

Abstract
There have been advances in personalized therapy directed by molecular profiles in lung adenocarcinoma, but not in lung squamous cell carcinoma (SCC). The lack of actionable driver oncogenes in SCC has restricted the use of small‐molecule inhibitors. Here, we show that SCC cell lines displayed differential sensitivities to belinostat, a pan‐histone deacetylase inhibitor. Phosphoproteomic analysis of belinostat‐treated SCC cells revealed significant downregulation of the MAPK pathway, along with the induction of apoptosis. In cisplatin‐resistant cells that demonstrated aberrant MAPK activation, combined treatment with belinostat significantly inhibited cisplatin‐induced ERK phosphorylation and exhibited strong synergistic cytotoxicity. Furthermore, belinostat transcriptionally upregulated the F‐box proteins FBXO3 and FBXW10, which directly targeted son of sevenless (SOS), an upstream regulator of the MAPK pathway, for proteasome‐mediated degradation. Supporting this, suppression of SOS/ERK pathway by belinostat could be abrogated by inhibiting proteasomal activity either with bortezomib or with siRNA knockdown of FBXO3/FBXW10. Taken together, these preclinical data offer a novel understanding of the epigenetic mechanism by which belinostat exerts its cytotoxicity and supports the combination with cisplatin in clinical settings for chemorefractory SCC tumors.
Keywords: histone deacetylase inhibitors, lung squamous cell carcinoma, MAPK inhibition, ubiquitin‐proteasome system
Abbreviations
- CI
combination index
- FBXO3
F‐box protein 3
- FBXW10
F‐box and WD repeat domain containing 10
- HDAC
histone deacetylases
- IC50
half‐maximum inhibitory concentrations
- MAPK
mitogen‐activated protein kinases
- NSCLC
non‐small‐cell lung cancer
- PARP
poly(ADP‐ribose) polymerase
- PI
propidium iodide
- PXD101
belinostat
- RT‐qPCR
quantitative real‐time polymerase chain reaction
- SCC
squamous cell carcinoma
- Scr
scrambled control siRNA
- SOS
son of sevenless
- STAT3
signal transducer and activator of transcription 3
- UBE2C
ubiquitin‐conjugating enzyme E2 C
1. Introduction
Modification of histones through epigenetic dysregulation is an important oncogenic mechanism and has been exploited for cancer drug development. Histone deacetylases (HDAC) are reported to be overexpressed or aberrantly recruited by oncoproteins (Minucci and Pelicci, 2006), which subsequently leads to cancer initiation and progression (Glozak and Seto, 2007; Hess‐Stumpp, 2005). Studies involving the knockdown of HDAC1 have shown that loss of HDAC1 expression blocked mitosis in tumor cells (Senese et al., 2007), while HDAC3 silencing reduces the growth of colon cancer cells (Wilson et al., 2006). Furthermore, inhibition of HDACs sensitizes both chemotherapy and small‐molecule inhibitors in drug‐resistant cancer cells (Bangert et al., 2011; Chen et al., 2013; Sharma et al., 2010). HDAC inhibitors have been developed clinically, of which romidepsin, vorinostat, and belinostat have received regulatory approval for the treatment of cutaneous and peripheral T‐cell lymphoma (Mann et al., 2007; O'Connor et al., 2015; Piekarz et al., 2009). These studies emphasize the critical role of HDACs in tumor cells and implicate the potential of HDAC inhibitor as a potent antineoplastic agent.
However, single‐agent activity of HDAC inhibitors has been disappointing in solid tumors, likely contributed by significant clinical toxicities that preclude continuous exposure and dose escalation to adequate concentrations required for activity (Lane and Chabner, 2009). Moreover, unlike tyrosine kinase‐targeting agents, there is no available biomarker to select patients who are likely to respond to HDAC inhibitors treatment. Despite this, HDAC inhibitors still pose to be an interesting therapeutic regime when used in combination, especially in lung cancer. Vorinostat has been shown to improve clinical response when added to both squamous and nonsquamous non‐small‐cell lung cancer (NSCLC) patients treated with paclitaxel and carboplatin, despite not significantly improving the progression‐free and overall survival rates (Ramalingam et al., 2010). Moreover, metastatic lung cells could be resensitized to EGFR inhibitors with HDAC inhibitors (Witta et al., 2006). However, the role of HDAC inhibitors in tumors lacking actionable driver oncogenes, such as lung squamous cell carcinoma (SCC), has not yet been explored.
Belinostat (PXD101) is a hydroxamic acid‐type pan‐HDAC inhibitor that has demonstrated strong antineoplastic activity in vitro (Lin et al., 2013; Plumb et al., 2003; Qian et al., 2006). Importantly, when compared to other HDAC inhibitors, belinostat seemingly produces tolerable adverse effects, while hematological toxicity is rare (Gimsing et al., 2008; Ramalingam et al., 2009; Steele et al., 2008; Yeo et al., 2012). Indeed, belinostat has progressed to late‐stage clinical development in multiple hematologic and solid malignancies (Gimsing et al., 2008; Mackay et al., 2010; Ramalingam et al., 2009). Despite the mixed outcome obtained from these clinical evaluations, the well‐tolerable adverse effects of belinostat have encouraged the study of combination regimens.
The current study highlights the therapeutic efficacy of belinostat in lung SCC cells and characterizes the downstream molecular signaling in belinostat‐treated cells. We have previously identified activation of MAPK/ERK as a mechanism of resistance to cisplatin (Kong et al., 2015). The increase in ERK phosphorylation upon cisplatin treatment was shown to be regulated by son of sevenless (SOS) upstream. The current study demonstrates the promising chemosensitizing characteristic of belinostat in lung SCC through the suppression of MAPK activity. We further studied the mechanistic actions of HDAC inhibition by analyzing the global gene expression profiling, and identified the possible role of ubiquitin‐proteasome pathway in the regulation of SOS/MAPK signaling.
2. Materials and methods
2.1. Cell lines and reagents
All lung cell lines were obtained directly from the American Type Culture Collection (ATCC, Manassas, VA, USA). Lung SCC lines H226, H2170, H520, H596, and ChaGo‐k‐1 were maintained in RMPI‐1640 (Nacalai Tesque), Calu‐1 in McCoy's 5a (Nacalai Tesque, Kyoto, Japan), SK‐MES‐1 in DMEM (Nacalai Tesque), SW900 in Leibovitz’s medium (Gibco, Life Technologies, Carlsbad, CA, USA), H1869 in ACL‐4 (Gibco, Life Technologies), and H2066 in HITES medium (Gibco, Life Technologies). Normal lung fibroblast cells (MRC‐5, IMR‐90, and WI‐38) were cultured in EMEM (Gibco, Life Technologies). All media were supplemented with 10% fetal bovine serum, 2 mm l‐glutamine, 100 μg·mL−1 streptomycin, and 100 U·mL−1 penicillin. ACL‐4 and HITES were further supplemented with additional nutrients as recommended by the ATCC. All cell lines were authenticated with GenePrint® 10 System (Promega, Fitchburg, WI, USA).
For inhibitor studies, belinostat (PXD101), bortezomib (Velcade), GDC0879, PD0325901, RDEA119, and GSK1120212 were obtained from Selleck Chemicals (Houston, TX, USA); cisplatin from Hospira (Lake Forest, IL, USA); MG‐132 from Sigma (St. Louis, MO, USA); and cetuximab was from Merck (Darmstadt, Germany).
2.2. Cell viability assay and synergism analysis
Cells were cultured (2000–4000 cells/well) on microtiter culture plates and treated with various concentrations of compounds for 72 h. At assay endpoint, cells were incubated with CellTiter 96® AQueous One Solution (MTS) solution (Promega) at 37 °C for 3 h. Absorbance was measured at 490 nm. All data points were set up with three replicates for each experiment. Percent cell proliferation was calculated relative to DMSO control‐treated cells.
The sigmoidal dose–response curve fitting for belinostat and the half‐maximum inhibitory concentration (IC50) were calculated by graphpad prism software (GraphPad Software, La Jolla, CA, USA). Combination index (CI) for belinostat and cisplatin was calculated based on Loewe additivity equation (Chou and Talalay, 1984), using the drug concentrations that resulted in the inhibition of cell viability between 50% and 80%, and tabulated at ED50‐80.
2.3. Proteome profiler antibody array
Whole‐cell lysates from vehicle‐ or belinostat‐treated cells were prepared 48 h after drug incubation and normalized according to protein concentrations. Cell lysates (250 μg) were mixed with biotinylated detection antibodies, and Proteome Profiler Phospho‐MAPK arrays (R&D Systems, Minneapolis, MN, USA) were performed. Chemiluminescence signal detection was performed, and densitometric data were analyzed with imagej (NIH, Bethesda, MD, USA). The analyses were performed by subtracting the background density signal, and positive control signals were used as internal control. Comparisons were made between the vehicle‐ and belinostat‐treated cells and expressed in fold change to vehicle control group.
2.4. Total protein extraction and western blotting
Vehicle‐ or compound‐treated cells were lysed 48 h after drug incubation with Cell Lytic buffer (Sigma; 150 mm NaCl, 0.41% bicine, 2% EDTA) supplemented with complete protease inhibitor cocktail (Roche, Basel, Switzerland) and phosphatase inhibitor cocktail (Roche). Protein concentration was quantified with BCA assay, and normalized samples were resolved with Bio‐Rad SDS/PAGE system on 8–12% protein gels. Signal detection was performed with chemiluminescence detection system (GE Healthcare, Chicago, IL, USA).
Poly(ADP‐ribose) polymerase (PARP), caspase 3, p‐ERK1/2 (Thr202/Tyr204), total ERK1/2, p‐p38 (Thr180/Tyr182), total p38, p‐BRAF (Ser445), p‐MEK1/2 (Ser217/221), total MEK1/2, SOS1, acetyl‐H3 (Lys9/14), p‐STAT3 (Ser727), total STAT3, UBE2C, HDAC1, HDAC2, HDAC3, HDAC4, HDAC5, Sirt1, β‐actin, and HRP‐conjugated anti‐rabbit were obtained from Cell Signaling (Danvers, MA, USA); SOS2 was purchased from Abcam (Cambridge, UK); FBXO3 was purchased from Sigma Aldrich (St. Louis, MO, USA); FBXW10 was purchased from Novus Biologicals (Littleton, CO, USA).
2.5. Apoptosis assay
Annexin‐V and propidium iodide (PI) staining was carried out according to the manufacturer's protocol with slight modifications (Thermo Fisher, Waltham, MA, USA). Briefly, 1–1.5 × 105 cells were treated with belinostat for 48 h. On day of assay, both floating and adherent cells were collected. Annexin‐V‐APC (Thermo Fisher) was used as a probe for apoptosis, while PI (BD Biosciences, Franklin Lakes, NJ, USA) as an indicator for dead cells. Cells were stained in 100 μL Annexin‐V binding buffer containing both Annexin‐V‐APC and PI in the dark for 15 min at room temperature. The staining profile was collected and analyzed using LSR II Flow Cytometer (BD Biosciences).
2.6. Data preprocessing of Affymetrix microarray gene expression
Microarray gene expression data of eight pairs of control belinostat‐treated lung cancer cell lines were performed on Affymetrix Gene 1.0 ST and are deposited in Gene Expression Omnibus with the accession ID GSE85979. Robust multichip average normalization was performed using Affymetrix Powertool v1.18.0 (Santa Clara, CA, USA). All arrays were checked for quality: dabg, PM_MEAN, POS_vs_NEG_AUC, and passed the recommended quality criteria.
2.7. RT2 profiler and quantitative real‐time polymerase chain reaction (RT‐qPCR)
Total RNA was prepared using RNeasy according to the manufacturer's protocol (Qiagen, Venlo, Netherlands). Reverse transcription PCR for the conversion of cDNA was performed according to the recommended protocols for Taqman ® (Applied Biosystems, Foster City, CA, USA) and RT2 First Strand kit (SABiosciences, Venlo, Netherlands), respectively.
The human Ubiquitination Pathway RT2 Profiler PCR array (SABiosciences) was used to assess the regulation of ubiquitin‐proteasome‐related genes upon belinostat treatment. The expressions of 84 key genes associated with the ubiquitination pathway were quantified according to the manufacturer's protocol. Data shown represent the mean of two replicates and were normalized to multiple housekeeping genes. qPCR was performed using either SYBR Green or Taqman ® system, and the primer sequences are listed in Table S1. GAPDH was applied as housekeeping gene.
2.8. Anchorage‐independent soft agar assay
Soft agar was mixed with culture media to form multiple agar layers: a bottom layer with 0.6% agar; a middle layer with 0.36% agar and resuspended with 5000–10 000 cells; and a top layer with complete media containing belinostat, cisplatin, or belinostat / cisplatin combination at various doses. Colonies were allowed to form for 2–4 weeks. On assay endpoint, viable colonies were stained with MTT solutions (5 mg·mL−1 in PBS) at 37 °C for 4 h. Images of each well were acquired with Epson V330 Photo scanner. The number and size of the colonies were analyzed and quantified using imagej (NIH). Percentage cell colony formation was calculated relative to DMSO control‐treated cells.
2.9. RNA interference
For gene knockdown, Stat3 was obtained from Ambion (Thermo Fisher Scientific, Waltham, MA, USA). FBXO3 siRNA (sequence: 5′‐GACGAUUAUCGAUGUUCAUTT‐3′), FBXW10 siRNA (sequence: 5′‐CUCCGGUCUAUAUCCGAAATT‐3′), and AllStar scrambled control siRNA (scr siRNA) were obtained from Qiagen. Transfection (50 nm siRNA for each target in each reaction) was conducted with JetPRIME reagent (Polyplus Transfection, Strasbourg, France).
2.10. Xenograft studies
All in vivo studies adhered to the Institutional Animal Care and Use Committee (IACUC) guidelines on animal use and handling. Calu‐1 xenograft model was established and maintained in 8‐ to 10‐week‐old female SCID mice. In brief, 10 × 106 Calu‐1 cells in 100 μL of PBS were injected subcutaneously into the flanks of each mice. Treatment began when the tumor sizes reached approximately 200 mm3; the mice were assigned into four stratified groups based on average tumor volume: vehicle (1% w/v polysorbate 80), cisplatin, belinostat, belinostat + cisplatin (n = 5 animals/10 tumors per group). Cisplatin was provided in 0.9% NaCl and given at 4 mg·kg−1 by intraperitoneal (ip), once weekly for 3 weeks. Belinostat was formulated in 30% (w/v) Captisol and 30% (w/v) PEG300 at 40 mg·kg−1 and given via PO (orally), 5 days a week, until mice were sacrificed. Tumor growth was monitored and measurements were taken three times weekly (tumor volume was estimated by [length × width × width × 3.14159/6]). Bodyweight at sacrifice and tumor samples were collected upon reaching experiment endpoint when mice were sacrificed 3 weeks after treatment commenced.
2.11. Histone extraction
Vehicle‐ or belinostat‐treated cells were lysed 48 h after drug incubation with Triton extraction buffer (PBS containing 1% Triton X‐100 (v/v), 10 mm Tris/HCl, 50 mm sodium bisulfite, 100 mm MgCl, and 8.6% sucrose pH 6.3) and subjected to Dounce homogenization. Cell nuclei were collected by centrifugation, and pellets were washed several times with Triton extraction buffer. The histones were acid‐extracted for an hour at 4 °C with 0.4 N sulfuric acid. Next, the supernatants were collected with centrifugation and mixed with acetone overnight at −20 °C. Finally, histones were pelleted at 14 000 rpm, air‐dried at room temperature, and dissolved in water.
2.12. Statistical analysis
All experiments were conducted three times unless stated otherwise. Results are presented as mean ± SD. Statistical analysis for the comparison between two groups was conducted using Student's t‐test, while comparisons between multiple groups were made using ANOVA. All tests were two‐sided, and the significance level was set at P < 0.05.
3. Results
3.1. Characterizing the antitumor effects of belinostat in lung SCC cells
To determine the sensitivity of HDAC inhibitors in SCC cells, we screened a panel of 10 lung SCC cells and three normal fibroblast cells against belinostat to determine the half‐maximum inhibitory concentrations (IC50). All three lung fibroblast cells were shown to have similar toxicity to belinostat (IC50 ~ 3 μm). In order to avoid normal tissue toxicity, we defined lung SCC cells with IC50 < 3 μm as belinostat sensitive, while those above this cutoff as belinostat resistant (Fig. 1A). Interestingly, lung SCC cells (H2066, SW900, H2170, Calu‐1, H520, and SK‐MES‐1), which we previously demonstrated to exhibit decreased sensitivity to cisplatin, were highly sensitive to belinostat (Kong et al., 2015). The reciprocally inverse sensitivity among belinostat and cisplatin suggested the potential for synergistic combination. Our combination index (CI) analysis revealed that belinostat synergized with cisplatin in most cell lines, with the most pronounced synergy observed in SK‐MES‐1, Calu‐1, and H520 cells (CI < 0.6) (Fig. 1B). However under anchorage‐independent growth, belinostat showed additive effect when used in combination with cisplatin (Fig. S1A), with average CI (ED50‐ED80) in between 0.82 and 1.19 (Fig. S1B). Taken together, these results suggest that belinostat regulates chemosensitivity in lung SCC cells, and are consistent with previous reports on HDAC inhibitors (Ramalingam et al., 2010).
Figure 1.
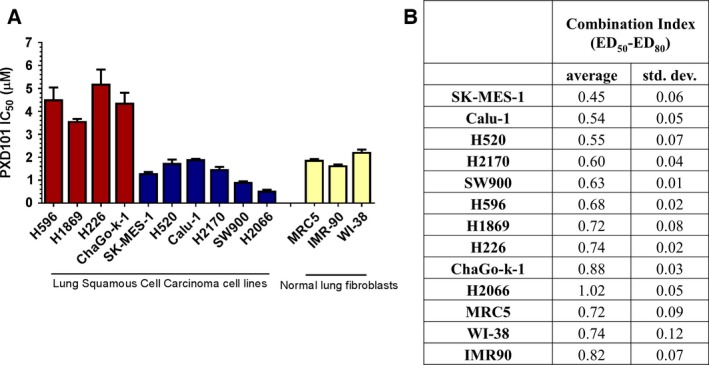
Belinostat demonstrates efficacy in lung squamous cell carcinoma (SCC) cells. (A) Lung SCC cell lines and normal lung fibroblast cell lines were treated with belinostat (PXD101) for 72 h, and cell viability was determined with CellTiter assay. Data are represented as mean IC 50 ± SD (n = 3). (B) Lung SCC cell lines and normal lung fibroblast cell lines were treated with belinostat (PXD101) and cisplatin for 72 h using constant ratio at IC 50 ( PXD 101)/IC 50 ( CDDP ) combination, and cell viability was determined with CellTiter assay. The combination index (CI) values of belinostat and cisplatin between effective doses of 50–80% were calculated from isobologram analysis. Values were shown as average CI ± SD (n = 3). Combination index of CI < 0.8 indicates synergism, 0.8 < CI < 1.2 indicates additive effect, and CI > 1.2 indicates antagonism.
3.2. Suppression of MAPK signaling is associated with sensitivity to belinostat
HDAC inhibitors are epigenetic modulators that often induce growth inhibition in cancer cells. To investigate the antitumor mechanisms in the context of lung SCC, we examined the transcriptomic and kinomic changes in belinostat‐treated cells. Gene expression profiling was conducted to compare the significantly altered genes after exposure to belinostat. However, transcriptomic analyses offered little insight into the mechanism of action of belinostat, as gene ontology and pathway enrichment analyses could not identify differentially regulated signaling pathway across the belinostat‐sensitive (H2066, SW900, H2170, and Calu‐1) and belinostat‐resistant (H226, H596, ChaGo‐k‐1, and H1869) cells (data not shown).
Next, phosphokinases arrays covering multiple crucial phosphoproteins were performed on belinostat‐sensitive H520 and Calu‐1 cells (Fig. 2A). The signal intensities of each pair of duplicate targets were quantified, and the expression levels in belinostat‐treated cells were compared to the DMSO control. Across these two cell lines, perturbations were observed in the phosphoprotein expressions of p38, ERK1/2, JNK, GSK3α, AMPK, AKT, TOR, c‐JUN, RSK, CHK‐2, PRAS40, STAT2, STAT3, STAT5, and STAT6 (Fig. 2B). Interestingly, belinostat consistently suppressed the phosphorylation of MAP kinases (p38, ERK, and JNK), while it increased STAT3 phosphorylation, in both Calu‐1 and H520 cells (Fig. 2B).
Figure 2.
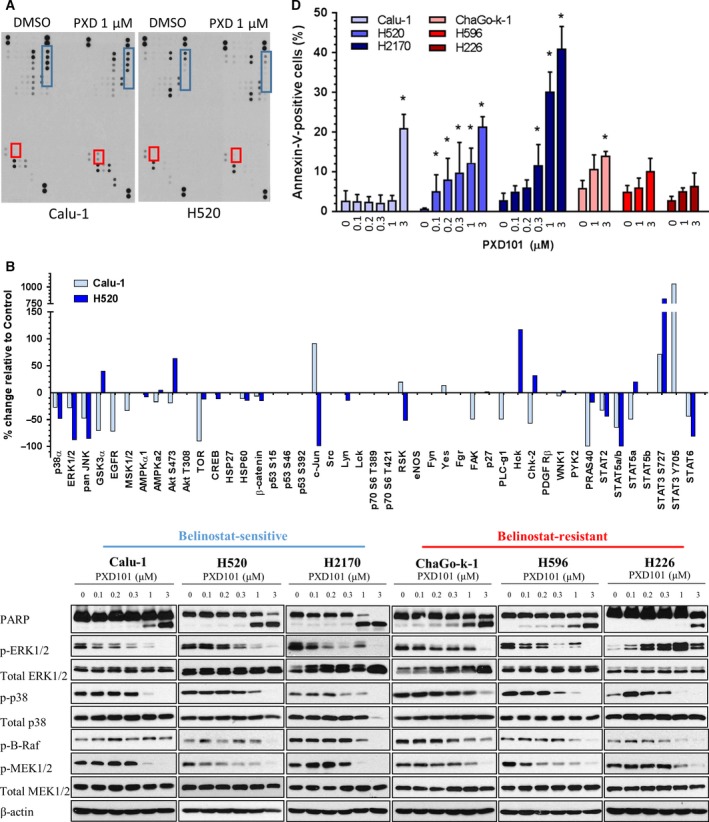
Belinostat suppresses MAPK signaling and triggers apoptosis in lung squamous cell carcinoma (SCC). (A) Calu‐1 and H520 cells were treated with vehicle or 1 μm belinostat (PXD101) for 48 h with the protein lysates harvested for phosphokinase profiling. Dots representing consistently elevated targets are highlighted in red (STAT3), and suppressed kinases are highlighted in blue (p38, ERK1/2, JNK). (B) Densitometry analysis on the phosphokinase profiling was performed as described in Materials and methods. The average values of the significantly regulated duplicate were shown. (C) Cell lysates were harvested from lung SCC cells after treatment with increasing doses of belinostat (0.1, 0.2, 0.3, 1, 3 μm). Immunoblotting was performed to evaluate the changes in phosphorylated protein levels of the targets identified in 1D (ERK1/2, p38, B‐Raf, MEK1/2) as well as PARP. β‐Actin shown as loading control. (D) Lung SCC cells were treated with vehicle or belinostat (0.1, 0.2, 0.3, 1, 3 μm for Calu‐1, H520, and H2170; 1, 3 μm for ChaGo‐k‐1, H596, and H226) and stained with Annexin‐V/propidium iodide (PI). The percentage of Annexin‐V‐positive cells was shown as mean ± SD. *P < 0.05.
We sought to verify the changes in kinase phosphorylation by western blotting in independent cell lysates of belinostat‐sensitive (Calu‐1, H520, H2170) and belinostat‐resistant cell lines (H226, H596, ChaGo‐k‐1). SCC cells were treated with increasing doses of belinostat that fall within the clinically achievable plasma concentration (Yeo et al., 2012). Of note, belinostat treatment abrogated the phosphorylation of ERK1/2 and p38 in all SCC cells, except for the highly resistant H226 cells (Fig. 2C). Consistently, upstream kinases of ERK1/2, MEK1/2, and B‐Raf were inhibited by increasing doses of belinostat. As activation of the MAP kinase pathway limits the induction of apoptosis, we looked to see whether apoptosis was induced in belinostat‐treated cells. As expected, treatment with belinostat increased PARP cleavage, a marker of apoptosis (Fig. 2C, top). Furthermore, belinostat dose‐dependent treatment significantly increased Annexin‐V‐positive populations in belinostat‐sensitive cells (Calu‐1, H520, and H2170), but not in belinostat‐resistant cells (ChaGo‐k‐1, H596, and H226) (Fig. 2D).
The proapoptotic Bim protein has been well characterized to be negatively regulated by MAPK signaling (Luciano et al., 2003). We next asked whether Bim is involved in belinostat‐induced apoptosis, and showed that belinostat significantly elevated Bim expression in belinostat‐sensitive cells (Calu‐1, H520), but to a lesser extent in H226 cells (Fig. S4A). Accordingly, silencing of Bim partially reduced apoptosis in belinostat‐treated Calu‐1 and H520 cells, as shown by the decrease in Annexin‐V‐positive cells (Fig. S4B). Collectively, these data suggest that treatment with belinostat induces cell death in lung SCC cells, possibly through the suppression of MAPK signaling and Bim regulation.
Interestingly, STAT3 phosphorylation was enhanced in belinostat‐treated Calu‐1 cells (Fig. S2A), likely through the increase in transcripts of STAT3‐related cytokines (Fig. S2B). This is in concordance with data from a recent study showing that MEK inhibition triggers autocrine activation of JAK/STAT3 pathway in oncogene‐addicted cells as a positive feedback mechanism (Lee et al., 2014). Likewise, treatment with the MEK inhibitors (PD0325901, RDEA119, and trametinib), but not the B‐Raf inhibitor (GDC0879) or the EGFR inhibitor (cetuximab), significantly suppressed ERK activation and enhanced STAT3 phosphorylation in Calu‐1 cells (Fig. S2C). However, in the context of SCC cells, STAT3 activation likely does not provide a significant survival advantage as STAT3 silencing did not augment belinostat‐induced PARP cleavage in Calu‐1 cells (Fig. S2D). Nevertheless, these observations strengthen our claim that belinostat treatment attenuates MAPK signaling, which in return triggers positive STAT3 activation as a feedback mechanism.
3.3. Belinostat blocks SOS/MAPK activation in cisplatin‐resistant cells
Son of sevenless is a guanine nucleotide exchange factor that regulates the activation of Ras‐GTPases and subsequently induces MAPK signaling (Gureasko et al., 2008; Rogge et al., 1991). It has been shown previously that SOS upregulation leads to cisplatin resistance in lung SCC cells through MAPK/ERK pathway activation (Kong et al., 2015). As belinostat demonstrated strong inhibition of MAPK signaling, we therefore investigated the expression status of SOS upon exposure to belinostat. We observed suppression of SOS proteins, particularly SOS2, in all SCC cell lines irrespective of their belinostat sensitivity (Fig. 3A). We therefore postulated that tumor cells with aberrant activation of MAPK signaling, such as those resistant to cisplatin, will be sensitized by belinostat. Cisplatin was used at a concentration (10 μm) below the maximum tolerated dose (van Hennik et al., 1987). Accordingly, we showed that combined treatment with 1 μm belinostat significantly suppressed cisplatin‐induced ERK1/2 phosphorylation in SCC, particularly H520 cell line (Fig. 3B). Furthermore, combined treatment with cisplatin and belinostat resulted in increased apoptosis as demonstrated by the augmentation in PARP and caspase 3 cleavage (Fig. 3B), together with potentiation in Annexin‐V‐positive cells (Fig. 3C). It is of note that with 1 μm belinostat, synergistic cytotoxicity was more significant in H520 cells. This potentiation in cytotoxicity positively correlated with the extent of p‐ERK1/2 suppression and was associated with downregulation of both SOS1 and SOS2 upon combined treatment with cisplatin and belinostat (Fig. 3D). Collectively, these data suggest a potential chemosensitization effect of belinostat through the inhibition of cisplatin‐induced SOS/MAPK activation in lung SCC cells.
Figure 3.
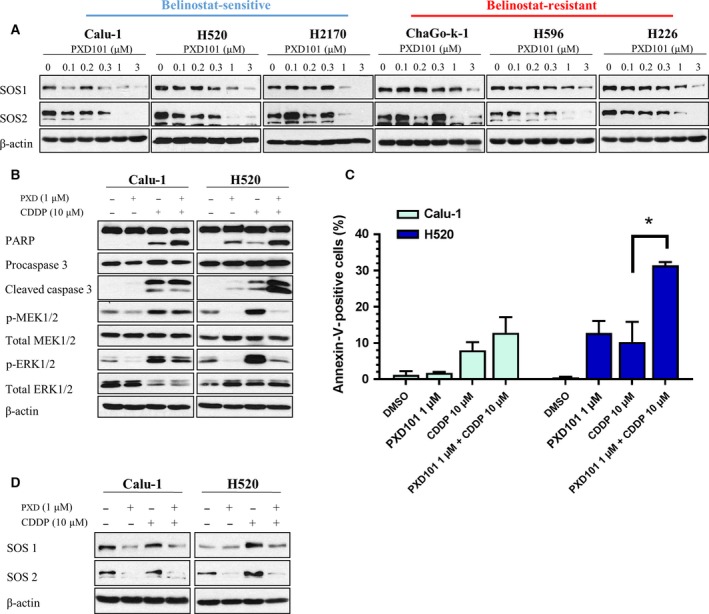
Suppression of MAPK signaling by belinostat is associated with downregulation of SOS and leads to synergistic cytotoxicity in cisplatin‐resistant cells. (A) Cell lysates were harvested from lung SCC cells after treatment with increasing doses of belinostat (PXD101) (0.1, 0.2, 0.3, 1, 3 μm) to evaluate the changes in SOS1 and SOS2. The combinatory effects of belinostat (1 μm) and cisplatin (10 μm) on (B) PARP, caspase 3, MEK, and ERK; and (D) SOS1 and SOS2 were examined with immunoblotting, (C) while the effects on apoptosis were evaluated with Annexin‐V/propidium iodide (PI). β‐Actin shown as loading control. Data were shown as mean ± SD. *P < 0.05.
To further elucidate the molecular underpinnings of SOS downregulation through HDAC inhibition by belinostat, we hypothesize that histone acetylation by belinostat regulates transcriptional events leading to SOS downregulation. Belinostat significantly induced acetylation of histone H3, as early as 6 h post‐treatment (Fig. 4, top). This observation was accompanied by the downregulation of SOS and decreased phosphorylation of MAP kinases within 48 h after belinostat treatment in both Calu‐1 and H520 cells (Fig. 4, bottom). In concordance with the reduction in acetylated H3, the inhibitory effects on MAPK signaling faded at 72 h post‐treatment in Calu‐1 cells. These temporal changes indicate the possibility of epigenetic‐regulated post‐transcriptional events that could impact the dynamics of SOS/MAPK signaling.
Figure 4.
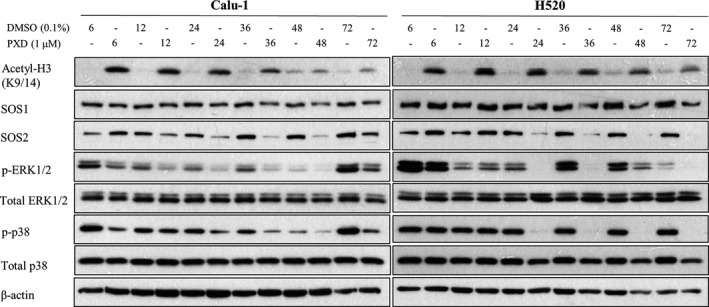
Belinostat induces acetylation of histone H3 and downregulates MAPK pathway progressively. Kinetic studies of belinostat (PXD101) treatment (1 μm) were performed over 72 h (6, 12, 24, 36, 48, 72 h) in Calu‐1 and H520 cells. The effects on acetylated histone H3, SOS1, SOS2, ERK, and p38 were evaluated with immunoblotting. β‐Actin shown as loading control.
3.4. Induction of the ubiquitin ligases FBXO3 and FBXW10 in response to belinostat treatment in lung SCC
We first investigated the possibility of transcriptional perturbations through histone acetylation induced by belinostat to explain SOS downregulation. However, SOS1 and SOS2 mRNA expressions were not reduced following belinostat treatment (Fig. 5A). An alternative mechanism of SOS downregulation involving proteasomal degradation was explored. Through global gene expression analysis of belinostat‐treated cells, we derived gene sets to determine the possible involvement of ubiquitin‐proteasome pathway in the suppression of SOS in belinostat‐treated cells. Gene sets comprising ubiquitin‐related genes (657) as annotated by Molecular Signature Database (Msigdb.v5.0) were compiled and mapped to transcriptional changes in SCC cell lines induced by exposure to belinostat for 8 h. Expression values were derived relative to the DMSO control samples. The transcriptomic profiles of both belinostat‐tolerant (H226, H596, ChaGo‐k‐1, H1869) and belinostat‐sensitive (Calu‐1, H2170, SW900, H2066) cells were mapped to the curated and computational gene sets (GSEA c2, c4, and c5) (Table S2). Among the genes that appeared in the Affymetrix Gene 1.0ST platform, we filtered out and retained those that were regulated by a log2 fold change of > 0.5 in at least one cell line, and performed supervised cluster analysis (Fig. 5B). Interestingly, an ubiquitin‐related gene signature was significantly affected upon HDAC inhibition in both sensitive and resistant cell lines with 391 altered genes, but no apparent pattern that could be correlated with drug response. Of these, we identified clusters of genes that were consistently upregulated (54) or downregulated (91) upon belinostat treatment across all SCC cells. There was no significant perturbation of SOS1 and SOS2 in SCC cells within the same transcriptomic dataset, except for SW900 (Fig. 5C), which is consistent with the qPCR analysis shown in Fig. 5A.
Figure 5.
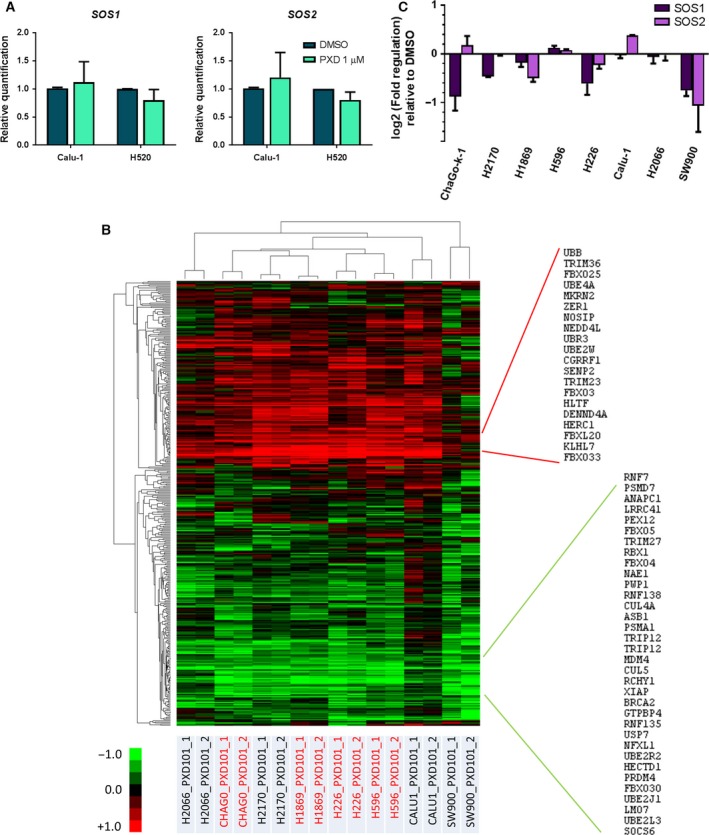
Ubiquitin‐related genes, but not SOS 1 and 2, are transcriptionally regulated by belinostat in lung squamous cell carcinoma (SCC) cells. (A) Real‐time quantitative PCR (qPCR) was conducted to quantify the mRNA expression of SOS1 and SOS2 after vehicle or belinostat (PXD101) (1 μm) treatment. Data were represented as average relative quantification ± SD (n = 3). (B) Heat map represents the gene expression of altered genes (fold change > 1.5) identified by Affymetrix microarrays across all lung SCC cells (H596, H1869, H226, and ChaGo‐k‐1, Calu‐1, H2170, SW900, and H2066) treated with belinostat at respective IC 50. Consistently upregulated genes were highlighted in red, and downregulated genes in green. (C) Expressions of both SOS1 and SOS2 from the same array were tabulated in the bar chart as average fold regulation ± SD.
We verified the clusters of gene panels derived from the microarray using RT2 Profiler PCR Array specifically targeting the expression of well‐established ubiquitination‐related genes encoding for E1, E2, and E3 enzymes. The gene expression changes were measured in Calu‐1 cells 24 h after belinostat treatment, when the inhibition of SOS/MAPK signaling was first observed (Fig. 4). The significantly altered genes were identified from the array (Fig. 6A) and validated by qPCR in both Calu‐1 and H520 cells (Fig. 6B). Among these genes, FBXO3, FBXW10, and UBE2C were identified as commonly altered genes (Fig. 6B). Moreover, exposure to 1 μm belinostat increased protein expressions of FBXO3 and FBXW10, but decreased UBE2C, in both Calu‐1 and H520 cells, providing concordance to the RT2 gene expression profile (Fig. 6C). Similar observations were made in belinostat‐resistant H226 cells (Fig. 6C). Taken together, the observations suggest a significant mechanism of functional protein regulation that occurs through epigenetic modulation of ubiquitin‐proteasome pathways by belinostat treatment. Moreover, these results may explain the downregulation of MAPK signaling through ubiquitination and degradation of SOS proteins.
Figure 6.
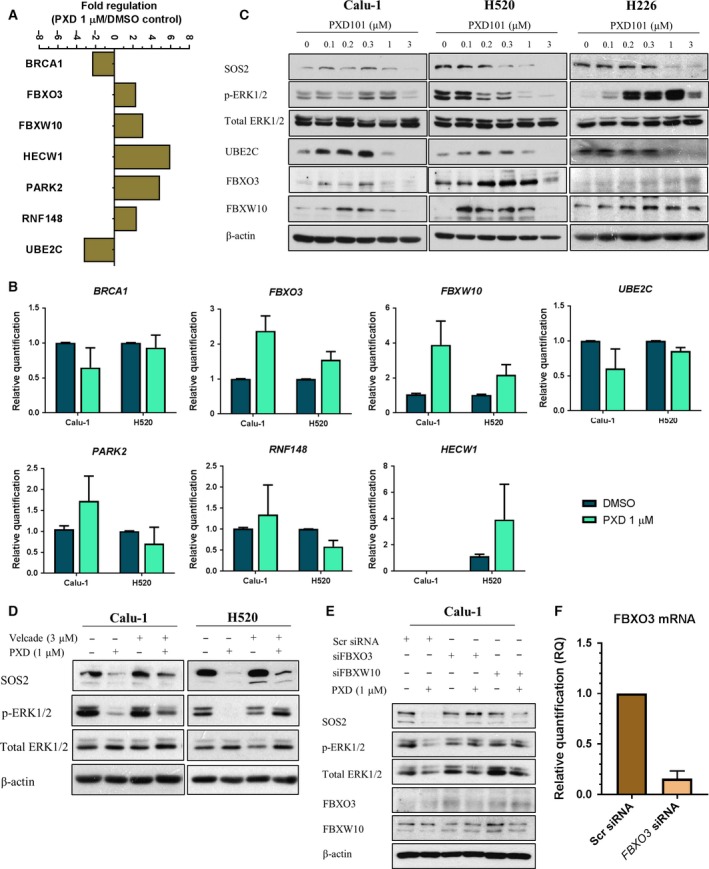
Regulation of ubiquitin gene expression profile upon belinostat treatment is associated with suppression of SOS/MAPK and could be rescued by bortezomib treatment. (A) Calu‐1 cells were treated with vehicle or belinostat (PXD101) (1 μm). Total mRNA transcript levels of 84 key ubiquitination genes were assessed with RT2 Profiler PCR Array. Data were presented as bar chart representing the relative value of the significantly altered genes (> twofolds) as compared to vehicle (n = 1). (B) The significantly altered genes identified from RT2 Profiler were verified with real‐time quantitative PCR (qPCR) in Calu‐1 and H520 cells treated with vehicle or belinostat (PXD101) (1 μm). Data were represented as average relative quantification ± SD (n = 3). (C) The genes regulated in the similar manner identified from qPCR were verified with immunoblotting in Calu‐1, H520, and H226 cells after treatment with vehicle or belinostat (1 μm). (D) Immunoblotting was performed to evaluate the effects of bortezomib (3 μm) on SOS2 and p‐ERK1/2 in belinostat‐treated cells. (E) Immunoblots show the effects of scrambled (Scr), FBXO3, or FBXW10 siRNA knockdown on affecting the modulation of belinostat on SOS/MAPK in Calu‐1 cells. (F) Knockdown efficiency of FBXO3 in Calu‐1 cells was confirmed with RT‐qPCR (n = 2). Fifty nanomolar siRNA was used per transfection. β‐Actin shown as loading control.
To further validate this hypothesis, expression of SOS1/2 and ERK phosphorylation was studied after belinostat treatment after pharmacological inhibition of proteasomal degradation by bortezomib. The kinetic studies for bortezomib have been previously reported, with strong dose‐dependent inhibition on 20S proteasome activity observed within 24 h after treatment (Adams et al., 1999). Here, we combined 3 μm bortezomib in belinostat‐treated cells and demonstrated a rescue in the SOS2 expression and prevented ERK1/2 dephosphorylation (Fig. 6D). Importantly, similar results were observed in Calu‐1 cells transfected with siRNA targeting either FBXO3 or FBXW10 (Fig. 6E,F). These results strengthen our claim that suppression of SOS/MAPK is associated with ubiquitin‐mediated proteasomal degradation pathway following belinostat treatment.
4. Discussion
The mechanism of synergistic activity of HDAC inhibitor with chemotherapy is poorly understood. Several mechanisms have been proposed, including upregulation of tumor suppressor genes like p53, as well as inhibition of the aggresome (Condorelli et al., 2008; Mishima et al., 2015). Aberrations in the transcriptome of tumor cells are often controlled by chromatin remodeling and histone modifications (Glozak and Seto, 2007; Hess‐Stumpp, 2005). HDACs are often inappropriately expressed in cancer cells, with contentious roles in the development of oncogenesis. For instance, class II HDACs have been shown to repress oncogenes, and the reduced expression of HDAC5 and HDAC10 favored malignant progression in NSCLC (Osada et al., 2004). On the contrary, high expression of HDAC1 has been linked with poor prognosis in patients with lung adenocarcinoma (Minamiya et al., 2011). However, which HDACs are coregulated to augment cytotoxicity to cancer cells, particularly here in lung SCC tumors, remains elusive. We propose that belinostat synergizes cisplatin in platinum‐resistant SCC cells through upregulation of genes that regulate ubiquitin‐mediated proteasomal degradation leading to disruptions of key survival signaling molecules.
We initially screened a panel of 10 SCC cells and revealed a range of sensitivities to belinostat. Increasing doses of belinostat induced apoptosis in SCC cells, which is consistent with previous observations analyzing the effects of other HDAC inhibitors (Moore et al., 2004; Zhang et al., 2004). It is important to note that these differential sensitivities were not concordant with basal expression of HDACs, apart from HDACs 3 and 6 (Fig. S3); hence, alternative mechanisms were postulated to be responsible for the efficacy of belinostat in respective SCC cells. Through a phosphoproteomic profiling screen, we found that belinostat has a strong inhibitory effect on MAPK signaling (ERK1/2 and p38). Mechanistic studies demonstrated that various MAP kinases (MEK and B‐Raf) exhibited decreased phosphorylation levels, and expression of SOS was reduced following belinostat exposure. In contrast, STAT3 activity was enhanced. Lee et al. (2014) have previously demonstrated that MEK inhibition induces activation of STAT3 through a feedback mechanism, therefore supporting a strong mechanistic link between HDAC inhibition and MAPK signaling pathways (Vultur et al., 2014).
The expanding understanding on the chromatin architecture has described the post‐translational modifications of histone tails as a targeted therapeutic approach. HDAC inhibitors are known to augment the acetylation of lysine residues associated with histones and promote a relaxed chromatin structure to perturb gene transcriptions as a mean of shifting equilibrium toward proapoptosis (Moore et al., 2004; Struhl, 1998). We hypothesize that this could indirectly affect expression of SOS. While HDAC inhibitors have been reported to resensitize erlotinib‐resistant cells by transcriptionally reducing EGFR expression (Chen et al., 2013; Liffers et al., 2016), SOS mRNA levels were found to be unchanged following the addition of belinostat even though belinostat dose dependently induced acetylation of histones H3 and H4 (Fig. S5). However, overall SOS1 and SOS2 protein expression was reduced in belinostat‐treated cells, suggesting that belinostat may regulate SOS protein expression through a post‐transcriptional event. The lack of consistency between the protein and mRNA expressions of SOS prompted us to investigate more closely the transcriptional perturbations elicited by belinostat in SCC cells. In doing so, we identified a potential role of the ubiquitin‐proteasome pathway in these effects.
Collectively, transcriptional profiling of microarray data with subsequent RT‐qPCR validation showed enhanced expressions of a number of ubiquitin‐proteasome genes including members of the F‐box protein family (FBXW10, FBXO3) and E3 ligase components (FBXO3, BRCA1), as well as reduced expression of the ubiquitin‐conjugating enzyme (UBE2C), after belinostat treatment. The role of F‐box proteins has been implicated as an important component in tumor progression and development due to their direct regulation on various oncogenic signaling, and pertinently, they have been demonstrated to be epigenetically regulated. For instance, FBXW10 gene is found to be hypermethylated in clear cell renal cell carcinoma (Wang et al., 2015), while FBXO3 influences TGF‐β signaling by targeting SMURF1 for proteasomal degradation (Li et al., 2015). This epigenetic regulation of the ubiquitin‐proteasome pathway relevant to oncogenesis affords therapeutic opportunity by HDAC inhibition and possibly DNA methylation inhibitors, and our work in lung SCC cells demonstrates this. We further validated this using both chemical and genetic means whereby the depletion of SOS by belinostat was prevented through the addition of the proteasomal inhibitor bortezomib or siRNA knockdown of FBXO3 and FBXW10. However, it is important to note that combination therapies using inhibitors of proteasomes and HDACs are currently being explored (Bhatt et al., 2013; Pei et al., 2004). Similarly, we demonstrate that, despite observing the partial restoration of SOS/MAPK at early time point, prolonged exposure to bortezomib/belinostat combination leads to cell death (data not shown), which is likely due to the inhibition of aggresome formation in cells treated with bortezomib (Mishima et al., 2015). Nonetheless, we showed that silencing of either FBXO3 or FBXW10 could partially rescue cell viability under exposure to 1 μm belinostat in H520, but not in H226 cells, thus highlighting the role of F‐box protein in facilitating the cytotoxicity of belinostat (Fig. S6). Further studies will be required to elucidate the crosstalk between HDAC inhibition and the proteasome system.
Platinum‐based chemotherapies remain as the standard‐of‐care treatment for the past decades in lung SCC (Wang and Lippard, 2005), while small‐molecule kinase inhibitors have demonstrated limited efficacy to date, thus emphasizing the urgent need for a more effective treatment strategy for this disease. The present study shows that a belinostat/cisplatin combination is broadly effective across a panel of lung SCC cell lines, with a strong inhibition of MAPK and SOS. Our group has previously described the significance of SOS/MAPK activation in cisplatin resistance (Kong et al., 2015). On the basis of this study, we speculate that belinostat treatment promotes ubiquitin‐proteasome activity in SCC cells; however, cisplatin‐resistant cells that exploit SOS/MAPK activation for survival are more susceptible to belinostat treatment. The strong synergistic cytotoxicity indicates the potential of belinostat as a chemosensitizing agent especially in the treatment for chemorefractory tumors. Nonetheless, clinical trials on HDAC inhibitors have yet to achieve superior progress in advanced solid malignancies (Bradley et al., 2009; Mackay et al., 2010), largely due to the poor bioavailability and potency (Elaut et al., 2007). Our preliminary xenograft studies have shown a lack of antitumor activity when a tolerable dose of belinostat (40 mg·kg−1) was used singly or in combination with cisplatin (Fig. S7A–D). This could be reasoned by the poor pharmacokinetics of belinostat, as shown by the failure to induce histone H3 acetylation in the tumor mass (Fig. S7E). Therefore, the clinical application of belinostat still requires much fine tuning, both in terms of drug delivery and in terms of therapeutic window.
5. Conclusion
In summary, we have described a novel mechanism of belinostat sensitivity against lung SCC, a disease in which small‐molecule inhibitors have mostly failed due to the lack of actionable driver oncogenes. This mechanism involves the disturbance of the expressions of ubiquitin‐related proteins, which influences the activity of key survival signals, leading to an increase in cellular apoptosis. In the context of SCC, belinostat treatment triggers the proteasomal degradation of SOS proteins and downregulates the downstream MAPK signaling. Interestingly, HDAC inhibitors have been previously implicated in the depletion of mutant p53 through the transcriptional induction of MDM2, an E3 ubiquitin ligase that negatively regulates p53, instead of directly affecting TP53 transcription (Blagosklonny et al., 2005). Likewise, a similar finding was earlier reported whereby a tyrosine kinase inhibitor, CI‐1033, significantly enhanced ubiquitination in HER2 molecule together with the inhibition of kinase activity (Citri et al., 2002). These reports, along with our own experimental findings discussed in this manuscript, highlight the use of compounds that enhance drug‐induced ubiquitin modification to augment antineoplastic effects and warrant the use of these inhibitors in further studies. Nevertheless, the lack of efficacy in preclinical xenograft models suggests that additional work is needed for clinical development of belinostat in SCC tumors.
Author contributions
LRK, SCL, WJC, and BCG conceived and designed the project. LRK, WRO, and CL acquired the data. LRK, TZT, HH, and PJE analyzed and interpreted the data. LRK, WJC, PJE, and BCG wrote and/or reviewed the manuscript. BCG supervised the study.
Supporting information
Fig. S1. Belinostat and cisplatin inhibits colony formation in lung squamous cell carcinoma (SCC) cells.
Fig. S2. Belinostat treatment induces STAT3 signaling in Calu‐1 cells.
Fig. S3. Expressions of HDACs and sirtuin‐1 in normal lung and squamous cell carcinoma (SCC) cell lines.
Fig. S4. Belinostat increases Bim expression in lung SCC cells.
Fig. S5. Belinostat treatment induces acetylation of both Histone H3 and H4.
Fig. S6. Knockdown of FBXO3 and FBXW10 partially reduces belinostat sensitivity in H520 cells.
Fig. S7. Lack of in vivo efficacy of belinostat and cisplatin in lung squamous cell carcinoma (SCC) xenograft.
Table S1. Sequences for primers used in real‐time quantitative PCR.
Table S2. Computational and curated gene sets for ubiquitination‐related genes (GSEA c2, c4, and c5).
Acknowledgements
This work was funded by the Singapore Ministry of Health's National Medical Research Council (NMRC/CSA/021/2010) to Boon Cher Goh, the National Research Foundation Singapore, and the Singapore Ministry of Education under its Research Centres of Excellence initiatives.
References
- Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S and Elliott PJ (1999) Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 59, 2615–2622. [PubMed] [Google Scholar]
- Bangert A, Häcker S, Cristofanon S, Debatin KM and Fulda S (2011) Chemosensitization of glioblastoma cells by the histone deacetylase inhibitor MS275. Anticancer Drugs 22, 494–499. [DOI] [PubMed] [Google Scholar]
- Bhatt S, Ashlock BM, Toomey NL, Diaz LA, Mesri EA, Lossos IS and Ramos JC (2013) Efficacious proteasome/HDAC inhibitor combination therapy for primary effusion lymphoma. J Clin Invest 123, 2616–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV, Trostel S, Kayastha G, Demidenko ZN, Vassilev LT, Romanova LY, Bates S and Fojo T (2005) Depletion of mutant p53 and cytotoxicity of histone deacetylase inhibitors. Cancer Res 65, 7386–7392. [DOI] [PubMed] [Google Scholar]
- Bradley D, Rathkopf D, Dunn R, Stadler WM, Liu G, Smith DC, Pili R, Zwiebel J, Scher H and Hussain M (2009) Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): trial results and interleukin‐6 analysis: a study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer 115, 5541–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MC, Chen CH, Wang JC, Tsai AC, Liou JP, Pan SL and Teng CM (2013) The HDAC inhibitor, MPT0E028, enhances erlotinib‐induced cell death in EGFR‐TKI‐resistant NSCLC cells. Cell Death Dis 4, e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC and Talalay P (1984) Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22, 27–55. [DOI] [PubMed] [Google Scholar]
- Citri A, Alroy I, Lavi S, Rubin C, Xu W, Grammatikakis N, Patterson C, Neckers L, Fry DW and Yarden Y (2002) Drug‐induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J 21, 2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli F, Gnemmi I, Vallario A, Genazzani AA and Canonico PL (2008) Inhibitors of histone deacetylase (HDAC) restore the p53 pathway in neuroblastoma cells. Br J Pharmacol 153, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elaut G, Rogiers V and Vanhaecke T (2007) The pharmaceutical potential of histone deacetylase inhibitors. Curr Pharm Des 13, 2584–2620. [DOI] [PubMed] [Google Scholar]
- Gimsing P, Hansen M, Knudsen LM, Knoblauch P, Christensen IJ, Ooi CE and Buhl‐Jensen P (2008) A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol 81, 170–176. [DOI] [PubMed] [Google Scholar]
- Glozak MA and Seto E (2007) Histone deacetylases and cancer. Oncogene 26, 5420–5432. [DOI] [PubMed] [Google Scholar]
- Gureasko J, Galush WJ, Boykevisch S, Sondermann H, Bar‐Sagi D, Groves JT and Kuriyan J (2008) Membrane‐dependent signal integration by the Ras activator Son of sevenless. Nat Struct Mol Biol 15, 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hennik MB, van der Vijgh WJ, Klein I, Elferink F, Vermorken JB, Winograd B and Pinedo HM (1987) Comparative pharmacokinetics of cisplatin and three analogues in mice and humans. Cancer Res 47, 6297–6301. [PubMed] [Google Scholar]
- Hess‐Stumpp H (2005) Histone deacetylase inhibitors and cancer: from cell biology to the clinic. Eur J Cell Biol 84, 109–121. [DOI] [PubMed] [Google Scholar]
- Kong LR, Chua KN, Sim WJ, Ng HC, Bi C, Ho J, Nga ME, Pang YH, Ong WR, Soo RA et al (2015) MEK inhibition overcomes cisplatin resistance conferred by SOS/MAPK pathway activation in squamous cell carcinoma. Mol Cancer Ther 14, 1750–1760. [DOI] [PubMed] [Google Scholar]
- Lane AA and Chabner BA (2009) Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 27, 5459–5468. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ and Settleman J (2014) Drug resistance via feedback activation of Stat3 in oncogene‐addicted cancer cells. Cancer Cell 26, 207–221. [DOI] [PubMed] [Google Scholar]
- Li D, Xie P, Zhao F, Shu J, Li L, Zhan Y and Zhang L (2015) F‐box protein Fbxo3 targets Smurf1 ubiquitin ligase for ubiquitination and degradation. Biochem Biophys Res Commun 458, 941–945. [DOI] [PubMed] [Google Scholar]
- Liffers K, Kolbe K, Westphal M, Lamszus K and Schulte A (2016) Histone deacetylase inhibitors resensitize EGFR/EGFRvIII‐overexpressing, erlotinib‐resistant glioblastoma cells to tyrosine kinase inhibition. Target Oncol 11, 29–40. [DOI] [PubMed] [Google Scholar]
- Lin SF, Lin JD, Chou TC, Huang YY and Wong RJ (2013) Utility of a histone deacetylase inhibitor (PXD101) for thyroid cancer treatment. PLoS One 8, e77684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G and Auberger P (2003) Phosphorylation of Bim‐EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene 22, 6785–6793. [DOI] [PubMed] [Google Scholar]
- Mackay HJ, Hirte H, Colgan T, Covens A, MacAlpine K, Grenci P, Wang L, Mason J, Pham PA, Tsao MS et al (2010) Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur J Cancer 46, 1573–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann BS, Johnson JR, Cohen MH, Justice R and Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T‐cell lymphoma. Oncologist 12, 1247–1252. [DOI] [PubMed] [Google Scholar]
- Minamiya Y, Ono T, Saito H, Takahashi N, Ito M, Mitsui M, Motoyama S and Ogawa J (2011) Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 74, 300–304. [DOI] [PubMed] [Google Scholar]
- Minucci S and Pelicci PG (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6, 38–51. [DOI] [PubMed] [Google Scholar]
- Mishima Y, Santo L, Eda H, Cirstea D, Nemani N, Yee AJ, O'Donnell E, Selig MK, Quayle SN, Arastu‐Kapur S et al (2015) Ricolinostat (ACY‐1215) induced inhibition of aggresome formation accelerates carfilzomib‐induced multiple myeloma cell death. Br J Haematol 169, 423–434. [DOI] [PubMed] [Google Scholar]
- Moore PS, Barbi S, Donadelli M, Costanzo C, Bassi C, Palmieri M and Scarpa A (2004) Gene expression profiling after treatment with the histone deacetylase inhibitor trichostatin A reveals altered expression of both pro‐ and anti‐apoptotic genes in pancreatic adenocarcinoma cells. Biochim Biophys Acta 1693, 167–176. [DOI] [PubMed] [Google Scholar]
- O'Connor OA, Horwitz S, Masszi T, Van Hoof A, Brown P, Doorduijn J, Hess G, Jurczak W, Knoblauch P, Chawla S et al (2015) Belinostat in patients with relapsed or refractory peripheral T‐cell lymphoma: results of the pivotal phase II BELIEF (CLN‐19) study. J Clin Oncol 33, 2492–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada H, Tatematsu Y, Saito H, Yatabe Y, Mitsudomi T and Takahashi T (2004) Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int J Cancer 112, 26–32. [DOI] [PubMed] [Google Scholar]
- Pei XY, Dai Y and Grant S (2004) Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res 10, 3839–3852. [DOI] [PubMed] [Google Scholar]
- Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, Zain J, Prince HM, Leonard JP, Geskin LJ et al (2009) Phase II multi‐institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T‐cell lymphoma. J Clin Oncol 27, 5410–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, La Thangue NB and Brown R (2003) Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol Cancer Ther 2, 721–728. [PubMed] [Google Scholar]
- Qian X, LaRochelle WJ, Ara G, Wu F, Petersen KD, Thougaard A, Sehested M, Lichenstein HS and Jeffers M (2006) Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol Cancer Ther 5, 2086–2095. [DOI] [PubMed] [Google Scholar]
- Ramalingam SS, Belani CP, Ruel C, Frankel P, Gitlitz B, Koczywas M, Espinoza‐Delgado I and Gandara D (2009) Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J Thorac Oncol 4, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam SS, Maitland ML, Frankel P, Argiris AE, Koczywas M, Gitlitz B, Thomas S, Espinoza‐Delgado I, Vokes EE, Gandara DR et al (2010) Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first‐line therapy of advanced non‐small‐cell lung cancer. J Clin Oncol 28, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogge RD, Karlovich CA and Banerjee U (1991) Genetic dissection of a neurodevelopmental pathway: Son of sevenless functions downstream of the sevenless and EGF receptor tyrosine kinases. Cell 64, 39–48. [DOI] [PubMed] [Google Scholar]
- Senese S, Zaragoza K, Minardi S, Muradore I, Ronzoni S, Passafaro A, Bernard L, Draetta GF, Alcalay M, Seiser C et al (2007) Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol 27, 4784–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA et al (2010) A chromatin‐mediated reversible drug‐tolerant state in cancer cell subpopulations. Cell 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele NL, Plumb JA, Vidal L, Tjørnelund J, Knoblauch P, Rasmussen A, Ooi CE, Buhl‐Jensen P, Brown R, Evans TR et al (2008) A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin Cancer Res 14, 804–810. [DOI] [PubMed] [Google Scholar]
- Struhl K (1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 12, 599–606. [DOI] [PubMed] [Google Scholar]
- Vultur A, Villanueva J, Krepler C, Rajan G, Chen Q, Xiao M, Li L, Gimotty PA, Wilson M, Hayden J et al (2014) MEK inhibition affects STAT3 signaling and invasion in human melanoma cell lines. Oncogene 33, 1850–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li J, Gu J, Yu J, Guo S, Zhu Y and Ye D (2015) Abnormal methylation status of FBXW10 and SMPD3, and associations with clinical characteristics in clear cell renal cell carcinoma. Oncol Lett 10, 3073–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D and Lippard SJ (2005) Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 4, 307–320. [DOI] [PubMed] [Google Scholar]
- Wilson AJ, Byun DS, Popova N, Murray LB, L'Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH and Mariadason JM (2006) Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem 281, 13548–13558. [DOI] [PubMed] [Google Scholar]
- Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita M et al (2006) Restoring E‐cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 66, 944–950. [DOI] [PubMed] [Google Scholar]
- Yeo W, Chung HC, Chan SL, Wang LZ, Lim R, Picus J, Boyer M, Mo FK, Koh J, Rha SY et al (2012) Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: a multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J Clin Oncol 30, 3361–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD, Gillespie SK, Borrow JM and Hersey P (2004) The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria‐dependent apoptosis of melanoma cells. Mol Cancer Ther 3, 425–435. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Belinostat and cisplatin inhibits colony formation in lung squamous cell carcinoma (SCC) cells.
Fig. S2. Belinostat treatment induces STAT3 signaling in Calu‐1 cells.
Fig. S3. Expressions of HDACs and sirtuin‐1 in normal lung and squamous cell carcinoma (SCC) cell lines.
Fig. S4. Belinostat increases Bim expression in lung SCC cells.
Fig. S5. Belinostat treatment induces acetylation of both Histone H3 and H4.
Fig. S6. Knockdown of FBXO3 and FBXW10 partially reduces belinostat sensitivity in H520 cells.
Fig. S7. Lack of in vivo efficacy of belinostat and cisplatin in lung squamous cell carcinoma (SCC) xenograft.
Table S1. Sequences for primers used in real‐time quantitative PCR.
Table S2. Computational and curated gene sets for ubiquitination‐related genes (GSEA c2, c4, and c5).