

Abstract
Deterioration of pancreatic beta-cells plays a critical role in the development of type 2 diabetes. Among the various stressors contributing to these deleterious effects, glucotoxicity and superoxides have been proposed as major players. In this context, the mitochondrial uncoupling protein UCP2 is regularly associated with the stress response. In the present study, we tested the effects of UCP2 upregulation in mouse islets with beta-cell specific overexpression of UCP2 (RIP-UCP2). Islets were subjected to both chronic glucotoxicity (7 days at 30 mM glucose) and acute oxidative stress (200 µM H2O2 for 10 min). Increased UCP2 expression did not alter mitochondrial potential and ATP generation but protected against glucotoxic effects. Glucose-stimulated insulin secretion was altered by both glucotoxicity and oxidative stress, in particular through higher basal insulin release at non-stimulatory glucose concentrations. The secretory response to glucose stimulation was partially preserved in beta-cells overexpressing UCP2. The higher rate of cell death induced by chronic high glucose exposure was lower in RIP-UCP2 islets. Finally, superoxide production was reduced by high glucose, both under acute and chronic conditions, and not modified by UCP2 overexpression. In conclusion, upregulation of UCP2 conferred protective effects to the stressed beta-cell through mechanisms not directly associated with superoxide production.
Keywords: Glucotoxicity, Oxidative stress, UCP2, Insulin secretion, Beta-cell, Pancreatic islet
Graphical abstract
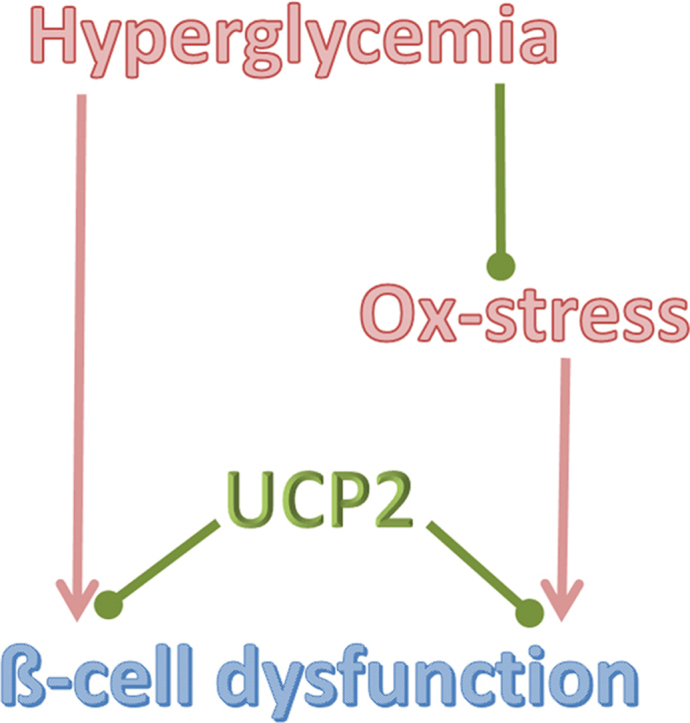
Highlights
-
•
UCP2 upregulation protects pancreatic ß-cells against glucotoxicity.
-
•
High glucose reduces superoxide production in pancreatic islets.
-
•
UCP2 upregulation does not change superoxide production.
-
•
UCP2 upregulation protects ß-cells against oxidative stress.
1. Introduction
Type 2 diabetes, characterized by hyperglycemia, develops as a consequence of significant loss of functional ß-cells secondary to chronic exposure to stressful pathophysiological conditions, such as high glucose [1], [2], [3], [4], [5], [6]. Such conditions, recognized long ago and referred to as glucotoxicity [7], [8], [9], were then proposed to be mediated by oxidative stress that would be induced by the overload of glucose metabolism in the ß-cell [10], [11], [12], [13]. Oxidative stress has been shown to directly disrupt metabolism-secretion coupling in insulin secreting cells [14], [15], [16]. However, the putative contribution of oxidative stress to the etiology of diabetes remains debated [17], as well as the contribution of high glucose to the generation of reactive oxygen species (ROS) [18], [19], [20].
Another controversy is associated with UCP2, a mitochondrial protein sharing 59% homology with the uncoupling protein UCP1 [21]. UCP2 was originally proposed to induce proton leakage dissipating the mitochondrial proton motive force [21]. Some studies reported that overexpression of UCP2 in insulin-secreting cells increases respiration [22] and limits ATP production and glucose-stimulated insulin secretion [23], [24], while others observed no alteration of mitochondrial coupling [25], [26]. Thus, different maneuvers changing UCP2 expression in pancreatic ß-cells reported by various groups did not raise a consensus on the function of UCP2, neither at the cellular level (reviewed in [27]) nor in animal models (reviewed in [28]). An extensive review of the available studies on UCP2 in the ß-cell highlighted the lack of evidence for a significant mitochondrial proton leak contributed by UCP2 [27]. Interestingly, a recent study has shown that UCP2 is in fact a mitochondrial C4-metabolite transporter [29]. Such a function could establish a dissipative proton circuit [27], compatible with some kind of mild uncoupling activity through the reverse transport of protons.
Whatever the exact function of UCP2, it has consistently been reported to be upregulated in endocrine cells as a stress response [15], [30], [32], [33], [34]. Therefore, UCP2 may confer some protection, although it is rather challenging to dissociate effects of induced UCP2 from other concomitant stress responses. Upregulation of UCP2 before exposure to the stressors is one way to address this question. This has been achieved previously using cytokines as stress inducer, showing that upregulation of UCP2 prevents further cytokine-induced ß-cell death through the suppression of ROS production [26].
In the present study, we investigated the putative effects of upregulation of UCP2 in mouse ß-cells subjected to either chronic glucotoxicity or acute oxidative stress. In order to do so, we used pancreatic islets isolated from transgenic mice with ß-cell specific overexpression of UCP2 (RIP-UCP2) exposed for 7 days to high glucose in the culture medium. We also studied the link between glucotoxicity and oxidative stress in general and the glucose contribution to ROS production in particular. The results show that high glucose impairs ß-cell function without promoting superoxide production and that UCP2 upregulation partially protects against both glucotoxicity and oxidative stress.
2. Material and methods
2.1. Animals and in vivo experiments
Generation and analysis of transgenic mice with ß-cell specific overexpression of UCP2 (RIP-UCP2) have been described previously [26]. Mice from in-house breeding (CMU-zootechnie, Geneva, Switzerland) of 2–5 months of age were age-matched with C57BL/6 J (wild-type, WT) control mice. We followed the principles of laboratory animal care and the study was approved by the responsible ethics committee. Glucose tolerance tests were performed upon intraperitoneal injection of D-glucose (2 g/kg) after overnight fasting. Blood samples were taken at time 0 and 15 min and glucose levels were determined using Accu-Check Aviva glucometer (Roche Diagnostics). Insulin, glucagon, and leptin were measured using Luminex xMAP™ technology and commercially available kits (Bio-Plex Pro Diabetes Assays, Biorad and Milliplex Mouse Metabolic Magnetic Bead Panel, Millipore).
2.2. Isolation and culture of pancreatic islets
Pancreatic islets were isolated from WT and RIP-UCP2 mice matched for sex and age by collagenase digestion followed by histopaque-10771 (Sigma-Aldrich, St.-Louis, MO) gradient centrifugation as described before [35]. Isolated islets were cultured overnight free-floating in RPMI-1640 (Life Technologies, Grand Island, NY) supplemented with 10 mM HEPES, 10% (v/v) heat-inactivated fetal calf serum, 100 U/mL penicillin, 100 µg/mL streptomycin; then hand-picked for experiments [35].
2.3. Glucotoxicity induction
Glucotoxicity was induced by culturing isolated islets at 30 mM glucose (glucotoxicity), compared with standard 11 mM (control), for 7 days at 37 °C in the presence of 5% CO2 in serum-free RPMI-1640 medium containing 5 g/L bovine serum albumin (BSA, Sigma-Aldrich). The 11 mM glucose condition served as control since, unlike human islets, rodent islets are commonly cultured at this concentration and because it corresponds to non-fasting euglycemia (200 mg/dL = 11 mM) for most mouse strains [36]. Medium was refreshed every 2 days to restore consumed glucose.
2.4. Oxidative stress induction
Oxidative stress (ox-stress) was induced by transiently challenging WT and RIP-UCP2 islets with H2O2 as described previously [14], [15]. Briefly, cultured islets were maintained for 45 min in 2.8 mM glucose before exposure to a single acute oxidative stress (200 μM H2O2, Sigma-Aldrich), which was neutralized after 10 min by adding catalase (100units/mL, Sigma-Aldrich) to the medium. Islets were then washed in the same medium and either immediately collected after the ox-stress for immunoblotting and secretion assay or further cultured in standard RPMI-1640 medium (11 mM glucose) for a 3-day recovery period before experiments.
2.5. Immunoblotting
Protein extracts from mouse islets treated as described were subjected to electrophoresis on a 12% polyacrylamide gel, electro-transferred onto nitrocellulose membrane, and blocked with 3% BSA in PBS (1.4 mM KH2PO4, 8 mM Na2HPO4, 140 mM NaCl and 2.7 mM KCl at pH 7.3). Membranes were then incubated overnight at 4 °C with different antibodies: goat anti-human polyclonal antibody to UCP2 (1:1000, #6527, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and mouse monoclonal anti-actin (1:5000, #4700, Sigma-Aldrich) in PBS containing 3% BSA and 0.05% Tween-20. After washing 3 times with PBS supplemented with 0.05% Tween-20, membranes were incubated with a horseradish peroxidase (HRP)-conjugated donkey anti-goat (1:10000, #2056, Santa Cruz) or anti-mouse antibody (1:5000, NA931, Amersham Biosciences, UK) for 1hr at room temperature. After washes, the immunoreactivity was visualized by SuperSignal West Pico Chemiluminescent Substrate system (Pierce Biotechnology, Inc., Rockford, IL) and Molecular Imager ChemiDoc XRS system (Bio-Rad, Hercules, CA) controlled by Quantity One 1-D (Bio-Rad) analysis software.
2.6. Mitochondrial membrane potential
Following the culture period, islets from the different groups were maintained for 30 min at 2.8 mM glucose in KRBH buffer (KRBH, 135 mM NaCl, 3.6 mM KCl, 5 mM NaHCO3, 0.5 mM NaH2PO4, 0.5 mM MgCl2, 1.5 mM CaCl2, and 10 mM HEPES at pH 7.4) containing 0.1% BSA (KRBH/BSA), washed, and then pre-incubated for 30 min in KRBH/BSA containing 2.8 mM glucose and 10 µg/mL rhodamine-123 (Life Technologies). The mitochondrial membrane potential was monitored at 37 °C in ImageXpress Micro Widefield High Content Screening system (Molecular Devices, Sunnyvale, CA) with excitation and emission filters set as 490 and 530 nm, respectively. During experiments, glucose was raised from low 2.8 mM to stimulatory 22.8 mM to induce hyperpolarization of the mitochondrial membrane. Complete mitochondrial membrane potential was revealed by the addition of 1 µM of the protonophore carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP, Sigma-Aldrich). Fluorescence intensity of single islet was recorded and analyzed with MetaXpress High Content Image Acquisition and Analysis Software 2.0 (Molecular Devices). Data were obtained from at least 3 independent islet preparations; normalized to signals obtained at low glucose before stimulation.
2.7. Cellular ATP generation
Cellular ATP levels were measured in pancreatic islets transduced with ATeam adenovirus expressing the fluorescence resonance energy transfer (FRET)-based ATP indicator [37]. Practically, on the 5th day of glucotoxicity induction, islets were transduced with ATeam adenovirus for 90 min in the presence of either 11 mM or 30 mM glucose and then maintained in corresponding culture condition for 36hr. Acquisition of ATeam fluorescence of islets was conducted within the next 12hr. Islets were kept at 2.8 mM glucose and then stimulated with 22.8 mM glucose, followed by addition of 2 mM azide. Data obtained from islets of at least 3 mice were analyzed as FRET signal (as assessed by the emission ratio YFP/CFP) and normalized to the azide response reflecting the mitochondrial contribution to ATP generation.
2.8. Cellular calcium levels
Cytosolic [Ca2+] changes were monitored as ratiometric measurements of Fura-2 fluorescence. Isolated mouse islets were cultured on glass coverslips treated with poly-L-lysine (Sigma-Aldrich) and placed in a thermostatic chamber (Harvard Apparatus, Holliston, MA) before incubation with 2 µM Fura-2/acetoxymethyl ester (AM) for 60 min. After washing, Fura-2 fluorescence of a single islet was imaged with alternate 340/380 nm excitation and 510 nm emission using an Axiovert S100 TV through a 40 × 1.3 NA oil immersion objective (Carl Zeiss GmbH, Jena, Germany) as described [38]. After the 22.8 mM glucose stimulation, 1 μM thapsigargin was added to evaluate the contribution of endoplasmic reticulum to cytosolic [Ca2+] changes. Data obtained from islets of at least 3 mice were presented both as 340/380 ratio and normalized to cytosolic [Ca2+] at low glucose.
2.9. Insulin secretion
Both WT and RIP-UCP2 mouse islets were pre-incubated in KRBH/BSA at 2.8 mM glucose for 1hr. Then, batches of 10 islets per group were hand-picked and treated with either low 2.8 mM or stimulatory 22.8 mM glucose for an incubation period of 1hr. At the end of this period, supernatants were collected for the measurement of secreted insulin and islets pellets were resuspended in cold acid-ethanol to determine insulin content [38]. Insulin concentrations were determined by radioimmunoassay (Linco Research Inc., St. Charles, MO).
2.10. Cell death
Following the 7-day culture period, groups of 20 islets in different culture conditions were hand-picked and washed once with KRBH. Quantification of cytoplasmic nucleosomes, as readout for apoptosis, was performed with the Cell Death Detection ELISA kit (Roche, Basel, Switzerland). Data were expressed as absorbance at 405 nm subtracted by the background absorbance at 490 nm [39].
2.11. Measurement of superoxide production
Superoxide production was detected by dihydroethidine (DHE, Sigma-Aldrich) fluorescence assay as described [25], [40]. DHE (also known as hydroethidine) reacts with superoxide radical anion to form a fluorescent product [40], widely used for measurements of intracellular oxidative species [41], [42]. We measured superoxide levels both directly at the end of the 7-day glucotoxicity culture period and in response to acute (1hr) glucose stimulation. For the former, at the end of the 7-day glucotoxicity period, groups of 20 islets were hand-picked and incubated with 25 μM DHE for 1hr. For the latter, groups of 20 islets were pre-incubated for 1hr at 2.8 mM glucose in KRBH/BSA, followed by 1hr incubation with 25 μM DHE in the presence of 2.8 mM or 22.8 mM glucose. Fluorescence was measured in a thermostated plate reader (Fluostar Optima; BMG Labtechnologies, Offenburg, Germany) for 60 min with excitation at 544 nm and emission at 590 nm [25].
2.12. Statistics
Results are shown as mean±SE from at least 3 independent experiments. ANOVA and two-tailed paired or unpaired t-tests as well as the Mann–Whitney test were used for statistical analysis. A value of p < 0.05 was considered as statistically significant.
3. Results
3.1. Prolonged exposure to high glucose does not modify UCP2 protein levels
In human islets, we previously reported that mRNA levels of UCP2 are elevated by high glucose culture conditions, although this effect is not translated at the protein level [31]. In mouse islets, exposure to 30 mM glucose for 7 days did not change UCP2 protein levels compared to islets cultured at standard 11 mM glucose that corresponds to non-fasting euglycemia in mice [36], see Fig. 1A. As expected, islets isolated from RIP-UCP2 mice exhibited increased UCP2 expression, maintained after the 7-day culture at either 11 mM or 30 mM glucose (Fig. 1A). Accordingly, potential changes observed in RIP-UCP2 islets under glucotoxic conditions in this study should be associated primarily with the transgenic overexpression of UCP2 rather than glucose-induced alteration of UCP2 protein levels. At the end of the 7-day culture period, both WT and RIP-UCP2 islets exposed to 30 mM glucose partially lost integrity, showing some cell debris in the petri dishes (Fig. 1B). Of note, overexpression of UCP2 in ß-cells does neither change their insulin content nor the metabolic homeostasis of these transgenic mice when maintained in standard conditions, as reported previously [26]. In particular, plasma parameters (glucose, insulin, glucagon, and leptin) of RIP-UCP2 mice following a glucose challenge were similar to WT mice used in the present study (Table 1).
Fig. 1.

UCP2 over-expression in ß-cells of islets exposed to glucotoxic conditions. WT and RIP-UCP2 islets were collected for analysis 7 days after culture at either 11 mM glucose (11 mM) or 30 mM glucotoxic condition (30 mM). (A) UCP2 protein levels, normalized to ß-actin, were assessed by immunoblotting. Bar-graph shows means ± SE of 3 independent experiments, p < 0.05 versus WT. (B) binocular observation of islets at the end of the culture period.
Table 1.
Plasma parameters following a glucose challenge and weight of RIP-UCP2 mice.
| Glucose | Insulin | Glucagon | Leptin | Body mass | |
|---|---|---|---|---|---|
| (mM) | (pg/mL) | (pg/mL) | (pg/mL) | (g) | |
| WT (t0) | 5.2 ± 0.8 | 531 ± 120 | 41.5 ± 8.4 | 156 ± 64 | 27.7 ± 2.0 |
| WT (t15) | 21.9 ± 7.2 | 1037 ± 123 | 41.7 ± 8.6 | 166 ± 70 | |
| RIP-UCP2 (t0) | 5.5 ± 0.8 | 662 ± 111 | 55.7 ± 11.1 | 112 ± 20 | 29.9 ± 1.5 |
| RIP-UCP2 (15) | 24.5 ± 3.7 | 865 ± 93 | 51.8 ± 9.8 | 116 ± 24 |
The intraperitoneal glucose challenge was performed on 4-month old mice using 2 g of glucose per kg of body weight. Blood samples were collected from tail vein at time 0 (t0) and 15 min after injection (t15). Values are means ± SE, n = 5.
3.2. Overexpression of UCP2 partially preserved mitochondrial function altered by glucotoxicity
Originally, UCP2 was described as a mitochondrial uncoupler, resulting in controversies because of the lack of reliable demonstration of such an effect [27], while recent data ascribed to UCP2 the role of a C4-metabolite transporter [29]. In this context, we measured the mitochondrial membrane potential of RIP-UCP2 islets under glucotoxic conditions. Under standard culture conditions, WT and RIP-UCP2 islets responded similarly to glucose stimulation, substantiating the lack of detectable uncoupling properties of UCP2 (Fig. 2A) and consistent with the reported preservation of oxygen consumption rate in RIP-UCP2 islets [26]. Exposure of islets for 7 days to 30 mM glucose resulted in higher resting potential of the mitochondrial membrane at low 2.8 mM glucose, both in WT and RIP-UCP2 islets. This was revealed by the FCCP-mediated collapse of the mitochondrial potential, displaying a higher amplitude in glucotoxic conditions compared with standard culture (Fig. 2B). Such a resting hyperpolarization of the mitochondrial membrane might be contributed by an elevated metabolic flux secondary to glycogen mobilization. Indeed, it has been shown that culture of insulin-secreting cells at high glucose promotes glycogen stores and that subsequent incubation at low glucose induces glycogenolysis [43]. Regarding the acute glucose response, mitochondria from WT islets of the glucotoxic conditions were further hyperpolarized upon glucose stimulation, although the response was markedly reduced compared with WT islets from the standard culture (Fig. 2C). In RIP-UCP2 islets, the response to glucose-induced hyperpolarization was conserved, showing preservation of the acute effects of glucose stimulation in UCP2 overexpressing islets from the glucotoxic condition (Fig. 2D).
Fig. 2.

Effects of UCP2 over-expression on mitochondrial membrane potential in islets exposed to glucotoxic conditions. At the end of the 7-day culture period at either standard 11 mM glucose (11) or glucotoxic condition (30), WT and RIP-UCP2 islets were assayed for mitochondrial membrane potential. (A) WT and RIP-UCP2 islets from the standard 11 culture conditions were monitored first at low 2.8 mM Glc followed by acute stimulation with 22.8 mM Glc (Glucose). (B) WT and RIP-UCP2 islets from either standard 11 or glucotoxic 30 culture condition were monitored at low 2.8 mM Glc before complete depolarization of the mitochondrial membrane induced by the addition of 1 µM FCCP. (C, D) WT (C) and RIP-UCP2 (D) islets from either standard 11 or glucotoxic 30 culture condition were monitored first at low 2.8 mM Glc followed by acute stimulation with 22.8 mM Glc (Glucose). Values are means from 3 independent experiments (n = 11–14 islets); *p < 0.05 wt 30 versus WT 11, #p < 0.05 RIP-UCP2 30 versus RIP-UCP2 11.
Hyperpolarization of the mitochondrial membrane drives ATP generation, which was then measured in live cells by FRET. Following standard culture conditions, RIP-UCP2 islets had similar basal mitochondrion-dependent ATP levels compared with WT islets, although acute glucose-induced ATP generation was slightly enhanced in islets overexpressing UCP2 versus WT islets (Fig. 3A-B). The 7-day glucotoxic treatment significantly increased basal cellular ATP contributed by the mitochondria in both WT and RIP-UCP2 islets, as revealed by the levels of the azide-sensitive FRET signal when compared with standard cultures (Fig. 3A, see solid versus dashed lines respectively). Following the culture period in glucotoxic conditions, the magnitude of ATP generation upon acute glucose stimulation was not altered in WT islets versus standard culture, while the acute response of RIP-UCP2 islets was returned to WT fold changes (Fig. 3B). Therefore, neither glucotoxic culture nor UCP2 overexpression inhibited ATP generation in mouse islets.
Fig. 3.

Effects of UCP2 over-expression on cellular ATP levels in islets exposed to glucotoxic conditions. At the end of the 7-day culture period at either standard glucose (11 mM) or glucotoxic condition (30 mM), WT and RIP-UCP2 islets transduced with Ad-ATEAM were pre-incubated for 1 h at 2.8 mM glucose before ATP monitoring by FRET, first at low glucose (2.8 mM) before acute stimulation with 22.8 mM (Glucose). Overall mitochondrial contribution to ATP levels was determined by further addition of 2 mM of the mitochondrial poison azide. (A) Signals were acquired every 15 s and kinetic data were normalized to the azide response for WT (orange lines) and RIP-UCP2 (blue lines) islets cultured in standard (11, dashed lines) or glucotoxic (30, solid lines) conditions. (B) Corresponding quantifications of glucose-stimulated ATP rise shown as fold changes over basal levels. (A) values are means ± SE; *p < 0.05 wt 30 versus WT 11 (orange*) and RIP-UCP2 30 versus RIP-UCP2 11 (blue*), #p < 0.05 RIP-UCP2 30 versus WT 30; (B) *p < 0.05 RIP-UCP2 30 versus RIP-UCP2 11; #p < 0.05 RIP-UCP2 11 versus WT 11. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3. UCP2 overexpression did not restore impaired [Ca2+] responses induced by glucotoxicity
Following standard culture conditions, WT and RIP-UCP2 islets exhibited similar robust elevations of cytosolic [Ca2+] in response to an acute stimulation with 22.8 mM glucose, further elevated by the addition of thapsigargin mobilizing Ca2+ from the endoplasmic reticulum (Fig. 4A and B). After 7 days of culture in glucotoxic conditions, the cytosolic [Ca2+] response to acute glucose stimulation was reduced by 48.0% in WT islets and by 43.2% in islets overexpressing UCP2 (Fig. 4B). Despite these blunted [Ca2+] elevations, thapsigargin was not able to further increase cytosolic [Ca2+] (Fig. 4A). These data indicate that UCP2 is not playing a major role in cellular [Ca2+] homeostasis.
Fig. 4.

Effects of UCP2 over-expression on cellular [Ca2+] changes in islets exposed to glucotoxic conditions. At the end of the 7-day culture period at either standard glucose (11 mM) or glucotoxic condition (30 mM), cellular [Ca2+] changes were monitored in WT and RIP-UCP2 as ratiometric measurements of Fura-2 fluorescence. Islets were first kept at low 2.8 mM glucose before raising glucose to 22.8 mM (Glucose), as indicated. Finally, 1 µM thapsigargin was added to release Ca2+ from the endoplasmic reticulum. Signals were acquired every 10 s. Data were calculated as 340/380 ratio from an average of islets from 3 mice in each condition (A) and then normalized to low glucose levels (B). Values are means ± SE; *p < 0.05 wt 30 versus WT 11 (orange*) and RIP-UCP2 30 versus RIP-UCP2 11 (blue*), #p < 0.05 RIP-UCP2 11 versus WT 11. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.4. Overexpression of UCP2 partially preserved the secretory response and cell integrity of islets under glucotoxicity
After 7 days of culture in standard culture conditions, WT islets responded to 22.8 mM glucose stimulation with an 18.9-fold (p < 0.05) increase in insulin secretion versus insulin release at 2.8 mM low glucose (Fig. 5A). Overexpression of UCP2 in RIP-UCP2 islets from standard culture did not affect insulin release at low glucose, although the secretory response to stimulatory 22.8 mM glucose (15.3-fold, p < 0.05) was slightly reduced (−37%, p < 0.05) compared with the one observed in WT islets.
Fig. 5.

Effects of UCP2 over-expression on insulin secretion and cell death in islets exposed to glucotoxic conditions. (A) Glucose stimulated insulin secretion from WT and RIP-UCP2 islets was tested at the end of the 7-day culture period at either standard glucose (11 mM Glc) or glucotoxic condition (30 mM Glc). Both WT and RIP-UCP2 islets were pre-incubated at 2.8 mM glucose for 1hr and then insulin release was measured from islets kept at 2.8 mM glucose (Low) or stimulated by 22.8 mM glucose (Stim) for 1hr. *p < 0.05 versus corresponding Low of the same genotype and the same culture condition, #p < 0.05 versus corresponding WT in the same culture condition, &p < 0.05 versus corresponding secretion assay condition from the 11 mM Glc culture islets. (B) Islet cell apoptosis was determined in cell lysates by quantification of cytoplasmic nucleosomes in WT and RIP-UCP2 islets collected at the end of the 7-day culture period. **p < 0.01, *p < 0.05 versus corresponding genotype of 11 mM Glc; #p < 0.05 versus WT of 30 mM Glc.
Following 7 days of glucotoxic culture conditions (30 mM glucose), WT islets exhibited a dramatic 12.4-fold increase of insulin release at non-stimulatory glucose concentration (2.8 mM) versus WT islets from standard culture (Fig. 5A). As a consequence, the secretory response to 22.8 mM glucose stimulation was reduced to 6.5-fold in WT islets under glucotoxic conditions, although in absolute values insulin secretion was enhanced versus standard culture. RIP-UCP2 islets from the glucotoxicity culture had lower insulin release at low glucose compared with WT islets from the same high glucose culture condition (−56%, p < 0.05). Following culture under glucotoxic conditions, glucose-stimulated insulin secretion from RIP-UCP2 islets was 8.9-fold (p < 0.05) higher than the counterpart at low glucose from the same islet group. Therefore, overexpression of UCP2 partially prevented the elevation of non-stimulated insulin output induced by glucotoxic conditions.
Islet cell integrity was then assessed by the quantification of cytoplasmic nucleosomes as a readout for apoptosis. In WT islets, a 7-day high glucose exposure resulted in 4.8-fold increase in cell death versus WT islets from standard culture (p < 0.001, Fig. 5B). RIP-UCP2 islets exhibited similar apoptotic rates in standard culture compared with WT islets cultured the same way. Under glucotoxic conditions, there was a 3.1-fold increase of cell death in RIP-UCP2 islets, i.e. 27% less than in WT islets (p < 0.05, Fig. 5B). This shows that UCP2 overexpression conferred partial protection of islet cells exposed to chronic high glucose.
3.5. Effects of UCP2 overexpression and of acute oxidative stress on UCP2 levels and insulin secretion
Oxidative stress has regularly been postulated to be induced by glucotoxic conditions [4] and UCP2 to be in turn upregulated in response to oxidative stress [15]. In this context, we tested the secretory response of ß-cells overexpressing UCP2 following an acute oxidative stress without the exposure to glucotoxicity and putative confounding factors related to the associated altered metabolic activation. Regarding expression of UCP2, protein levels were not changed by transient oxidative stress (200 µM H2O2 for 10 min), neither right after the acute stress nor 3 days post stress corresponding to mid-term glucotoxic cultures (Fig. 6A-B). Similarly, the levels of UCP2 overexpression in RIP-UCP2 islets were not modified by the same oxidative stress and following the same recovery periods (Fig. 6A-B).
Fig. 6.

Effects of UCP2 over-expression and of oxidative stress on UCP2 levels and insulin secretion. Isolated WT and RIP-UCP2 islets were subjected to transient oxidative stress (200 µM H2O2 for 10 min). Then, islets were either collected immediately (Acute ox-stress, A, C) or after a 3-day recovery period (Post ox-stress 3 days, B, C). (A, B) UCP2 protein levels, normalized to ß-actin, were assessed by immunoblotting. Bar-graphs show averages of 3 individual experiments. (C) Insulin secretion over a 1 h incubation at either 2.8 mM glucose (Low) or stimulatory 22.8 mM glucose (Stim) was determined both directly after the 10 min transient ox-stress (Acute stress) and 3 days after H2O2 exposure (3 days after stress), and compared with islets not exposed to H2O2 (No stress). *p < 0.05 versus corresponding Low of the same genotype in the same condition; ##p < 0.01 versus corresponding condition in No stress; &, p < 0.05 versus corresponding WT of the same stress.
Glucose-stimulated insulin secretion evoked by 22.8 mM glucose was totally blunted immediately after transient exposure to oxidative stress, both in WT and RIP-UCP2 islets (Fig. 6C). Following a 3-day culture period post acute oxidative stress, WT islets partially recovered their secretory response to glucose stimulation (2.04-fold, p < 0.05). At the same time, RIP-UCP2 islets exhibited much higher glucose-stimulated insulin secretion (5.08-fold, p < 0.05), showing beneficial effects of UCP2 overexpression.
3.6. Glucose stimulation reduced superoxide production that was not changed by UCP2 overexpression
UCP2 has been proposed as a stress protein, potentially playing a role as a negative feedback limiting the production of ROS by mitochondria [44]. In the present study, the increased cell death observed after 7 days of glucotoxic conditions (Fig. 5B) was not associated with higher ROS levels measured as intracellular superoxide-sensitive hydroethidine fluorescence [40]. Indeed, superoxide production was even lower in WT islets cultured at 30 mM glucose versus standard conditions (−44%, p < 0.05, Fig. 7A). UCP2 overexpression did not change ROS levels in RIP-UCP2 islets cultured either at 11 mM or 30 mM glucose compared with WT islets.
Fig. 7.

Effects of UCP2 over-expression on superoxide generation in islets exposed to glucotoxic conditions. At the end of the 7-day culture period at either standard glucose (11 mM Glc) or glucotoxic condition (30 mM Glc), superoxide generation was measured in WT and RIP-UCP2 islets either immediately at the end of the 7-day culture (A) or following further glucose challenge (B). (A) superoxide levels from control (11 mM Glc) and glucotoxic (30 mM Glc) conditions in both WT and RIP-UCP2 islets. *p < 0.05 versus WT from the 11 mM Glc culture condition. (B) superoxide levels were measured in WT and RIP-UCP2 islets following further acute incubation at 2.8 mM glucose (Low) and 22.8 mM glucose (Stim) after 1hr pre-incubation at 2.8 mM glucose. **p < 0.01, *p < 0.05 versus corresponding Low of the same genotype.
After the 7-day culture period, islets were challenged with stimulatory glucose (22.8 mM) for measurements of acute (1 h) ROS generation. In all groups, we observed a marked reduction of superoxide production upon glucose stimulation versus low 2.8 mM glucose (Fig. 7B). Neither the culture conditions (11 mM versus 30 mM glucose) nor did UCP2 levels (WT versus RIP-UCP2 islets) modulate the production of superoxides. Therefore, the protective effects of UCP2 in ß-cells were not mediated by a reduction of ROS.
4. Discussion
Expression of UCP2 in insulin-secreting cells has been reported to be induced by stress conditions, in particular upon oxidative stress [15], [32] and glucotoxicity [31], [33]. However, these observations were based on measurements of mRNA levels. When both protein and mRNA of UCP2 were quantified in human islets treated with high glucose, only UCP2 transcript was shown to be upregulated as opposed to its translation product [31]. Accordingly, in the present study mouse islets exposed for 7 days to 30 mM glucose did not change protein levels of UCP2. The observed discrepancy between mRNA and protein levels of UCP2 indicates a tight control of UCP2 expression at the translational level, as reported previously in different cell types, including insulin-secreting cells [45], [46]. Here, we imposed higher UCP2 protein levels in ß-cells of RIP-UCP2 islets, while stress conditions did not change these levels, neither in WT nor in RIP-UCP2 islets.
Exposure of mouse islets to high 30 mM glucose over a period of 7 days induced glucotoxic effects, as revealed by the increased rate of apoptosis and dysfunction of the subsisting ß-cells. In particular, non-stimulated insulin release was markedly increased by glucotoxic conditions, resulting in lower secretory response in terms of fold increase upon glucose stimulation, as reported previously using similar experimental setup [33]. Overexpression of UCP2 in ß-cells partially prevented such an elevation of insulin release at low glucose induced by glucotoxic conditions, preserving to some extent metabolism-secretion coupling.
The opposite maneuver consisting in the abrogation of UCP2 expression results in similar protective effects against glucotoxicity [33]. This apparent paradox could be explained by differences in the models, i.e. global UCP2 knockout used previously [33] versus ß-cell specific overexpression in the present study. Indeed, UCP2 has been shown to be highly expressed in another endocrine cell type composing the pancreatic islet, namely the α-cell, and suppression of UCP2 in α-cells lowers glucagon release at low glucose [47]. Consistent with reciprocal regulation of insulin and glucagon secretion [48], alteration of the response of α-cells lacking UCP2 modifies insulin release in stressed islets [49]. Different studies have reported that global ablation of UCP2 partially preserves the ß-cell function and reduces the α-cell response, effects associated with the attenuation of the diabetic state in mice fed a high-fat diet [50], in mice with streptozotocin-induced diabetes [51], or in ob/ob mice [52]; although some of the reported effects might be contributed by the genetic background [53]. Another hypothesis for the observed preservation of ß-cell function under glucotoxic conditions when UCP2 is either suppressed [33] or overexpressed (present study) might be a bell-shape effect in terms of expression levels. This effect can be triggered by hormetic adaptive response to chronic stresses [54]. In this regard, UCP2 would be implicated in the mitohormetic response of the ß-cell [25].
Glucotoxicity has been postulated to induce an oxidative stress that would in turn injure the ß-cell [10], [11], [12], [13]. This represents an attractive concept since ß-cells are sensitive to ROS attacks [14], [15], [16]. However, other studies using either insulinoma cells or primary ß-cells have shown that glucose stimulation reduces superoxide production; an effect observed after both acute (1 h) [19], [20] and semi-chronic (2–3 days) [19], [31] glucose exposures. In the present study, we observed lower superoxide levels upon acute glucose stimulation. Similarly, following 7 days of high glucose exposure, mouse islets generated less ROS than islets cultured in standard medium. This might be the result of an adaptive stress response with upregulation of antioxidant enzymes, such as superoxide dismutase as reported previously [15]. Accordingly, one cannot rule out increased superoxide anions upon chronic glucotoxic conditions, blunted by enhanced activity of adaptive antioxidant defenses. By itself, overexpression of UCP2 in ß-cells did not change superoxide generation, both at low and stimulatory glucose concentrations, as well as following the glucotoxic treatment. The absence of UCP2 in ß-cells has previously been associated with higher ROS levels [49]. Therefore, we cannot exclude an effect of UCP2 on the production of superoxide anions relying on a threshold level of UCP2 limiting, although not abrogating, ROS production. This highlights the need for accurate assessment of UCP2 expression when studying its relationship with stressors.
Partial hepatic mitochondrial uncoupling has recently been shown to reverse metabolic syndrome in rats with type 2 diabetes [55]. In the present study, despite possible mild uncoupling activity [27], [45], overexpression of UCP2 in RIP-UCP2 islets did not significantly alter the hyperpolarization of the mitochondria and its coupled ATP generation, in agreement with previous observations made in insulinoma cells [25], [26]. However, the hyperpolarization of the mitochondrial membrane that was altered by glucotoxic conditions was partially preserved by the overexpression of UCP2. We previously reported that in response to glucotoxic stress, human islets increase mRNA levels of UCP2 and of two others mitochondrial metabolite transporters, i.e. the dicarboxylate carrier DIC and the aspartate-glutamate carrier AGC [31]. In keeping with the newly uncovered properties of UCP2 [29], it is striking to note that this set of carriers are all C4-transporters (malate and oxaloacetate for UCP2, malate for DIC, and aspartate for AGC) [56]. This might be indicative of the protective effects of UCP2 overexpression on glucose-stimulated mitochondrial activation, optimizing metabolite flux in chronically overloaded ß-cells by nutrients. Specifically, upregulation of UCP2 would promote transport of C4 metabolites out of mitochondria, in particular the TCA cycle intermediates malate and oxaloacetate [29], thereby reducing chronic glucose oxidation upon glucotoxic conditions. Similar mechanism has been recently uncovered in pancreatic ß-cells through the identification of glycerol-3-phosphate phosphatase, mediating protective leak out of glycolytic intermediates under glucotoxic conditions [57].
In conclusion, increasing expression of UCP2 in ß-cells protected against glucotoxic conditions. Cell death was reduced and the secretory response to glucose stimulation was partially preserved. UCP2 overexpression had no effects on superoxide generation that was in fact reduced by high glucose. Therefore, UCP2 confers protective effects to the stressed ß-cell through mechanisms not directly associated with ROS production. Future work should delineate the complex interplay between glucose metabolism and superoxide levels.
Conflict of interest
There is no conflict of interest in this study.
Acknowledgement
Authors thank Françoise Assimacopoulos-Jeannet (Geneva) for providing RIP-UCP2 mice and Thierry Brun (Geneva) for stimulating discussions. This work was supported by the State of Geneva and the Swiss National Science Foundation (31003A_146984 to P.M.).
References
- 1.Butler A.E., Janson J., Bonner-Weir S., Ritzel R., Rizza R.A., Butler P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 2.Robertson R.P., Harmon J., Tran P.O., Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(Suppl 1):S119–S124. doi: 10.2337/diabetes.53.2007.s119. [DOI] [PubMed] [Google Scholar]
- 3.Poitout V., Amyot J., Semache M., Zarrouki B., Hagman D., Fontes G. Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys. Acta. 2010;1801:289–298. doi: 10.1016/j.bbalip.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bensellam M., Laybutt D.R., Jonas J.C. The molecular mechanisms of pancreatic beta-cell glucotoxicity: recent findings and future research directions. Mol. Cell Endocrinol. 2012;364:1–27. doi: 10.1016/j.mce.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Ashcroft F.M., Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell. 2012;148:1160–1171. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee M.S. Role of islet beta cell autophagy in the pathogenesis of diabetes. Trends Endocrinol. Metab. 2014;25:620–627. doi: 10.1016/j.tem.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 7.DeFronzo R.A. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37:667–687. doi: 10.2337/diab.37.6.667. [DOI] [PubMed] [Google Scholar]
- 8.Leahy J.L. Natural history of beta-cell dysfunction in NIDDM. Diabetes Care. 1990;13:992–1010. doi: 10.2337/diacare.13.9.992. [DOI] [PubMed] [Google Scholar]
- 9.Rossetti L., Giaccari A., DeFronzo R.A. Glucose toxicity. Diabetes Care. 1990;13:610–630. doi: 10.2337/diacare.13.6.610. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka Y., Tran P.O., Harmon J., Robertson R.P. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc. Natl. Acad. Sci. USA. 2002;99:12363–12368. doi: 10.1073/pnas.192445199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robertson R.P., Harmon J., Tran P.O., Tanaka Y., Takahashi H. Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes. 2003;52:581–587. doi: 10.2337/diabetes.52.3.581. [DOI] [PubMed] [Google Scholar]
- 12.Tang C., Han P., Oprescu A.I., Lee S.C., Gyulkhandanyan A.V., Chan G.N., Wheeler M.B., Giacca A. Evidence for a role of superoxide generation in glucose-induced beta-cell dysfunction in vivo. Diabetes. 2007;56:2722–2731. doi: 10.2337/db07-0279. [DOI] [PubMed] [Google Scholar]
- 13.Wu L., Nicholson W., Knobel S.M., Steffner R.J., May J.M., Piston D.W., Powers A.C. Oxidative stress is a mediator of glucose toxicity in insulin-secreting pancreatic islet cell lines. J. Biol. Chem. 2004;279:12126–12134. doi: 10.1074/jbc.M307097200. [DOI] [PubMed] [Google Scholar]
- 14.Maechler P., Jornot L., Wollheim C.B. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J. Biol. Chem. 1999;274:27905–27913. doi: 10.1074/jbc.274.39.27905. [DOI] [PubMed] [Google Scholar]
- 15.Li N., Brun T., Cnop M., Cunha D.A., Eizirik D.L., Maechler P. Transient oxidative stress damages mitochondrial machinery inducing persistent beta-cell dysfunction. J. Biol. Chem. 2009;284:23602–23612. doi: 10.1074/jbc.M109.024323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brun T., Scarcia P., Li N., Gaudet P., Duhamel D., Palmieri F., Maechler P. Changes in mitochondrial carriers exhibit stress-specific signatures in INS-1Ebeta-cells exposed to glucose versus fatty acids. PLoS One. 2013;8:e82364. doi: 10.1371/journal.pone.0082364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolb H., Eizirik D.L. Resistance to type 2 diabetes mellitus: a matter of hormesis? Nat. Rev. Endocrinol. 2011;8:183–192. doi: 10.1038/nrendo.2011.158. [DOI] [PubMed] [Google Scholar]
- 18.Futamura M., Yao J., Li X., Bergeron R., Tran J.L., Zycband E., Woods J., Zhu Y., Shao Q., Maruki-Uchida H., Goto-Shimazaki H., Langdon R.B., Erion M.D., Eiki J., Zhou Y.P. Chronic treatment with a glucokinase activator delays the onset of hyperglycaemia and preserves beta cell mass in the Zucker diabetic fatty rat. Diabetologia. 2012;55:1071–1080. doi: 10.1007/s00125-011-2439-3. [DOI] [PubMed] [Google Scholar]
- 19.Martens G.A., Cai Y., Hinke S., Stange G., Van de Casteele M., Pipeleers D. Glucose suppresses superoxide generation in metabolically responsive pancreatic beta cells. J. Biol. Chem. 2005;280:20389–20396. doi: 10.1074/jbc.M411869200. [DOI] [PubMed] [Google Scholar]
- 20.Sarre A., Gabrielli J., Vial G., Leverve X.M., Assimacopoulos-Jeannet F. Reactive oxygen species are produced at low glucose and contribute to the activation of AMPK in insulin-secreting cells. Free Radic. Biol. Med. 2012;52:142–150. doi: 10.1016/j.freeradbiomed.2011.10.437. [DOI] [PubMed] [Google Scholar]
- 21.Fleury C., Neverova M., Collins S., Raimbault S., Champigny O., Levi-Meyrueis C., Bouillaud F., Seldin M.F., Surwit R.S., Ricquier D., Warden C.H. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997;15:269–272. doi: 10.1038/ng0397-269. [DOI] [PubMed] [Google Scholar]
- 22.Hong Y., Fink B.D., Dillon J.S., Sivitz W.I. Effects of adenoviral overexpression of uncoupling protein-2 and −3 on mitochondrial respiration in insulinoma cells. Endocrinology. 2001;142:249–256. doi: 10.1210/endo.142.1.7889. [DOI] [PubMed] [Google Scholar]
- 23.Chan C.B., De Leo D., Joseph J.W., McQuaid T.S., Ha X.F., Xu F., Tsushima R.G., Pennefather P.S., Salapatek A.M., Wheeler M.B. Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes. 2001;50:1302–1310. doi: 10.2337/diabetes.50.6.1302. [DOI] [PubMed] [Google Scholar]
- 24.Chan C.B., MacDonald P.E., Saleh M.C., Johns D.C., Marban E., Wheeler M.B. Overexpression of uncoupling protein 2 inhibits glucose-stimulated insulin secretion from rat islets. Diabetes. 1999;48:1482–1486. doi: 10.2337/diabetes.48.7.1482. [DOI] [PubMed] [Google Scholar]
- 25.Li N., Stojanovski S., Maechler P. Mitochondrial hormesis in pancreatic beta cells: does uncoupling protein 2 play a role? Oxid. Med. Cell Longev. 2012;2012:740849. doi: 10.1155/2012/740849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Produit-Zengaffinen N., Davis-Lameloise N., Perreten H., Becard D., Gjinovci A., Keller P.A., Wollheim C.B., Herrera P., Muzzin P., Assimacopoulos-Jeannet F. Increasing uncoupling protein-2 in pancreatic beta cells does not alter glucose-induced insulin secretion but decreases production of reactive oxygen species. Diabetologia. 2007;50:84–93. doi: 10.1007/s00125-006-0499-6. [DOI] [PubMed] [Google Scholar]
- 27.Nicholls D.G. The pancreatic beta-cell: a bioenergetic perspective. Physiol. Rev. 2016;96:1385–1447. doi: 10.1152/physrev.00009.2016. [DOI] [PubMed] [Google Scholar]
- 28.Supale S., Li N., Brun T., Maechler P. Mitochondrial dysfunction in pancreatic beta cells. Trends Endocrinol. Metab. 2012;23:477–487. doi: 10.1016/j.tem.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 29.Vozza A., Parisi G., De Leonardis F., Lasorsa F.M., Castegna A., Amorese D., Marmo R., Calcagnile V.M., Palmieri L., Ricquier D., Paradies E., Scarcia P., Palmieri F., Bouillaud F., Fiermonte G. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA. 2014;111:960–965. doi: 10.1073/pnas.1317400111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lameloise N., Muzzin P., Prentki M., Assimacopoulos-Jeannet F. Uncoupling protein 2: a possible link between fatty acid excess and impaired glucose-induced insulin secretion? Diabetes. 2001;50:803–809. doi: 10.2337/diabetes.50.4.803. [DOI] [PubMed] [Google Scholar]
- 31.Brun T., Li N., Jourdain A.A., Gaudet P., Duhamel D., Meyer J., Bosco D., Maechler P. Diabetogenic milieus induce specific changes in mitochondrial transcriptome and differentiation of human pancreatic islets. Hum. Mol. Genet. 2015;24:5270–5284. doi: 10.1093/hmg/ddv247. [DOI] [PubMed] [Google Scholar]
- 32.Li L.X., Skorpen F., Egeberg K., Jorgensen I.H., Grill V. Uncoupling protein-2 participates in cellular defense against oxidative stress in clonal beta-cells. Biochem Biophys. Res. Commun. 2001;282:273–277. doi: 10.1006/bbrc.2001.4577. [DOI] [PubMed] [Google Scholar]
- 33.Krauss S., Zhang C.Y., Scorrano L., Dalgaard L.T., St-Pierre J., Grey S.T., Lowell B.B. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J. Clin. Investig. 2003;112:1831–1842. doi: 10.1172/JCI19774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Urbano F., Filippello A., Di Pino A., Barbagallo D., Di Mauro S., Pappalardo A., Rabuazzo A.M., Purrello M., Purrello F., Piro S. Altered expression of uncoupling protein 2 in GLP-1-producing cells after chronic high glucose exposure: implications for the pathogenesis of diabetes mellitus. Am. J. Physiol. Cell Physiol. 2016;310:C558–C567. doi: 10.1152/ajpcell.00148.2015. [DOI] [PubMed] [Google Scholar]
- 35.Carobbio S., Ishihara H., Fernandez-Pascual S., Bartley C., Martin-Del-Rio R., Maechler P. Insulin secretion profiles are modified by overexpression of glutamate dehydrogenase in pancreatic islets. Diabetologia. 2004;47:266–276. doi: 10.1007/s00125-003-1306-2. [DOI] [PubMed] [Google Scholar]
- 36.Hayashi K., Kojima R., Ito M. Strain differences in the diabetogenic activity of streptozotocin in mice. Biol. Pharm. Bull. 2006;29:1110–1119. doi: 10.1248/bpb.29.1110. [DOI] [PubMed] [Google Scholar]
- 37.Karaca M., Frigerio F., Migrenne S., Martin-Levilain J., Skytt D.M., Pajecka K., Martin-del-Rio R., Gruetter R., Tamarit-Rodriguez J., Waagepetersen H.S., Magnan C., Maechler P. GDH-dependent glutamate oxidation in the brain dictates peripheral energy substrate distribution. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Li N., Li B., Brun T., Deffert-Delbouille C., Mahiout Z., Daali Y., Ma X.J., Krause K.H., Maechler P. NADPH oxidase NOX2 defines a new antagonistic role for reactive oxygen species and cAMP/PKA in the regulation of insulin secretion. Diabetes. 2012;61:2842–2850. doi: 10.2337/db12-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frigerio F., Brun T., Bartley C., Usardi A., Bosco D., Ravnskjaer K., Mandrup S., Maechler P. Peroxisome proliferator-activated receptor alpha (PPARalpha) protects against oleate-induced INS-1E beta cell dysfunction by preserving carbohydrate metabolism. Diabetologia. 2010;53:331–340. doi: 10.1007/s00125-009-1590-6. [DOI] [PubMed] [Google Scholar]
- 40.Zhao H., Kalivendi S., Zhang H., Joseph J., Nithipatikom K., Vasquez-Vivar J., Kalyanaraman B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic. Biol. Med. 2003;34:1359–1368. doi: 10.1016/s0891-5849(03)00142-4. [DOI] [PubMed] [Google Scholar]
- 41.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic. Biol. Med. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 42.Kalyanaraman B., Dranka B.P., Hardy M., Michalski R., Zielonka J. HPLC-based monitoring of products formed from hydroethidine-based fluorogenic probes--the ultimate approach for intra- and extracellular superoxide detection. Biochim Biophys. Acta. 2014;1840:739–744. doi: 10.1016/j.bbagen.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersson L.E., Nicholas L.M., Filipsson K., Sun J., Medina A., Al-Majdoub M., Fex M., Mulder H., Spegel P. Glycogen metabolism in the glucose-sensing and supply-driven beta-cell. FEBS Lett. 2016;590:4242–4251. doi: 10.1002/1873-3468.12460. [DOI] [PubMed] [Google Scholar]
- 44.Arsenijevic D., Onuma H., Pecqueur C., Raimbault S., Manning B.S., Miroux B., Couplan E., Alves-Guerra M.C., Goubern M., Surwit R., Bouillaud F., Richard D., Collins S., Ricquier D. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 45.Hurtaud C., Gelly C., Chen Z., Levi-Meyrueis C., Bouillaud F. Glutamine stimulates translation of uncoupling protein 2mRNA. Cell Mol. Life Sci. 2007;64:1853–1860. doi: 10.1007/s00018-007-7039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Azzu V., Affourtit C., Breen E.P., Parker N., Brand M.D. Dynamic regulation of uncoupling protein 2 content in INS-1E insulinoma cells. Bba-Bioenerg. 2008;1777:1378–1383. doi: 10.1016/j.bbabio.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diao J., Allister E.M., Koshkin V., Lee S.C., Bhattacharjee A., Tang C., Giacca A., Chan C.B., Wheeler M.B. UCP2 is highly expressed in pancreatic alpha-cells and influences secretion and survival. Proc. Natl. Acad. Sci. USA. 2008;105:12057–12062. doi: 10.1073/pnas.0710434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooperberg B.A., Cryer P.E. Insulin reciprocally regulates glucagon secretion in humans. Diabetes. 2010;59:2936–2940. doi: 10.2337/db10-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robson-Doucette C.A., Sultan S., Allister E.M., Wikstrom J.D., Koshkin V., Bhattacharjee A., Prentice K.J., Sereda S.B., Shirihai O.S., Wheeler M.B. Vol. 60. 2011. Beta-cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion; pp. 2710–2719. (Diabetes). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joseph J.W., Koshkin V., Zhang C.Y., Wang J., Lowell B.B., Chan C.B., Wheeler M.B. Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes. 2002;51:3211–3219. doi: 10.2337/diabetes.51.11.3211. [DOI] [PubMed] [Google Scholar]
- 51.Lee S.C., Robson-Doucette C.A., Wheeler M.B. Uncoupling protein 2 regulates reactive oxygen species formation in islets and influences susceptibility to diabetogenic action of streptozotocin. J. Endocrinol. 2009;203:33–43. doi: 10.1677/JOE-09-0117. [DOI] [PubMed] [Google Scholar]
- 52.Zhang C.Y., Baffy G., Perret P., Krauss S., Peroni O., Grujic D., Hagen T., Vidal-Puig A.J., Boss O., Kim Y.B., Zheng X.X., Wheeler M.B., Shulman G.I., Chan C.B., Lowell B.B. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 53.Pi J., Bai Y., Daniel K.W., Liu D., Lyght O., Edelstein D., Brownlee M., Corkey B.E., Collins S. Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic beta-cell function. Endocrinology. 2009;150:3040–3048. doi: 10.1210/en.2008-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calabrese E.J., Bachmann K.A., Bailer A.J., Bolger P.M., Borak J., Cai L., Cedergreen N., Cherian M.G., Chiueh C.C., Clarkson T.W., Cook R.R., Diamond D.M., Doolittle D.J., Dorato M.A., Duke S.O., Feinendegen L., Gardner D.E., Hart R.W., Hastings K.L., Hayes A.W., Hoffmann G.R., Ives J.A., Jaworowski Z., Johnson T.E., Jonas W.B., Kaminski N.E., Keller J.G., Klaunig J.E., Knudsen T.B., Kozumbo W.J., Lettieri T., Liu S.Z., Maisseu A., Maynard K.I., Masoro E.J., McClellan R.O., Mehendale H.M., Mothersill C., Newlin D.B., Nigg H.N., Oehme F.W., Phalen R.F., Philbert M.A., Rattan S.I., Riviere J.E., Rodricks J., Sapolsky R.M., Scott B.R., Seymour C., Sinclair D.A., Smith-Sonneborn J., Snow E.T., Spear L., Stevenson D.E., Thomas Y., Tubiana M., Williams G.M., Mattson M.P. Biological stress response terminology: integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicol. Appl. Pharmacol. 2007;222:122–128. doi: 10.1016/j.taap.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 55.Perry R.J., Zhang D., Zhang X.M., Boyer J.L., Shulman G.I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science. 2015;347:1253–1256. doi: 10.1126/science.aaa0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brun T., Maechler P. Beta-cell mitochondrial carriers and the diabetogenic stress response. Biochim Biophys. Acta. 1863;2016:2540–2549. doi: 10.1016/j.bbamcr.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 57.Mugabo Y., Zhao S., Seifried A., Gezzar S., Al-Mass A., Zhang D., Lamontagne J., Attane C., Poursharifi P., Iglesias J., Joly E., Peyot M.L., Gohla A., Madiraju S.R., Prentki M. Identification of a mammalian glycerol-3-phosphate phosphatase: role in metabolism and signaling in pancreatic beta-cells and hepatocytes. Proc. Natl. Acad. Sci. USA. 2016;113:E430–E439. doi: 10.1073/pnas.1514375113. [DOI] [PMC free article] [PubMed] [Google Scholar]