultured in high serum growth media for proliferation below 60% confluence, and induced to differentiate in low serum differentiation media when the cells reached 99% confluence. The growth media was F-10 media-based (Gibco), and contained 20% fetal bovine serum (Gibco), 1% penicillin/streptomycin (Gibco), rhFGF (10 ng/mL, Promega), and dexamethasone (1 μM, Sigma). The differentiation media was also F-10 media-based (Gibco), and contained 1% horse serum (Gibco), 1% penicillin/streptomycin (Gibco), insulin (10 μg/mL, Sigma), and transferrin (10 μg/mL, Sigma). HEK293T cells used for lentivirus production were obtained from ATCC (CRL-11268) and were cultured in DMEM supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Gibco). Cells were cultured at 37°C and 5% CO2. For myoblast cells, fresh growth media was switched every other day, and cells were split when they reached 50% confluence at ratios of 1:2 or 1:4. For HEK293T cells, cells were split twice per week at a 1:10 ratio.
Primary myoblast culture
Early passages of primary myoblasts isolated from individuals MB135 (female) and MB2401 (male) were kind gifts obtained from Dr. Laurie Snider and Dr. Stephen J. Tapscott. Primary myoblasts were cultured under the same conditions as the myoblast cell lines described above. Primary myoblasts were fed with fresh growth media every day. These primary cells can only be maintained for 10 passages.
Microbe strains
NEB® 5-alpha Competent E. coli (High Efficiency) were obtained from NEB (C2987H). Cells were stored at -80°C and grown in LB medium at 37°C. NEB® 5-alpha cells were used for cloning expression plasmids. NEB® Stable Competent E. coli (High Efficiency) were obtained from NEB (C3040H). Cells were stored at -80°C and grown in LB medium at 30°C. NEB® Stable cells were used for production of lentivirus constructs.
METHOD DETAILS
siRNA transfection
siUPF1 #1 (Dharmacon, ON-TARGETplus siRNA J-011763-07), siControl #1 (Dharmacon, ON-TARGETplus negative control siRNA D-001810-03), siUPF1 #2 (Ambion, Silencer Select siRNA s11928), and siControl #2 (Ambion, Silencer Select negative control siRNA 4390843) were transfected with Lipofectamine RNAiMAX (Life Technologies). For transfection in a six-well plate format, 10 nM of siRNA and 6 μL of Lipofectamine RNAiMAX was added on top of cells at 60% confluence in each well. The cells would reach 99% confluence in two days and then be ready for differentiation.
Immunofluorescence microscopy
To assay cell cycle exit by BrdU incorporation, cells were incubated with BrdU-containing media (1 mM/mL, Life Technologies) at 37°C for 1 hour, and then fixed with 4% paraformaldehyde (Electron Microscopy Science) at 4°C for 30 min. To permeabilize fixed cells and break open genomic DNA structure, cells were first incubated with 0.5% TritonX100-containing PBS, and then with 1N HCl, 2N HCl and 0.1M Borate buffer. For labeling myogenic markers, cells were first fixed with 4% paraformaldehyde at room temperature for 10 min, and then permeabilized with 0.5% TritonX100-containing PBS. To immunolabel BrdU and myogenic markers after the above preparations, cells were blocked with PBS containing 1% BSA, incubated with primary antibodies in blocking buffer, washed with PBS, and then incubated with secondary antibodies. Antibodies used here were anti-BrdU (Invitrogen, 33900), anti-MYOG (Santa Cruz, M225), anti-MHC (R&D systems, MF20), secondary anti-mouse-FITC (BD Pharmingen), and secondary anti-rabbit-TRITC (BD Pharmingen). Immunofluorescently labeled cells were viewed and imaged with a ZEISS Axiophot fluorescence microscope. Pictures of ≥ 8 random fields were taken and then analyzed and quantified with ImageJ (Fiji).
RNA and protein extraction
For UPF1 knockdown and overexpression experiments, cells were lysed in TRIzol (Invitrogen) and RNA and protein were extracted in parallel according to the manufacturer’s protocol. Extracted RNA was then cleaned up using the Qiagen RNeasy Mini Kit. Extracted protein pellets were resuspended in SDS-Tris buffer (5% SDS and 0.5M Tris base) and briefly sonicated. For protein collection only, cells were lysed with RIPA buffer (Cell Signaling) supplemented with protease inhibitor tablets (Fisher Scientific), and then briefly sonicated. Soluble protein was then collected and measured by the bicinchoninic acid (BCA) assay (Thermo).
Western blotting
After determining protein concentrations by the BCA assay, 5 or 10 μg of total protein per sample was used for LI-COR system-based Western blotting. After transfer, the nitrocellulose membrane was blocked in the Odyssey® blocking buffer. Primary and 800CW IRDye-conjugated secondary antibody incubations were also carried out in the Odyssey® blocking buffer. Primary antibodies used were anti-UPF1 (Abcam, ab86057), anti-α-tubulin (Sigma, T9026), anti-MYOD (Thermo, 5.8A, MA5-12902, for knockdown or overexpression experiments; Abcam, ab126726, for MB135-Tet-UPF1S124A/N138A/T139A related experiments), anti-Ubiquitin (Boston Biochem, A-104), anti-FLAG (Thermo, FG4R, MA1-91878), anti-H3 (Abcam, ab1791) and anti-HA (Thermo, 2-2.2.14, 26183).
Real-time qPCR
1 μg of purified RNA per sample was converted into cDNA with the SuperScriptIII First-Strand System (Life Technologies). cDNA was diluted at a 1:50 ratio and used as the template for qPCR. In the final reaction, diluted cDNA was mixed with the 2X Power SYBR Green Master Mix (Life Technologies), together with relevant PCR primers. Primer information for qPCR is provided in Table S1.
Lentivirus construction and production
A UPF1 sequence-verified cDNA clone was ordered from Dharmacon (clone ID 5555509). The UPF1 cDNA was then amplified and cloned into the Dox-inducible lentiviral vector pCW57.1 (Addgene #41393) using the Gibson Assembly Cloning Kit (NEB). Due to high GC content within the first 500 bp of UPF1 cDNA, several attempts to introduce an N-terminal FLAG-tag via PCR primer overhangs failed. The FLAG-tag sequence was therefore introduced after the ATG start codon by replacing the first 381 bp of the UPF1 coding sequence with an in-frame, codon-optimized (lower GC content) gBlock DNA fragment containing the FLAG-tag (IDT). This lentiviral pCW57-FLAG-UPF1wt plasmid was later used as a template to introduce the S124A/N138A/T139A mutations with PCR primers containing mismatched nucleotides that altered the original codons to GCT (Alanine). To produce lentivirus, the lentiviral plasmids pCW57-FLAG-UPF1WT or pCW57-FLAG-UPF1S124A/N138A/T139A, together with the pCMV-VSV-G envelope (Addgene #8454) and the psPAX2 packaging (Addgene #12260) plasmids, were co-transfected into HEK293T cells. For each 10 cm plate of cells, 10 μg lentiviral plasmid, 2 μg envelope plasmid, and 2 μg packaging plasmid was used for calcium phosphate-based transfection. 16 hours after transfection, fresh media (DMEM with 10% cosmic calf serum, Gibco) containing 1 mM sodium butyrate (Sigma) was changed. 24 hours after changing the media, virus-containing media was collected, filtered through 0.45 μM filters, and concentrated by centrifuging at 5000 rpm at 4°C for 24 hours. The virus pellet was then resuspended in plain RPMI media (Gibco), aliquoted, and stored at -80°C. Lentiviral particle preparations were titrated by infecting MB135 cells with a serial dilution of virus and selecting with puromycin (2 ng/mL) for 7 days. Primer information for cloning is provided in Table S1.
Transgenic UPF1 cell line generation
MB135-Tet-UPF1WT and MB135-Tet-UPF1S124A/N138A/T139A cells were generated by transducing MB135 myoblasts with the relevant lentivirus at very low MOI. Starting the day after infection, cells were selected with puromycin (2 ng/mL). After single colonies formed on the low MOI-transduced plates, clones were picked, placed into 24-well plates using sterile cloning cylinders (Sigma, C7983), and expanded. All Dox induction was carried out at 1 μg/mL.
RNA-seq library preparation
4 μg of total RNA was collected from 54-1 cells transfected with siUPF1 #1 or a non-targeting siControl #1 two days post-differentiation and used to prepare RNA-seq libraries. Paired-end, poly(A)-selected, unstranded libraries were prepared using the Illumina TruSeq protocol with modifications to select for DNA fragments of length 100-400 bp by varying the bead-to-library ratios (Agencourt AMPure XP beads, Beckman Coulter). Size-selected DNA fragments were then amplified by PCR for 15 cycles and separated on a 2% agarose gel. DNA fragments of length 300 bp were cut out and gel purified using the Qiagen MinElute gel extraction kit. Barcoded RNA-seq libraries were sequenced on the Illumina HiSeq 2500 machine, resulting in ~100 million paired-end 2 × 49 bp reads per sample.
RNA-seq read mapping
Reads were mapped to the genome as previously described (Feng et al., 2015). Briefly, the steps involved were: (1) Map reads to the UCSC hg19 (NCBI GRCh37) genome assembly with Bowtie v1.0.0 (Langmead et al., 2009) and RSEM v.1.2.4 (B. Li and Dewey, 2011) with the ‘--v2’ argument. RSEM was called with the arguments ‘--bowtie-m 100 --bowtie-chunkmbs 500 --calc-ci --output-genome-bam’. (2) Filter out aligned reads with mapq scores of 0 or splice junction overhangs shorter than 6 bp. (3) Align the remaining unaligned reads with TopHat v2.0.8b (Trapnell et al., 2009) invoked with the arguments ‘--bowtie1 --read-mismatches 3 --read-edit-dist 2 --no-mixed --no-discordant --min-anchor-length 6 --splice-mismatches 0 --min-intron-length 10 --max-intron-length 1,000,000 --min-isoform-fraction 0.0 --no-novel-juncs --no-novel-indels --raw-juncs’. (4) Filter TopHat alignments as in step 2. (5) Merge aligned reads reported by RSEM and TopHat.
Gene expression measurement and Gene Ontology enrichment analysis
Gene expression was quantified using RSEM as above. Gene expression values were normalized with the TMM method (Robinson and Oshlack, 2010), with the scaling factor calculated using protein-coding transcripts only. Genes exhibiting ≥ 1.5-fold up-regulation with a Bayes factors ≥ 100 in the UPF1 KD sample compared to the control KD were identified, where Bayes factors were computed using Wagenmaker’s framework (Wagenmakers et al., 2010). Gene Ontology analysis was performed by comparing up-regulated genes to all protein-coding genes with the GOseq method (Young et al., 2010), using the ‘Wallenius’ approximation to correct for gene length bias. False discovery rates were then corrected using the Benjamini-Hochberg approach.
UPF1 RING domain conservation estimate
HHblits (Remmert et al., 2012) was used to generate multiple sequence alignments by querying the following fragment of UPF1 against the UniProt database (UniProt Consortium, 2012): KDLPIHACSYCGIHDPACVVYCNTSKKWFCNGRGNTSGSHIVNHLVRAKCKEVTLHKD GP. The e-value cutoff used was 1E-10 with four iterations. The generated alignment file was then input into WebLogo (Crooks et al., 2004) to create the plot of sequence conservation.
Actinomycin D chase, MG132 treatment and CHX chase experiments
To measure MYOD mRNA half-life at day 0 (following transfection of 54-1 cells with siControl #1 or siUPF1 #1), actinomycin D (ActD, Sigma, 2.5μg/mL) was added to cells to inhibit transcription. After 0, 0.5, 1, 2, 4, 8 and12 hours of ActD treatment, cells were collected with TRIzol (Invitrogen), RNA was extracted, and qPCR was carried out with primers specific to mature MYOD mRNA. To inhibit the 26S proteasome, MG132 (10μM) was added to MB135-Tet-UPF1WT cells 12 hours post-Dox induction. After 8 hours of MG132 treatment, identical numbers of cells were collected in RIPA buffer (Cell Signaling) supplemented with protease inhibitor tablets (Fisher Scientific), protein was extracted, and immunoblots for MYOD were performed. To measure MYOD protein levels in MB135-Tet-UPF1WT or MB135-Tet-UPF1S124A/N138A/T139A cells that were or were not treated with Dox to induce transgenic UPF1 expression, cycloheximide (CHX, 100μg/mL) was added to cells 12 hours post-Dox induction to inhibit translation. After 0, 2, 4, 6 and 8 hours of CHX treatment, identical numbers of cells were collected in RIPA buffer (Cell Signaling) supplemented with protease inhibitor tablets (Fisher Scientific), protein was extracted, and immunoblots for MYOD were performed.
CDC34-HA expression plasmid construction, mammalian cell transfection and co-immunoprecipitation experiments
A CDC34 sequence-verified cDNA clone was ordered from Dharmacon (clone ID 4103120). The CDC34 cDNA was cut out with restriction enzymes EcoRI/XhoI (NEB) and cloned into the mammalian expression vector pUB6/V5-His A (ThermoFisher Scientific) via ligation. A C-terminal HA-tag was introduced via PCR primer overhangs to generate the pUB6-CDC34-HA construct for transfection. Primer information for cloning is provided in Table S1. To assay UPF1 interaction with CDC34, MB135-Tet-UPF1WT and MB135-Tet-UPF1S124A/N138A/T139A cells were transfected with the pUB6-CDC34-HA construct 48 hours prior to induction of transgenic UPF1 expression by adding or not adding Dox for 12 hours. Two 15 cm plates of cells at ~60% confluence were used per condition. Each 15cm plate was transfected with 45 μg of DNA using the Lipofectamine 3000 Reagent (ThermoFisher Scientific). To collect cell lysates for FLAG immunoprecipitation, cells were trypsinized, washed twice with PBS, and cross-linked with DSP (ThermoFisher Scientific). Cross-linking was terminated by quenching with 20mM Tris-Cl pH 7.4 in PBS. After one more PBS wash, cells were lysed in 1mL pre-chilled NET-2 lysis buffer (50mM Tris-Cl pH 7.4, 150 mM NaCl and 0.05% NP-40) supplemented with phosphatase and protease inhibitor tablets (Fisher Scientific), followed by brief sonication (3 × 10 s) and incubation on ice (15 min). Cell lysates were collected after removing cell debris by centrifugation (10,000g at 4C for 15 min) and protein concentrations were determined with the Qubit® Protein Assay Kit (Fisher Scientific). 50 μL of lysates per sample were saved as inputs. To pull-down the transgenic FLAG-tagged UPF1, 40 μL of anti-FLAG M2 Magnetic Beads (Sigma) was combined with 1.2 mg of lysates and incubated overnight with rotation at 4C. To collect IP eluates, the protein-antibody-beads complexes were washed three times with pre-chilled NET-2 buffer, and then directly eluted by suspending beads in 2X SDS sample buffer. 20 μg of inputs and half of IP eluates per sample were used for the downstream immunoblotting experiments.
shRNA expressing lentivirus production
Lentiviral plasmids expressing either a non-targeting control shRNA (GCGCGATAGCGCTAATAATTT, Sigma-Aldrich) or a UPF1 3’UTR-targeting shRNA (GCATCTTATTCTGGGTAATAA, TRCN0000022254) were obtained as kind gifts from Dr. Omar Abdel-Wahab. Similar to concentrated lentivirus production above, to produce fresh lentivirus for efficient shRNA delivery and endogenous UPF1 knock-down, HEK293T cells on each 10cm plates, were co-transfected with 10 μg lentiviral plasmid, 2 μg envelope plasmid, and 2 μg packaging plasmid. 16 hours after transfection, fresh myoblast growth media (F-10 (Gibco) with 20% fetal bovine serum (Gibco), 1% penicillin/streptomycin (Gibco), rhFGF(10 ng/mL, Promega), and dexamethasone (1 μM, Sigma)) was changed onto HEK293T cells. 24 hours later, virus-containing myoblast growth media was collected, filtered through 0.45 μM filters, mixed with Polybrene (8 μg/mL, Sigma), and added onto myoblasts ready for shRNA experiments.
UPF1, MYOD, and ubiquitin co-immunoprecipitation experiments
Interactions between UPF1 and MYOD and MYOD ubiquitination were assayed as follows. 24 hours before collection, MB135-Tet-UPF1WT and MB135-Tet-UPF1S124A/N138A/T139A cells were or were not treated with Dox to induce transgenic UPF1; 6 hours before collection, cells were or were not treated with MG132 (Sigma, 10 μM) to inhibit the proteasome. Two 15 cm plates of cells at ~60% confluence were used per condition. To collect cell lysates for co-immunoprecipitation, cells were first trypsinized and washed three times with PBS, and then lysed in 1 mL pre-chilled NP40 cell lysis buffer (Fisher Scientific) supplemented with phosphatase and protease inhibitor tablets (Fisher Scientific), followed by brief sonication (3 x 10 s) and incubation on ice (15 min). Cell lysates were collected after removing cell debris by centrifugation (10,000g at 4C for 15 min), and protein concentrations were determined with the Qubit® Protein Assay Kit (Fisher Scientific). To prepare antibody-coupled magnetic beads, the following antibodies were used: anti-UPF1 (Abcam, ab86057), anti-MYOD (Abcam, ab126726), and anti-Ubiquitin (BostonBiochem, A-104). 1.5 μg of antibody was incubated with 50 μL protein G-coupled Dynabeads® (Fisher Scientific) for each IP. For each IP, 0.2 mg of lysates from each condition were added to the 1.5 μg antibody-coupled 50 μL beads and incubated overnight with rotation at 4C. To collect IP eluates, the protein-antibody-beads complexes were washed three times, transferred to new tubes to avoid elution of unspecific proteins bound to the tube wall, and then eluted in 20 μL of elution buffer by incubating with rotation for 2 min at room temperature. One third of IP eluates per sample were used for the downstream immunoblotting experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
As described above, differentially expressed genes were identified from the RNA-seq using a Bayesian statistical framework. Bayes factors were computed using Wagenmaker’s framework (Wagenmakers et al., 2010). Gene Ontology (GO) enrichment analyses for differentially expressed genes were performed using the GOseq method to correct for biases introduced by transcript length and expression (Young et al., 2010). False discovery rates for GO enrichment analyses were corrected using the Benjamini-Hochberg approach.
Statistical analyses for experimental data, including qPCR data (three replicates performed per experiment) and immunofluorescence microscopy data (eight to ten fields analyzed per experiment), were performed as follows. We computed p-values for a difference in distribution between two samples (e.g., control versus UPF1 KD) with a two-tailed t-test. The resulting p-values were graphically illustrated in figures with asterisks as described in figure legends.
Error bars in bar plots illustrating qPCR results represent the estimated mean ± standard deviation. Box plots illustrating immunofluorescence microscopy analyses represent the second and the third quartiles with two different shaded boxes, wherein the black dot indicates the mean and the error bars indicate the maximum and minimum over the measured data.
DATA AND SOFTWARE AVAILABILITY
Experimental data and plasmids
Raw experimental data from this study has been deposited in Mendeley (http://dx.doi.org/doi:10.17632/z3ff59vm4w.1). Relevant plasmids are available through Addgene (https://www.addgene.org/Robert_Bradley/).
Supplementary Material
Highlights.
UPF1 knockdown accelerates myogenesis, while UPF1 overexpression slows myogenesis
UPF1 promotes MYOD protein decay but does not affect MYOD mRNA stability
UPF1 has E3 ubiquitin ligase activity that targets MYOD protein for degradation
Mutating UPF1’s E3 ligase domain reverses UPF1’s repressive role in myogenesis
Acknowledgments
We thank Sergey Ovchinnikov for help with interpreting the UPF1 domain structure, Melissa Conerly for advice with MYOD antibodies, and Stephen Tapscott and Laurie Snider for project feedback and a gift of primary myoblasts. This research was supported by the Ellison Medical Foundation AG-NS-1030-13 (RKB), NIH/NINDS P01 NS069539 (RKB), and the FSH Society FSHS-22014-01 (SJ).
Footnotes
AUTHOR CONTRIBUTIONS:
QF, SJ, and RKB designed the experiments. QF performed the experiments and created the figures. QF, SJ, and RKB analyzed the data. QF and RKB wrote the paper.
Accession codes. FASTQ files from the RNA-seq experiments have been deposited in NCBI’s GEO database (accession number GSE87679).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balistreri G, Horvath P, Schweingruber C, Zünd D, McInerney G, Merits A, Mühlemann O, Azzalin C, Helenius A. The host nonsense-mediated mRNA decay pathway restricts Mammalian RNA virus replication. Cell Host Microbe. 2014;16:403–411. doi: 10.1016/j.chom.2014.08.007. [DOI] [PubMed] [Google Scholar]
- Barberan-Soler S, Lambert N, Zahler A. Global analysis of alternative splicing uncovers developmental regulation of nonsense-mediated decay in C. elegans. RNA. 2009;15:1652–1660. doi: 10.1261/rna.1711109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Armache J-P, Jarasch A, Anger AM, Villa E, Sieber H, Motaal BA, Mielke T, Berninghausen O, Beckmann R. Structure of the no-go mRNA decay complex Dom34-Hbs1 bound to a stalled 80S ribosome. Nat Struct Mol Biol. 2011;18:715–720. doi: 10.1038/nsmb.2057. [DOI] [PubMed] [Google Scholar]
- Bengtson MH, Joazeiro CAP. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 2010;467:470–473. doi: 10.1038/nature09371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21:301–307. doi: 10.1038/nsmb.2780. [DOI] [PubMed] [Google Scholar]
- Brandman O, Hegde RS. Ribosome-associated protein quality control. Nat Struct Mol Biol. 2016;23:7–15. doi: 10.1038/nsmb.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li G-W, Zhou S, King D, Shen PS, Weibezahn J, Dunn JG, Rouskin S, Inada T, Frost A, Weissman JS. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 2012;151:1042–1054. doi: 10.1016/j.cell.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannan KW, Jin W, Huelga SC, Banks CAS, Gilmore JM, Florens L, Washburn MP, Van Nostrand EL, Pratt GA, Schwinn MK, Daniels DL, Yeo GW. SONAR Discovers RNA-Binding Proteins from Analysis of Large-Scale Protein-Protein Interactomes. Mol Cell. 2016;64:282–293. doi: 10.1016/j.molcel.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno IG, Karam R, Huang L, Bhardwaj A, Lou CH, Shum EY, Song H-W, Corbett MA, Gifford WD, Gecz J, Pfaff SL, Wilkinson MF. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol Cell. 2011;42:500–510. doi: 10.1016/j.molcel.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Mourão A, Gutsche I, Gehring NH, Hentze MW, Kulozik A, Kadlec J, Sattler M, Cusack S. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J. 2009;28:2293–2306. doi: 10.1038/emboj.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conerly ML, Yao Z, Zhong JW, Groudine M, Tapscott SJ. Distinct Activities of Myf5 and MyoD Indicate Separate Roles in Skeletal Muscle Lineage Specification and Differentiation. Dev Cell. 2016;36:375–385. doi: 10.1016/j.devcel.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CAP. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Snider L, Jagannathan S, Tawil R, van der Maarel SM, Tapscott SJ, Bradley RK. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. elife. 2015;4:e04996. doi: 10.7554/eLife.04996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flury V, Restuccia U, Bachi A, Mühlemann O. Characterization of phosphorylation- and RNA-dependent UPF1 interactors by quantitative proteomics. J Proteome Res. 2014;13:3038–3053. doi: 10.1021/pr5002143. [DOI] [PubMed] [Google Scholar]
- Gardner LB. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol. 2008;28:3729–3741. doi: 10.1128/MCB.02284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloggnitzer J, Akimcheva S, Srinivasan A, Kusenda B, Riehs N, Stampfl H, Bautor J, Dekrout B, Jonak C, Jiménez-Gómez JM, Parker JE, Riha K. Nonsense-mediated mRNA decay modulates immune receptor levels to regulate plant antibacterial defense. Cell Host Microbe. 2014;16:376–390. doi: 10.1016/j.chom.2014.08.010. [DOI] [PubMed] [Google Scholar]
- Gong C, Kim YK, Woeller CF, Tang Y, Maquat LE. SMD and NMD are competitive pathways that contribute to myogenesis: effects on PAX3 and myogenin mRNAs. Genes Dev. 2009;23:54–66. doi: 10.1101/gad.1717309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt JA, Robertson AD, Burge CB. Global analyses of UPF1 binding and function reveals expanded scope of nonsense-mediated mRNA decay. Genome Res. 2013;23:1636–1650. doi: 10.1101/gr.157354.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi J, Perry RL, Asakura A, Rudnicki MA. MyoD induces myogenic differentiation through cooperation of its NH2- and COOH-terminal regions. J Cell Biol. 2005;171:471–482. doi: 10.1083/jcb.200502101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Furlan A, Gonzalez-Hilarion S, Leroy C, Gruenert DC, Tulasne D, Lejeune F. Caspases shutdown nonsense-mediated mRNA decay during apoptosis. Cell Death Differ. 2015;22:1754–1763. doi: 10.1038/cdd.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlec J, Guilligay D, Ravelli RB, Cusack S. Crystal structure of the UPF2-interacting domain of nonsense-mediated mRNA decay factor UPF1. RNA. 2006;12:1817–1824. doi: 10.1261/rna.177606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam R, Lou C-H, Kroeger H, Huang L, Lin JH, Wilkinson MF. The unfolded protein response is shaped by the NMD pathway. EMBO Rep. 2015;16:599–609. doi: 10.15252/embr.201439696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krom YD, Dumonceaux J, Mamchaoui K, Hamer, den B, Mariot V, Negroni E, Geng LN, Martin N, Tawil R, Tapscott SJ, van Engelen BGM, Mouly V, Butler-Browne GS, van der Maarel SM. Generation of isogenic D4Z4 contracted and noncontracted immortal muscle cell clones from a mosaic patient: a cellular model for FSHD. Am J Pathol. 2012;181:1387–1401. doi: 10.1016/j.ajpath.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroha K, Ando K, Nakagawa R, Inada T. The Upf factor complex interacts with aberrant products derived from mRNAs containing a premature termination codon and facilitates their proteasomal degradation. J Biol Chem. 2013;288:28630–28640. doi: 10.1074/jbc.M113.460691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroha K, Tatematsu T, Inada T. Upf1 stimulates degradation of the product derived from aberrant messenger RNA containing a specific nonsense mutation by the proteasome. EMBO Rep. 2009;10:1265–1271. doi: 10.1038/embor.2009.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci USA. 2003;100:189–192. doi: 10.1073/pnas.0136770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Shi Y, Wang P, Guachalla LM, Sun B, Joerss T, Chen Y-S, Groth M, Krueger A, Platzer M, Yang Y-G, Rudolph KL, Wang Z-Q. Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. EMBO J. 2015;34:1630–1647. doi: 10.15252/embj.201489947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Karam R, Zhou Y, Su F, Ji Y, Li G, Xu G, Lu L, Wang C, Song M, Zhu J, Wang Y, Zhao Y, Foo WC, Zuo M, Valasek MA, Javle M, Wilkinson MF, Lu Y. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med. 2014;20:596–598. doi: 10.1038/nm.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci USA. 1999;96:11364–11369. doi: 10.1073/pnas.96.20.11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou CH, Shao A, Shum EY, Espinoza JL, Huang L, Karam R, Wilkinson MF. Posttranscriptional Control of the Stem Cell and Neurogenic Programs by the Nonsense-Mediated RNA Decay Pathway. Cell Rep. 2014;6:748–764. doi: 10.1016/j.celrep.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Plank T-D, Su F, Shi X, Liu C, Ji Y, Li S, Huynh A, Shi C, Zhu B, Yang G, Wu Y, Wilkinson MF, Lu Y. The nonsense-mediated RNA decay pathway is disrupted in inflammatory myofibroblastic tumors. J Clin Invest. 2016;8:3058–3062. doi: 10.1172/JCI86508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14:1050–1061. doi: 10.1038/embor.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 2015;16:665–677. doi: 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- Martin L, Gardner LB. Stress-induced inhibition of nonsense-mediated RNA decay regulates intracellular cystine transport and intracellular glutathione through regulation of the cystine/glutamate exchanger SLC7A11. Oncogene. 2014;34:4211–4218. doi: 10.1038/onc.2014.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain DR, Pan Q, Reilly PT, Elia AJ, McCracken S, Wakeham AC, Itie-Youten A, Blencowe BJ, Mak TW. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci USA. 2010;107:12186–12191. doi: 10.1073/pnas.1007336107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta. 2014;1843:47–60. doi: 10.1016/j.bbamcr.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW, Maquat LE. Attenuation of nonsense-mediated mRNA decay facilitates the response to chemotherapeutics. Nat Commun. 2015;6:6632. doi: 10.1038/ncomms7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW-L, Maquat LE. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu Rev Genet. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramage HR, Kumar GR, Verschueren E, Johnson JR, Dollen Von J, Johnson T, Newton B, Shah P, Horner J, Krogan NJ, Ott M. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol Cell. 2015;57:329–340. doi: 10.1016/j.molcel.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger LLC. The PyMOL Molecular Graphics System [Google Scholar]
- Shao S, Malsburg von der K, Hegde RS. Listerin-dependent nascent protein ubiquitination relies on ribosome subunit dissociation. Mol Cell. 2013;50:637–648. doi: 10.1016/j.molcel.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Eyler DE, Green R. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science. 2010;330:369–372. doi: 10.1126/science.1192430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14:369–381. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider L, Geng LN, Lemmers RJLF, Kyba M, Ware CB, Nelson AM, Tawil R, Filippova GN, van der Maarel SM, Tapscott SJ, Miller DG. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song A, Wang Q, Goebl MG, Harrington MA. Phosphorylation of nuclear MyoD is required for its rapid degradation. Mol Cell Biol. 1998;18:4994–4999. doi: 10.1128/mcb.18.9.4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Araki Y, Ohya Y, Sakuno T, Hoshino S-I, Kontani K, Nishina H, Katada T. Upf1 potentially serves as a RING-related E3 ubiquitin ligase via its association with Upf3 in yeast. RNA. 2008;14:1950–1958. doi: 10.1261/rna.536308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ, Davis RL, Thayer MJ, Cheng PF, Weintraub H, Lassar AB. MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science. 1988;242:405–411. doi: 10.1126/science.3175662. [DOI] [PubMed] [Google Scholar]
- Thoren LA, Nørgaard GA, Weischenfeldt J, Waage J, Jakobsen JS, Damgaard I, Bergström FC, Blom AM, Borup R, Bisgaard HC, Porse BT. UPF2 is a critical regulator of liver development, function and regeneration. PLoS ONE. 2010;5:e11650. doi: 10.1371/journal.pone.0011650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR, Hetzer MW. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell. 2013;154:971–982. doi: 10.1016/j.cell.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–5. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295:2262–2264. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- Verma R, Oania RS, Kolawa NJ, Deshaies RJ. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. elife. 2013;2:e00308. doi: 10.7554/eLife.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenmakers E-J, Lodewyckx T, Kuriyal H, Grasman R. Bayesian hypothesis testing for psychologists: a tutorial on the Savage-Dickey method. Cogn Psychol. 2010;60:158–189. doi: 10.1016/j.cogpsych.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Wang D, Zavadil J, Martin L, Parisi F, Friedman E, Levy D, Harding H, Ron D, Gardner LB. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol Cell Biol. 2011;31:3670–3680. doi: 10.1128/MCB.05704-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Mönch K, Thoren LA, Nielsen FC, Jacobsen SEW, Nerlov C, Porse BT. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev. 2008;22:1381–1396. doi: 10.1101/gad.468808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.