

Abstract
This review aims to provide a summary of current knowledge of host genetic effects on human immunodeficiency virus (HIV) disease. Mapping of simple single nucleotide polymorphisms (SNP) has been largely successful in HIV, but more complex genetic associations involving haplotypic or epigenetic variation, for example, remain elusive. Mechanistic insights explaining SNP associations are incomplete, but continue to be forthcoming. The number of robust immunogenetic correlates of HIV is modest and their discovery mostly predates the genome-wide era. Nevertheless, genome-wide evaluations have nicely validated the impact of HLA and CCR5 variants on HIV disease, and importantly, made clear the many false positive associations that were previously suggested by studies using the candidate gene approach. We describe how multiple HIV outcome measures such as acquisition, viral control, and immune decline have been studied in adults and in children, but that collectively these identify only the two replicable loci responsible for modifying HIV disease, CCR5, and HLA. Recent heritability estimates in this disease corroborate the modest impact of genetic determinants and their oligogenic nature. While the mechanism of protection afforded by genetic variants that diminish CCR5 expression is clear, new aspects of HLA class I-mediated protection continue to be uncovered. We describe how these genetic findings have enhanced insights into immunobiology, been clinically translated into CCR5 antagonists, allowed prioritization of antigens for vaccination efforts, and identified targets for genome-editing interventions. Finally, we describe how studies of genetically complex parts of the genome using new tools may begin revealing additional correlates.
Electronic supplementary material
The online version of this article (doi:10.1007/s00251-017-1000-z) contains supplementary material, which is available to authorized users.
Keywords: HIV, Host genetics, HLA, CCR5, Immunology
Introduction
Nearly 78 million people have been infected by HIV, primarily through sexual acquisition (about 80%), and 35 million have died from HIV-1 and related diseases. HIV infects infants through mother-to-child-transmission (MTCT) and adults through bloodborne or sexual transmission. The HIV epidemic has affected subpopulations in heterogeneous fashions, with rates varying by age, sex, predominant mode of exposure (intravenous or sexual), and geographic location. The overall risk of transmission of HIV is thought to be low, roughly 0.18% per sex act, but varies considerably from 0.02 to 1.6% in published meta-analyses (Boily et al. 2009). Similarly, level of viremia during acute HIV infection, setpoint viral load, rate of CD4+ T cell depletion, and eventual rates of progression to AIDS all vary widely. For example, while some individuals have been observed to develop profound CD4+ T cell lymphopenia and AIDS within 2 years, others remain AIDS-free for up to 15 years following HIV seroconversion (van der Helm et al. 2014; Mlisana et al. 2014).
Since the sequencing of the human genome in 2001 (Lander et al. 2001), large-scale genetic and genomic studies have played an increasingly important role in delineating the pathogenesis of human disease. Understanding the variable risk and course of HIV infection in vivo by studying host genetics has been instrumental to understanding the immunopathogenesis of HIV. Moreover, it has complemented the in vitro understanding of the biology of HIV infection and replication that led to development of antiretroviral drugs, which are the cornerstone of HIV prevention and therapy.
Studies of host genetic correlates have reflected the wide-ranging nature of possible HIV outcome measures over the course of HIV infection, beginning with acquisition through disease progression and development of opportunistic infection and AIDS. Similarly, these studies reflect the heterogeneity in populations affected by HIV. Hundreds of studies have been conducted into the genetic determinants of HIV acquisition and disease progression. Older studies, referred to as candidate gene studies, were locus or gene-centric, while larger recent studies have searched for correlates across the genome using genome-wide genotyping or whole exome/genome sequencing in an unbiased, “agnostic” manner. In total, five genome-wide association studies (GWAS) of HIV acquisition (Johnson et al. 2015; Lingappa et al. 2011; Petrovski et al. 2011; Joubert et al. 2010; McLaren et al. 2013) and 11 of markers of HIV disease course have been reported (Lingappa et al. 2011; Fellay et al. 2009; Fellay et al. 2007; Herbeck et al. 2010; H. I. V. C. S. International 2010; Le Clerc et al. 2009; Limou et al. 2009; Pelak et al. 2011; Pelak et al. 2010; Troyer et al. 2011; Wei et al. 2015). Combining most of these studies, a meta-analysis in individuals of European ancestry of HIV acquisition (McLaren et al. 2013) and HIV viremia (McLaren et al. 2015) has also been reported. A range of candidate gene and GWA studies of HIV-related traits have been reported, such as atherosclerosis (Shrestha et al. 2010), response to antiretroviral drugs (Chantarangsu et al. 2011; Irvin et al. 2011; Leger et al. 2014; Lehmann et al. 2015), and blood counts in HIV-infected or -exposed patients (Ferreira et al. 2010; Moore et al. 2015; Ramsuran et al. 2011). Similarly HIV-associated diseases have also been interrogated, such as HIV-dementia (Levine et al. 2012) and hepatitis C (Ulveling et al. 2016). Although we do not discuss each study in detail here, Supplementary Table 1 provides a recent extract from the GWAS catalog for all HIV and related traits. These modern studies, in which confounding due to population structure can be adequately adjusted for, have been illuminating in refuting candidate gene findings likely to have been false-positive associations and in confirming two loci that were identified in candidate gene studies as important in modifying HIV outcomes: those encoding CCR5, a co-receptor for HIV entry, and human leukocyte antigen (HLA) class I, the protein system responsible for antigen presentation to immune cells. These GWAS have also been instrumental in generating estimates of the relative contribution of host and viral genetic variation in HIV. Accordingly, we review this literature briefly, and then turn attention to the two well-validated genetic correlates of HIV acquisition and HIV disease outcome. We specifically highlight questions, which, in our view, remain unanswered with regard to the mechanism of HLA effects in HIV. We anticipate further progress in these areas and discuss additional future prospects in this review.
The relative role of host and viral genetic variation in HIV
Classically, twin and family studies have been used to infer the host genetic contribution to disease risk. Such studies have been exceedingly challenging to perform in HIV either with regard to HIV acquisition or HIV disease progression due to poor sample sizes, although one very enlightening sibling study has been reported, emphasizing the importance of the major histocompatibility complex (MHC) in disease outcome (Kroner et al. 1995). A second important determinant of disease course, unique to diseases caused by a polymorphic pathogen, is the relative importance of pathogen variation. While estimates of the host genetic contribution to HIV acquisition have not been reported (but likely to be modest), estimates of the genetic contribution to HIV control after infection suggest that around one third of the variance in setpoint viremia can be attributed to viral variation (Fraser et al. 2014). Modern approaches that estimate the host polygenic heritability using genome-wide genotyping data have recently been reported in HIV. In a meta-analysis of 6315 individuals for whom setpoint viremia was recorded, ~25% of variance in setpoint viremia was attributable to common host genetic variation, and the majority of this could be explained by variation in the HLA or CCR5 loci leaving around 5.5% of variance attributable to variation outside of these two regions (McLaren et al. 2015). Moreover, Bartha et al. (2013, 2017) demonstrated in studies, where viral and host genetic variation were both available, that almost all of the effect of the HLA region on setpoint viremia is mediated through its pressure on viral genetic variation, and that human variants only in the HLA region and not elsewhere in the genome are associated with specific viral mutations. These findings corroborate earlier findings of HLA-mediated selection of viral variants in vivo (Brumme et al. 2007; Carlson and Brumme 2008). Taken together, these studies are striking in demonstrating that the CCR5 and HLA loci explain the majority of host genetic effects on HIV viral control. Larger studies may yet yield additional rare variants that alter disease, but more extensive coverage through whole-genome studies has not as yet yielded new insights (Pelak et al. 2011). Variants with population-specific effects may be identified, as is suggested by early GWAS in Asian populations, for example (Wei et al. 2015). Strikingly, the genetic mapping of HIV is amongst the most complete of any complex disease, as there are few other diseases where the majority of variance in genetically mediated modification of disease course has been mapped and attributed to specific genes. Efforts in HIV genetic research have therefore focused on delineating the nature of CCR5 or HLA-mediated protection and finding therapeutic opportunities to exploit this.
Variants in CCR5 that diminish expression of a co-receptor used for cell entry by HIV protect from acquisition and reduce rates of disease progression
Genetic variants in CCR5, a chemokine receptor physiologically responsible for binding RANTES, MIP-1α, and MIP-1β, affect HIV acquisition risk and disease progression rates (Fig. 1). The observation that homozygosity for a 32-base pair deletion in the coding region of CCR5 (CCR5Δ32) accounted for reduced HIV infection risk was independently reported by three groups (Dean et al. 1996; Liu et al. 1996; Samson et al. 1996) within months of the identification of CCR5 as an important co-receptor for HIV viral entry (Choe et al. 1996; Deng et al. 1996; Doranz et al. 1996; Dragic et al. 1996; Rucker et al. 1996; Alkhatib et al. 1997). The frequency of this variant is the highest amongst people of European descent in whom ~10% of individuals carry a copy of the CCR5Δ32 allele, intermediate amongst those of Middle Eastern and Asian descent (2–5%), and near absent amongst individuals of African ancestry (Martinson et al. 1997). Although initial studies noting a gradient in allele frequency in Europe (Lucotte and Mercier 1998) led to conjecture about likelihood of selection for this variant (Galvani and Slatkin 2003; Novembre et al. 2005; Stephens et al. 1998), other studies conclusively demonstrated that the deletion allele was present in prehistoric humans (Hummel et al. 2005), and has been subject to neutral evolution (Sabeti et al. 2005).
Fig. 1.
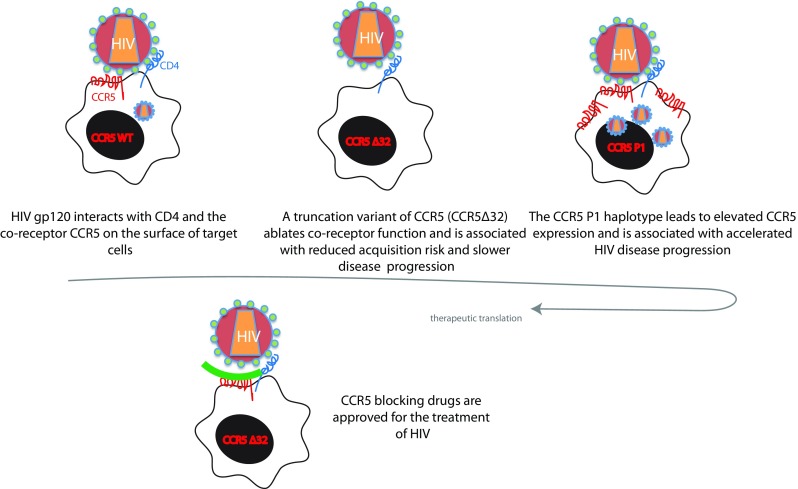
CCR5 is a key co-receptor for HIV entry. CCR5Δ32 individuals in whom CCR5 expression is lost have reduced HIV acquisition risk and disease progression while individuals with the CCR5P1 haplotype have higher CCR5 expression and accelerated disease progression. These and other observations led to drugs that block CCR5, which are licensed for the treatment of HIV
The protective effect of CCR5Δ32 has been confirmed in studies of children (Misrahi et al. 1998) and extended to demonstrate that children heterozygous for the variant also experience slower disease progression. Independent variants in the promotor region of CCR5 (known as the CCR5P1 haplotype) were observed to accelerate HIV disease progression (Martin et al. 1998), and this allele was shown to result in higher expression of CCR5 (McDermott et al. 1998). Genome-wide genotyping arrays do not universally contain variants that tag the CCR5Δ32 allele, but careful meta-analyses of published GWAS have readily replicated the effect of CCR5Δ32 on reducing HIV acquisition and slowing disease progression, and the effect of CCR5P1 on increasing HIV viremia (McLaren et al. 2013; McLaren et al. 2015). These meta-analyses also suggest that there may yet be additional signals in the region, not attributable to CCR5Δ32 or CCR5P1 that modify HIV control. These findings highlight the fact that the locus around CCR5 encodes several other chemokine receptors (Mummidi et al. 1998), including CCL3L1 (Dolan et al. 2007; Gonzalez et al. 2005) and CCR2 (Smith et al. 1997), which have been implicated in HIV control.
Variation in HLA class I mediates altered HIV control through multiple mechanisms
Soon after the first description of HIV, cytotoxic T lymphocytes (CTL) targeting specific HIV antigens were identified in HIV-infected patients (Walker et al. 1987; Nixon et al. 1988). CTL were known, by this stage, to recognize target cells through virus-derived antigens presented by MHC class I molecules (Zinkernagel and Doherty 1974), known in humans as HLA. Contemporaneously, the exceptional polymorphism in HLA class I and class II was becoming more amenable to study at greater scale and at higher resolution using PCR methods to complement classical serological typing. The HLA loci on chromosome 6 are the most polymorphic genes of the human genome. HLA-A, -B, and -C (the class I genes) and HLA-DR, -DP, and -DQ (the class II genes) encode surface proteins that present host and pathogen-derived peptides to immune cells. The exceptional allelic diversity is mirrored by diversity in regulation of gene expression, and interaction with other components of the antigen presentation machinery and with other receptors. There is extensive linkage disequilibrium amongst HLA genes, particularly between HLA-B and -C genes in class I, and -DR and -DQ in class II.
The identification of CTL specific for HIV (Walker et al. 1987; Nixon et al. 1988) raised the possibility that the highly polymorphic HLA class I region may be involved in modifying HIV disease on the basis that such variation may alter CTL recognition. HLA alleles have been associated with more than 100 other diseases including most autoimmune and many infectious diseases, such as malaria and influenza (Matzaraki et al. 2017). Discoveries made in HIV have had broad implications for understanding immunity against viruses in general, as the HLA class I locus is the most robust correlate of HIV disease outcome in humans and has been widely replicated across settings and in GWAS. Here, we review several important themes that relate to the association between polymorphism in the HLA class I genes and HIV disease course (which has been the subject of other detailed reviews). Next, we highlight important, unanswered research questions.
The influence of HLA alleles on HIV disease progression includes both protective and harmful effects
Figure 2 shows the estimated effect of each HLA-A, -B, and -C allele from a stepwise logistic regression model comparing HIV controllers to HIV non-controllers (Bashirova et al. 2014). HLA class II alleles have vastly smaller effects than class I alleles, rarely reach significance after correction for multiple comparisons, and have been less well replicated. As shown, HLA-B*57, B*27, B*58:01, B*51, B*13, and B*81:01 are amongst the most reproducibly protective correlates of HIV. In contrast, HLA-B*58:02 and HLA-B*35Px alleles are associated with accelerated HIV disease progression. In addition, there is a wide range of alleles with intermediate effects. While some of the allelic effects on HIV appear reproducible across ethnic groups, there is likely considerable heterogeneity of effect by age, as is suggested by comparing effects in adults vs. children (Adland et al. 2015). For some alleles, temporal variation of the effect following seroconversion (Gao et al. 2005) has also been reported.
Fig. 2.
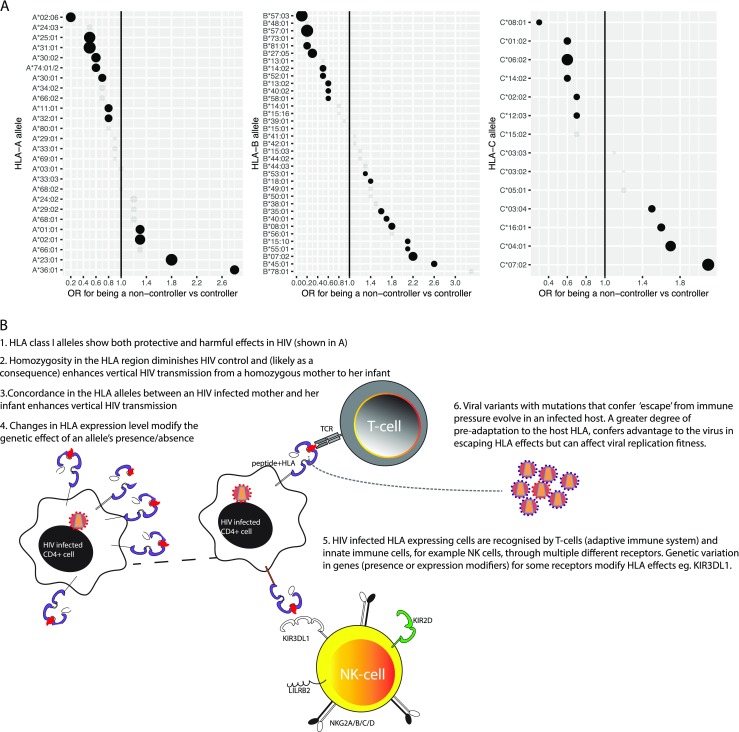
a The effect of HLA alleles on HIV vary across a continuum from harmful to protective effects and b involve a wide-variety of different mechanisms in mediating their effect. Data in a are derived from Bashirova et al. (2014)
Reduced diversity in HLA is associated with poor HIV outcomes
Homozygosity at class I alleles has consistently been associated with poor HIV outcomes, including higher viremia, increased risk of transmission of HIV from an HLA-homozygote mother to her infant (likely due to higher viremia and higher likelihood of greater mother-child concordance), and accelerated HIV disease progression (Kroner et al. 1995; Carrington et al. 1999; Tang et al. 1999; Mackelprang et al. 2008). These data are consistent with a model in which the greater diversity of peptides presented to immune cells in heterozygotes is associated with a broader range of immune responses and better control, whereas homozygosity at the HLA loci constrains the set of peptides that can be presented. While homozygosity for HLA-Bw4 has been reported to be associated with enhanced HIV viremic control and reduced disease progression (Flores-Villanueva et al. 2001), this is almost certainly due to the fact that the most protective HLA class I alleles, which generally bind distinct HIV epitopes, carry the Bw4 epitope.
HLA concordance between HIV-discordant transmitting pairs is associated with elevated risk
Infants who share the same allele as their mother are at increased risk for acquiring HIV (Mackelprang et al. 2008; MacDonald et al. 1998; Polycarpou et al. 2002). This may have to do with the level of HIV adaptation to the HLA type carried by the infant.
Non-classical mechanisms play a role in HLA-allelic associations with HIV
The major mechanism through which HLA alleles have been shown to mediate their effects in HIV is the presentation of specific HIV-derived peptides to CTL, resulting in killing of HIV-infected target cells. This mechanism is incontrovertibly supported by functional studies that demonstrate HLA-restricted CTL targeting. However, variation in HLA expression level also appears to have a substantial effect. In particular, the effect of HLA-C alleles on HIV is inversely associated with the expression level of the allotypes they encode (Thomas et al. 2009; Apps et al. 2013). This finding was initially suggested by the identification of a genome-wide significant variant (rs924942) near HLA-C that was associated with HIV control (Fellay et al. 2007), a variant later shown to tag HLA-C expression in individuals of European descent. While rs924942 is highly unlikely to have any direct effect on HLA-C expression, in Caucasians, it marks variation in a microRNA-binding site in the 3′ UTR of HLA-C, which differentially affects HLA-C expression levels in an allele-dependent manner (Kulkarni et al. 2011; Kulkarni et al. 2013). It also marks an Oct1 binding site that has been shown to account for up to 35% of the differential expression level of HLA-C (Vince et al. 2016).
HLA class I alleles interact with other receptors; notably the killer cell immunoglobulin-like receptors (KIR) and leukocyte immunoglobulin-like receptors (LILR), further diversifying the function of HLA class I
The leukocyte receptor complex (LRC) on chromosome 19, and in particular the KIR locus, demonstrates pronounced structural and allelic variation. The KIR locus encodes anywhere from 9 to 16 surface receptors expressed on natural killer and T cells, which interact with HLA molecules to transduce either inhibitory or activating signals. Specific HLA allotypes interact with specific KIR; and in some cases, the peptide presented by the HLA allotype modifies this effect (See a previous review for further details (Martin and Carrington 2013)). The combination of HLA-B alleles that encode the Bw4 epitope with isoleucine at position 80 (Bw4-80I) and presence of KIR3DS1 is associated with enhanced HIV control (Martin et al. 2002). Similarly, the combination of HLA-B and specific KIR3DL1 alleles shows a gradient of effects on HIV, where HLA-B Bw4 alleles with high expression KIR3DL1 alleles show synergistic protective effects (Martin et al. 2007). Recent evidence has also demonstrated a role for LILR binding to HLA as a mediator of HLA effects against HIV. In this study, the strength of HLA-B binding to LILRB2, an inhibitory receptor expressed on myeloid and lymphoid cells that binds differentially to HLA-B in an allotype-dependent manner (Jones et al. 2011), is inversely associated with HIV viral control (Bashirova et al. 2014). HLA-B*35Px alleles in particular have been suggested to mediate their adverse impact on HIV through enhanced interaction and hence inhibition of LILRB2 expressing dendritic cells (Huang et al. 2009).
Focal viral sequence variation is a footprint of immune pressure
Evolution of viral variants that escape the targeted immune pressure mediated by HLA-restricted responses shows mutations specifically in epitopes presented by HLA. In a “genome-to-genome” analysis, Bartha et al. demonstrate that all of the variants in the HIV genome that are significantly associated with host genetic variants are also associated with variants in the HLA region (Bartha et al. 2013). Each of the major HIV proteins demonstrates HLA-associated selection pressure, although the pressure varies by protein (Carlson et al. 2012). These “escape variants” are enriched for localization within CTL epitopes, with 29% of all HLA-associated viral variants localizing within ±3 bp of an optimally described CTL epitope. Mutations that escape the CTL response occur most frequently at HLA anchor residues (1.8-fold enrichment) (Carlson et al. 2012). Just as HLA concordance between transmitting pairs has been observed epidemiologically to have adverse consequences, transmission of a virus even amongst HLA-discordant partners that is pre-adapted to the recipient HLA alleles is associated with impaired viremic control (Carlson et al. 2016). Not only does the HLA impact on HIV have an effect on the disease course in any given individual but also it shapes features of the epidemic at the population level. Increased immune pressure in individuals has been reported to reduce the virulence of the virus at a population level (Juarez-Molina et al. 2014; Payne et al. 2014; Herbeck et al. 2012; Kawashima et al. 2009). However, no clear or substantial decrease in HIV virulence has been observed since the epidemic began; and from the host standpoint, the detrimental effect of viral adaptation to the host HLA type appears greater than the beneficial effect associated with a loss in viral fitness (Carlson et al. 2016). Finally, although not as well replicated, carriage of specific KIR alleles is associated with variation at specific sites in HIV (Alter et al. 2011; Holzemer et al. 2015).
Outstanding research questions in HLA-HIV biology
What is the mechanism for the observed harmful effects of some HLA alleles? Protection conferred by certain HLA alleles is generally attributed to the HIV epitopes they recognize, though multiple characteristics of each allele are likely to explain their effects. HLA associations with susceptibility or impaired HIV control, on the other hand, are more difficult to explain although, once again, their overall effects are likely due to a combination of determinants. It is clear, however, that some alleles have detrimental effects and are not merely associated with relative impaired effects. For example, carriage of even one HLA-B*35 allele is associated with worse outcomes than being homozygous for any other HLA-B allele (Carrington et al. 1999). HLA associations with impaired clinical outcomes are common in autoimmune diseases, presumably due to involvement of that allele in instigating or maintaining autoimmune responses, but how this may be mediated in HIV is not immediately obvious. For some alleles, clear evidence of interaction with inhibitory LILRB2 receptors has been demonstrated, but this is unlikely to explain the full impact of an allele on poor outcome after HIV infection. Understanding this may yield new approaches for ameliorating the harmful effects of some HLA alleles.
Are HLA class II alleles reproducibly and independently associated with HIV control? A distinguishing feature of the nature of association in HIV compared to other diseases is the relative paucity of class II associations that predominate in autoimmune and other viral diseases. Although a handful of class II associations have been reported (Kroner et al. 1995; Ranasinghe et al. 2013; Julg et al. 2011), the extensive linkage disequilibrium in the class II region and relatively smaller sample sizes of class II-focused studies have made unequivocal demonstration of their role challenging. Even the largest GWAS have provided no indication that class II variation accounts for differential outcomes after HIV infection, though confounding circumstances may hamper identification of such effects (McLaren and Carrington 2015). Large studies in which HLA class I and linkage disequilibrium effects can be accounted for and where class II is typed directly may yield definitive results.
Are the effects of HLA alleles purely additive, or do some have synergistic effects, and how does this provide insights into mechanism? The observation that certain pairs of HLA class I alleles together may target sites that result in profound crippling of HIV (Dahirel et al. 2011; Ferguson et al. 2013) suggests that at least some combinations of alleles may have non-additive effects. The delineation of rules governing the effect of advantageous and deleterious features of HLA alleles when carried together requires large sample sizes, but if achievable, may inform the conditions under which non-additive effects on HIV control may be present and suggest epitopes that may be beneficial targets in vaccine design.
What additional non-classical mechanisms account for the association of HLA alleles with HIV control? Elevated HLA-C expression is robustly associated with enhanced HIV control, but the precise nature of this, including the relevance of KIR-mediated HLA-C recognition, remains unresolved. Understanding these effects may also help identify whether use of HLA-C/KIR blocking therapies, such as lirilumab may be useful in HIV. Although HLA-B alleles do not vary to any clear extent in mRNA expression (Ramsuran et al. 2017), recent studies demonstrate how HLA-B alleles vary in their impact on NK cell responses to HIV-infected cells (Merino et al. 2013). HLA-B alleles can be broadly split into two groups based on polymorphism encoding either a methionine (M) or threonine in the leader peptide of all HLA-B alleles. Horowitz et al. (2016) recently demonstrated that the ancestral M encoding variant almost invariably occurs on haplotypes with HLA-Bw6 alleles and HLA-C alleles of the C1 group. They demonstrate that this results in profound differences in NK cell responses in vitro, and the effect of this variant in vivo may be an exciting avenue for further research. Finally, HLA-A expression levels vary substantially (Ramsuran et al. 2015) and whether such variation has any impact on HIV, as was shown for HLA-C, is an intriguing question.
Do KIR-HLA interactions involving KIR2D and HLA-C modify HIV outcomes in epidemiologic analyses (as do KIR3DL1/S1 plus HLA-Bw4), and if so how? The interaction between HLA and KIR is incontrovertible, yet the in vivo effects of this complex interaction have been challenging to study because of the marked polymorphism in the two regions. Viral sequence footprints indicative of KIR-mediated pressure and in vitro evidence of an effect on HIV responses are suggestive of further epidemiological associations and in-depth examination of these two regions together in large datasets where there is power to control for confounding by known HLA/KIR effects may prove fruitful. Moreover, the originally reported HLA-Bw4-80I/KIR3DS1 associations deserve re-appraisal in light of the evidence that KIR3DS1 binds HLA-F (Burian et al. 2016; Garcia-Beltran et al. 2016), and current lack of clarity on whether KIR3DS1 may also bind to HLA-Bw4 epitopes in vivo under specific circumstances, such as in the presence of specific peptides (O'Connor et al. 2015).
Immunogenetics of HIV on the horizon
A crucial caveat to the extant knowledge on genetic modifiers of HIV is that genetically complex regions are systematically under-represented. For example, the evidence for KIR-HLA interactions undoubtedly suggests that additional effects are likely to exist, but are not yet identified because they are neither tagged by single-nucleotide variants in GWAS studies nor have they been fully characterized. For example, aside from KIR3DL1, allelic typing of KIR loci has not been widely performed, but rather, the field has concentrated primarily on associations at the gene presence/absence level. New, in-depth typing methods for the KIR locus will facilitate progress in this regard (Norman et al. 2016). Similarly, variation in the immunoglobulin locus and TCR loci, which are fiendishly complex, may be fruitful, as these loci may have substantial effects in HIV given the known importance of binding and neutralizing antibody responses in HIV.
The relative absence of genetic correlates of disease outside of HLA and CCR5 juxtaposes against the extensive evidence for a slew of host proteins involved in HIV pathogenesis. This may be due in some cases to their lack of functional polymorphism or to the exquisite redundancy that is characteristic of many of these biological systems. Also, pairs of variants that affect functionally linked components may be expected to show epistatic associations, and these would not be identified by classical GWAS techniques. Examples where HLA in combination with KIR3DS1, KIR3DL1, and LILRB2 are highly suggestive that such genetic effects do occur at some frequency. We anticipate that careful analysis of such pairs (or higher order combinations), guided by the extensive biological characterization of host restriction or dependency factors identified by siRNA (Bushman et al. 2009; Zhu et al. 2014) and in newly published CRISPR screens (Park et al. 2017), will be fruitful.
A third exciting prospect is the use of Mendelian randomization approaches to validate or refute non-genetic correlates of disease. For example, immune activation has long been linked to HIV acquisition, is associated with HIV disease progression, and has some mechanistic basis for being causally involved in these associations (Sereti and Altfeld 2016). However, clinical trial efforts to modify immune activation have not had a profound effect on HIV (Bandera et al. 2017). To distinguish whether such efforts have been unsuccessful due to an insufficient ability to reduce immune activation, or whether immune activation is an epiphenomenon of HIV disease, Mendelian randomization studies may be fruitful. Using genetic correlates of immune activation as an instrument could allow inference as to the causal role of immune activation in HIV disease. This is done by specifically identifying genetic variants associated with immune activation, and then testing whether these variants are associated with HIV outcomes to the extent predicted by the effect of the variant on immune activation and the reported association between activation and outcomes (Didelez and Sheehan 2007). The major advantage of such an approach is its use of genetic variables, which are randomly inherited and subject to fewer confounders. Such efforts have been immensely powerful, for example, in refuting HDL as a target for cardiovascular preventive therapy. Studies have demonstrated that HDL cholesterol levels are unlikely to be causally linked to cardiovascular outcomes (Voight et al. 2012) because variants that robustly associate with HDL cholesterol are not associated with outcomes.
Concluding remarks
Our present understanding of genetically mediated control of HIV acquisition and disease progression stands amongst the most advanced of any polygenic disorder, and this holds true in comparison to malaria or tuberculosis, the two other leading infectious pandemics of our time. The two loci originally identified by candidate gene studies in the 1990s (CCR5 and HLA) remain the only replicable and robust correlates of HIV acquisition or of disease progression. The observation that homozygotes for the CCR5∆32 null mutation are highly resistant to HIV infection and that heterozygotes for this mutation progress more slowly to disease has led to targeted therapy in the form of approved CCR5 antagonists such as maraviroc, contributed to the only HIV cure in an adult recipient of a CCR5Δ32 allogeneic stem cell transplant (Hutter et al. 2009), and has been leading targets for genome-editing strategies in HIV (Tebas et al. 2014). Although direct translation of HLA findings into patient care is less straightforward, the understanding of the central role of HLA-restricted responses and the findings that stemmed from this, such as the importance of gag-specific responses in vivo, is playing an important role in antigen prioritization in HIV vaccine development. Nevertheless, the nature of how HLA variation affects HIV continues to be expanded upon. Modern genetics provides a powerful tool to determine causal pathways involved in HIV control, and discoveries regarding the many layers of HLA effects on HIV disease have and will continue to have major relevance to our understanding of human disease in general.
Electronic supplementary material
Summary of findings of GWAS studies of HIV outcomes or HIV-related outcomes extracted from the GWAS catalog (https://www.ebi.ac.uk/gwas/). (XLSX 74 kb).
Acknowledgements
This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s00251-017-1000-z) contains supplementary material, which is available to authorized users.
References
- Adland E, et al. Discordant impact of HLA on viral replicative capacity and disease progression in pediatric and adult HIV infection. PLoS Pathog. 2015;11:e1004954. doi: 10.1371/journal.ppat.1004954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhatib G, Locati M, Kennedy PE, Murphy PM, Berger EA. HIV-1 coreceptor activity of CCR5 and its inhibition by chemokines: independence from G protein signaling and importance of coreceptor downmodulation. Virology. 1997;234:340. doi: 10.1006/viro.1997.8673. [DOI] [PubMed] [Google Scholar]
- Alter G, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature. 2011;476:96. doi: 10.1038/nature10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apps R, et al. Influence of HLA-C expression level on HIV control. Science. 2013;340:87. doi: 10.1126/science.1232685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandera A, Colella E, Rizzardini G, Gori A, Clerici M. Strategies to limit immune-activation in HIV patients. Expert Rev Anti-Infect Ther. 2017;15:43. doi: 10.1080/14787210.2017.1250624. [DOI] [PubMed] [Google Scholar]
- Bartha I, et al. A genome-to-genome analysis of associations between human genetic variation, HIV-1 sequence diversity, and viral control. elife. 2013;2:e01123. doi: 10.7554/eLife.01123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartha I, et al. Estimating the respective contributions of human and viral genetic variation to HIV control. PLoS Comput Biol. 2017;13:e1005339. doi: 10.1371/journal.pcbi.1005339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashirova AA, et al. LILRB2 interaction with HLA class I correlates with control of HIV-1 infection. PLoS Genet. 2014;10:e1004196. doi: 10.1371/journal.pgen.1004196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boily MC, et al. Heterosexual risk of HIV-1 infection per sexual act: systematic review and meta-analysis of observational studies. Lancet Infect Dis. 2009;9:118. doi: 10.1016/S1473-3099(09)70021-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumme ZL, et al. Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1. PLoS Pathog. 2007;3:e94. doi: 10.1371/journal.ppat.0030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burian A, et al. HLA-F and MHC-I open conformers bind natural killer cell Ig-like receptor KIR3DS1. PLoS One. 2016;11:e0163297. doi: 10.1371/journal.pone.0163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman FD, et al. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 2009;5:e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JM, Brumme ZL. HIV evolution in response to HLA-restricted CTL selection pressures: a population-based perspective. Microbes Infect. 2008;10:455. doi: 10.1016/j.micinf.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Carlson JM, et al. Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J Virol. 2012;86:13202. doi: 10.1128/JVI.01998-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JM, et al. Impact of pre-adapted HIV transmission. Nat Med. 2016;22:606. doi: 10.1038/nm.4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington M, et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283:1748. doi: 10.1126/science.283.5408.1748. [DOI] [PubMed] [Google Scholar]
- Chantarangsu S, et al. Genome-wide association study identifies variations in 6p21.3 associated with nevirapine-induced rash. Clinical Infectious Diseases: an Official Publication of the Infectious Diseases Society of America. 2011;53:341. doi: 10.1093/cid/cir403. [DOI] [PubMed] [Google Scholar]
- Choe H, et al. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135. doi: 10.1016/S0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- Dahirel V, et al. Coordinate linkage of HIV evolution reveals regions of immunological vulnerability. Proc Natl Acad Sci U S A. 2011;108:11530. doi: 10.1073/pnas.1105315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273:1856. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- Deng H, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Didelez V, Sheehan N. Mendelian randomization as an instrumental variable approach to causal inference. Stat Methods Med Res. 2007;16:309. doi: 10.1177/0962280206077743. [DOI] [PubMed] [Google Scholar]
- Dolan MJ, et al. CCL3L1 and CCR5 influence cell-mediated immunity and affect HIV-AIDS pathogenesis via viral entry-independent mechanisms. Nat Immunol. 2007;8:1324. doi: 10.1038/ni1521. [DOI] [PubMed] [Google Scholar]
- Doranz BJ, et al. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149. doi: 10.1016/S0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- Dragic T, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- Fellay J, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellay J, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5:e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AL, et al. Translating HIV sequences into quantitative fitness landscapes predicts viral vulnerabilities for rational immunogen design. Immunity. 2013;38:606. doi: 10.1016/j.immuni.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MA, et al. Quantitative trait loci for CD4:CD8 lymphocyte ratio are associated with risk of type 1 diabetes and HIV-1 immune control. Am J Hum Genet. 2010;86:88. doi: 10.1016/j.ajhg.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Villanueva PO, et al. Control of HIV-1 viremia and protection from AIDS are associated with HLA-Bw4 homozygosity. Proc Natl Acad Sci U S A. 2001;98:5140. doi: 10.1073/pnas.071548198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser C, et al. Virulence and pathogenesis of HIV-1 infection: an evolutionary perspective. Science. 2014;343:1243727. doi: 10.1126/science.1243727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvani AP, Slatkin M. Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta32 HIV-resistance allele. Proc Natl Acad Sci U S A. 2003;100:15276. doi: 10.1073/pnas.2435085100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, et al. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat Med. 2005;11:1290. doi: 10.1038/nm1333. [DOI] [PubMed] [Google Scholar]
- Garcia-Beltran WF, et al. Open conformers of HLA-F are high-affinity ligands of the activating NK-cell receptor KIR3DS1. Nat Immunol. 2016;17:1067. doi: 10.1038/ni.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- H. I. V. C. S. International et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Helm JJ, et al. Characterisation of long-term non-progression of HIV-1 infection after seroconversion: a cohort study. The lancet HIV. 2014;1:e41. doi: 10.1016/S2352-3018(14)70016-5. [DOI] [PubMed] [Google Scholar]
- Herbeck JT, et al. Multistage genomewide association study identifies a locus at 1q41 associated with rate of HIV-1 disease progression to clinical AIDS. The Journal of infectious diseases. 2010;201:618. doi: 10.1086/649842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbeck JT, et al. Is the virulence of HIV changing? A meta-analysis of trends in prognostic markers of HIV disease progression and transmission. AIDS. 2012;26:193. doi: 10.1097/QAD.0b013e32834db418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzemer A, et al. Selection of an HLA-C*03:04-restricted HIV-1 p24 gag sequence variant is associated with viral escape from KIR2DL3+ natural killer cells: data from an observational cohort in South Africa. PLoS Med. 2015;12:e1001900. doi: 10.1371/journal.pmed.1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz A, et al. Class I HLA haplotypes form two schools that educate NK cells in different ways. Science Immunology. 2016;1:eaag1672. doi: 10.1126/sciimmunol.aag1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, et al. HLA-B*35-Px-mediated acceleration of HIV-1 infection by increased inhibitory immunoregulatory impulses. J Exp Med. 2009;206:2959. doi: 10.1084/jem.20091386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel S, Schmidt D, Kremeyer B, Herrmann B, Oppermann M. Detection of the CCR5-Delta32 HIV resistance gene in Bronze Age skeletons. Genes Immun. 2005;6:371. doi: 10.1038/sj.gene.6364172. [DOI] [PubMed] [Google Scholar]
- Hutter G, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- Irvin MR, et al. Genes linked to energy metabolism and immunoregulatory mechanisms are associated with subcutaneous adipose tissue distribution in HIV-infected men. Pharmacogenet Genomics. 2011;21:798. doi: 10.1097/FPC.0b013e32834b68f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EO, et al. Novel genetic locus implicated for HIV-1 acquisition with putative regulatory links to HIV replication and infectivity: a genome-wide association study. PLoS One. 2015;10:e0118149. doi: 10.1371/journal.pone.0118149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DC, et al. HLA class I allelic sequence and conformation regulate leukocyte Ig-like receptor binding. J Immunol. 2011;186:2990. doi: 10.4049/jimmunol.1003078. [DOI] [PubMed] [Google Scholar]
- Joubert BR, et al. A whole genome association study of mother-to-child transmission of HIV in Malawi. Genome medicine. 2010;2:17. doi: 10.1186/gm138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez-Molina CI, et al. Impact of HLA selection pressure on HIV fitness at a population level in Mexico and Barbados. J Virol. 2014;88:10392. doi: 10.1128/JVI.01162-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julg B, et al. Possession of HLA class II DRB1*1303 associates with reduced viral loads in chronic HIV-1 clade C and B infection. The Journal of infectious diseases. 2011;203:803. doi: 10.1093/infdis/jiq122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima Y, et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature. 2009;458:641. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroner BL, et al. Concordance of human leukocyte antigen haplotype-sharing, CD4 decline and AIDS in hemophilic siblings. Multicenter Hemophilia Cohort and Hemophilia Growth and Development Studies. AIDS. 1995;9:275. doi: 10.1097/00002030-199509030-00009. [DOI] [PubMed] [Google Scholar]
- Kulkarni S, et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;472:495. doi: 10.1038/nature09914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S, et al. Genetic interplay between HLA-C and MIR148A in HIV control and Crohn disease. Proc Natl Acad Sci U S A. 2013;110:20705. doi: 10.1073/pnas.1312237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Le Clerc S, et al. Genomewide association study of a rapid progression cohort identifies new susceptibility alleles for AIDS (ANRS Genomewide Association Study 03) The Journal of infectious diseases. 2009;200:1194. doi: 10.1086/605892. [DOI] [PubMed] [Google Scholar]
- Leger PD, et al. Genome-wide association study of peripheral neuropathy with D-drug-containing regimens in AIDS Clinical Trials Group Protocol 384. J Neurovirol. 2014;20:304. doi: 10.1007/s13365-014-0235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann DS, et al. Genome-wide association study of virologic response with efavirenz-containing or abacavir-containing regimens in AIDS clinical trials group protocols. Pharmacogenet Genomics. 2015;25:51. doi: 10.1097/FPC.0000000000000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, et al. Genome-wide association study of neurocognitive impairment and dementia in HIV-infected adults. American Journal of Medical Genetics. Part B. Neuropsychiatric Genetics: the Official Publication of the International Society of Psychiatric Genetics. 2012;159B:669. doi: 10.1002/ajmg.b.32071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limou S, et al. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02) The Journal of Infectious Diseases. 2009;199:419. doi: 10.1086/596067. [DOI] [PubMed] [Google Scholar]
- Lingappa JR, et al. Genomewide association study for determinants of HIV-1 acquisition and viral set point in HIV-1 serodiscordant couples with quantified virus exposure. PLoS One. 2011;6:e28632. doi: 10.1371/journal.pone.0028632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367. doi: 10.1016/S0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- Lucotte G, Mercier G. Distribution of the CCR5 gene 32-bp deletion in Europe. Journal of Acquired Immune Deficiency Syndromes and Human Retrovirology: Official Publication of the International Retrovirology Association. 1998;19:174. doi: 10.1097/00042560-199810010-00011. [DOI] [PubMed] [Google Scholar]
- MacDonald KS, et al. Mother-child class I HLA concordance increases perinatal human immunodeficiency virus type 1 transmission. The Journal of infectious diseases. 1998;177:551. doi: 10.1086/514243. [DOI] [PubMed] [Google Scholar]
- Mackelprang RD, et al. Maternal HLA homozygosity and mother-child HLA concordance increase the risk of vertical transmission of HIV-1. The Journal of Infectious Diseases. 2008;197:1156. doi: 10.1086/529528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MP, Carrington M. Immunogenetics of HIV disease. Immunol Rev. 2013;254:245. doi: 10.1111/imr.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MP, et al. Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science. 1998;282:1907. doi: 10.1126/science.282.5395.1907. [DOI] [PubMed] [Google Scholar]
- Martin MP, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31:429. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- Martin MP, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet. 2007;39:733. doi: 10.1038/ng2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinson JJ, Chapman NH, Rees DC, Liu YT, Clegg JB. Global distribution of the CCR5 gene 32-basepair deletion. Nat Genet. 1997;16:100. doi: 10.1038/ng0597-100. [DOI] [PubMed] [Google Scholar]
- Matzaraki V, Kumar V, Wijmenga C, Zhernakova A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017;18:76. doi: 10.1186/s13059-017-1207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott DH, et al. CCR5 promoter polymorphism and HIV-1 disease progression. Multicenter AIDS Cohort Study (MACS) Lancet. 1998;352:866. doi: 10.1016/S0140-6736(98)04158-0. [DOI] [PubMed] [Google Scholar]
- McLaren PJ, Carrington M. The impact of host genetic variation on infection with HIV-1. Nat Immunol. 2015;16:577. doi: 10.1038/ni.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren PJ, et al. Association study of common genetic variants and HIV-1 acquisition in 6300 infected cases and 7200 controls. PLoS Pathog. 2013;9:e1003515. doi: 10.1371/journal.ppat.1003515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren PJ, et al. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc Natl Acad Sci U S A. 2015;112:14658. doi: 10.1073/pnas.1514867112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino AM, et al. Dimorphic HLA-B signal peptides differentially influence HLA-E- and natural killer cell-mediated cytolysis of HIV-1-infected target cells. Clin Exp Immunol. 2013;174:414. doi: 10.1111/cei.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misrahi M, et al. CCR5 chemokine receptor variant in HIV-1 mother-to-child transmission and disease progression in children. French Pediatric HIV Infection Study Group. JAMA. 1998;279:277. doi: 10.1001/jama.279.4.277. [DOI] [PubMed] [Google Scholar]
- Mlisana K, et al. Rapid disease progression in HIV-1 subtype C-infected South African women. Clinical Infectious Diseases: an Official Publication of the Infectious Diseases Society of America. 2014;59:1322. doi: 10.1093/cid/ciu573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CB, et al. Phenome-wide association study relating pretreatment laboratory parameters with human genetic variants in AIDS Clinical Trials Group protocols. Open forum infectious diseases. 2015;2:ofu113. doi: 10.1093/ofid/ofu113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummidi S, et al. Genealogy of the CCR5 locus and chemokine system gene variants associated with altered rates of HIV-1 disease progression. Nat Med. 1998;4:786. doi: 10.1038/nm0798-786. [DOI] [PubMed] [Google Scholar]
- Nixon DF, et al. HIV-1 gag-specific cytotoxic T lymphocytes defined with recombinant vaccinia virus and synthetic peptides. Nature. 1988;336:484. doi: 10.1038/336484a0. [DOI] [PubMed] [Google Scholar]
- Norman PJ, et al. Defining KIR and HLA class I genotypes at highest resolution via high-throughput sequencing. Am J Hum Genet. 2016;99:375. doi: 10.1016/j.ajhg.2016.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J, Galvani AP, Slatkin M. The geographic spread of the CCR5 Delta32 HIV-resistance allele. PLoS Biol. 2005;3:e339. doi: 10.1371/journal.pbio.0030339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor GM, et al. Peptide-dependent recognition of HLA-B*57:01 by KIR3DS1. J Virol. 2015;89:5213. doi: 10.1128/JVI.03586-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park RJ, et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat Genet. 2017;49:193. doi: 10.1038/ng.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne R, et al. Impact of HLA-driven HIV adaptation on virulence in populations of high HIV seroprevalence. Proc Natl Acad Sci U S A. 2014;111:E5393. doi: 10.1073/pnas.1413339111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelak K, et al. Host determinants of HIV-1 control in African Americans. The Journal of Infectious Diseases. 2010;201:1141. doi: 10.1086/651382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- K. Pelak, Shianna, K., Ge, D., and Goldstein, D, Paper presented at the The American Society of Human Genetics (ASHG), Montreal., 2011
- Petrovski S, et al. Common human genetic variants and HIV-1 susceptibility: a genome-wide survey in a homogeneous African population. AIDS. 2011;25:513. doi: 10.1097/QAD.0b013e328343817b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polycarpou A, et al. Association between maternal and infant class I and II HLA alleles and of their concordance with the risk of perinatal HIV type 1 transmission. AIDS Res Hum Retrovir. 2002;18:741. doi: 10.1089/08892220260139477. [DOI] [PubMed] [Google Scholar]
- Ramsuran V, et al. Duffy-null-associated low neutrophil counts influence HIV-1 susceptibility in high-risk South African black women. Clinical Infectious Diseases: an Official Publication of the Infectious Diseases Society of America. 2011;52:1248. doi: 10.1093/cid/cir119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsuran V, et al. Epigenetic regulation of differential HLA-A allelic expression levels. Hum Mol Genet. 2015;24:4268. doi: 10.1093/hmg/ddv158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsuran V, et al. Sequence and phylogenetic analysis of the untranslated promoter regions for HLA class I genes. J Immunol. 2017;198:2320. doi: 10.4049/jimmunol.1601679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranasinghe S, et al. Association of HLA-DRB1-restricted CD4(+) T cell responses with HIV immune control. Nat Med. 2013;19:930. doi: 10.1038/nm.3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucker J, et al. Regions in beta-chemokine receptors CCR5 and CCR2b that determine HIV-1 cofactor specificity. Cell. 1996;87:437. doi: 10.1016/S0092-8674(00)81364-1. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, et al. The case for selection at CCR5-Delta32. PLoS Biol. 2005;3:e378. doi: 10.1371/journal.pbio.0030378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson M, et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- Sereti I, Altfeld M. Immune activation and HIV: an enduring relationship. Curr Opin HIV AIDS. 2016;11:129. doi: 10.1097/COH.0000000000000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha S, et al. A genome-wide association study of carotid atherosclerosis in HIV-infected men. AIDS. 2010;24:583. doi: 10.1097/QAD.0b013e3283353c9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MW, et al. Contrasting genetic influence of CCR2 and CCR5 variants on HIV-1 infection and disease progression. Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC), ALIVE Study. Science. 1997;277:959. doi: 10.1126/science.277.5328.959. [DOI] [PubMed] [Google Scholar]
- Stephens JC, et al. Dating the origin of the CCR5-Delta32 AIDS-resistance allele by the coalescence of haplotypes. Am J Hum Genet. 1998;62:1507. doi: 10.1086/301867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, et al. HLA class I homozygosity accelerates disease progression in human immunodeficiency virus type 1 infection. AIDS Res Hum Retrovir. 1999;15:317. doi: 10.1089/088922299311277. [DOI] [PubMed] [Google Scholar]
- Tebas P, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R, et al. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat Genet. 2009;41:1290. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyer JL, et al. Genome-wide association study implicates PARD3B-based AIDS restriction. The Journal of infectious diseases. 2011;203:1491. doi: 10.1093/infdis/jir046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulveling D, et al. A new 3p25 locus is associated with liver fibrosis progression in human immunodeficiency virus/hepatitis C virus-coinfected patients. Hepatology. 2016;64:1462. doi: 10.1002/hep.28695. [DOI] [PubMed] [Google Scholar]
- Vince N, et al. HLA-C level is regulated by a polymorphic Oct1 binding site in the HLA-C promoter region. Am J Hum Genet. 2016;99:1353. doi: 10.1016/j.ajhg.2016.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, et al. Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet. 2012;380:572. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BD, et al. HIV-specific cytotoxic T lymphocytes in seropositive individuals. Nature. 1987;328:345. doi: 10.1038/328345a0. [DOI] [PubMed] [Google Scholar]
- Wei Z, et al. Genome-wide association studies of HIV-1 host control in ethnically diverse Chinese populations. Scientific reports. 2015;5:10879. doi: 10.1038/srep10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, et al. Comprehensive identification of host modulators of HIV-1 replication using multiple orthologous RNAi reagents. Cell Rep. 2014;9:752. doi: 10.1016/j.celrep.2014.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkernagel RM, Doherty PC. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature. 1974;248:701. doi: 10.1038/248701a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of findings of GWAS studies of HIV outcomes or HIV-related outcomes extracted from the GWAS catalog (https://www.ebi.ac.uk/gwas/). (XLSX 74 kb).