

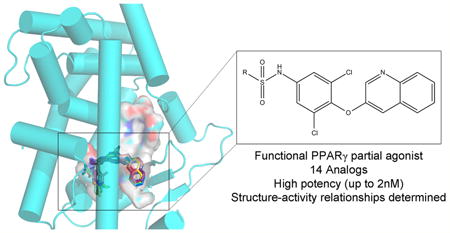
Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) is a nuclear receptor central to fatty acid and glucose homeostasis. PPARγ is the molecular target for type 2 diabetes mellitus (T2DM) therapeutics TZDs (thiazolidinediones), full agonists of PPARγ with robust antidiabetic properties, which are confounded with significant side effects. Partial agonists of PPARγ, such as INT131 (1), have displayed similar insulin-sensitizing efficacy as TZDs, but lack many side effects. To probe the structure-activity relationship (SAR) of the scaffold 1, we synthesized 14 analogs of compound 1 which revealed compounds with higher transcriptional potency for PPARγ and identification of moieties of the scaffold 1 key to high transcriptional potency. The sulfonamide linker is critical to activity, substitutions at position 4 of the benzene ring A were associated with higher transcriptional activity, substitutions at position 2 aided in tighter packing and activity, and the ring type and size of ring A affected the degree of activity.
Graphical abstract
Introduction
The peroxisome proliferator-activated receptor γ (PPARγ) is a transcription factor and member of the multidomain ligand-modulated nuclear receptor superfamily. PPARγ performs its function in part through heterodimerization with the nuclear receptor Retinoid X Receptor α (RXRα) and binding to DNA response elements in the proximal promoter region of target genes to regulate their expression. PPARγ target genes include proteins involved in peripheral insulin sensitivity, adipogenesis, fatty acid uptake and storage, glucose homeostasis, and metabolism of lipids and carbohydrates.1–4 The array of PPARγ target genes makes this receptor essential for normal insulin sensitivity and proper regulation of blood glucose. For instance, dominant negative partial loss of function mutations in PPARγ causes severe insulin resistance and is often accompanied by the onset of type 2 diabetes (T2DM).5 Adipose depots secrete various cytokines and fat cell specific hormones called adipokines, including adiponectin, adipsin, and resistin, many of which are under direct or indirect transcriptional control of PPARγ.6 These proteins modulate insulin sensitivity of muscle, liver, and adipose depots. An imbalance in this process leads to the development of insulin resistance and eventually type 2 diabetes.
The key role of PPARγ in metabolism has made it an appealing target for therapeutics of type 2 diabetes. The thiazolidinedione (TZD) class of PPARγ modulators, rosiglitazone (Avandia, GlaxoSmithKline) and Pioglitazone (Actos, Takeda), bind tightly within the ligand binding pocket of PPARγ and fully agonize the receptor by driving the interaction of the receptor with transcriptional coactivator proteins. Although TZDs afford robust insulin sensitization and normalization of blood glucose in T2DM patients, treatment with TZDs has been linked to an array of adverse side effects which has significantly reduced their utility. TZD side effects include weight gain, increased adipogenesis, renal fluid retention, loss of bone density, congestive heart failure, and plasma volume expansion leading to hemodilution.1,7,8 The exact causes of edema and myocardial infarction exhibited by some patients using TZD antidiabetics have not yet been elucidated to date. Given that PPARγ is a promiscuous binder of low affinity fatty acids and other metabolic signaling molecules, one possibility is that side effects arise from disruption of the natural signaling processes by a very high affinity agonist such as rosiglitazone, hyperactivation of target genes yet unidentified, or off-target interactions. However, ligands of PPARγ that only partially agonize the receptor have been shown to have reduced side-effect profiles in preclinical species and in some cases in clinical trials, yet they maintain robust insulin-sensitizing properties.9,10 These observations suggest that insulin sensitization can be separated from some if not all of the adverse effects associated with TZDs. Previous structural analysis of partial agonists of PPARγ demonstrated that their unique transcriptional output is attributed to distinct binding mechanisms,1,11 suggesting that structural properties of PPARγ ligands can govern their therapeutic index as T2DM therapeutics.
The PPARγ compound 2,4-dichloro-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)benzenesulfonamide (1; INT131) is a highly potent ligand of PPARγ, with a Ki of 10 nM in ligand-displacement direct binding assays, sufficient to displace rosiglitazone from the ligand binding pocket.11 1 is highly potent in cell based transcriptional activation assays, with an EC50 value of 4 nM and a maximal transcriptional activation of reporter genes of approximately 30% as compared to rosiglitazone. Distinct coregulatory recruitment profiles of 1, as compared to full agonists such as rosiglitazone, have been shown through cell based functional assays.11 In vivo studies in rodents have shown that 1 lowers blood glucose levels by over 30% with only a 0.3 mg/kg dose compared to the 3 mg/kg dose required for a similar effect using rosiglitazone. Significantly, 1 showed less total weight gain, heart weight gain, and lung weight gain than those observed with rosiglitazone administration, supporting the idea that 1 leads to less adipogenesis and fluid retention/edema. Adipocyte differentiation was not enhanced in cultured cells treated with 1, in contrast to significant induction with rosiglitazone treatment. Phase 1 studies with 1 (4 studies) showed favorable pharmacokinetic and pharmacodynamic properties.4 Phase 2a studies have demonstrated through a multicenter, double blind, placebo controlled study with T2DM patients that 1 is generally well tolerated while displaying antidiabetic effects;12,13 however, there are no publications available related to phase 2b or phase 3 studies of this compound.
While partial agonists such as 1 have shown great promise as insulin sensitizers, their mechanism of action has been less forthcoming. Antidiabetic effects have been shown to be correlated to blocking of phosphorylation of the receptor at position Ser273 of the ligand binding domain (LBD) by CDK5-activated ERK. Blockage of pS273 normalizes PPARγ target genes that are repressed in the diabetic and obese state. 7,14 However, the structural mechanism of this phenomenon is not well understood.
PPARγ is a multidomain protein containing a highly conserved DNA binding domain and a structurally conserved LBD. The LBD of PPARγ is comprised of 13 α-helices (H1 – H12 and H2′) and a small β-sheet. The binding pocket within the LBD is large enough (approximately 1200 Å3) to accommodate binding of a wide range of structurally distinct ligands, and the exact nature of the endogenous ligand remains controversial.15 Within the LBD is the Activation Function 2 (AF2) surface, which is formed by helices 3–5 and helix 12 and is important for ligand-dependent cofactor binding.1,2,16 The TZD headgroup of full agonists such as rosiglitazone has been shown to stabilize helix 12 and the AF2 by means of a tight hydrogen bond network, allowing for coactivator binding, as well as making hydrophobic contacts with H3 and stabilizing hydrogen bonding with the β-sheet. Interestingly, partial agonists of the receptor have been shown to require no stabilization of helix 12 for transcriptional activation.1
In order to better define the specific chemical epitopes of 1 responsible for high transcriptional activity, we developed a SAR platform correlating chemistry, potency based on activity, and protein structure. Here we present 14 analogs of 1. These analogs were tested for their ability to transcribe target genes (potency) in a cellular reporter assay, allowing us to compare activity to structure by means of EC50 values. An X-ray crystal structure was obtained for one of the analogs, 4-bromo-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)-2,5-difluorobenzenesulfonamide (10), bound to the PPARγ ligand binding domain to a resolution of 2.2 Å. The binding mode of the other 13 analogs was deciphered by means of in silico docking methods. The analogs displayed a wide range of activity from an EC50 of 2 nM to no binding at all, allowing us to define several important moieties in the structure–activity of the compound 1 scaffold. These analogs allowed us to define the sulphonamide linker, positions 2 and 4 of benzene ring A, as well as the nature and position of the substituents in ring A as being most important for defining high potency.
Results and Disussion
Design and Activity of Compound 1 Analogs
Compound 1 is a 514 Da sulfonamide comprised of three major aromatic moieties, denoted A, B, and C (Figure 1). A is a 2,4 di-Cl benzene linked by a sulphonamide to a 3,5-di-Cl aniline moiety (with aniline nitrogen as atom 1) denoted B. Ring system C is a quinoline moiety joined to B by an ether linker. The cocrystal structure of 1 bound to PPARγ has been solved,11 which shows that, unlike TZDs, 1 does not contact and stabilize helix 12 but instead wraps around helix 3 to stabilize helix 3 and the β-sheet region11 (Figure 2). The ligand forms two hydrogen bonds with Tyr327 of helix 5, which donates a hydrogen to the S=O group of 1 and accepts a hydrogen bond from the sulfonamide N–H in the center of the compound. A displaced parallel π–π interaction between the headgroup ring (ring A) of 1 and Phe363 of helix 7 situated 3.7 Å away further promotes ligand binding. The apolar region generated by Ile341, Cys285, Gly284, and Phe363 is also important for its interaction with the aromatic rings of 1.
Figure 1.

Chemical composition of partial agonist 1. The compound is comprised of three major moieties, denoted A, B, and C. The potential substitution positions on ring A are numbered.
Figure 2.

Crystal structure of 10 bound to PPARγ LBD. (A) Ribbons diagram of the PPARγ LBD (green) in complex with 10 (blue sticks). (B) Comparison of 10 (blue sticks) binding mode to 1 (yellow sticks), with the main scaffold in the same position and some similar hydrogen bonds formed (1; PBD: 3FUR).11 (C) Superimposition of 10 and 1 in the region contacting the β-sheet and H3.
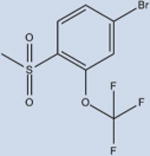
Previous structure–activity data has indicated that the composition of the C ring system of 1 had profound effects on CYP450 inhibition (CYP3A4). Specifically, the presence of a pyridine ring in the C position of 1 led to potent CYP3A4 inhibition (IC50 1 nM → 1 μM).4 However, changing the C ring from pyridine to quinoline showed the highest affinity PPARγ direct binding with significantly reduced CYP3A4 inhibition. Additionally, Cl substitution of positions R3 and R5 of the B ring aniline demonstrated the highest affinity PPARγ direct binding and transactivation.4 In order to define the structure–activity relationship of compound 1 analogs, we focused on altering the substitution of the A ring given the need for a quinoline group in the C ring for drug metabolism reasons and the need for a dichloro-aniline ring at the B position for affinity reasons. The purpose of our SAR is to precisely define what chemical epitopes of 1 and, conversely, what regions of the PPARγ LBD need to be stabilized for optimum affinity as well as transactivation. Additionally, given the appeal of antagonists of PPARγ as therapeutic agents, compound 1 analogs with reduced transactivation potential are likely to be required for an attractive therapeutic profile with a very low or no incidence of side effects.
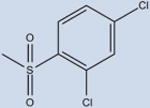
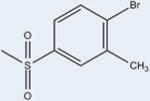
Fourteen analogs of 1 (as well as 1) were synthesized with various alterations of the A ring (Table 1) including one previously reported compound, (4-bromo-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)-2-(trifluoromethoxy)-benzenesulfonamide, described in our assays as compound 3.4 Synthesis was performed following reported protocols as in the Experimental Section.4 Substitutions of the benzene ring included Br, F, CF3, O–CH3, as well as CH3 at varying positions. The benzene ring was substituted for rings of a different nature in two of the compounds: a naphthalene ring system (N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)-naphthalene-2-sulfonamide (12) and a thiophene ring N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)thiophene-2-sulfonamide (15). These substituents were chosen to probe the effect of having a ring other than benzene in the A ring position of 1. All compounds contained a sulfonamide linker to the A ring with the exception of N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)-thiophene-2-carboxamide (7), containing an amide linker to the A ring in order to probe the effect on binding and activity of substitution at this position.
Table 1. Structure Activity of Compound 1 Analogsa.
| Formula | Compound | EC50 (nM) n=3 | Trans-activation (%)* n=2 | Purity (%) |
|---|---|---|---|---|
|
1 (INT131) | 170±10 | 24±4 | >95 |
|
2 | 600±61 | 29±6 | >99 |
|
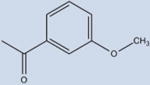
3 | 4±1 | 21±9 | >99 |
|
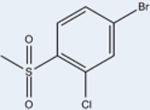
4 | 134±13 | 27±8 | 85 |
|
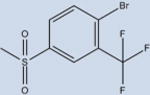
5 | 957±499 | 18±7 | 97 |
|
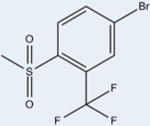
6 | 94±10 | 34±10 | 93 |
|
7 | ND | 2±1 | >99 |
|
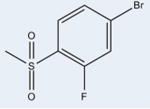
8 | 2±1 | 13±7 | 88 |
|
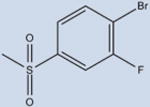
9 | 169±14 | 9±5 | 94 |
|
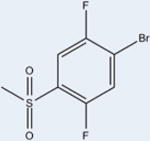
10 | 131±6 | 10±6 | >98 |

|
11 | 151±20 | 11±6 | 96 |
|
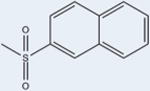
12 | 4289±289 | 23±12 | 90 |
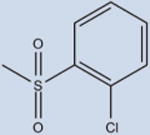
|
13 | 5810±945 | 19±11 | 99 |
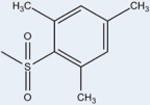
|
14 | 3314±2923 | 14±6 | >99 |
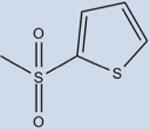
|
15 | ND | 14±8 | >99 |
Each data point in the EC50 dose response was repeated in triplicate, and standard deviation was derived from these values. In addition, two biological replicates were performed to ensure reproducibility of the data. ND means not determined.
All compounds were tested for activity against PPARγ in a cell based transcriptional reporter assay to measure both potency (EC50) as well as the maximal transcriptional output as normalized to the model full agonist rosiglitazone (transactivation = 100%). Potencies of the compounds ranged from 2 nM (8) to no activity (7 and 15). EC50 and maximal transactivation values can be found in Table 1 for all compounds. The maximum transactivation level for 1 was 24%, and our analogs displayed maximum transactivation levels in the range 2–34%, indicating that all compounds were partial agonists.
Structural Analysis of Compound 1 Analogs in Complex with PPARγ
In order to probe the structural basis of 1 analog potency, we performed cocrystallization experiments of compound 1 analogs with PPARγ. Cocrystals were only obtained for 4-bromo-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)-2,5-difluorobenzenesulfonamide (10) in complex with the PPARγ LBD. The PPARγ LBD bound to 10 was solved to a resolution of 2.2 Å, and the phase problem was overcome by molecular replacement. Data processing and refinement statistics can be found in Table 2. The asymmetric unit contained two subunits of PPARγ (homodimer), conforming to the canonical PPARγ LBD fold. The 10 bound LBD structure revealed high global similarity to previously solved structures with a 0.84 Å RMSD with the LBD from the full agonist rosiglitazone bound structure (over 256 Cα atoms, PDB: 2PRG), a 0.69 Å RMSD with the LBD from the apo structure (over 258 Cα atoms, PDB: 1PRG), and a 0.88 Å RMSD with the partial agonist 1 bound LBD (over 254 Cα atoms, PDB: 3FUR).11,17 A ribbons diagram of the 10 bound LBD can be seen in Figure 2A. The 10 ligand was easily visible in the electron density and modeled into the difference Fourier electron density. A reduced model bias electron density map of 10 can be viewed in Figure S1.
Table 2. Crystallographic Dataa.
| Parameter | Compound (10) |
|---|---|
| Space group | C2 |
| Cell dimensions a, b, c (Å) | 93.1, 62.2, 118.9 |
| Monoclinic angle β (deg) | 102.2 |
| X-ray source | Synchrotron: APS 22-ID |
| Wavelength (Å) | 1.0 |
| Resolution range (Å) | 50–2.24 |
| Last shell (Å) | 2.32–2.25 |
| Rmerge (%) | 0.051 (0.178) |
| Observations | 146759 (13592) |
| Unique reflections | 31321 (2946) |
| Mean (I)/σ(I) | 15.3 (5.9) |
| Completeness | 98.0 (99.7) |
| Multiplicity | 4.7 (4.6) |
| Structure refinement | |
| Resolution range (Å) | 45.5–2.24 |
| Rwork (%) | 0.2039 |
| Rfree (%) | 0.2555 |
| Total number of | |
| Non-hydrogen atoms | |
| Protein atoms | 4139 |
| Ligand atoms | 64 |
| Water molecules | 159 |
| RMSD | |
| Bond length (Å) | 0.007 |
| Bond angle (deg) | 0.878 |
| B-factors (Å2) | |
| Overall | 61.2 |
| Average protein atoms | 61.3 |
| Average ligand atoms | 63.2 |
| Average solvent | 58.8 |
| Ramachandran statistics | |
| Most favored regions (%) | 96.5 |
| Allowed regions (%) | 3.29 |
| Disallowed regions (%) | 0.19 |
Values in parentheses are for the highest resolution shell.
Compound 10 occupies the ligand binding pocket of PPARγ and is centered at and bends around helix 3; the scaffold location of 10 is similar to 1 (Figure 2B and 2C). Several weak electrostatic interactions are formed between the sulfonamide linker of 10 and residues of or near the AF2. One oxygen atom of the sulfonamide moiety of 10 is in weak hydrogen bonding distance to the Tyr327 side chain oxygen atom (3.5 Å), the Lys367 side chain nitrogen atom (2.9 Å), and the His449 side chain nitrogen atom (3.5 Å). This leaves Tyr473 of the AF2 helix 12 unstabilized and not within hydrogen bonding distance to His449 (4.6 Å), as seen in full agonist bound structures. Tyr327 accepts a hydrogen atom from the sulfonamide N–H in the center of the 10 compound to form a 3.2 Å hydrogen bond analogous to 1 (2.8 Å). A comparable set of π–π interactions between the A ring of 10 and Phe363 exists and likely contributes to ligand binding affinity. 10 engages in hydrophobic contacts with Ile341 (β sheet), Cys285 (H3), and Gly284 (H3), similarly to 1 (Figure 2C). 10 contains a Br atom at position 4 of the A ring (in contrast to a Cl in 1) which is in weak halogen bonding distance to the main chain nitrogen atom of Phe282 (3.7 Å).
In silico docking of all other ligands (those other than 10) shown to bind to the PPARγ LBD was carried out using a Monte Carlo method in the Molsoft ICM Software suite to identify the binding mechanisms of these compounds. As a positive control, 1 was docked into the PPARγ LBD and compared to the experimentally derived X-ray crystal structure. The binding mode of the two structures was nearly identical, and a superimposition of the two molecules is located in Figure S2. Docking of the 1 analogs revealed a similar overall binding mode for them all, with the C and B rings exhibiting very high positional similarity with only minor differences in the A rings due to substitution. A superimposition of the ligands docked to the receptor can be found in Figure 3.
Figure 3.

Superimposition of docked 1 analogs. The ligands (colored sticks) that have been shown to bind to PPARγ were docked in silico to the PPARγ LBD receptor (blue ribbons). The scaffold of the ligands binds in a similar position, with slight variations at ring A due to substitutions at that location.
The Sulfonamide Moiety Is Essential for Binding to PPARγ
The A ring of 1 is located in the hydrophobic pocket between H3 and H7, making not only hydrophobic interactions with Cys285 of H3 but more importantly several critical π–π interactions with Phe363. The A ring of 1 is connected to the 1 scaffold via the S atom of the sulfonamide linker. The sulfur atom displays tetrahedral geometry, which places the connected A ring in the ideal position to interact with Phe363 in a stacking manner (Figure 2B).
To probe if the sulfonamide linker of 1 is necessary for activity, we synthesized 7. Table 1 shows that 7 shows no significant transactivation activity and contains an amide linker instead of the sulfonamide of the other ligands. This substitution makes the atom which connects the A ring to the compound 7 scaffold, C of the C=O, in a planar Sp2 hybridized state. As shown in Figure 4, this places the A ring in a location closer to the AF2 surface and incapable of making the favorable π–π interactions with Phe363 as well as the favorable hydrophobic interactions in the surrounding pocket. The sulfonamide moiety absent in 7 also means that there is no atom in appropriate distance to act as a hydrogen bond donor or acceptor with the side chain of Tyr327. Taken together, our data thus suggests that the A ring linker geometry, and to a lesser degree interaction with Tyr327, is essential for significant receptor binding and activity. This is critical for potential drug design, as the inclusion of a sulfonamide moiety will substantially promote compound binding to PPARγ.
Figure 4.

Substitution of a sulfonamide for an amide linker has detrimental effects on the capability of 7 to bind to PPARγ. Shown is a superimposition of the 7 structure (white) with the 1 structure (green). The carbon of the C=O in 7 (white sticks) is confined to a planar conformation which prevents the A ring of 7 from making favorable π–π interactions with Phe363 as well as hydrophobic interactions with residues of the binding pocket.
Higher Affinity Is Achieved with Br at Position 4 of Benzene Ring A
The ligands with the nine highest potencies (2–957 nM) all have a Br atom at position 4 of the A ring (4-bromo-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)-3-methyl-benzenesulfonamide (2), 3, 4-bromo-2-chloro-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)benzenesulfonamide (4), 4-bromo-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)-3-(trifluoromethyl)benzenesulfonamide (5), 4-bromo-N-(3,5-di-chloro-4-(quinolin-3-yloxy)phenyl)-2-(trifluoromethyl)-benzenesulfonamide (6), 4-bromo-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)-2-fluorobenzenesulfonamide (8), 4-bromo-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)-3-fluorobenzenesul-fonamide (9), 10, and 4-bromo-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)benzenesulfonamide (11)). In addition, the five lowest affinity ligands (3314 nM-no significant activity) all lack Br at this position. Superimposition of Br-containing compound 1 analogs at different positions in the A ring can be seen in Figure 5. Structural analysis reveals that the Br atom is located in nearly identical positions in all of these structures. This places the Br atom within distance to form a halogen bond with the backbone nitrogen atom of Phe282 (Figure 5) and allows for better packing within the ligand binding pocket through van der Waals interactions. The halogen bond is indeed weak (4 Å), and improved activity from substitution at this site with Br is perhaps associated with tighter packing of the pocket in this region. This is clearly an important observation for ongoing drug design efforts with 1, which would therefore ideally incorporate Br at this position or place a hydrogen bonding partner extending from position 4 of the A ring in proximity to the backbone nitrogen atom of Phe282.
Figure 5.

Superimposition of ligands (colored sticks) containing a 4-Br substitution in benzene ring A reveal very similar positions of the bromine atom within the binding pocket of PPARγ (green ribbons). A bromine at position 4 of ring A results in higher affinity of the ligand. The presence of a Br atom in this position enables a weak hydrogen bond with the backbone nitrogen of Phe282 (shown as dashes).
Tight Packing within the Ligand Binding Pocket Is Associated with Higher Activity
Examination of space filling representations of the compound 1 analogs within the ligand binding pocket of PPARγ shows that the highest affinity ligands are distinguished from lower affinity ligands by means of tighter molecular packing. The five ligands (3, 4, 6, 8, and 10) with the highest affinities (2–957 nM) present a substituent at position 2 on benzene ring A. These substituents contribute steric bulk to the ligand and more tightly occupy the space of the binding pocket between helices 3 and 7, increasing the strength and number of van der Waals interactions. Substitutions at position 2 of the benzene ring A of these compounds include –F, –CF3, –Cl, and –OCF3. van der Waals volumes for these groups range from 13 Å3 to 100 Å3, with a general trend of the smaller van der Waals volume substitutions correlating with the lower activity. As shown in Figure 6A, position 2 of the benzene ring A is most amenable to substitution for improved packing, such that compounds with substitutions on other positions do not pack as favorably in the binding pocket (Figure 6B) and induce lower activation rates of the receptor. Despite 3 and 6 having very similar structures (–OCF3 versus –CF3), 3 is significantly more potent than 6 because the addition of the oxygen enables the trifluoro group to pack deeper in the binding pocket between helices 3, 7, and 11 (Figure 6C). Furthermore, this extension also enables two of the fluorine atoms to be within halide bond distance (both approximately 3 Å) of the side chain nitrogen atom of Gln286, which further increases the compound's affinity for PPARγ.
Figure 6.

Effective space filling within the PPARγ LBD binding pocket has been shown to correlate with higher affinities. Displayed are superimpositions of the compound 1 analogs (colored sticks) bound to PPARγ (ribbons, green or blue), and a surface representation of the ligand binding pocket (gray surface). Ligands with substitutions at (A) position 2 of ring A have better packing within the pocket and higher affinities than (B) those which have substitutions at other positions. Ligands with substitutions at position 2 include 3 (red), 4 (orange), 6 (green), and 8 (cyan). Ligands with additional substitutions at positions 3 or 5 have lower affinities and include 2 (yellow), 5 (pale orange), 9 (teal), 11 (purple), 2-chloro-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)benzenesulfonamide (13) (light blue), 14 (turquoise), 15 (lilac), and 10 (dark blue). (C) The oxygen linker in the trifluoro substitution of 3 enables it to reach further into the binding pocket than 6 and confer a better lock-and-key fit in addition to forming additional hydrogen bonds with Gln286. (D) The naphthalene moiety of 12 (pink) extends as far into the binding pocket as other ligands, as shown by comparison with 9 (teal), 10 (dark blue), and 11 (purple).
Divergence from Aromatic Benzene Ring A Does Not Favor High Activity
Two of the compound 1 analogs (12 and 15) differ from the otherwise conserved A-ring containing a single aromatic benzene. These two ligands have very low potencies (4289 nM and ND, respectively). This suggests that the six-membered aromatic ring is optimal for PPARγ activity. 12 has a naphthalene moiety, sizably enhancing its hydrophobicity at this position as compared to the other analogs (Figure 7). Despite having many more interactions with hydrophobic residues of the binding pocket, its potency is poor, with an EC50 of 4289 nM. This may be due to the nature of the aromatic system. Rings B and C of 12 are in nearly identical positions to the B and C rings of 1 and other analogs (Figure 3), suggesting that differences in activity lie solely in the A ring. Additionally, the naphthalene moiety of 12 is in a parallel displaced stacking interaction with Phe363 positioned similarly to ring A of the other analogs. While the naphthalene group is larger than the benzene ring A of the other analogs, it packs well without clashing into the hydrophobic pocket of the LBD between H3 and H7 (Figure 6D). Although 12 can form stacking interactions with Phe363, the activity is much lower than those of the other compound 1 analogs, implying that differences in affinity may lie in the absence of substitutions on the naphthalene group. 12 is the only compound that lacks a substituent on the A ring. The Hunter/Sanders model has demonstrated that electron withdrawing substituents, such as the halides, diminish electron density in the π cloud of the ring, leading to enhanced π-stacking.18 This is consistent with our data in which the electron withdrawing substitutions of the A ring, such as F, Br, and Cl, show stronger activity. In agreement with this hypothesis is compound N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)-2,4,6-trimethylbenzenesulfonamide (14), which is substituted with no electron withdrawing groups, only methyl groups (electron donating), which also shows diminished activity as compared to the analogs with electron withdrawing substitutions.
Figure 7.

Naphthalene substitution at position A of 12 enables extensive hydrophobic contacts contributed by helices 3 and 7. Shown is a superimposition of the 1 (yellow sticks) and 12 (purple sticks) compounds bound to the PPARγ LBD (green ribbons). The naphthalene moiety of 12 (purple) is more hydrophobic than other ligands, enabling unique hydrophobic interactions with the PPARγ binding pocket.
Compound 15, where the benzene ring was replaced by a thiophene moiety, showed no data in terms of potency for the receptor. Importantly, 15 does not make π–π interactions with Phe363, with the component thiophene known to be less aromatic than benzene, which provides less availability for π–π interaction, likely decreasing binding affinity. It could be postulated that the interaction with Phe363 favors higher affinity ligands. Critically, structural analysis also demonstrated 15 does not pack as well in the PPARγ LBD due to the lack of substitution patterns characteristic of the higher-affinity compounds. This includes a halide at position 4, and steric bulk in positions 2 or 3. The five-membered thiophene ring does not pack as tightly in the hydrophobic pocket as the other analogs due to lack of substitution as well as the smaller, five-membered ring size. This, taken together, suggests that a single aromatic benzene forms the best scaffold for ring A.
Substitutions at Benzene Ring A of Compound 1 Have Modest Effects on the Degree of Agonism
Given the promise of PPAR antagonists as a novel class of antidiabetics with little or no side effects, decreasing the degree of agonism of 1 would be ideal. There is some variation in the level of transactivation of the receptor among all of the compound 1 analogs we surveyed, as maximum transactivation rates were in the range 2–34%. While this is a similar transactivation rate from the compound 1 parent compound (24% maximal transactivation), further decreases in trans-activation will require more SAR efforts. Our data suggests that modifying the ligands at the A ring moiety affects only their affinities for the receptor, and not their level of agonism. Comparing the binding mechanisms of the current compounds with full agonists demonstrates that they bind in different positions, and this governs their level of transcriptional output. The consistent binding mechanisms of the partial agonists can be attributed to the presence of the conserved regions of rings B and C. It is important for design of T2DM therapeutics using the compound 1 scaffold to include these moieties, so that the compound will have high affinity binding while not fully agonizing the receptor. Lessons from other scaffolds have revealed that reducing agonism can be achieved through destabilization of the AF2 surface and H12. Hence, future compound 1 based therapeutics with lessoned transcriptional activation may be achieved through substitution of the quinolone ring extending to H12 to form destabilizing contacts.
Conclusion
We have described here the synthesis of 14 analogs of the antidiabetic compound 1 by means of chemical alteration of the A ring. This study includes protein structural studies which, when combined with transcriptional activation assays (which measure potency of the compound 1 analogs), allow the derivation of precise structure–activity relationships of these compounds. This SAR study defined the effects that substituents of the A ring, the position of substituents on the A ring, and the type of ring at position A have on transcriptional activity and, more importantly, how these substituents affect interaction of the compound 1 scaffold with the PPARγ receptor. The SAR of compound 1 analogs revealed seven ligands with increased potency for PPARγ. These maintained the sulfonamide moiety and a bromine atom at position 4 on the aromatic benzene ring A. They differ from 1 in their substitutions, which enable better lock-and-key fitting in the binding pocket of PPARγ, mediated by the presence of a bulky substitution at position 2 of benzene A. Additionally, the SAR data demonstrates the importance of a sulfonamide linker as well as a substituted, 6-membered benzyl ring in position A. Taken together, these results present a clearer picture of the molecular mechanism of 1 and how future drug discovery efforts for new therapeutics targeting PPARγ can be tailored for higher potency.
Experimental Section
Transactivation Assay
4.5 μg of human GAL4-PPARγ-Hinge-LBD, 4.5 μg of 5× multimerized UAS-luciferase reporter, and 27 μL of X-treme Gene 9 transfection reagent were cotransfected into HEK293T cells (ATCC; cat. no. CRL-3216) and grown in serum-free UAS-LUC were cotransfected into cells as a control. Cells were grown for 18 h at 37 °C in a 5% CO2 incubator, after which they were plated in quadruplicate in white 384-well plates (PerkinElmer) at a density of 10,000 cells in each well. Cells were treated with either DMSO only or the compound of interest (doses from 169 pM to 10 μM) after replating. Cells were treated with Brite Lite Plus (PerkinElmer) after 18 h incubation and read in a 384-well Luminescence PerkinElmer EnVision Multilabel plate reader. Fold change of treated cells over DMSO-treated control cells was plotted to define EC50 values. GraphPad Prism was used for plotting and statistical analysis including error bars. Each data point in the EC50 dose response was repeated in triplicate, and standard error of mean (S.E.M.) was derived from these values. In addition, two biological replicates were performed to ensure reproducibility of the data.
Protein Purification
The PPARγ ligand binding domain (residues 205–477), including an N-terminal hexa-histidine tag, was encoded in the pET11 expression vector. The expression vector was transformed into E. coli BL21(DE3). Cells were grown at 37 °C in LB media containing 50 μg/mL ampicillin until an optical density of 0.5 was reached. Cells were induced at 16 °C for 18 h, harvested by centrifugation, resuspended in buffer A (20 mM Tris 8.0, 0.5 M NaCl, 10 mM imidazole, and 2 mM BME), and stored at −80 °C. Cells were lysed by three passes through a French Press. Cell lysate was clarified by centrifugation at 10,000g for 1 h and applied to a 5 mL His-Trap FF crude column (GE Healthcare), washed with 100 mL buffer A, and eluted with 25 mL buffer B (20 mM Tris 8.0, 0.5 M NaCl, 250 mM imidazole, and 2 mM BME). The elution fractions were pooled and dialyzed to buffer C (20 mM Tris 8.0, 10 mM NaCl, and 1 mM DTT) using a 10,000 molecular weight cutoff dialysis bag (Spectrum Laboratories, Inc.) for 18 h. The PPARγ sample was applied to a HiPrep 26/60 Sephacryl S-300 HR size-exclusion column and eluted at 1 mL/min over one column volume. Fractions containing purified PPARγ were concentrated to 10 mg/mL using a 10,000 molecular weight cutoff centripetal concentrator (Millipore). Protein used in crystallization trials was used fresh and without freeze/thaw (within 3 days of preparation).
Crystallization/Data Processing
Crystallization trials were carried out with all compounds, but only compound 10 produced diffracting cocrystals. For complex formation, a sample of 10 mg/mL PPARγ LBD was mixed with 10 (5 mM final concentration 10) and incubated on ice for 30 min. Prior to crystallization, the sample was clarified by centrifugation at 10,000g at 4 °C for 10 min and the supernatant was extracted for use in crystallization trials. Crystals were formed using the vapor diffusion method by mixing 1 μL of PPARγ-10 complex with 1 μL of well solution in an Intelli-plate (Art Robbins) using a sitting drop style plate. The well solution consisted of 75 μL of 2 M ammonium sulfate. Crystallization trials were conducted at 289 K. Cubic crystals of approximately 150 μm in each dimension appeared after 3 days. The well solution containing 15% ethylene glycol was used as a cryoprotectant. Crystals were harvested using a cryo-loop (Hampton Research) and flash cooled to 100 K. All data was collected at 100 K. Data was collected at APS beamline 22-ID. 450 images were collected at 0.5° oscillations (225° data total) at 0.6 s exposure time per image. Data was processed using iMosflm19 and scaled in aimless20 to a resolution of 2.2 Å. Resolution cutoff was determined by use of the CC1/2 criteria.21 PDB: 3FUR, stripped of ligands and water molecules, was used as a search model, and phases were obtained by molecular replacement in Phaser.22 Initial difference Fourier maps revealed clear electron density for 10, which was modeled manually. Refinement, including TLS refinement, was carried out in Phenix23 with multiple rounds of manual rebuilding carried out in Coot.24 Refinement was completed when R-factors converged. Unsuccessful soaks of PPARγ apo crystals were diffracted at the Australian Synchrotron beamline MX1 and MX2.25
Docking
The ICM Molsoft suite26 was used to dock compound 1 analogs into the PPARγ LBD structure. PDB 3FUR with ligands removed was used as the starting model for docking. The PPARγ LBD structure was prepared for docking by protonation, deletion of water molecules, and energy minimization by means of the ICM force field and distance dependent dielectric potential with an RMS gradient of 0.1. PocketFinder within ICM was used to define the ligand binding pocket and was consistent with previously published X-ray structures. Default settings within the ICM docking module were used with a rectangular box centered at the LBD with a grid spacing of 0.5 Å. The top ranked docking for each ligand was chosen for interpretation, as the conformations were very consistent with scaffold placement of 1 and 10 in the X-ray crystal structure.
Supplementary Material
Acknowledgments
We would like to thank the staff at SER-CAT beamline for assistance with data collection. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. We would like to thank the staff at beamlines MX1 and MX2 of the Australian Synchrotron for help with data collection of the PPAR soaks (This work was supported by CAP6919 and CAP8221 of the AS). This work was funded in part by NIH NIDDK DK105825 PI: Griffin.
Abbreviations
- PPAR
peroxisome proliferator-activated receptor
- TZD
thiazolidinedione
- SAR
structure–activity relationship
- RXR
retinoid X receptor
- T2DM
Type II Diabetes Mellitus
Footnotes
ORCID: Andrew D. Abell: 0000-0002-0604-2629
John B. Bruning: 0000-0002-6919-1824
Notes: The authors declare no competing financial interest.
The PDB code for PPARγ in complex with 10 is 5TTO. The authors will release the atomic coordinates and the experimental data upon article publication.
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.6b01727.
Additional figures (S1 and S2) illustrating sample electron density and comparison of the docking to structures derived from experimental X-ray. Details of compounds synthesis, including H1 NMR, MS, and HPLC analysis. (PDF)
Structure of PPARγ in complex with 10 (PDB)
Molecular formula strings (CSV)
References
- 1.Bruning JB, Chalmers MJ, Prasad S, Busby SA, Kamenecka TM, He Y, Nettles KW, Griffin PR. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–1271. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 2.Kroker AJ, Bruning JB. Review of the structural and dynamic mechanisms of PPARgamma partial agonism. PPAR Res. 2015;2015:816856. doi: 10.1155/2015/816856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Marrewijk LM, Polyak SW, Hijnen M, Kuruvilla D, Chang MR, Shin Y, Kamenecka TM, Griffin PR, Bruning JB. SR2067 Reveals a unique kinetic and structural signature for PPARgamma partial agonism. ACS Chem Biol. 2016;11:273–283. doi: 10.1021/acschembio.5b00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taygerly JP, McGee LR, Rubenstein SM, Houze JB, Cushing TD, Li Y, Motani A, Chen JL, Frankmoelle W, Ye G, Learned MR, Jaen J, Miao S, Timmermans PB, Thoolen M, Kearney P, Flygare J, Beckmann H, Weiszmann J, Lindstrom M, Walker N, Liu J, Biermann D, Wang Z, Hagiwara A, Iida T, Aramaki H, Kitao Y, Shinkai H, Furukawa N, Nishiu J, Nakamura M. Discovery of INT131: a selective PPARgamma modulator that enhances insulin sensitivity. Bioorg Med Chem. 2013;21:979–992. doi: 10.1016/j.bmc.2012.11.058. [DOI] [PubMed] [Google Scholar]
- 5.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O'Rahilly S. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 6.Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR, Spiegelman BM. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, Marciano DP, Cameron MD, Laznik D, Jurczak MJ, Schurer SC, Vidovic D, Shulman GI, Spiegelman BM, Griffin PR. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wakabayashi K, Hayashi S, Matsui Y, Matsumoto T, Furukawa A, Kuroha M, Tanaka N, Inaba T, Kanda S, Tanaka J, Okuyama R, Wakimoto S, Ogata T, Araki K, Ohsumi J. Pharmacology and in vitro profiling of a novel peroxisome proliferator-activated receptor gamma ligand, Cerco-A. Biol Pharm Bull. 2011;34:1094–1104. doi: 10.1248/bpb.34.1094. [DOI] [PubMed] [Google Scholar]
- 9.Berger JP, Petro AE, Macnaul KL, Kelly LJ, Zhang BB, Richards K, Elbrecht A, Johnson BA, Zhou G, Doebber TW, Biswas C, Parikh M, Sharma N, Tanen MR, Thompson GM, Ventre J, Adams AD, Mosley R, Surwit RS, Moller DE. Distinct properties and advantages of a novel peroxisome proliferator-activated protein [gamma] selective modulator. Mol Endocrinol. 2003;17:662–676. doi: 10.1210/me.2002-0217. [DOI] [PubMed] [Google Scholar]
- 10.DePaoli AM, Higgins LS, Henry RR, Mantzoros C, Dunn FL. Can a selective PPARgamma modulator improve glycemic control in patients with type 2 diabetes with fewer side effects compared with pioglitazone? Diabetes Care. 2014;37:1918–1923. doi: 10.2337/dc13-2480. [DOI] [PubMed] [Google Scholar]
- 11.Motani A, Wang Z, Weiszmann J, McGee LR, Lee G, Liu Q, Staunton J, Fang Z, Fuentes H, Lindstrom M, Liu J, Biermann DH, Jaen J, Walker NP, Learned RM, Chen JL, Li Y. INT131: a selective modulator of PPAR gamma. J Mol Biol. 2009;386:1301–1311. doi: 10.1016/j.jmb.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 12.Higgins LS, Mantzoros CS. The development of INT131 as a selective PPARgamma modulator: approach to a safer insulin sensitizer. PPAR Res. 2008;2008:936906. doi: 10.1155/2008/936906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGee LR, Rubenstein SM, Houze JB. Discovery of AMG131: a selective modulator of PPARγ. Proceedings of the 231st American Chemical Society National Meeting. 2006 [Google Scholar]
- 14.Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, Shulman GI, Gygi SP, Spiegelman BM. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature. 2015;517:391–395. doi: 10.1038/nature13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puhl AC, Bernardes A, Silveira RL, Yuan J, Campos JL, Saidemberg DM, Palma MS, Cvoro A, Ayers SD, Webb P, Reinach PS, Skaf MS, Polikarpov I. Mode of peroxisome proliferator-activated receptor gamma activation by luteolin. Mol Pharmacol. 2012;81:788–799. doi: 10.1124/mol.111.076216. [DOI] [PubMed] [Google Scholar]
- 16.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 17.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 18.Wheeler SE. Understanding substituent effects in noncovalent interactions involving aromatic rings. Acc Chem Res. 2013;46:1029–1038. doi: 10.1021/ar300109n. [DOI] [PubMed] [Google Scholar]
- 19.Powell HR, Johnson O, Leslie AG. Autoindexing diffraction images with iMosflm. Acta Crystallogr, Sect D: Biol Crystallogr. 2013;69:1195–203. doi: 10.1107/S0907444912048524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr, Sect D: Biol Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD. Automated structure solution with the PHENIX suite. Methods Mol Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 24.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 26.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.