

Abstract
Replication-defective (RD) recombinant simian virus 40 (SV40)-based gene delivery vectors hold a great potential for clinical applications because of their presumed non-immunogenicity and capacity to induce immune tolerance to the transgene products in humans. However, the clinical use of SV40 vectors has been hampered by the lack of a packaging cell line that produces replication-competent (RC) free SV40 particles in the vector production process. To solve this problem, we have adapted the current SV40 vector genome used for the production of vector particles and generated a novel Vero-based packaging cell line named SuperVero that exclusively expresses the SV40 large T antigen. SuperVero cells produce similar numbers of SV40 vector particles compared to the currently used packaging cell lines, albeit in the absence of contaminating RC SV40 particles. Our unique SV40 vector platform named SVac paves the way to clinically test a whole new generation of SV40-based therapeutics for a broad range of important diseases.
Keywords: SV40 viral vectors, SuperVero, SVac, packaging cells
Introduction
After small molecules and therapeutic proteins, gene therapy will be the next wave of medicines potentially capable of curing today’s major diseases. Key to the success of gene therapy will be the safe and efficient delivery of the therapeutic genes into cells of the affected tissue or organ target cells of a patient’s body. Viruses evolve to efficiently transfer and express their genes into host cells. This ability renders them ideally suited for use as gene delivery vectors. Among the viral vector systems currently used for gene therapy, lentiviral (LV) and adeno-associated viral (AAV) vectors are the most popular because they transduce a wide variety of tissues and mediate long-term expression when administered to animals.1, 2, 3 For both vectors it has been shown that such replication-defective (RD) viral vectors are non-immunogenic or tolerogenic in hosts that are naive to the cognate virus.4, 5, 6, 7, 8 After successful preclinical and clinical trials, the first LV and AAV vector-based therapeutics have now reached the market.9 However, both LV and AAV vector systems have shortcomings that limit their application.
LV vectors integrate randomly in the host genome, and thus increase the risk of insertional mutagenesis.10 Moreover, LV vector particles are highly instable, rapidly degraded when administered in vivo, and the development of stable packaging cell lines has remained challenging.11 For these reasons, LV vectors are mainly used for the ex vivo transduction of leukocytes and/or their progenitors to treat blood-related genetic disorders, lysosomal storage diseases, or cancer. AAV vectors are mainly used for in vivo gene therapies. However, the majority of the human population encountered wild-type AAV together with its helper virus (adenovirus, causing the common cold) and developed a strong humoral and cellular immune memory for the AAV capsid proteins. Clinical studies using recombinant AAV vectors revealed that administration of vector particles elicits innate and adaptive immune responses against the viral and transgene-encoded proteins in the vast majority of treated patients, leading to decreasing expression levels of the therapeutic transgenes over time and elimination of the transduced cells from the body, compromising re-administration of the vector. The few treated patients who showed long-term transgene expression most likely have never been infected with AAV, and thus were immunologically naive to the RD vector used in the study. For these reasons, the efficacy of AAV vector-based in vivo gene therapies is limited.12, 13
RD simian virus 40 (SV40) vectors could be an attractive alternative to AAV vectors for clinical gene therapy.14 SV40 is a polyomavirus with icosahedral capsids of 45 nm in size containing a 5.25 kb-long circular double-stranded DNA. The virus strictly replicates in its natural host, macaques, where it causes chronic asymptomatic infections. SV40 particles enter infected cells via the caveolar-endosomal route, but in contrast with other viruses are able to avoid the lysosomal degradation, thereby evading exposure to the host immune system.15, 16, 17, 18, 19, 20, 21 RD SV40 vectors have been generated by deleting the coding region of the two early non-structural proteins named large T antigen (LTag) and small T antigen (STag), giving 2.7 kb of space for cloning the transgene encoding the therapeutic protein or RNA. SV40 vectors transduce a wide range of cell types in vivo, and their therapeutic potential has been demonstrated in animal models of human disease.22, 23, 24, 25, 26 Because humans can be considered naive to SV40,27, 28 it is expected that RD SV40 vectors are non-immunogenic or tolerogenic when applied in clinical settings. The non-immunogenicity in humans and capacity to induce immune tolerance to transgene proteins render SV40 vectors highly attractive for use in gene replacement and immunotherapies.
However, the translation of SV40 vectors to the clinic has been hampered by the lack of a packaging cell line that does not accumulate detectable amounts of replication-competent (RC) SV40 particles during vector production.29, 30, 31, 32 To date, two cell lines have been mainly used for the production of SV40 vectors: COS-1 and COS-7. COS cell lines were generated by transformation of monkey CV1 cells with SV40 DNA.29 Passaging of SV40 vectors in COS cells, however, results in the appearance of wild-type SV40 particles. This most likely occurs by sequence homology-dependent recombination between the chromosomally inserted SV40-specific DNA sequences and episomally replicating vector-specific DNA sequences.33
Other cell lines that express the SV40 T antigens in trans are COT18 and CMT4. Both CV1-derived cell lines were generated using SV40 DNA in which the T antigens were expressed under transcriptional control of the mouse metallothionein promoter. Because the complete early region of the SV40 genomic DNA is present in the chromosomal DNA, the ability that RC virus contaminants emerge during the vector production process has remained. Moreover, the vector yields of both packaging cell lines are not increased compared with those of COS cells.33, 34, 35
Polyomavirus-based virus-like particle (VLP) vector systems have been developed to prevent the occurrence of RC virus particles in the vector preparations. In vitro-generated VLPs consisting of circular double-stranded DNA encapsidated with polyomavirus VP1 lack VP2 and VP3 in the capsids and histones covering the encapsidated DNA molecules. Although these particles display a higher packaging capacity, the absence of VP2/VP3 in the VLPs has a negative impact on their entry in the nucleus.36
In order to overcome the generation of wild-type virus contaminants, we optimized the SV40 vector genome used to initiate the production of vector particles and generated a safe and efficient Vero-based SV40 vector packaging cell line named SuperVero. The SuperVero cells solely express the viral LTag and accumulate vector particles at high titers, comparable with those obtained in the conventional packaging cell lines COS-1 and COS-7. Because RD SV40 vectors are safe, highly efficient for gene delivery, and non-immunogenic/tolerogenic in humans, our improved SV40 vector platform named SVac holds promising qualities for developing effective treatments for the major diseases of our time.37
Results
Construction of SV40 Vector Destination pSVac and Derivative Vector Expression Plasmids
In order to improve the efficacy and versatility of the SV40 vector system, we modified vector plasmid pSL-PL38 in a series of steps yielding SV40 destination plasmid pSVac (Figure 1) that is used to initiate the production of recombinant SV40 particles. The low copy number bacterial backbone of pSL-PL (pBR322) was replaced with a high copy number backbone (pBluescript SK−). The residual 3′-terminal LTag coding sequences present in pSL-PL were removed, and a Gateway gene cassette derived from pEF5/FRT/V5-DEST was introduced in the early region of the SV40 vector DNA, providing a versatile method to introduce transgenes encoding therapeutic proteins or RNAs into the vector destination plasmid by LR clonase-mediated recombination between pSVac DNA and that of an entry plasmid harboring a transgene resulting in vector expression plasmids. The bovine growth hormone (BGH) polyadenylation (pA) signal originating from pEF5/FRT/V5-DEST was cloned downstream of the Gateway cassette to facilitate transient expression studies by transfecting target cells with DNA from vector expression plasmids. Unique AscI and SpeI restriction sites were introduced between the SV40 early promoter and the Gateway cassette to facilitate the introduction of tissue-specific promoters. We generated a series of SV40 expression plasmids containing different transgenes by Gateway recombination. The resulting expression plasmids encode the jellyfish green fluorescent protein (pSVGFP), the human activated blood clotting factor VII (pSVFVIIa), and a short hairpin RNA specific for the firefly luciferase (pSVshLuc).
Figure 1.

Schematic Representation of the SV40 Vector Destination Plasmid pSVac
The Gateway gene cassette comprising the ccdB and the chloramphenicol resistance (CmR) genes present in pSVac is substituted by the transgene using Gateway recombination yielding an SV40 vector expression plasmid.
For the generation of SV40 vector particles, the prokaryotic backbone of pSVac needs to be removed from the SV40 vector sequences. For this purpose, NotI restriction sites and loxP recombination sites were introduced between the Gateway cassette and BGH pA signal and between the SV40 pA signal and the pBluescript SK− bacterial backbone. Circular vector DNA used as starting material for the production of SV40 vector particles in packaging cells was generated by digesting a vector expression plasmid with NotI followed by re-circularization of the vector DNA using T4 DNA ligase. Alternatively, circular vector DNA can be generated by homologous recombination at the loxP sites using Cre recombinase.
Generation of a New SV40 Vector Packaging Cell Line
The SV40 T antigens are required for virus DNA replication and expression of the late gene encoding the viral capsid proteins. In order to verify whether the LTag by itself is sufficient for the generation of SV40 vector particles, we generated the expression plasmids pHY338 and pHY359 as described under Materials and Methods. Expression plasmid pHY338 encodes both STag and LTag; plasmid pHY359 exclusively encodes LTag (Figure 2A). Vero cells were selected as acceptor cells for both expression plasmids because these cells are permissive for SV40 and because Vero constitutes an accepted cell culture platform for vaccine production.39 Vero cells grown in serum-free cell culture medium were transfected with either pHY338 or pHY359 DNA, and cells were cultured for 1 month in serum-free culture medium supplemented with puromycin. Puromycin-resistant cell clones were subsequently transduced with SVLuc particles produced in COS-1 cells31 at an MOI of 100. As a positive control, COS-1 cells were transduced with SVLuc particles at the same MOI. Three days after transduction, the amount of SVLuc particles present in the culture medium of the transduced cells was determined by quantitative real-time PCR. The qPCR assay has been developed in such a way that only packaged vector genomes are measured (Figure 2B). Both the pHY338 and pHY359-transfected Vero cells produced vector particles at approximately 30% of the amount of vector particles produced in COS-1 cells. To verify that the vector particles produced in the transfected Vero cells are biologically active, we subsequently used the culture media from the SVLuc-transduced Vero and COS-1 cells for transducing fresh COS-1 cells. Three days after transduction, crude cell lysates were prepared and the luciferase activity was determined in these lysates by luminescence. Figure 2C shows that the transfected Vero cells produced vector particles that induced luciferase expression when transferred to COS-1 cells. These experiments demonstrated that the presence of SV40 LTag in Vero cells is sufficient for producing SV40 vector particles.
Figure 2.

Vero Cells Stably Transfected with pHY359 Support the Production of SVLuc Vector Particles
(A) Schematic representation of the expression plasmids pHY338 and pHY359. Plasmid pHY338 contains the SV40 genomic T antigen sequence encoding both LTag and STag. Plasmid pHY359 contains the LTag cDNA sequence encoding LTag only. In both plasmids, transgene expression is driven by the EF-1α promoter. Both plasmids contain the puromycin resistance (PuroR) gene under transcriptional control of the CMVie promoter and BGH pA signal. (B) Ratio between the number of SVLuc particles produced in Vero cells transfected with pHY338 and pHY359 plasmid DNA and those produced in COS-1 cells transduced with SVLuc particles. (C) Luminescence of COS-1 cells transduced with SVLuc particles produced in transfected Vero or COS-1 cells. (D) Luminescence of COS-1 cells transduced with SVLuc particles produced in stably transfected Vero cell clones. COS-1 cells were used as positive control.
In order to generate a new Vero-based packaging cell line for the production of SV40 vector particles, we cultured cells transfected with pHY359 DNA in the presence of puromycin for 1 month. Individual puromycin-resistant cell colonies were picked and sub-cultured in culture medium with puromycin. More than 100 puromycin-resistant colonies were obtained and subsequently tested for their capacity to produce SV40 vector particles using the same assay as described above. One cell colony produced comparable amounts of SVLuc particles to COS-1 cells (Figure 2D). Subsequently, six subclones of this cell colony were generated by limited dilution of the puromycin-resistant cells and tested for their capacity to produce vector particles using the same assay. The cell clone that produced the largest amount of vector particles, denoted SuperVero, was expanded in serum-free culture medium without puromycin and used for generating a research cell bank of 50 vials that is stored at −150°C. For all further studies, vials of the research cell bank were thawed and the cells were cultured and expanded in serum-free glutamine-supplemented OptiPRO medium.
Molecular Analysis of the SuperVero Packaging Cell Line
In order to confirm the presence of a functional LTag-encoding gene in the chromosomal DNA of SuperVero cells and to investigate whether the cells harbor contaminating viruses, the transcriptome of SuperVero cells was analyzed by Massive Parallel Sequencing (MP-Seq).40 More than 1.6 million different cDNA sequences from SuperVero poly-A-positive RNAs were generated. In parallel, more than 800,000 different cDNA sequences were generated from poly-A-positive RNAs isolated from reference cell line Vero ATCC CCL-81.
Among the SuperVero cDNA sequences, 202 cDNA reads mapped to the LTag gene present in the genomic DNA of the cells (see Table 1). SV40 cDNA sequences specific for other parts of the SV40 genome were absent in SuperVero cells. As expected, Vero cells do not contain SV40-specific mRNAs (Table 1).
Table 1.
Comparison of the cDNA Reads of SuperVero and Vero Cells to a Viral Sequence Database
| Original Accession No. Identified | SuperVero (1,673,145 Total Reads) |
Vero ATCC CCL-81 (806,262 Total Reads) |
Reference Genome Accession No. | Taxonomy | ||
|---|---|---|---|---|---|---|
| Total No. of ViralDB Wins | Total No. of Reads Mapped to Refseqa | Total No. of ViralDB Wins | Total No. of Reads Mapped to Refseqa | |||
| NC_001669.1 | 202b | 238b | 0 | 0 | NC_001669 | simian virus 40, complete genome |
| AC146999.1 | 63 | 97 | 0 | 14 | NC_006273 | human herpesvirus 5 |
| EU410304.1 | 23 | 21 | 13 | 3 | NC_006998 | vaccinia virus GLV-1h68 |
| X03922.1 | 17 | 97 | 0 | 14 | NC_006273 | human herpesvirus 5 |
| HM143845.1 | 12 | 0 | 6 | 0 | M11841 | simian retrovirus 1 |
| U85506.1 | 9 | 18 | 10 | 14 | U85505 | simian endogenous retrovirus |
| Z54175.1 | 7 | 10 | 5 | 5 | NC_001461 | bovine viral diarrhea virus 1 |
| AF104029.1 | 6 | 1 | 6 | 0 | NC_002032 | bovine viral diarrhea virus 2 |
| FM212572.1 | 4 | 5,330 | 1 | 2,926 | AB047240 | human endogenous retrovirus |
| JN134185.1 | 3 | 87 | 7 | 18 | JN134185 | simian endogenous retrovirus SRVcae |
| AF290913.1 | 1 | 0 | 0 | 1 | U13766 | murine leukemia virus |
| M15805.1 | 1 | 64 | 0 | 21 | M15805 | feline sarcoma virus |
A single read may map to multiple positions of the reference sequence and each mapping is counted independently.
202 cDNA reads mapped to the LTag gene present in the genomic DNA of the cells.
The 202 LTag-specific cDNA reads of SuperVero cells were aligned and are homologous to the DNA sequence of pHY359 used to generate the cell line. Sequence variants were not present among the 202 sequenced cDNAs (Figure 3A).
Figure 3.

SuperVero Cells Contain pHY359 DNA and Express SV40 LTag
(A) Schematic representation of the cDNA reads from the MP-seq analysis corresponding with the SV40 LTag mRNA expressed in SuperVero cells. (B and C) Detection of the LTag protein in SuperVero cells (B) by western blot and (C) by immunocytochemistry using a mouse monoclonal antibody specific to the amino-terminal region of the LTag and STag. Vero cells are used as a negative control. COS-1 cells are used as a positive control expressing both LTag and STag. (D) Densitometric analysis of the western blot bands shown in (B). The graph represents the ratio between the density units of LTag and α-tubulin, used as an internal loading control.
The remaining cDNA reads obtained from the SuperVero cells with the MP-Seq method were compared with a viral sequence database (ViralDB) that contains 964,619 sequences extracted from the complete GenBank nucleotide sequence database.40 The most significant homologies found in the comparison are shown in Table 1. In the SuperVero sample, cDNAs were found that show homology to human herpesvirus 5 DNA. This homology corresponds with a short transcribed region in the cytomegalovirus immediate early (CMVie) promoter that drives transcription of the pac puromycin-resistance gene.
All other sequence homologies found between cDNAs from the SuperVero cells and the ViralDB are also found when the cDNAs from the Vero ATCC CCL-81 cells were compared with the sequences in the database. The MP-Seq study has shown that the SuperVero cell line is not contaminated with known viruses during its generation from Vero ATCC CCL-81 cells.
The presence of the LTag protein in SuperVero cells was subsequently confirmed by western blot analysis. Both LTag and STag proteins can be detected using an antibody specific for the N-terminal part of the LTag protein. As expected, COS-1 cells accumulate both LTag and STag, whereas SuperVero cells express only LTag (Figure 3B). Immunocytochemistry of the SuperVero and COS-1 cells further confirmed the presence of the LTag in the nucleus of the cells (Figure 3C). A densitometric analysis of the bands on the western blots revealed that the relative amount of LTag in SuperVero cells was slightly lower than that in COS-1 cells (Figure 3D).
The SV40 late gene encoding the capsid proteins is exclusively expressed in cells permissive for SV40 that accumulate the viral LTag. This implies that the viral capsid proteins are produced only in the packaging cells during vector manufacturing, but absent in (human) target cells after transduction. The transcriptional silencing of the SV40 late gene in cells lacking the viral LTag was verified by transfecting SuperVero and HeLa cells with pSVGFP DNA. After 3 days, the presence of GFP, LTag, and the major viral capsid protein VP1 in both cell types was checked on western blots using monoclonal antibodies specific for GFP, LTag, and VP1, respectively. A monoclonal antibody specific for GAPDH was included as a protein loading control. Only SuperVero cells constitutively produce SV40 LTag. In both pSVGFP-transfected SuperVero and HeLa cells, GFP was detected. However, VP1 accumulated only in pSVGFP-transfected SuperVero cells, demonstrating that the SV40 late gene is not expressed in target cells lacking SV40 LTag (Figure 4).
Figure 4.
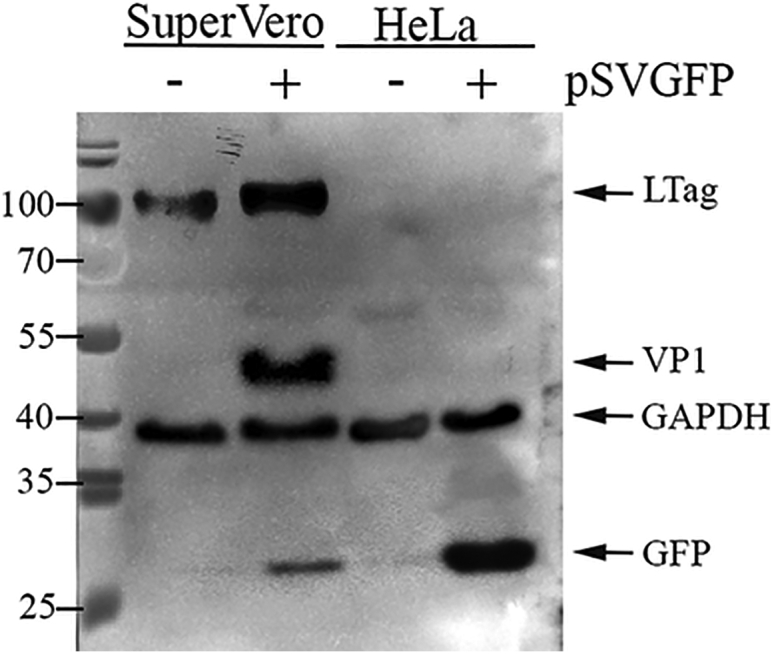
The SV40 Late Gene Is Only Expressed in SuperVero Cells during the Production of Vector Particles and Not in Human Target Cells
Detection of GFP, SV40 LTag, and major capsid protein VP1 in cell lysates from SuperVero and HeLa cells transfected with pSVGFP DNA by western blot analysis using antibodies specific for GFP, LTag, and VP1. An anti-GAPDH antibody was used as a loading control.
Production of SV40 Vector Particles in SuperVero Cells
When COS-1 cells cultured in medium with fetal bovine serum and SuperVero cells cultured in serum-free medium are transduced with SV40 vector particles at an MOI of 400 vector particles per cell, both cell lines secreted comparable numbers of vector particles in the culture medium (Figure 5A). The capacity of SuperVero cells to produce consistent amounts of SV40 particles was further tested by passaging vector particles in fresh SuperVero cells and measuring the number of vector particles after each passage. In SuperVero cells transduced with SVLuc particles obtained at subsequent passages, consistently 108–109 vector particles per milliliter cell culture are found in the cell culture medium (Figure 5B). Moreover, approximately the same numbers of vector particles remain inside the cells and are not harvested after production (data not shown). The transduction efficacy (potency) of the produced vector particles after each passage was determined using a modified tissue culture infectious dose 50 (TCID50) assay. Because SV40 vector particles do not lyse transduced cells, we used SVLuc as a reporter vector and used luminescence as a readout in this limited dilution assay. These experiments revealed that the vector particles quantified by qPCR (Figure 5B) remain fully functional after passaging them in SuperVero cells as determined by TCID50 (Figure 5C). Furthermore, defective interfering vector particles do not accumulate during the vector production process as demonstrated by the stability of the TCID50 titer over subsequent passaging (Figure 5C).
Figure 5.

SuperVero Cells Consistently Accumulate Large Amounts of Functional SV40 Vector Particles
(A) Determination of the number of SVLuc particles produced in COS-1 and SuperVero cells using qPCR. (B) Determination of the number of SVLuc particles after passaging in SuperVero cells. (C) Determination of the potency of SVLuc particles after passaging in SuperVero cells. (D) Determination of the number of SV40 vector particles expressing different transgenes produced in SuperVero cells. Error bars represent SD, n = 3.
We next produced SVLuc, SVGFP, SVFVIIa, and SVshLuc vector batches in SuperVero cells. The vector yields obtained after three passages consistently ranged between 108 and 109 particles per milliliter cell culture, demonstrating the robustness of the vector production process (Figure 5D).
SVLuc particles produced in SuperVero cells were stored for different periods of time at different temperatures. The numbers of vector particles were determined by qPCR (Figure 6A), whereas the potency of the particles was determined using the TCID50 assay (Figure 6B). Independent of the storage temperature, the amounts of vector particles did not significantly decrease after 6 months. These experiments demonstrate that SV40 vector particles stored at 4°C or at lower temperatures remain fully functional for at least 6 months.
Figure 6.

SV40 Vector Particles Remain Functional after Storage for 6 Months at 4°C or Lower Temperatures
SVLuc particles were stored at +4°C, −20°C, −80°C, and −150°C. (A) The number of vector particles was determined by qPCR. (B) The potency of the vector particles was determined by the TCID50 analysis.
Detection of RC SV40 Particles
It has been found that the production of SV40 vectors in COS cells results in the emergence of RC SV40 particles.33 In order to sort out whether the virus contamination problem is solved by using the SuperVero cell line (Figure 7), we designed a set of primers to detect RC SV40 particles by qPCR (Figure 8).
Figure 7.

Production of SV40 Vectors in SuperVero and COS-1 Cells
The production of SV40 particles in COS-1 cells (right side) results in the emergence of RC SV40 particles because of homologous recombination between chromosomal inserted SV40 sequences and episomally replicating SV40 DNA sequences. The generation of RC variants is prevented in SuperVero cells (left side) harboring chromosomally inserted SV40 LTag DNA sequences without sequence overlap to the episomally replicating SV40 vector DNA sequences.
Figure 8.

Primers and Probes Used for the qPCR Analysis to Detect RC and RD SV40 Vector Particles
(A) Schematic representation of the SV40 genome showing the position of the primers and probe for the detection of RC SV40 particles. (B) The sequences of primers and probes used in the qPCR analysis.
In all the samples tested so far using these primers, including samples from highly concentrated vector preparations and samples from subsequent vector passages (up to 10), the number of RC SV40 particles remained below the detection limit of the assay. This means that after passaging the vector particles in SuperVero cells, RC SV40 particles certainly do not overgrow the RD vector particles.
Discussion
SV40 gene delivery vectors have shown therapeutic efficacy in numerous preclinical studies,22, 23, 24, 25, 41 and because of their predicted non-immunogenic/tolerogenic potential in humans these vectors are an attractive alternative to the currently used vector systems for developing safe and efficient gene therapies. To date, however, SV40 vectors have not been tested in clinical trials because an efficient packaging cell line that does not accumulate RC SV40 particles during the vector production process is lacking. Here we report on the development of an SV40-based gene delivery platform, which we named SVac, that is well suited for producing RD SV40 vector particles free of contaminating RC revertants, and thus eligible for use in clinical gene therapy studies.
We developed a versatile SV40 vector destination plasmid (pSVac) that allows the evaluation of promoters, transgenes, and other regulatory elements for safety and efficacy in animals and humans. Vero cells were selected for the generation of a new packaging cell line because this cell line is permissive to SV40 and is recommended by the World Health Organization for vaccine production for human use.42
RD SV40 vector genomes lack the T antigen region. In order to produce SV40 vector particles, T antigen must be provided by the packaging cell line. The SV40 early gene encodes LTag and STag. LTag is a replicase-associated protein required for viral DNA replication and for activation of the viral late promoter.43 STag inhibits cellular protein synthesis from capped messenger RNAs (a process denoted host shutoff), thereby promoting the synthesis of viral capsid proteins.44 Simultaneous expression of LTag and STag results in immortalization of primary mammalian cells, transformation of established mammalian cell lines, and induction of tumors in immuno-compromised young-born hamsters. However, it has been shown that expression of the individual T antigens in mammalian cells does not lead to immortalization, transformation, or tumor induction.45 We here show for the first time that expression of the LTag in the absence of STag in Vero cells is sufficient for the replication and packaging of SV40 vector particles. A Vero-based SV40 packaging cell line denoted SuperVero was generated that expresses the viral LTag, and thus lacks the viral oncogene and oncoproteins. Because sequence overlap between chromosomally inserted SV40 LTag coding sequences and episomally replicating SV40 vector sequences is lacking, the emergence of RC particles during the vector production process is highly unlikely.
We demonstrate that the LTag in SuperVero cells activates transcription from the viral late promoter, resulting in the accumulation of capsid proteins during the vector production process. However, in target cells lacking SV40 LTag transduced with an RD SV40 vector, only the transgene is expressed from the viral early promoter.
From the previous attempts to generate an SV40 vector packaging cell line it was found that the amount of the LTag is directly correlated with the number of SV40 vector particles,33, 34 but inversely correlated with cell viability.33 This could be the reason why we had to screen more than a hundred independent puromycin-resistant clones to identify one cell clone that is capable of producing vector particles to levels comparable with those in COS cells. Remarkably, this cell clone accumulates LTag to levels similar to COS-1 cells. Apparently there is a narrow margin of LTag expression tolerated by Vero cells that supports SV40 vector replication. We initiated experiments to further increase the amount of LTag in the packaging cells during the production process to even improve the vector yields.
The SV40 vector particles remain fully functional after passaging them in SuperVero cells, indicating that defective interfering particles do not accumulate during the production process. Furthermore, the produced vector particles are highly stable upon storage at 4°C or lower temperatures. The major advantage of SuperVero packaging cells above COS cells for producing RD SV40 vector particles is the absence of contaminating RC SV40 particles during the vector production process.32
The circular vector DNA molecules used in our studies to start the production of SV40 vector particles in SuperVero cells are similar, with the same genetic elements, to those used in previous studies to produce vector particles in COS cells. Because both Vero and CV1 cell lines are derived from kidney tissue obtained from African green monkeys (Cercopithecus aethiops),46 it is assumed that the in vivo transduction efficacy of SV40 vector particles produced in a Vero-derived packaging cell line is similar to that of particles produced in CV1-derived packaging cell lines such as COS. Indeed, ongoing in vivo studies with SVGFP and SVLuc particles produced in SuperVero cells show high transduction rates and transgene expression levels of different organs and/or tissues depending on the mode of administration (unpublished data).
Overall, RD SV40 vectors are efficient, non-immunogenic/tolerogenic in humans, highly stable, safe to use, and can be produced in large quantities in SuperVero packaging cells. The generation of the SVac platform described here paves the way for developing the next wave of medicines. SV40 vectors expressing functional gene products can be used in gene replacement therapies to treat genetic disorders such as, for example, low-density lipoprotein receptor deficiency, primary hyperoxaluria type 1, or hemophilia. In addition, SV40 vectors expressing T cell receptors (TCRs) or chimeric antigen receptors (CARs) can be used as vaccines to treat cancer. Last but not least, because of their capacity to induce immune tolerance to the transgene products in humans, SV40 vectors expressing primary self-antigens of degenerative/autoimmune diseases can be used as reverse vaccines to treat the major diseases of our time, such as neurodegenerative and psychiatric diseases,37 atherosclerotic cardiovascular disease, diabetes mellitus, arthritis, and chronic obstructive pulmonary disease (COPD).
Materials and Methods
Plasmid Construction
Expression plasmid pHY338 was constructed using a pBluescript SK− (Stratagene; Agilent Technologies) backbone by adding a gene cassette comprising the SV40 T antigen coding sequence (NCBI reference sequence NC_001669.1) flanked by the EF-1α promoter and the BGH pA signal (EF-1α-T antigen-BGH pA) and a gene cassette comprising the pac puromycin resistance gene flanked by the human CMVie promoter and the BGH pA (CMVie-pac-BGH pA).
Expression plasmid pHY359 exclusively expressing the SV40 LTag was generated by deletion of the large intron from the T antigen coding sequence by fusion PCR from plasmid pHY338 as follows. First, with forward primer 5′-GCAGGCTACCATGGATAAAGTTTTAAACAGAGAG-3′ homologous to the 5′ end of the LTag coding sequence and reverse primer 5′-CCCATTCATCAGTTCCATAGGTTGGAATCTCAGTTGCATCCCAGAAGCCTCCAAAG-3′ covering the intron splice site, a first PCR fragment was generated comprising the 5′ end of the LTag sequence. Second, with forward primer 5′-CTTTGGAGGCTTCTGGGATGCAACTGAGATTCCAACCTATGGAACTGATGAATGGG-3′ covering the intron splice site and reverse primer 5′-AGGAATGTTGTACACCATGCATTTTAAAAAGTC-3′ covering the NsiI site in the center of the LTag coding domain, a second PCR fragment was generated. Third, the two PCR fragments were used to perform a fusion PCR using the two external primers. The generated PCR fragment comprising the 5′ end of the LTag coding sequence without the large intron was cloned into the NcoI-NsiI sites of expression plasmid pHY338 yielding expression plasmid pHY359.
Cell Line Generation
Vero cells (ATTC, CCL-81) were cultured in serum-free OptiPRO (GIBCO, Thermo Fisher Scientific) medium supplemented with 4 mM glutamine (GIBCO, Thermo Fisher Scientific). The pBluescript SK− backbone was removed from the pHY359 DNA, and Vero cells were transfected with the DNA fragment containing the pac and LTag gene cassettes using ExGen 500 in vitro Transfection Reagent (BIOMOL) following the manufacturer’s instructions. Two days after transfection, the cells were divided into culture medium containing 2 μg/mL puromycin (Sigma-Aldrich) and kept under the antibiotic pressure for 1 month.
Construction of SV40 Destination Vector Plasmid pSVac
DNA of pSL-PL was subjected to PCR using primers 5′-CGGGATCCAGACATGATAAGATACATTG-3′ and 5′-ATAGTTTAGCGGCCGCAATGAATGCAATTGTTGTTGTTAACTTG-3′. The resulting PCR fragment comprising the SV40-pA signal was cloned into pBluescript SK− yielding pAM002. Plasmid pEF5/FRT/5-DEST (Invitrogen) DNA was subjected to PCR using primers 5′-CCGCTCGAGTTGCGGCCGCTGTGCCTTCTAGTTGCCAGCCATC-3′ and 5′-GGTACCATAGAGCCCACCGCATCCCCAGCATGCC-3′. The resulting PCR fragment comprising the BGH pA was cloned into pAM002, resulting in pAM003. The two complementary oligonucleotides 5′-GGCCGCTTTATTAATTAAGCCCT GCAGGTTGTTTAAACTTGGCGCGCCTTAT-3′ and 5′-CGATAAGGCGCGCCAAGTTTAAACAACCTGCAGGGCTTAATTAATAAAGC-3′ were annealed, digested with Notl and CIaI, and cloned into pAM003, yielding pAM004. Plasmid pSL-PL DNA containing the SV40 origin and late region was digested with CIaI and BamHI, and cloned into pAM004, resulting in pAM005. Plasmid pEF5/FRT/5-DEST DNA was subjected to PCR using primers 5′-TGGCGCGCCTATAGGGAGACCCAAGCTGGCTAG-3′ and 5′- CAATCATACCGTTTAAACGAACCGCGGGCCCTCTAGAC-3′. The resulting PCR fragment comprising the ccdB Gateway and the chloramphenicol resistance (CmR) gene cassettes flanked by AttR1 and AttR2 recombination sites was cloned into pAM005, resulting in SV40 destination vector plasmid pSVac.
SV40 Vector Production
Vector batches were produced according to Vera et al.31 In brief, after digestion of SV40 expression plasmid DNA with NotI to remove the bacterial backbone, vector DNA was isolated from agarose gels and subsequently re-circularized using T4 ligase (NEB). SuperVero cells growing in roller bottles (growth area: 850 cm2; Greiner Bio One) to 20%–70% confluence in OptiPRO serum-free media containing 4 mM l-glutamine (5% carbon dioxide, 37°C) were transfected with the re-circularized vector DNA. Three days posttransfection, the culture medium containing the vector particles was collected and replaced by fresh medium. In order to further increase the number of vector particles, we used the pooled vector harvests for at least two subsequent transduction rounds. In each transduction round, SuperVero cells were transduced with 400 SV40 vector genomes per cell. Three days posttransduction, the vector particle-containing culture medium was collected and replaced by fresh media. Vector harvests were clarified and concentrated by ultracentrifugation, and stored at 4°C or other temperatures as indicated.
SV40 Vector Quantification
The concentration of the vector particles in the produced vector batches was determined by quantitative real-time PCR. Samples were treated with DNase I (Sigma-Aldrich) to remove naked DNA. The vector DNA was isolated from particles by incubating the samples at 37°C for 1 hr in a proteinase K (Sigma-Aldrich) working solution (2.75 mg/mL in 8.6 mM Tris-HCl, 86 mM NaCl, 0.43% w/v SDS, pH 8). Magnetic beads (MagneSil Blue; Promega) were used to selectively bind vector DNA. Isolated DNA was used as a template for amplification of the VP2 gene region using AmpliTaq Gold (Thermo Fisher Scientific) polymerase and primers and probes shown in Figure 8B. The qPCR was set up according to the manufacturer’s recommendations (7300 Real-Time PCR System; Applied Biosystems) under the thermal cycling conditions of 2 min at 50°C and 10 min at 95°C, proceeding with 40 cycles of denaturation at 95°C for 15 s and annealing or extension at 60°C for 1 min. Data analyses were performed on the Applied Biosystems software.
The potency of SVLuc particles in the produced vector batches was determined using a modified TCID50 assay. This limited dilution assay quantifies the number of vector particles needed to transduce 50% of the target cells. In this assay, SVLuc particles were used as a reporter vector, and luminescence was used as a readout. Ten-fold serial dilutions of the SVLuc samples to be tested were added in quadruplicates to a 96-well cell culture plate containing 2 × 103 SuperVero cells in 100 μL of serum-free OptiPRO medium. Cells were exposed to the serial diluted vector and incubated in 5% carbon dioxide and at 37°C for 4 days. A dilution series of a standard SVLuc batch was added to the plates as a control. Intracellular luciferase was subsequently measured by luminescence. The estimated vector titer is the endpoint dilution at which 50% of the cell cultures (wells) are transduced by SVLuc and show luciferase expression. Vector titers were calculated according to the Spearman-Karber method and expressed as TCID50 per milliliter SuperVero cell culture (TCID50/mL).
Detection of RC SV40 Particles
The absence of RC SV40 revertants in the produced SV40 vector batches was confirmed by qPCR. Vector DNA was isolated from vector preparations using the PureLink RNA/DNA Mini Kit (Invitrogen). Isolated DNA was used as a template to amplify a fragment of 108 bp comprising the SV40 pA sequence and LTag coding region using the primers and probes shown in Figure 8. Plasmid pAM271 DNA harboring the wild-type SV40 genome was used as a positive control. The qPCR was set up according to the manufacturer (7300 Real-Time PCR System; Applied Biosystems) under the thermal cycling conditions of 2 min at 50°C and 10 min at 95°C, proceeding with 40 cycles of denaturation at 95°C for 15 s and annealing or extension at 60°C for 1 min. Data analyses were performed on the Applied Biosystems. Based on the signal obtained with the non-template control samples, we defined the limit of detection of the assay to be 60 RC SV40 particles per milliliter.
Luciferase Activity Measurements
Luciferase activity was measured from cells transduced with SVLuc by using the Luciferase Assay System (Promega). 200 μL of vector stock was used to transduce 1 × 104 cells in a 24-well plate format. All transductions were performed in triplicate. Three days after transduction, cells were harvested and luciferase activity was analyzed in a GloMax 96 Microplate Luminometer (Promega) according to the manufacturer’s recommendations.
Western Blot Analysis
Cells were lysed with SAM buffer (50 mM Tris-HCl, pH 6.8, 3% of SDS, 5% of β-mercapto-ethanol) containing protease inhibitor cocktail (SIGMAFAST Protease Inhibitor Tablets; Sigma-Aldrich), resolved by SDS-PAGE (10% polyacrylamide gels under reducing conditions) and electro-transferred to nitrocellulose membranes (Bio-Rad Laboratories). Membranes were blocked with 5% nonfat milk and probed with different primary antibodies for 1 hr at room temperature: (1) mouse anti-SV40 LTag 0.5 mg/mL (BD Biosciences), dilution 1:200; (2) rabbit anti-VP1 (Abcam), dilution 1:5,000; (2) mouse anti-GFP (Sigma-Aldrich), dilution 1:1,000; mouse anti-α-tubulin antibody (Sigma-Aldrich), dilution 1:5,000; mouse anti-GAPDH (Millipore), dilution 1:5,000. Finally, the membrane was hybridized with a goat anti-mouse or a goat anti-rabbit antibody conjugated with horseradish peroxidase (HRP) (Sigma-Aldrich), dilution 1:5,000. The blot was developed by chemiluminescence (Chemidoc MP Imaging System; Bio-Rad Laboratories). To quantify the amounts of protein, we scanned the blots by densitometry using the Image Lab software version 5.2.1 (Bio-Rad Laboratories).
Immunocytochemistry
SuperVero, Vero ATCC CCL-81, and COS-1 cells were cultured on coverslips and were fixed with 4% paraformaldehyde and blocked with 2% bovine serum albumin for 1 hr at room temperature. We incubated the cells overnight at 4°C with a solution of 2.5 μg/mL mouse antibody to the SV40 LTag diluted in PBS, 0.2% of Triton X-100. The secondary antibody (Alexa Fluor 488 donkey anti-goat; Life Technologies, Invitrogen) was incubated for 1 hr at room temperature diluted at 1:800 in PBS, 0.2% Triton X-100. Nuclear staining was performed with a solution of 5 μg/mL DAPI (Life Technologies, Invitrogen), and the slides were finally mounted using fluorescent mounting medium (DAKO, Agilent Technologies). Fluorescence microscopy images were acquired using a Leica DM6000B microscope (Leica Microsystem).
Statistical Analysis
All experiments were performed with a minimum of n = 3. All the statistical analyses were performed with the GraphPad Prism 5 software. We applied the two-tailed unpaired Student t test.
Author Contributions
Conceptualization, P.d.H., I.M., and M.G.T.; Methodology, P.d.H., I.M., and S.v.d.W.; Investigation, J.v.d.V., M.O., A.R., R.J.C.-G., and I.G.H.-G.; Writing – Original Draft, M.G.T., I.M., and P.d.H.; Writing – Review & Editing, M.G.T., I.M., and P.d.H.; Funding Acquisition, P.d.H.; Resources, P.d.H. and M.G.T.; Supervision, P.d.H., I.M., and M.G.T.
Conflicts of Interest
S.L. and B.V. are employed or sponsored by Amarna Therapeutics. The company is the owner of patents and patent applications on the production and use of polyomaviral vector particles.
Acknowledgments
Miguel G. Toscano is co-funded by the Torres Quevedo program PTQ-13-06051 from the Spanish Ministry of Economy, Industry and Competitiveness. We thank Dr. Katherine High and Dr. Paris Margaritis (Children’s Hospital of Philadelphia) for kindly providing us with the human activated factor VII cDNA and Dr. Piter Bosma (Academic Medical Center) and Dr. Benoit Gauthier (Center of Molecular Biology and Regenerative Medicine) for critically reading the manuscript. We also thank Dr. David Pozo (Center of Molecular Biology and Regenerative Medicine) for his help.
References
- 1.Jiang H., Lillicrap D., Patarroyo-White S., Liu T., Qian X., Scallan C.D., Powell S., Keller T., McMurray M., Labelle A. Multiyear therapeutic benefit of AAV serotypes 2, 6, and 8 delivering factor VIII to hemophilia A mice and dogs. Blood. 2006;108:107–115. doi: 10.1182/blood-2005-12-5115. [DOI] [PubMed] [Google Scholar]
- 2.Naldini L., Blömer U., Gallay P., Ory D., Mulligan R., Gage F.H., Verma I.M., Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 3.Herzog R.W., Hagstrom J.N., Kung S.H., Tai S.J., Wilson J.M., Fisher K.J., High K.A. Stable gene transfer and expression of human blood coagulation factor IX after intramuscular injection of recombinant adeno-associated virus. Proc. Natl. Acad. Sci. USA. 1997;94:5804–5809. doi: 10.1073/pnas.94.11.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perrin G.Q., Zolotukhin I., Sherman A., Biswas M., de Jong Y.P., Terhorst C., Davidoff A.M., Herzog R.W. Dynamics of antigen presentation to transgene product-specific CD4(+) T cells and of Treg induction upon hepatic AAV gene transfer. Mol. Ther. Methods Clin. Dev. 2016;3:16083–16088. doi: 10.1038/mtm.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobrzynski E., Herzog R.W. Tolerance induction by viral in vivo gene transfer. Clin. Med. Res. 2005;3:234–240. doi: 10.3121/cmr.3.4.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang P., Sun B., Osada T., Rodriguiz R., Yang X.Y., Luo X., Kemper A.R., Clay T.M., Koeberl D.D. Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine pompe disease. Hum. Gene Ther. 2012;23:460–472. doi: 10.1089/hum.2011.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sack B.K., Herzog R.W., Terhorst C., Markusic D.M. Development of gene transfer for induction of antigen-specific tolerance. Mol. Ther. Methods Clin. Dev. 2014;1:14013–14019. doi: 10.1038/mtm.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boisgerault F., Mingozzi F. The skeletal muscle environment and its role in immunity and tolerance to AAV vector-mediated gene transfer. Curr. Gene Ther. 2015;15:381–394. doi: 10.2174/1566523215666150630121750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Touchot N., Flume M. Early insights from commercialization of gene therapies in Europe. Genes (Basel) 2017;8:78. doi: 10.3390/genes8020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montini E., Cesana D., Schmidt M., Sanvito F., Ponzoni M., Bartholomae C., Sergi Sergi L., Benedicenti F., Ambrosi A., Di Serio C. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 11.Tomás, H.A., Rodrigues, A.F., Alves, P.M., and Coroadinha, A.S. (2013). Lentiviral gene therapy vectors: challenges and future directions. In Gene Therapy: Tools and Potential Applications, F. Martin, eds. (InTech), https://www.intechopen.com/books/gene-therapy-tools-and-potential-applications/lentiviral-gene-therapy-vectors-challenges-and-future-directions.
- 12.Hareendran S., Balakrishnan B., Sen D., Kumar S., Srivastava A., Jayandharan G.R. Adeno-associated virus (AAV) vectors in gene therapy: immune challenges and strategies to circumvent them. Rev. Med. Virol. 2013;23:399–413. doi: 10.1002/rmv.1762. [DOI] [PubMed] [Google Scholar]
- 13.Tse L.V., Moller-Tank S., Asokan A. Strategies to circumvent humoral immunity to adeno-associated viral vectors. Expert Opin. Biol. Ther. 2015;15:845–855. doi: 10.1517/14712598.2015.1035645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strayer D.S., Lamothe M., Wei D., Milano J., Kondo R. Generation of recombinant SV40 vectors for gene transfer. Methods Mol. Biol. 2001;165:103–117. doi: 10.1385/1-59259-117-5:103. [DOI] [PubMed] [Google Scholar]
- 15.Byun H., Gou Y., Zook A., Lozano M.M., Dudley J.P. ERAD and how viruses exploit it. Front. Microbiol. 2014;5:330. doi: 10.3389/fmicb.2014.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schelhaas M., Malmström J., Pelkmans L., Haugstetter J., Ellgaard L., Grünewald K., Helenius A. Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell. 2007;131:516–529. doi: 10.1016/j.cell.2007.09.038. [DOI] [PubMed] [Google Scholar]
- 17.Marsh M., Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian M., Cai D., Verhey K.J., Tsai B. A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection. PLoS Pathog. 2009;5:e1000465. doi: 10.1371/journal.ppat.1000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engel S., Heger T., Mancini R., Herzog F., Kartenbeck J., Hayer A., Helenius A. Role of endosomes in simian virus 40 entry and infection. J. Virol. 2011;85:4198–4211. doi: 10.1128/JVI.02179-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norkin L.C., Anderson H.A., Wolfrom S.A., Oppenheim A. Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J. Virol. 2002;76:5156–5166. doi: 10.1128/JVI.76.10.5156-5166.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelkmans L., Püntener D., Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science. 2002;296:535–539. doi: 10.1126/science.1069784. [DOI] [PubMed] [Google Scholar]
- 22.Sauter B.V., Parashar B., Chowdhury N.R., Kadakol A., Ilan Y., Singh H., Milano J., Strayer D.S., Chowdhury J.R. A replication-deficient rSV40 mediates liver-directed gene transfer and a long-term amelioration of jaundice in gunn rats. Gastroenterology. 2000;119:1348–1357. doi: 10.1053/gast.2000.19577. [DOI] [PubMed] [Google Scholar]
- 23.Vera M., Sobrevals L., Zaratiegui M., Martinez L., Palencia B., Rodríguez C.M., Prieto J., Fortes P. Liver transduction with a simian virus 40 vector encoding insulin-like growth factor I reduces hepatic damage and the development of liver cirrhosis. Gene Ther. 2007;14:203–210. doi: 10.1038/sj.gt.3302858. [DOI] [PubMed] [Google Scholar]
- 24.Sobrevals L., Rodriguez C., Romero-Trevejo J.L., Gondi G., Monreal I., Pañeda A., Juanarena N., Arcelus S., Razquin N., Guembe L. Insulin-like growth factor I gene transfer to cirrhotic liver induces fibrolysis and reduces fibrogenesis leading to cirrhosis reversion in rats. Hepatology. 2010;51:912–921. doi: 10.1002/hep.23412. [DOI] [PubMed] [Google Scholar]
- 25.Sobrevals L., Enguita M., Quiroga J., Prieto J., Fortes P. Insulin-like growth factor I (IGF-I) expressed from an AAV1 vector leads to a complete reversion of liver cirrhosis in rats. PLoS ONE. 2016;11:e0162955. doi: 10.1371/journal.pone.0162955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Louboutin J.-P., Chekmasova A.A., Marusich E., Chowdhury J.R., Strayer D.S. Efficient CNS gene delivery by intravenous injection. Nat. Methods. 2010;7:905–907. doi: 10.1038/nmeth.1518. [DOI] [PubMed] [Google Scholar]
- 27.Barbanti-Brodano G., Sabbioni S., Martini F., Negrini M., Corallini A., Tognon M. Simian virus 40 infection in humans and association with human diseases: results and hypotheses. Virology. 2004;318:1–9. doi: 10.1016/j.virol.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Garcea R.L., Imperiale M.J. Simian virus 40 infection of humans. J. Virol. 2003;77:5039–5045. doi: 10.1128/JVI.77.9.5039-5045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gluzman Y. SV40-transformed simian cells support the replication of early SV40 mutants. Cell. 1981;23:175–182. doi: 10.1016/0092-8674(81)90282-8. [DOI] [PubMed] [Google Scholar]
- 30.Oppenheim A., Peleg A. Helpers for efficient encapsidation of SV40 pseudovirions. Gene. 1989;77:79–86. doi: 10.1016/0378-1119(89)90361-2. [DOI] [PubMed] [Google Scholar]
- 31.Vera M., Prieto J., Strayer D.S., Fortes P. Factors influencing the production of recombinant SV40 vectors. Mol. Ther. 2004;10:780–791. doi: 10.1016/j.ymthe.2004.06.1014. [DOI] [PubMed] [Google Scholar]
- 32.Jasin M., de Villiers J., Weber F., Schaffner W. High frequency of homologous recombination in mammalian cells between endogenous and introduced SV40 genomes. Cell. 1985;43:695–703. doi: 10.1016/0092-8674(85)90242-9. [DOI] [PubMed] [Google Scholar]
- 33.Gerard R.D., Gluzman Y. New host cell system for regulated simian virus 40 DNA replication. Mol. Cell. Biol. 1985;5:3231–3240. doi: 10.1128/mcb.5.11.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arad U., Ben-Nun-Shaul O., El-Latif M.A., Nissim O., Oppenheim A. A new packaging cell line for SV40 vectors that eliminates the generation of T-antigen-positive, replication-competent recombinants. Virology. 2002;304:155–159. doi: 10.1006/viro.2002.1791. [DOI] [PubMed] [Google Scholar]
- 35.Kimchi-Sarfaty C., Ben-Nun-Shaul O., Rund D., Oppenheim A., Gottesman M.M. In vitro-packaged SV40 pseudovirions as highly efficient vectors for gene transfer. Hum. Gene Ther. 2002;13:299–310. doi: 10.1089/10430340252769815. [DOI] [PubMed] [Google Scholar]
- 36.Nakanishi A., Shum D., Morioka H., Otsuka E., Kasamatsu H. Interaction of the Vp3 nuclear localization signal with the importin alpha 2/beta heterodimer directs nuclear entry of infecting simian virus 40. J. Virol. 2002;76:9368–9377. doi: 10.1128/JVI.76.18.9368-9377.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Haan P., Klein H.C., ’t Hart B.A. Autoimmune aspects of neurodegenerative and psychiatric diseases: a template for innovative therapy. Front. Psychiatry. 2017;8:46. doi: 10.3389/fpsyt.2017.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de la Luna S., Martín J., Portela A., Ortín J. Influenza virus naked RNA can be expressed upon transfection into cells co-expressing the three subunits of the polymerase and the nucleoprotein from simian virus 40 recombinant viruses. J. Gen. Virol. 1993;74:535–539. doi: 10.1099/0022-1317-74-3-535. [DOI] [PubMed] [Google Scholar]
- 39.Barrett P.N., Mundt W., Kistner O., Howard M.K. Vero cell platform in vaccine production: moving towards cell culture-based viral vaccines. Expert Rev. Vaccines. 2009;8:607–618. doi: 10.1586/erv.09.19. [DOI] [PubMed] [Google Scholar]
- 40.Kolman J.L. Massively parallel sequencing for the detection of adventitious viruses. PDA J. Pharm. Sci. Technol. 2011;65:663–667. doi: 10.5731/pdajpst.2011.00836. [DOI] [PubMed] [Google Scholar]
- 41.Louboutin J.-P., Marusich E., Fisher-Perkins J., Dufour J.P., Bunnell B.A., Strayer D.S. Gene transfer to the rhesus monkey brain using SV40-derived vectors is durable and safe. Gene Ther. 2011;18:682–691. doi: 10.1038/gt.2011.13. [DOI] [PubMed] [Google Scholar]
- 42.Christian P., Deschamps M., Dhere R., Hutchens C., Lebron J., Knezevic I., Lambert S., Lewis A., Mallet L., Nandapalan P. World Health Organization; 2011. Recommendations for the Evaluation of Animal Cell Cultures as Substrates for the Manufacture of Biological Medicinal Products and for the Characterization of Cell Banks. [Google Scholar]
- 43.Cole C.N. Polyomaviridae: the viruses and their replication. In: Fields B.N., Knipe D.M., Howley P.M., editors. Fields Virology: 1997–2025. Lippincott-Raven Publishers; 1996. [Google Scholar]
- 44.Yu Y., Kudchodkar S.B., Alwine J.C. Effects of simian virus 40 large and small tumor antigens on mammalian target of rapamycin signaling: small tumor antigen mediates hypophosphorylation of eIF4E-binding protein 1 late in infection. J. Virol. 2005;79:6882–6889. doi: 10.1128/JVI.79.11.6882-6889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fahrbach K.M., Katzman R.B., Rundell K. Role of SV40 ST antigen in the persistent infection of mesothelial cells. Virology. 2008;370:255–263. doi: 10.1016/j.virol.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hronovský V., Plaisner V., Benda R. CV-1 monkey kidney cell line—a highly susceptible substrate for diagnosis and study of arboviruses. Acta Virol. 1978;22:123–129. [PubMed] [Google Scholar]