

Abstract
Subcomponent exchange transformed new high-spin FeII4L4 cage 1 into previously-reported low-spin FeII4L4 cage 2: 2-formyl-6-methylpyridine was ejected in favor of the less sterically hindered 2-formylpyridine, with concomitant high- to low-spin transition of the cage’s FeII centers. High-spin 1 also reacted more readily with electron-rich anilines than 2, enabling the design of a system consisting of two cages that could release their guests in response to combinations of different stimuli. The addition of p-anisidine to a mixture of high-spin 1 and previously-reported low-spin FeII4L6 cage 3 resulted in the destruction of 1 and the release of its guest. However, initial addition of 2-formylpyridine to an identical mixture of 1 and 3 resulted in the transformation of 1 into 2; added p-anisidine then reacted preferentially with 3 releasing its guest. The addition of 2-formylpyridine thus modulated the system’s behavior, fundamentally altering its response to the subsequent signal p-anisidine.
Stimuli-responsive container molecules,1 whose uptake and release of guests can be controlled through the application of external signals,2 are useful building blocks for molecular networks.3 The development of new stimuli-responsive behaviors1f,4 allows for these networks to increase in complexity, as cages may be addressed individually within mixtures,5 or signals may be passed between network members in order to construct complex responses.6 An ultimate goal is to approach the functional complexity exhibited by the signaling pathways in biological systems.
Structures prepared via subcomponent self-assembly7 can transform in response to external stimuli through the reversible reconfiguration of the dynamic covalent and coordinative bonds holding the structures together;8 examples include the rearrangements of a Schiff-base ligand7b and meso-helicates7e via aldehyde exchange, and the imine exchanges undergone by dynamic cages when an electron-rich amine substitutes an electron-poor amine residue.9
In this study, we envisaged a new approach whereby a chemical stimulus transforms a high-spin FeII4L4 cage into a low-spin analog through exchange of a more bulky aldehyde subcomponent for a less bulky one. Others10 and our group11 have reported FeII cages and helicates that undergo spin-crossover12 induced by heat and light. Mononuclear FeII complexes incorporating 2-formyl-6-methylpyridine undergo spin-crossover,13 attributed to a steric clash between methyl groups and adjacent pyridine rings destabilizing the low-spin state relative to the high-spin state.14 FeII mononuclear complexes are observed to preferentially incorporate 2-formylpyridine over 2-formyl-6-methylpyridine.14 Thus, alleviation of steric clash might drive exchange of 2-formylpyridine for 2-formyl-6-methylpyridine, transforming an FeII cage from high-spin to low-spin.
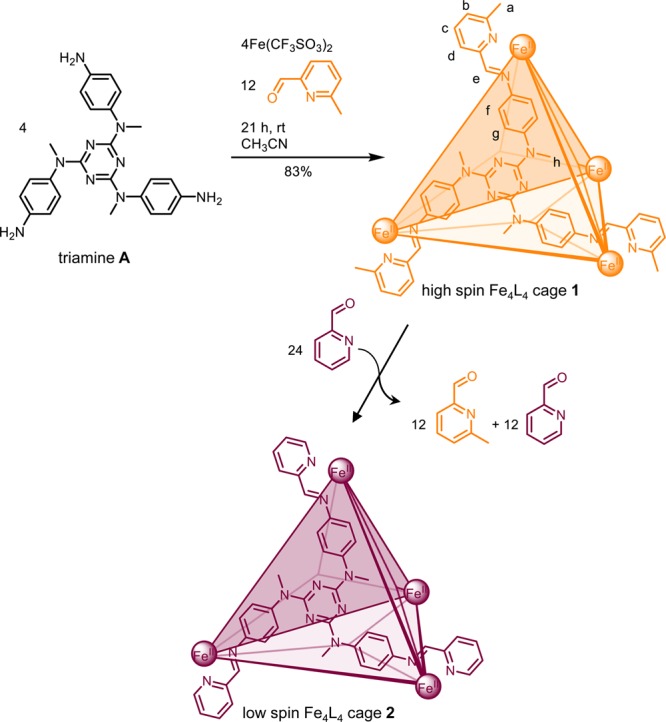
Here we report the self-assembly of high-spin FeII4L4 cage 1 incorporating 2-formyl-6-methylpyridine as a subcomponent. Cage 1 binds a variety of neutral guests in acetonitrile. Paramagnetic 1H NMR spectroscopy provides a sensitive means for detecting guest encapsulation and determining guest identity due to the isotropic shifts of the paramagnetic FeII centers.15 High-spin 1 did not transition to a low-spin state upon lowering the temperature to 268 K; however, the chemical stimulus 2-formylpyridine transformed high-spin 1 into previously reported low-spin FeII4L4 cage 2,16 as a consequence of aldehyde exchange. This transformation was used to set up a system such that either one of a pair of cages could be opened, and its guest released, following the addition of a different chemical stimulus, p-anisidine.
Cage 1 was prepared by reaction of iron(II) triflate, triamine A(16) and commercially available 2-formyl-6-methylpyridine (1:1:3) in CD3CN (Scheme 1). The [FeII4L4] composition was confirmed by high-resolution ESI-MS (Figure S4). Unlike previously reported low-spin 2,16 which contains 2-formylpyridine residues, the FeII centers of 1 are high-spin between 268 and 318 K (Figure S5). The 1H NMR spectrum of 1 has the eight proton signals expected for a T-symmetric structure spread over ca. 240 ppm (Figure S1), displaying Curie–Weiss behavior above 268 K (Figure S6).17 We infer the high-spin character of 1 to be a consequence of steric clash between methyl groups and adjacent pyridine rings.13,14
Scheme 1. Self-Assembly of High-Spin FeII4L4 Cage 1 and Transformation to Low-Spin Cage 2.
The paramagnetic signals of 1 were assigned following the methods employed by Raehm18 and Ward19 for paramagnetic CoII complexes. T1 relaxation times were measured and correlated to the distances between the paramagnetic center and proton according to the Solomon equation (Table S1, Figure S7).20 Cross-peaks observed in the COSY spectrum (Figure S3) and comparison to the calculated chemical shifts for a related high-spin FeII mononuclear complex21 provide additional support for our 1H NMR assignments and proposed solution structure of 1.
Host–guest studies revealed that cage 1 (2 mM) encapsulated a similar range of guests (10–15 equiv) to cage 2(16) in acetonitrile at 298 K, although guest uptake was faster (approaching equilibrium within 4 h) because high-spin FeII complexes have more labile N→FeII bonds than their low-spin analogs. In the 1H NMR spectra, each guest was bound in slow exchange, with shifts of both the cage and guest peaks observed upon encapsulation. Encapsulated guest signals were observed in all cases between −10 and −20 ppm and their T1 values were of a similar magnitude to the T1 value for proton h, reflecting the isotropic shifts experienced by the guests within the paramagnetic host cavity (Table S2).
Prospective guests for 1 were divided into three groups based upon the extent of host–guest complexation (Figure 1). Complete conversion to the host–guest complex was observed for guests that matched the size and shape of the cavity well, such as adamantane (Figures 1a, S8), whereas two sets of NMR signals corresponding to the empty cage and host–guest complex were observed for guests with a poorer match for the cavity, such as o-xylene (Figures 1b, S9). m- and p-Xylene bound in trace amounts (Figures S49–52) and hexafluorobenzene did not bind at all, which we attribute to unfavorable interactions between this electron deficient guest and the electron deficient cavity (Figure 1c). The relative binding affinities of the guests in Figure 1a were estimated from competition experiments (Figures S53–S66).
Figure 1.

Guests for cage 1 that a) bind strongly, b) bind weakly and c) are not observed to bind.
The paramagnetic FeII centers in cage 1 enabled the sensitive detection of guest encapsulation and straightforward discrimination of signals belonging to different isomers by 1H NMR spectroscopy. Encapsulated NMR signals are spread over a wider chemical shift range, thus reducing signal overlap and improving dispersion upon encapsulation.15b For strongly bound guests, such as adamantane, minor sets of encapsulated guest signals were observed alongside the major set of peaks corresponding to bound adamantane (Figure S14). GC–MS analysis of these guests (commercial claimed purity of 98+%) revealed trace impurities (Figures S10–13). We infer the minor NMR signals to correspond to these impurities encapsulated within cage 1. Thus, cage 1 can enable the detection of trace quantities of strongly binding guests within mixtures by NMR spectroscopy in a manner that would not be possible using diamagnetic hosts.
Similarly, trace cis-decalin could be sensed as an impurity in commercial trans-decalin (Figures S9, S65). Cage 1 also binds o-xylene over the other xylene isomers (Figure S66). The separation of xylene isomers from mixed hydrocarbons is costly and inefficient due to the close similarity in the physicochemical properties of the isomers.22 Host–guest complexation within 1 could thus enable the separation of o-xylene.23
Subcomponent exchange within cage 1, empty or with bound 1-fluoroadamantane (1-FA), proceeded in anhydrous CD3CN upon addition of 2-formylpyridine (24 equiv), resulting in a color change from orange to red-purple. The use of excess 2-formylpyridine was found to result in sharper 1H NMR spectra, rendering the process easier to follow. Released 2-formyl-6-methylpyridine was observed by 1H NMR spectroscopy with a concomitant decrease in the intensities of the peaks for cage 1 and appearance of new paramagnetic species after 16 h at 50 °C (Figures S69, S72). However, no NMR peaks corresponding to cage 2 (Figure S70) or its host–guest complex (Figure S73) were observed until 5% D2O (v/v) was added, resulting in a color change to dark purple (Figure S67). The requirement for water to complete the transformation implies that hydrolyzed species are intermediates during the exchange process, as reported by Hahn7b and Hooley.7e When no guest was present, the transformation was complete within 1 day at 50 °C (Figure S70) whereas the equilibration process was slower for the full cage, with kinetics depending on the amount of water added (Figure S75). In addition to the encapsulated 1-FA within 2, a putative intermediate encapsulated 1-FA species was observed in the 19F NMR spectrum during equilibration (Figure S74), suggesting that the guest remained bound during the transformation.
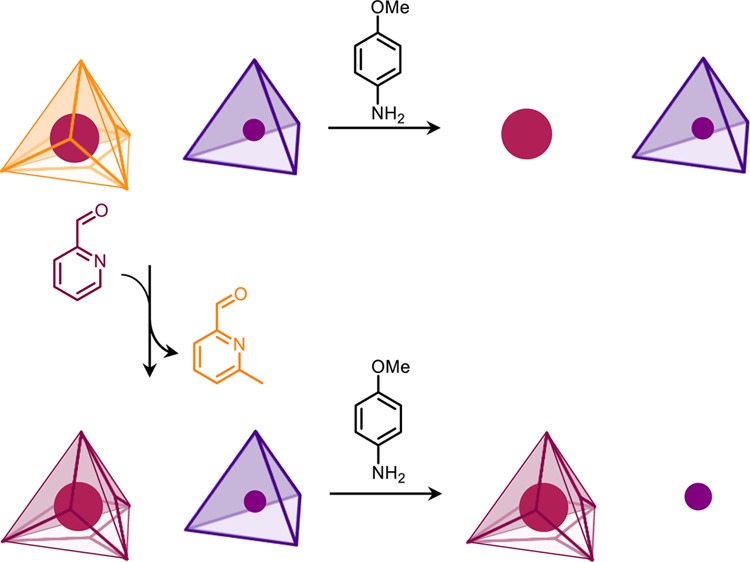
Selective cage breakdown and guest release could be triggered by the chemical signal p-anisidine, which opens cages through imine exchange.3a,24 As cage 1 was thermodynamically unstable with respect to 2 in the presence of 2-formylpyridine, we hypothesized that 1 might react more readily with p-anisidine and that a low-spin FeII4L6 cage 3(25) (Scheme 2) might react with p-anisidine at a rate intermediate between those of 1 and 2. This differential reactivity would allow for the functioning of the system shown in Scheme 2. A mixture of 1 and 3 would thus react with p-anisidine to selectively liberate the guest of 1, whereas initial treatment with 2-formylpyridine would result in a mixture of 2 and 3, whose reaction with p-anisidine would selectively liberate the guest of 3. Guests containing fluorine (BF4– for 3 and 1-FA for 1 and 2) were chosen so that guest release could be monitored by 19F NMR spectroscopy. Importantly, control experiments revealed there was no aldehyde or guest exchange between [BF4– ⊂ 3] and [1-FA ⊂ 1] and the aldehyde exchange process was unaffected by the presence of [BF4– ⊂ 3] (Figures S95–S97).
Scheme 2. Cage Disassembly and Guest Release Triggered by p-Anisidine.

High-spin FeII4L4 cage 1 reacted with p-anisidine in the presence of low-spin FeII4L6 cage 3, releasing the guest 1-fluoroadamantane (1-FA) and forming mononuclear complex 4. Following the transformation from high-spin cage 1 to low-spin cage 2, low-spin FeII4L6 cage 3 was instead broken down by p-anisidine releasing the guest BF4– and forming 5. Each transformation was performed in the presence of an excess of 1-FA (10 equiv per cage 1 or 2).
The selectivity of cage disassembly and guest release was first investigated for an equimolar mixture of [1-FA ⊂ 1] and [BF4– ⊂ 3]. As p-anisidine (24 equiv) was progressively added, the 1H NMR signals corresponding to [1-FA ⊂ 1] were observed to disappear whereas those for [BF4– ⊂ 3] remained (Figures S88–S89). Control titrations with each cage separately revealed p-anisidine to react with both cages, but more readily with [1-FA ⊂ 1] (Figures S91–S94). The addition of excess p-anisidine (24 equiv) to [1-FA ⊂ 1] thus resulted in its complete disassembly at room temperature in the presence of [BF4– ⊂ 3], releasing 1-FA (Figures S85–S87). The encapsulated 1-FA signal disappeared in the 19F NMR spectrum, whereas BF4– remained encapsulated within 3 (Figure S87).
In contrast, the selectivity of cage disassembly was inverted for an equimolar mixture of [1-FA ⊂ 2] and [BF4– ⊂ 3] due to the increased thermodynamic stability of face-capped compared with edge-bridged tetrahedra;24 only the NMR signals for [BF4– ⊂ 3] disappeared following progressive addition of p-anisidine (12 equiv) and heating (Figures S100–S101). Control experiments were also carried out for the individual cages (Figures S102–S105). The mixture of [1-FA ⊂ 2] and [BF4– ⊂ 3] resulting from aldehyde exchange reacted similarly with stoichiometric p-anisidine (12 equiv) upon heating (Figure S98), resulting in disassembly of [BF4– ⊂ 3] and release of BF4–, whereas 1-FA remained bound within the transformed cage 2 (Figure S99).
We have demonstrated the transformation of FeII centers in a FeII4L4 cage from high- to low-spin for the first time via selective aldehyde exchange. In the presence of low-spin FeII4L6 cage 3, this transformation was employed to switch the guest release outcome following application of the chemical signal p-anisidine, due to the difference in the relative reactivities of the high- and low-spin cages. Switching the spin state of a cage is thus a promising new strategy for signal transduction. Future work will focus upon integrating these processes into larger signaling networks.6
Acknowledgments
This work was funded by the European Research Council (695009) and the UK Engineering and Physical Sciences Research Council (EPSRC, EP/M01083X/1). We also thank the Cambridge Trusts for PhD funding (A.B.G.), the Cambridge Chemistry NMR staff and James Keeler for useful discussions, and Jenifer Mizen for preliminary studies on the microwave synthesis of a precursor of A. We also thank Cally Haynes, Julia Guilleme, Marion Kieffer, Felix Rizzuto and the EPSRC UK National Mass Spectrometry Facility at Swansea for collecting ESI-MS data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b01478.
Complete experimental procedures including full characterization, NMR data and ESI mass spectra (PDF)
Author Present Address
† Otto Diels-Institute of Organic Chemistry, Kiel University, Otto-Hahn-Platz 4, Kiel D-24098, Germany.
The authors declare no competing financial interest.
Supplementary Material
References
- a Cook T. R.; Stang P. J. Chem. Rev. 2015, 115, 7001–7045. 10.1021/cr5005666. [DOI] [PubMed] [Google Scholar]; b Frischmann P. D.; MacLachlan M. J. Chem. Soc. Rev. 2013, 42, 871–890. 10.1039/C2CS35337G. [DOI] [PubMed] [Google Scholar]; c Hasell T.; Cooper A. I. Nat. Rev. Mater. 2016, 1, 16053. 10.1038/natrevmats.2016.53. [DOI] [Google Scholar]; d Custelcean R. Chem. Soc. Rev. 2014, 43, 1813–1824. 10.1039/c3cs60371g. [DOI] [PubMed] [Google Scholar]; e Cook T. R.; Zheng Y.-R.; Stang P. J. Chem. Rev. 2013, 113, 734–777. 10.1021/cr3002824. [DOI] [PMC free article] [PubMed] [Google Scholar]; f McConnell A. J.; Wood C. S.; Neelakandan P. P.; Nitschke J. R. Chem. Rev. 2015, 115, 7729–7793. 10.1021/cr500632f. [DOI] [PubMed] [Google Scholar]
- a Han M.; Michel R.; He B.; Chen Y.-S.; Stalke D.; John M.; Clever G. H. Angew. Chem., Int. Ed. 2013, 52, 1319–1323. 10.1002/anie.201207373. [DOI] [PubMed] [Google Scholar]; b Wang S.; Sawada T.; Ohara K.; Yamaguchi K.; Fujita M. Angew. Chem., Int. Ed. 2016, 55, 2063–2066. 10.1002/anie.201509278. [DOI] [PubMed] [Google Scholar]; c Kishi N.; Akita M.; Yoshizawa M. Angew. Chem., Int. Ed. 2014, 53, 3604–3607. 10.1002/anie.201311251. [DOI] [PubMed] [Google Scholar]; d Gavette J. V.; Mills N. S.; Zakharov L. N.; Johnson C. A.; Johnson D. W.; Haley M. M. Angew. Chem., Int. Ed. 2013, 52, 10270–10274. 10.1002/anie.201302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Castilla A. M.; Ronson T. K.; Nitschke J. R. J. Am. Chem. Soc. 2016, 138, 2342–2351. 10.1021/jacs.5b13016. [DOI] [PubMed] [Google Scholar]; b Murase T.; Sato S.; Fujita M. Angew. Chem., Int. Ed. 2007, 46, 5133–5136. 10.1002/anie.200700793. [DOI] [PubMed] [Google Scholar]
- a Löffler S.; Lübben J.; Krause L.; Stalke D.; Dittrich B.; Clever G. H. J. Am. Chem. Soc. 2015, 137, 1060–1063. 10.1021/ja5130379. [DOI] [PubMed] [Google Scholar]; b Zheng Y.-R.; Lan W.-J.; Wang M.; Cook T. R.; Stang P. J. J. Am. Chem. Soc. 2011, 133, 17045–17055. 10.1021/ja207217t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Burke M. J.; Nichol G. S.; Lusby P. J. J. Am. Chem. Soc. 2016, 138, 9308–9315. 10.1021/jacs.6b05364. [DOI] [PubMed] [Google Scholar]; d Zhou X.-P.; Wu Y.; Li D. J. Am. Chem. Soc. 2013, 135, 16062–16065. 10.1021/ja4092984. [DOI] [PubMed] [Google Scholar]
- Riddell I. A.; Ronson T. K.; Clegg J. K.; Wood C. S.; Bilbeisi R. A.; Nitschke J. R. J. Am. Chem. Soc. 2014, 136, 9491–9498. 10.1021/ja504748g. [DOI] [PubMed] [Google Scholar]
- a Ray D.; Foy J. T.; Hughes R. P.; Aprahamian I. Nat. Chem. 2012, 4, 757–762. 10.1038/nchem.1408. [DOI] [PubMed] [Google Scholar]; b Kassem S.; Lee A. T. L.; Leigh D. A.; Markevicius A.; Solà J. Nat. Chem. 2016, 8, 138–143. 10.1038/nchem.2410. [DOI] [PubMed] [Google Scholar]
- a Ronson T. K.; Zarra S.; Black S. P.; Nitschke J. R. Chem. Commun. 2013, 49, 2476–2490. 10.1039/c2cc36363a. [DOI] [PubMed] [Google Scholar]; b Lewing D.; Koppetz H.; Hahn F. E. Inorg. Chem. 2015, 54, 7653–7659. 10.1021/acs.inorgchem.5b01334. [DOI] [PubMed] [Google Scholar]; c Bunzen H.; Nonappa; Kalenius E.; Hietala S.; Kolehmainen E. Chem. - Eur. J. 2013, 19, 12978–12981. 10.1002/chem.201302055. [DOI] [PubMed] [Google Scholar]; d Frischmann P. D.; Kunz V.; Stepanenko V.; Würthner F. Chem. - Eur. J. 2015, 21, 2766–2769. 10.1002/chem.201405866. [DOI] [PubMed] [Google Scholar]; e Wiley C. A.; Holloway L. R.; Miller T. F.; Lyon Y.; Julian R. R.; Hooley R. J. Inorg. Chem. 2016, 55, 9805–9815. 10.1021/acs.inorgchem.6b01644. [DOI] [PubMed] [Google Scholar]; f Yi S.; Brega V.; Captain B.; Kaifer A. E. Chem. Commun. 2012, 48, 10295–10297. 10.1039/c2cc35095e. [DOI] [PubMed] [Google Scholar]
- Meyer C. D.; Joiner C. S.; Stoddart J. F. Chem. Soc. Rev. 2007, 36, 1705–1723. 10.1039/b513441m. [DOI] [PubMed] [Google Scholar]
- a Hristova Y. R.; Smulders M. M. J.; Clegg J. K.; Breiner B.; Nitschke J. R. Chem. Sci. 2011, 2, 638–641. 10.1039/C0SC00495B. [DOI] [Google Scholar]; b Wood C. S.; Ronson T. K.; Belenguer A. M.; Holstein J. J.; Nitschke J. R. Nat. Chem. 2015, 7, 354–358. 10.1038/nchem.2205. [DOI] [PubMed] [Google Scholar]; c Neelakandan P. P.; Jiménez A.; Thoburn J. D.; Nitschke J. R. Angew. Chem., Int. Ed. 2015, 54, 14378–14382. 10.1002/ange.201507045. [DOI] [PubMed] [Google Scholar]
- a Ferguson A.; Squire M. A.; Siretanu D.; Mitcov D.; Mathoniere C.; Clerac R.; Kruger P. E. Chem. Commun. 2013, 49, 1597–1599. 10.1039/c3cc00012e. [DOI] [PubMed] [Google Scholar]; b Ren D.-H.; Qiu D.; Pang C.-Y.; Li Z.; Gu Z.-G. Chem. Commun. 2015, 51, 788–791. 10.1039/C4CC08041F. [DOI] [PubMed] [Google Scholar]; c Li L.; Saigo N.; Zhang Y.; Fanna D. J.; Shepherd N. D.; Clegg J. K.; Zheng R.; Hayami S.; Lindoy L. F.; Aldrich-Wright J. R.; Li C.-G.; Reynolds J. K.; Harman D. G.; Li F. J. Mater. Chem. C 2015, 3, 7878–7882. 10.1039/C5TC00991J. [DOI] [Google Scholar]; d Duriska M. B.; Neville S. M.; Moubaraki B.; Cashion J. D.; Halder G. J.; Chapman K. W.; Balde C.; Létard J.-F.; Murray K. S.; Kepert C. J.; Batten S. R. Angew. Chem., Int. Ed. 2009, 48, 2549–2552. 10.1002/anie.200805178. [DOI] [PubMed] [Google Scholar]; e Struch N.; Brandenburg J. G.; Schnakenburg G.; Wagner N.; Beck J.; Grimme S.; Lützen A. Eur. J. Inorg. Chem. 2015, 2015, 5503–5510. 10.1002/ejic.201501057. [DOI] [Google Scholar]; f Darawsheh M.; Barrios L. A.; Roubeau O.; Teat S. J.; Aromí G. Chem. - Eur. J. 2016, 22, 8635–8645. 10.1002/chem.201601080. [DOI] [PubMed] [Google Scholar]
- Bilbeisi R. A.; Zarra S.; Feltham H. L. C.; Jameson G. N. L.; Clegg J. K.; Brooker S.; Nitschke J. R. Chem. - Eur. J. 2013, 19, 8058–8062. 10.1002/chem.201300805. [DOI] [PubMed] [Google Scholar]
- a Brooker S. Chem. Soc. Rev. 2015, 44, 2880–2892. 10.1039/C4CS00376D. [DOI] [PubMed] [Google Scholar]; b Gutlich P.; Garcia Y.; Goodwin H. A. Chem. Soc. Rev. 2000, 29, 419–427. 10.1039/b003504l. [DOI] [Google Scholar]
- Schenker S.; Hauser A.; Wang W.; Chan I. Y. J. Chem. Phys. 1998, 109, 9870–9878. 10.1063/1.477681. [DOI] [Google Scholar]
- Schultz D.; Nitschke J. R. Angew. Chem., Int. Ed. 2006, 45, 2453–2456. 10.1002/anie.200504447. [DOI] [PubMed] [Google Scholar]
- a Terazzi E.; Rivera J.-P.; Ouali N.; Piguet C. Magn. Reson. Chem. 2006, 44, 539–552. 10.1002/mrc.1790. [DOI] [PubMed] [Google Scholar]; b Turega S.; Whitehead M.; Hall B. R.; Meijer A. J. H. M.; Hunter C. A.; Ward M. D. Inorg. Chem. 2013, 52, 1122–1132. 10.1021/ic302498t. [DOI] [PubMed] [Google Scholar]; c Drago R. S.; Zink J. I.; Richman R. M.; Perry W. D. J. Chem. Educ. 1974, 51, 464. 10.1021/ed051p464. [DOI] [Google Scholar]; d Weber B.; Walker F. A. Inorg. Chem. 2007, 46, 6794–6803. 10.1021/ic062349e. [DOI] [PubMed] [Google Scholar]; e Petzold H.; Djomgoue P.; Horner G.; Speck J. M.; Ruffer T.; Schaarschmidt D. Dalton Trans. 2016, 45, 13798–13809. 10.1039/C6DT01895E. [DOI] [PubMed] [Google Scholar]; f Hogue R. W.; Feltham H. L. C.; Miller R. G.; Brooker S. Inorg. Chem. 2016, 55, 4152–4165. 10.1021/acs.inorgchem.5b02851. [DOI] [PubMed] [Google Scholar]
- Bolliger J. L.; Ronson T. K.; Ogawa M.; Nitschke J. R. J. Am. Chem. Soc. 2014, 136, 14545–14553. 10.1021/ja5077102. [DOI] [PubMed] [Google Scholar]
- Landee C. P.; Turnbull M. M. J. Coord. Chem. 2014, 67, 375–439. 10.1080/00958972.2014.889294. [DOI] [Google Scholar]
- Amouri H.; Mimassi L.; Rager M. N.; Mann B. E.; Guyard-Duhayon C.; Raehm L. Angew. Chem., Int. Ed. 2005, 44, 4543–4546. 10.1002/anie.200500786. [DOI] [PubMed] [Google Scholar]
- Tidmarsh I. S.; Taylor B. F.; Hardie M. J.; Russo L.; Clegg W.; Ward M. D. New J. Chem. 2009, 33, 366–375. 10.1039/B816864D. [DOI] [Google Scholar]
- Solomon I. Phys. Rev. 1955, 99, 559–565. 10.1103/PhysRev.99.559. [DOI] [Google Scholar]
- Isley W. C.; Zarra S.; Carlson R. K.; Bilbeisi R. A.; Ronson T. K.; Nitschke J. R.; Gagliardi L.; Cramer C. J. Phys. Chem. Chem. Phys. 2014, 16, 10620–10628. 10.1039/c4cp01478b. [DOI] [PubMed] [Google Scholar]
- Lusi M.; Barbour L. J. Angew. Chem., Int. Ed. 2012, 51, 3928–3931. 10.1002/anie.201109084. [DOI] [PubMed] [Google Scholar]
- Sholl D. S.; Lively R. P. Nature 2016, 532, 435–437. 10.1038/532435a. [DOI] [PubMed] [Google Scholar]
- Jiménez A.; Bilbeisi R. A.; Ronson T. K.; Zarra S.; Woodhead C.; Nitschke J. R. Angew. Chem., Int. Ed. 2014, 53, 4556–4560. 10.1002/anie.201400541. [DOI] [PubMed] [Google Scholar]
- Clegg J. K.; Cremers J.; Hogben A. J.; Breiner B.; Smulders M. M. J.; Thoburn J. D.; Nitschke J. R. Chem. Sci. 2013, 4, 68–76. 10.1039/C2SC21486E. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.