

Abstract
The PhoP–PhoR two-component system is essential for the virulence of Mycobacterium tuberculosis (Mtb) and therefore represents a potential target for developing novel antituberculosis therapies. However, little is known about the mechanism by which this two-component system regulates the virulence. In this study, we demonstrated that a phoR mutant Mtb strain has phenotypes similar to those of a phoP mutant, suggesting that PhoP and PhoR work in the same pathway to regulate Mtb virulence. We determined the structure of the dimerization and histidine phosphotransfer (DHp) domain of PhoR to a 1.9 Å resolution. The structure revealed that the DHp domain is a dimer. Each subunit consists of two antiparallel α helices connected by a loop of five residues. The two subunits of the dimer fold into a four-helical bundle with a continuous hydrophobic core. The topology of the four-helical bundle is identical to the histidine kinases that are known to have a cis-autophosphorylation mechanism, suggesting that PhoR is likely to autophosphorylate in cis. The dimer is asymmetric, with one subunit having a greater bending angle than the other at the highly conserved proline residue five-residues downstream of the phosphorylation site histidine. This structural asymmetry of the dimer suggests the flexibility of the PhoR DHp domain, which is likely to be important for the signal transduction mechanism in controlling the autophosphorylation and phosphotransfer reactions and communicating with the upstream structure.
Introduction
Two-component regulatory systems (TCSs) are major signaling systems in bacteria and are essential for bacterial adaptation to the environments, including the ability to evade host immune response and to develop drug resistance in pathogenic bacteria. Because TCSs are absent from mammals, they are attractive targets for developing new antibiotics.1,2 The PhoP–PhoR TCS of Mycobacterium tuberculosis (Mtb) is essential for virulence,3 although the signals that this TCS responds to are still unknown. Tuberculosis (TB) remains as one of the most serious threats to public health because of the increasing prevalence of multidrug-resistant and extensive drug-resistant TB and the synergy of TB with HIV infection.4 In 2015, there were an estimated 10.4 million new cases of TB, and 1.8 million people died from TB, including 400 000 people who were also HIV positive.5
In the Mtb PhoP–PhoR TCS, PhoR is a transmembrane sensor histidine kinase (HK), and PhoP is a response regulator (RR) that affects the expression of more than 110 genes.6−8 Mutant strains of Mtb with the phoP gene or both phoPR genes disrupted are severely attenuated in macrophage and mice, and several studies have proposed to develop an attenuated live vaccine based on the phoP-null mutant strains.9,10 The phoPR genes are in a single operon, suggesting that the PhoPR proteins are likely to be an obligate cognate pair of TCS proteins.11 The gene encoding PhoR is located downstream of phoP, and the open-reading frames are separated by 45 base pairs.12 In vitro studies have demonstrated the ability of PhoR to phosphorylate PhoP.13 However, data of the phoR function in vivo are still lacking. PhoR is a membrane-associated protein with an extracytosolic sensory domain of 123 residues.3 This topological feature may make PhoR a more accessible target than the intracellular PhoP.
PhoR is expected to be a homodimer with a modular domain structure,3 each subunit consisting of two transmembrane helices flanking an extracytosolic sensor domain at the N-terminus, followed by a HAMP (found in HKs, adenyl cyclases, methyl-accepting chemotaxis proteins, and phosphatases) domain, a DHp (dimerization and histidine phosphotransfer) domain, and a CA (catalytic and ATP-binding) domain in the cytosol. Although it is well-established that the PhoPR TCS plays an important role in the virulence of Mtb, little data are available for the PhoR protein. However, some of the basic functions and signaling mechanisms can be inferred from studies of homologous HKs.14 HKs sense environmental signals through the sensor domain. The signals are propagated across the cellular membrane to the cytosolic portion through some still undefined conformational changes that turn on the kinase activity and autophosphorylate on the conserved histidine. The phosphoryl group is then transferred to a cognate RR, which then generates cellular responses, in most of the cases by regulating gene transcription. There are available structural information on several related HKs, such as EnvZ,15 DesK,16 and HK853.17 The overall sequence identity of PhoR to these HKs is low, that is, <30%. There are, however, well-conserved sequence motifs and a pattern of heptad repeats of hydrophobicity in the helical bundle domains.14
The DHp domain is the center of the HK function. It harbors the phosphorylation site histidine and interacts with the ATP-binding domain for autophosphorylation, interacts with the cognate RR for the phosphotransfer reaction to activate the RR, and receives signals through upstream domains from the external sensor domain to control the phosphorylation activities. The DHp domain makes up a large portion of the HK dimer interface. The DHp domain also harbors the binding site for the RR, and thus, its structure determines the fidelity of cognate pairs of HK and RR.17
In this work, we demonstrated the contribution of the PhoR HK to the roles of the PhoPR TCS in Mtb virulence. The results suggested that the PhoPR proteins are obligate cognate partners in a TCS. We presented the crystal structure of the DHp domain of PhoR, which revealed an asymmetric dimer of helical hairpins, suggesting the flexibility of the DHp domain that plays an important role in the signaling process of this TCS.
Results
PhoP and PhoR as an Obligate Cognate TCS Pair
To elucidate the role of PhoR in Mtb virulence, we constructed a phoR mutant ST163 (Figure 1a). Phenotypic comparisons were made to the H37Rv parental strain and the previously described phoP mutant ST100.7 Among the genes regulated by PhoP, a subset is involved in the synthesis of key cell wall components.3 A phoP mutant, therefore, has a compromised cell wall, resulting in its loss of acid fastness and sensitivity to the cell wall biosynthesis inhibitor vancomycin.7 Using the disk diffusion assay, we showed that the phoR and phoP mutants were similarly sensitive to vancomycin (Figure 1b), suggesting similar defects in the cell wall of both mutants. Wild-type-level resistance to vancomycin is restored in the complemented strain ST168.
Figure 1.

phoR mutant of Mtb H37Rv has the same phenotypes as the phoP mutant. (a) Schematic illustration of the phoR mutant design. DNA sequence (∼50 bp) encompassing His259 codon in the phoR gene was replaced by a DNA fragment containing a hygromycin-resistant gene. (b) Sensitivity of the phoP and phoR mutants to vancomycin. Disk diffusion assays were performed to compare the sensitivities of H37Rv, the phoP mutant (ST100),7 the phoR mutant (ST163), and the phoR-complemented strain (ST168) to vancomycin (800 ng/μL). Bars indicate the average zones of inhibition in millimeters from three independent experiments, and error bars indicate standard deviations. (c) Growth of Mtb strains in the mouse lung comparing strains of the wild-type H37Rv (solid line with diamonds), phoP mutant ST100 (solid line with squares), and phoR mutant ST163 (dashed line with triangles). (d) Growth of Mtb strains in the mouse lung comparing strains of the wild-type H37Rv (solid line with diamonds), phoR mutant ST163 (dashed line with triangles), and phoR mutant complemented with phoPR (ST184) (solid line with circles). For panels (c,d), each data point is the average of three experiments, and the error bar is the standard deviation.
To determine whether PhoR is important for virulence, as is PhoP, we infected C57BL/6 mice with the wild-type H37Rv, phoP mutant, phoR mutant, and phoR-complemented strains. The phoR and phoP mutants were similarly defective (∼2 log cfu below wild type) in the mouse lung (Figure 1c), and the ability to grow in the mouse lung was enhanced in the complemented strain, ST184, although not to wild-type levels (Figure 1d).
Our above data indicated that disruption of either phoP or phoR gave similar phenotypes in Mtb H37Rv, suggesting that PhoP and PhoR are an obligate TCS pair. Our data also showed that PhoR is as important as PhoP for Mtb virulence in mice and thus could serve as an antitubercular target.
Overall Structure of the PhoR DHp Domain
The PhoR DHp domain (designated as PhoRD) used in the structural study consisted of residues 240–310 of PhoR encoded in the Mtb genome.12 The polypeptide used in crystallization also included three extra residues GHM (single-letter codes for amino acids) from the fusion vector at the N-terminus and a mutation of Arg309 to Lys (to generate a HindIII site) to facilitate cloning. The purified protein has a size of a dimer in solution by gel filtration (Figure 2). We determined two structures, native and selenomethionine (SeMet)-labeled structures (Table 1). Both structures are essentially identical. The crystal structure contained two polypeptide chains in the asymmetric unit (designated as A and B). Each chain consists of two helices that formed a helical hairpin. Helix α2 is shorter than α1, with its C-terminal end reached to about one-third of α1 (Figure 3). The two polypeptides form a dimeric four-helical bundle. The N-terminal parts of α1 of the two subunits form a two-helix coiled coil. All residues, including those from the fusion vector, were modeled in the final refined structure. The extra residues from the cloning vector at the N-terminus extended the helix α1. The side chains of His259, the phosphorylation site, are solvent-exposed.
Figure 2.

PhoRD protein is a dimer in solution. The protein was loaded on a Superdex 75 column (GE Life Sciences, 10/300/GL) and eluted at 0.7 mL/min with a buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM EDTA. The column was calibrated with standards with molecular weights of 158 000, 44 000, 17 000, and 1350 Da (gel filtration standard, Bio-Rad), as shown in a straight line with square symbols. The molecular weight of a PhoRD dimer is 16 689 Da, based on its amino acid sequence.
Table 1. X-ray Diffraction Data and Refinement Statistics.
| data set | native | SeMet |
|---|---|---|
| wavelength (Å) | 1.0 | 0.9793 |
| resolution (Å)a | 50–2.02 (2.05–2.02) | 20–1.9 (1.93–1.90) |
| space group | P21 | P21 |
| cell dimension a (Å) | 23.25 | 23.22 |
| b (Å) | 74.53 | 74.59 |
| c (Å) | 44.54 | 45.47 |
| β (deg) | 96.64 | 97.31 |
| Rsym (%)a | 7.4 (22.5) | 17.6 (48.4) |
| Rpim (%)a | 4.3 (17.1) | 8.1 (34.5) |
| completeness (%)a | 82.6 (29.9) | 96.6 (81.5) |
| I/σa | 18.2 (2.5) | 14.1 (1.9) |
| Rworkb | 0.171 | 0.180 |
| Rfreeb | 0.217 | 0.212 |
| rmsd bond length (Å) | 0.015 | 0.027 |
| rmsd bond angle (deg) | 1.76 | 2.41 |
| no. protein atoms (av B, Å2) | 1166 (45.8) | 1166 (32.6) |
| no. other molecules (av B, Å2) | 1 K+ (41.0) | 1 K+ (30.4) |
| 3 ethylene glycol (62.9) | 2 ethylene glycol (54.9) | |
| 1 diethylene glycol (56.9) | 3 diethylene glycol (55.5) | |
| 1 triethylene glycol (58.3) | 1 triethylene glycol (53.1) | |
| 1 tetraethylene glycol (48.9) | 1 2-(2-methoxyethoxy)ethanol (50.9) | |
| 1 tetraethylene glycol (37.9) | ||
| no. water (av B, Å2) | 26 (57.09) | 26 (49.3) |
Values for the data in the highest resolution shell are shown in parentheses.
Rwork was calculated on the reflections used in the refinement; Rfree was calculated on a set of approximately 5% randomly chosen reflections that were never used for the refinement.
Figure 3.
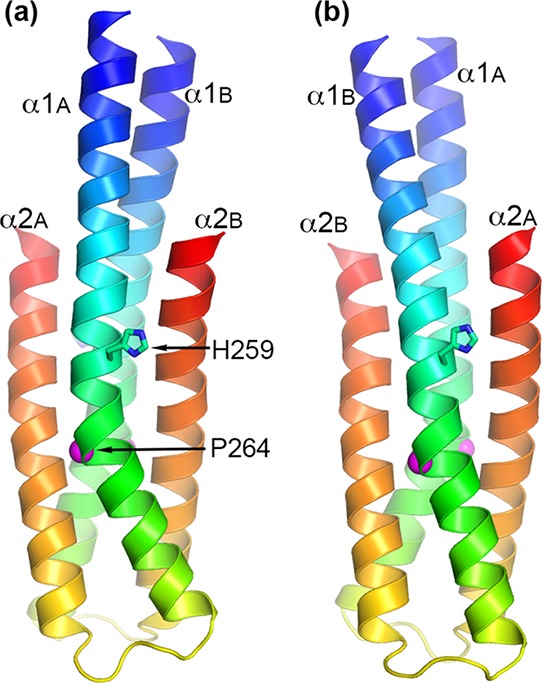
Cartoon representations of the PhoRD dimer structure. Panels (a,b) are views approximately 180° rotation from each other along a vertical axis. The two subunits are designated as A and B. Subunit A is in the front in (a), whereas it is in the back in (b). Each subunit is colored rainbow with N-terminus in blue and C-terminus in red. The phosphorylation site His259 is shown as sticks. The position of Pro264 is marked with a magenta sphere. Pro264 produces a kink in helix α1 below His259. This kink is more prominent in subunit A than in subunit B, making the dimer asymmetric.
The crystals were obtained from crystallization drops containing PEG 200 as a precipitant, and several linear molecules, such as ethylene glycol, diethylene glycol, triethylene glycol, 2-(2-methoxyethoxy)ethanol, and tetraethylene glycol, that fit with the electron density map were modeled (Table 1). These molecules could be impurities in PEG 200 or ordered fragments of longer polymers. One tetraethylene glycol molecule encircled a cation, which was bound near the carboxyl terminus of subunit A (Figure S1). The cation was modeled as a potassium ion (K+) because K+ was present in the crystallization buffer. The electron density fit well with a K+ ion with a B factor similar to the surrounding atoms. In a similar fashion as the tetraethylene glycol encircling the K+, a triethylene glycol surrounded the ammonium group of Lys309 of subunit A in a semicircle (Figure S1). The PEG fragments were bound at the crystal packing interfaces and therefore are unlikely to be physiologically relevant.
The structure determined from the native data set is essentially identical to that from the SeMet-derived crystal, except that the methionine residues have sulfur atoms in the place of the selenium atoms. Both structures had similar B factor profiles (Figure S2), reflecting the same crystal packing effect on the mobility of residues. The two structures were superimposable with an rmsd of main-chain atoms of 0.19 Å for all 148 residues in the dimer.
Differences between the Two PhoRD Subunits in the Dimer
In contrast to the identical dimer structures between the native and the SeMet crystals, the two subunits of each dimer were significantly different, forming an asymmetric dimer. Alignment of subunits A and B for all 74 residues gave an rmsd of main-chain atoms of 2.13 Å. Most differences were in helix α1, where the Pro264 residue, at one turn below His259, induced a kink that gave a different bending angle of the helix between the two subunits (Figure 3). The bending angle of α1 in subunit A is ∼28°, whereas that in subunit B is ∼5°. The two α2 helices were superimposable with an rmsd of 0.78 Å for main-chain atoms from residues 283–310 (see the amino acid sequence in Figure 4). Excluding the C-terminal three residues, which in subunit B had high B factors (Figure S2) and weak side-chain electron density, the rmsd of main-chain atoms of residues 284–307 was 0.41 Å (Figure 5c). The two halves of α1 each superimposed well between the two subunits when separately aligned, with an rmsd of 0.24 Å for the N-terminal half (residues Gly237 to His259, Figure 4) and an rmsd of 0.43 Å for the second half (residues Pro264 to Arg276) (Figure 5ab). The Pro264 residue introduces flexibility and allows the helix to bend. Alignment based on the second half of α1 allows most of α2 to be superimposable (Figure 5b) because this half of the structure contributes most of the intrasubunit interactions between α1 and α2. The difference between the two subunits results from the variation in the α1 bending angle.
Figure 4.

Sequence alignment of the DHp domains with known structures. Sequences were aligned based on the structures of the two helices and the position of the phosphorylation site histidine (in blue). Residues of the two helices are highlighted in yellow. The PhoRD sequence is shown on top with residues involved in intrasubunit interface in red, those in the dimer hydrophobic core underlined, and those involved in intersubunit hydrogen bonds or salt bridges in bold type. Residue numbers of PhoRD are labeled on top. HK853 is a HK of unknown function from Thermotoga maritima,18 EnvZ15 and CpxA19 are from Escherichia coli and sense osmotic and cellular envelope stress, respectively, DesK is a thermosensor from Bacillus subtilis,16 and LOV-HK is a light-sensing HK from Brucella abortus.20 DesK and LOV-HK are in other HK subfamilies and lack some conserved sequence architectures, including the absence of the conserved proline downstream of the phosphorylation site histidine.
Figure 5.
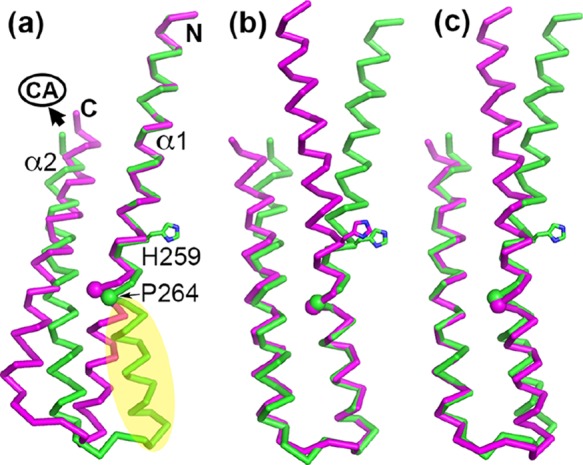
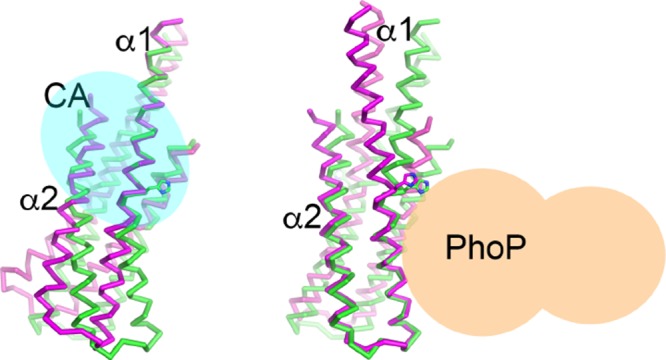
Structural alignments of the two subunits of the PhoRD dimer show that most of the differences resulted from the variation in the bending angle of helix α1. The alignments were based on (a) the N-terminal half of α1, (b) the second half of α1, and (c) helix α2. Subunit A is in green and subunit B in magenta. His259 side chain is shown in sticks. The position of Pro264 is marked with a sphere. In panel (a), the N and C termini are labeled. The C-terminus is where the CA domain is attached. Potential PhoP-binding site is marked in yellow.
Intrasubunit Interactions between the Two Helices of the Helical Hairpin
Interactions between α1 and α2 of a single subunit are hydrophobic. The sequence of PhoRD has a pattern of hydrophobic residues at every third or fourth positions (Figure 4). These hydrophobic side chains gather at the interface of the two helices to form a continuous hydrophobic core. Side chains participating in the interhelical hydrophobic interactions include Phe253, Ala257, Leu261, Pro264, Ile268, Phe271, and Tyr275 of α1 and Val283, Leu286, Ile290, Ala294, Met197, Leu300, Val301, Leu304, and Leu307 of α2 (Figure 4). The less-bent α1 in subunit B allows additional side chains of Met250 and Ala308 to be involved in the interhelical interactions, whereas the different linker conformation allows Leu274 side chain in subunit A to be a part of the interface. The hydrophobic part of the Arg289 side chain is also involved in the interhelical hydrophobic interactions. Most of the hydrophobic side chains of the intrasubunit interface are also involved in the dimer interface.
Dimer Interface Interactions
The interface between the two helical hairpins of the dimer covers ∼1600 Å2 surface area of each subunit, and the interface interactions include hydrophobic interactions, hydrogen bonds, and salt bridges (Table S1). The core of the interface is composed of hydrophobic side chains that hold the coiled coils together. Hydrogen bonds and salt bridges are on the periphery of this hydrophobic core. At the N-terminus above the junction of the four-helical bundle, the two α1 helices have intimate associations owing to the short side chain of Ala240 and Ala243 at the interface (Figure 4). The side chain of Met239, a residue from the expression vector, is also involved in this patch of hydrophobic dimer interface. At the junction, there are two hydrogen bonds between the side chains of Glu247 of one subunit and Ser246 of the other. These polar and charged interactions are likely to play a lesser role in the stability of the dimer, compared with the extensive hydrophobic core of the dimer.
At the four-helical bundle of the dimer, the interface is strongly hydrophobic and involves side chains of Met250, Ile254, Leu261, Leu265, Ile268, Ala272, Val283, Leu286, Leu287, Ile290, Ala294, Met297, Val301, Leu304, and Ala308 of both subunits, as well as additional side chains of Ala257 and Tyr275 of subunit A (Figure 4). Most of these side chains participate in the intrasubunit helical–helical interactions as described above. In addition to these hydrophobic interactions, there are hydrogen bonds and salt bridges between the two subunits. Arg276 of one subunit and Glu291 of the other, and likewise, Arg262 of one and Asp302 of the other, have charge–charge interactions and hydrogen bonds between side chains. The Arg276 side chain of subunit B has a hydrogen bond to the main-chain carbonyl oxygen of Gly284 of A. The Arg251 side chain of one subunit gives a hydrogen bond to the carbonyl oxygen atoms of Ala308 and Leu310 of the other. These strong interface interactions keep the four-helical bundle together to form a stable dimer structure.
Phosphorylation Site
The flexibility of the DHp domain changes the position of the phosphorylation site His259 relative to the rest of the structure but does not vary its nearby environment. The His259 side chain is surrounded by side chains of Thr255, Asp256, Glu260, Arg262, and Thr263 of the same subunit and those of Leu305 and Asp302 of the other subunit (Figure 6). Other than the potential hydrophobic interactions with Leu305, the His259 side chain does not have direct contacts with other side chains. Glu260 to Thr263 are well-conserved among the majority of HKs (Figure 4). An acidic side chain at the equivalent position of Glu260 is implicated to be a general base in the phosphotransfer reactions in CpxA and HK853.19,21
Figure 6.
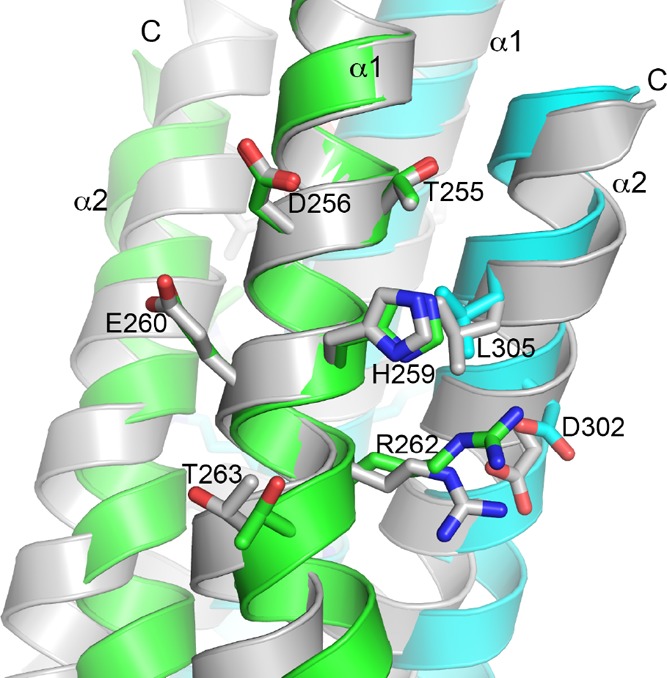
Phosphorylation site His259 and its nearby environment are similar in both subunits of the PhoRD dimer. To compare the two subunits around the His259 side chain, two dimers were superimposed by aligning the N-terminal half of α1 of subunit A from one dimer (colored in green for subunit A and cyan for subunit B) with that of subunit B from the other (shown in gray). The superposition is identical to that in Figure 4a. Side chains of His259 and its surrounding residues are shown in sticks.
Linker Topology of the Helical Hairpin
To deduce the likely mechanism of PhoR autophosphorylation, we compared its DHp structure with other representative structures with known autophosphorylation directionality (Figure 7). The topology of the β-hairpin loop in PhoRD is identical to that of HK85322 but is different from that of CpxA. CpxA has a right-handed helical bundle connectivity and autophosphorylates in trans,19 whereas HK853 has a left-handed connectivity and autophosphorylates in cis. Therefore, PhoR is likely to autophosphorylate in cis.
Figure 7.
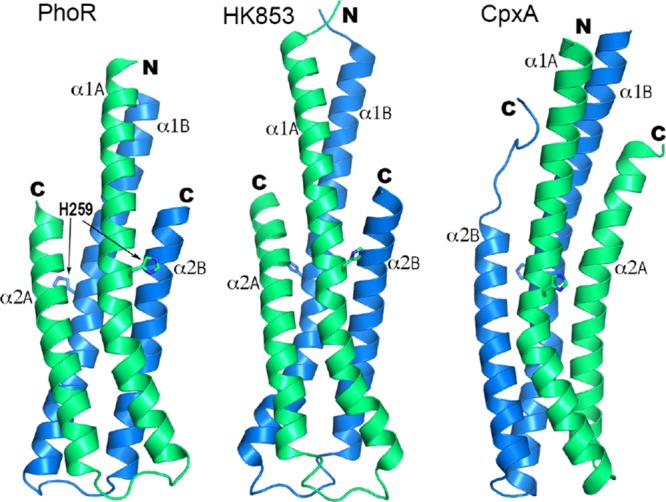
Comparison of the structure of the DHp domain of PhoR with those of HK853 (PDB id 2C2A) and CpxA (4BIX). The side chains of the phosphorylation site histidine are shown in sticks. The two α1 helices of each structure are in equivalent positions relative to each other. Subunits with the equivalent α1 helix are labeled and colored identically, subunit A in green and B in blue.
Discussion
We showed that the Mtb phoR mutant strain had phenotypes similar to the phoP mutant, indicating that disruption of either phoP or phoR affects the same biochemical pathway, thereby confirming that PhoP and PhoR work together in regulating the virulence of the pathogen. Our crystal structure of the DHp domain of PhoR revealed that in spite of the strong continuous hydrophobic core that holds the dimeric four-helical bundle together, the structure is flexible by varying the bending angle of helix α1. Also, the formation of an asymmetric dimer is likely to play a role in the signal transmission mechanism as suggested by studies of other HKs,19 and the helical connectivity topology predicts a cis-autophosphorylation mechanism.
Autophosphorylation of the HK dimer can go through one of the two possible mechanisms: trans, in which the CA domain of one subunit binds ATP and transfers the phosphoryl group to the DHp domain of another subunit; or cis, in which the CA and DHp domains involved in phosphotransfer belong to the same subunit of the dimer. Structural and biochemical data on HKs so far suggest that the directionality of autophosphorylation is determined by the topology of the DHp four-helical bundle.21,23 All known structures with a left-handed helical connectivity, for example, HK853, autophosphorylate in cis, and those with right-handed connectivity, for example, EnvZ24 and CpxA,19 autophosphorylate in trans. Although the linker between CA and DHp is flexible, it does not appear to affect the autophosphorylation directionality, likely because the length of the linker limits its reach and ability to have the specific interactions necessary for the autokinase reaction to only one of the phosphorylation sites. The left-handed helical connectivity of PhoRD suggests that PhoR is likely to autophosphorylate in cis.
The flexibility of the HK dimer is necessary to transmit the signals received by the sensor domain through conformational changes of the helical structures of transmembrane and intracellular domains to regulate the kinase activity. The proline residue that introduced a kink in helix α1 is highly conserved in HKs (Figure 4). Crystal structures of HKs that do not have this proline residue, such as DesK16 and LOV-HK,20 also show a kinked helix at the similar location. The crystal structures of CpxA19 and VicK25 also show significant asymmetry of the DHp domain, suggesting that the asymmetric conformation is the mechanism of regulation of the HK activities. Although there is no reported asymmetric HAMP domain structure, HAMP domains can vary their structure through helical rotation26 or a shift in register of coiled coils.27 Such structural changes in the HAMP domain can induce changes in the conformation of the DHp domain, including the dynamics of symmetry–asymmetry conversion.28 Our structural data of PhoR DHp alone are insufficient to conclude what role an asymmetric DHp dimer play in the signaling mechanism. However, the asymmetric dimer observed indicates the flexibility of α1, which has its mechanistic consequence. The phosphorylation site His259 is next to the bending pivot of α1 (Figure 5), where Pro264 disrupts the main-chain hydrogen bonds of the helix. Variation in the α1 bending angle in PhoR DHp changes the relative position between His259 and the C-terminal end of the DHp domain where the CA domain is attached (Figure 5a) and consequently modulates the interactions between the phosphorylation site and the CA domain, affecting the autokinase activity. Similarly, the α1 bending angle changes the relative position of the His259 side chain and the lower half of α1 (Figure 5a). The lower half of α1 is proposed to harbor the binding site for the cognate RR.17,29 Therefore, variation in the α1 bending angle will likely regulate the phosphotransfer and phosphatase activities. Phosphorylation of His259 will favor its interaction with the side chain of Arg262 (Figure 6) and thus bring the Arg262 side chain closer, thereby increasing the helix bending angle. Likewise, a bent α1 will be likely to facilitate phosphorylation of His259.
The PhoRD structure reported here enables further exploration of the PhoR signaling mechanism. The structure can guide mutagenesis studies to identify residues that are involved in binding the CA domain and those for binding PhoP. By selecting proper sites for labeling with fluorophores, one can design a FRET-based high-throughput system to screen for inhibitors to PhoR–PhoP binding interactions. Such inhibitors will disrupt the signaling pathway of this TCS and therefore attenuate the virulence of the bacilli.
Materials and Methods
Construction of phoR Mutant and Complemented Strains
To disrupt the phoR gene in the Mtb strain H37Rv, we deleted a region of the phoR gene surrounding the codon of the conserved phosphorylation-site histidine (His259) and inserted in its place a hygromycin-resistance cassette, a DNA fragment containing a hygromycin-resistant gene to disrupt the phoR gene, using a method similar to that for the construction of the phoP mutant ST100.7 The phoR mutant was designated as ST163. Two strategies were used for the complementation of the phoR mutant. For the in vitro assays, the wild-type phoR gene was introduced in trans via a multicopy mycobacterial expression plasmid, pMV261,30 under the control of the constitutive hsp promoter. This complemented strain was designated as ST168. Owing to concerns regarding the use of a multicopy plasmid in an in vivo context, a different approach was utilized for complementation in the mouse infection assay. In this case, the entire phoPR operon under its native promoter was introduced by means of an integrative plasmid, pMV306.kan,7 for insertion into the chromosomal attB site.31,32 This complemented strain is designated as ST184.
Mouse Infection Assays
Mice were infected by the aerosol method, as previously described.33Mtb strains were inoculated in liquid 7H9 medium and grown until the mid-log stage of growth. Female-specific pathogen-free C57BL/6 mice (6–8 week old) were infected with Mtb H37Rv, the phoP mutant ST100, the phoR mutant ST163, and the phoPR-complemented phoR mutant ST184, by the aerosol route with 10 mL of suspensions of the bacteria (1 × 105 per mL) using a nebulizer. At certain time points over a 12 week period, mice were sacrificed in triplicate and the whole organ homogenates were inoculated onto 7H10 solid media, using appropriate serial dilutions. After 3 weeks at 37 °C, colony counts were performed.
Growth Inhibition by Disk Zone Diffusion Assay
The ability of vancomycin to inhibit the growth of Mtb strains was assayed by the filter disk assay technique, as previously described.34 The disk assays used 6.5 mm paper disks (Scheicher and Schuell), and 10 μL of vancomycin at 800 μg/mL was spotted onto the disk. The assays were performed in triplicate.
Protein Expression and Purification
The PhoR dimerization
domain was expressed as a fusion protein with glutathione-S-transferase (GST). To construct the plasmid expressing
the fusion protein, the gst gene was PCR-amplified
using the pGEX-5X-1 plasmid (GE Healthcare) as a template and primers
gst_f, GCTGATTAAT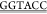 ATGTCCCCTATACTAGGTTATTGG,
and gst_r, GCTACATATGACCTTGGAAATAAAGATTTTCGCCACGACCTCGATCAGATCC.
The primer gst_r also contains a sequence encoding the TEV protease
cleavage site. The PCR fragment was inserted into the NdeI site of the pET28a plasmid (Novagen) to generate pGST. The correct
orientation of the insert was selected by restriction digestion with KpnI and HindIII, and the DNA sequence
was confirmed by sequencing. The DNA sequence encoding the PhoR DHp
domain (PhoRD, residues 240–310 of the PhoR protein encoded
in the Mtb genome) was PCR-amplified from the Mtb genomic DNA using the primers phoRD_f, CGGACATATGGCCGAAAAGGCCCGGGATTCAGAGG,
and phoRD_r, GCATCAAGCTTGGCAAGCAGCAGCAAATCGTCCACC. The phoRD sequence was inserted into pGST between NdeI and HindIII. The resulting plasmid, pGST–phoRD, produced
a fusion protein with the PhoRD polypeptide fused to the C-terminus
of GST through a linker that is cleavable by TEV protease.
ATGTCCCCTATACTAGGTTATTGG,
and gst_r, GCTACATATGACCTTGGAAATAAAGATTTTCGCCACGACCTCGATCAGATCC.
The primer gst_r also contains a sequence encoding the TEV protease
cleavage site. The PCR fragment was inserted into the NdeI site of the pET28a plasmid (Novagen) to generate pGST. The correct
orientation of the insert was selected by restriction digestion with KpnI and HindIII, and the DNA sequence
was confirmed by sequencing. The DNA sequence encoding the PhoR DHp
domain (PhoRD, residues 240–310 of the PhoR protein encoded
in the Mtb genome) was PCR-amplified from the Mtb genomic DNA using the primers phoRD_f, CGGACATATGGCCGAAAAGGCCCGGGATTCAGAGG,
and phoRD_r, GCATCAAGCTTGGCAAGCAGCAGCAAATCGTCCACC. The phoRD sequence was inserted into pGST between NdeI and HindIII. The resulting plasmid, pGST–phoRD, produced
a fusion protein with the PhoRD polypeptide fused to the C-terminus
of GST through a linker that is cleavable by TEV protease.
The plasmid pGST–phoRD was transformed into E. coli BL21(DE3) competent cells. The cells were grown at 37 °C in Luria–Bertani (LB) medium supplemented with 50 μg/mL kanamycin, and protein expression was induced by isopropyl β-d-1-thiogalactopyranoside (IPTG) (50 μM final concentration) either at 18 °C overnight or at 37 °C for 2 h. Expression at 18 °C yielded a small amount of soluble PhoRD protein. At 37 °C, the majority of PhoRD was in insoluble inclusion bodies. The inclusion bodies were solubilized in 8 M urea, 20 mM phosphate, pH 7.4, and 150 mM NaCl, and the protein was refolded by dialysis in 1 M urea, 20 mM phosphate, pH 7.4, and 150 mM NaCl. The protein was then passed through a HisTrap column, cleaved by TEV protease to remove the His-tagged GST, and then passed through the HisTrap column again, following a similar procedure detailed previously.35 The PhoRD protein thus purified was then passed through a Superdex 75 column (GE Healthcare). Both 18 and 37 °C expression yielded the identical protein from Ni-affinity and size-exclusion chromatography. However, refolding from the inclusion body had a yield that was >10-fold higher. Therefore, the refolded protein was used for protein crystallization.
SeMet-Labeled Protein Preparation
To obtain SeMet-labeled PhoRD (SeMet–PhoRD), M9 minimal media containing 50 μg/mL kanamycin was used in place of LB. A mixture of amino acids including lysine (100 mg), phenylalanine (100 mg), threonine (100 mg), isoleucine (50 mg), leucine (50 mg), valine (50 mg), and SeMet (60 mg) were added directly to each liter of culture at 15 min prior to induction.36 Cells were induced for protein expression with 50 μM IPTG for 3 h at 37 °C. Subsequent purification of SeMet–PhoRD was identical to that described above for the native protein.
Protein Crystallization
For crystallization, the protein was buffer-exchanged into 20 mM Tris, pH 8.0, 100 mM NaCl, and 1 mM EDTA by gel filtration, and it was concentrated to ∼6 mg/mL. Crystallization experiments were set up by mixing an equal volume of the protein with the precipitant. Both microbatch and vapor diffusion methods were performed. For microbatch crystallization, 1 μL of protein was mixed with 1 μL of crystallization solution, and the drops were covered with a 50/50 mixture of paraffin and silicon oil (Hampton Research, Aliso Viejo, California). For vapor diffusion experiments, 2 μL of protein was mixed with 2 μL of crystallization solution, and the drops were equilibrated against 0.5 mL of the crystallization solution in the reservoir. The crystallization plates were incubated at 20 °C. The native crystals were obtained from hanging-drop vapor diffusion, with the crystallization solution containing 0.1 M Na/K phosphate, pH 7.2, 2 mM EDTA, 0.2 M NaCl, and 35% PEG 200. The SeMet-labeled crystals were obtained by microbatch, with the crystallization solution containing 0.1 M Na/K phosphate, pH 7.2, 0.2 M KI, 2 mM EDTA, and 35% PEG 200.
X-ray Data Collection and Structural Determination
X-ray data were collected at National Synchrotron Light Source, Brookhaven National Laboratory. A set of data were collected on a native crystal at beamline X6A, and another set of single wavelength anomalous data were collected at beamline X25 on a SeMet-labeled crystal (Table 1). All crystals were in the same space group with identical cell parameters. There were two polypeptide chains per asymmetric unit, with a Matthews volume of ∼2.28 Å3/Da and a solvent content of 46%. The data were processed using HKL2000.37
The structure was determined using the single-wavelength anomalous diffraction phasing method with the SeMet data set. There are four methionine residues per PhoRD chain. Data output from HKL2000 was imported into the CCP4 format,38 and phases and initial maps were calculated with SHELXC/D/E.39 Automatic model building by ArpWarp40 built 140 out of a total of 148 residues in two polypeptide chains with an R factor of 0.287. The model was manually adjusted using COOT41 and refined using REFMAC.42 The native data set was phased by molecular replacement with Phaser43 and using the structure determined from the SeMet data set as the search model. The structural refinement statistics are listed in Table 1.
Acknowledgments
X-ray diffraction data were collected at beamlines X6A and X25, NSLS, Brookhaven National Laboratory, Upton, NY, USA. We thank Dr. Vivian Stojanoff and Dr. Jean Kakoncic for their assistance with the data collection. This work was supported by the National Institutes of Health grant R01GM079185 and the Uniformed Services University of the Health Sciences intramural grants R071IR and R0713018. The opinions or assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of Defense or the Uniformed Services University of the Health Sciences.
Glossary
Abbreviations
- CA
catalytic and ATP-binding domain
- cfu
colony-forming units
- DHp
dimerization and histidine phosphotransfer domain
- HK
histidine kinase
- Mtb
Mycobacterium tuberculosis
- PEG
polyethylene glycol
- RR
response regulator
- TB
tuberculosis
- TCS
two-component regulatory system
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00612.
Fragments of PEG identified in the crystal structure, plots of average B factors of main-chain atoms of the structural models from the SeMet and the native data, and analysis of interactions at the PhoRD dimer interface (PDF)
Author Present Address
§ D.X. is currently a student in the University of Maryland, College Park, Maryland.
Author Present Address
∥ Department of Pathology, New York University School of Medicine, New York, NY 10016 (M.B.R.).
The authors declare no competing financial interest.
Supplementary Material
References
- Gotoh Y.; Eguchi Y.0; Watanabe T.; Okamoto S.; Doi A.; Utsumi R. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr. Opin. Microbiol. 2010, 13, 232–239. 10.1016/j.mib.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Tang Y. T.; Gao R.; Havranek J. J.; Groisman E. A.; Stock A. M.; Marshall G. R. Inhibition of bacterial virulence: drug-like molecules targeting the Salmonella enterica PhoP response regulator. Chem. Biol. Drug Des. 2012, 79, 1007–1017. 10.1111/j.1747-0285.2012.01362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryndak M.; Wang S.; Smith I. PhoP, a key player in Mycobacterium tuberculosis virulence. Trends Microbiol. 2008, 16, 528–534. 10.1016/j.tim.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Dalton T.; Cegielski P.; Akksilp S.; Asencios L.; Caoili J. C.; Cho S.-N.; Erokhin V. V.; Ershova J.; Gler M. T.; Kazennyy B. Y.; Kim H. J.; Kliiman K.; Kurbatova E.; Kvasnovsky C.; Leimane V.; van der Walt M.; Via L. E.; Volchenkov G. V.; Yagui M. A.; Kang H. Prevalence of and risk factors for resistance to second-line drugs in people with multidrug-resistant tuberculosis in eight countries: a prospective cohort study. Lancet 2012, 380, 1406–1417. 10.1016/s0140-6736(12)60734-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . Global Tuberculosis Report 2016; World Health Organization: Geneva, 2016.
- Pérez E.; Samper S.; Bordas Y.; Guilhot C.; Gicquel B.; Martín C. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol. Microbiol. 2001, 41, 179–187. 10.1046/j.1365-2958.2001.02500.x. [DOI] [PubMed] [Google Scholar]
- Walters S. B.; Dubnau E.; Kolesnikova I.; Laval F.; Daffe M.; Smith I. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol. Microbiol. 2006, 60, 312–330. 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- Gonzalo-Asensio J.; Mostowy S.; Harders-Westerveen J.; Huygen K.; Hernández-Pando R.; Thole J.; Behr M.; Gicquel B.; Martín C. PhoP: a missing piece in the intricate puzzle of Mycobacterium tuberculosis virulence. PLoS One 2008, 3, e3496 10.1371/journal.pone.0003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambiar J. K.; Pinto R.; Aguilo J. I.; Takatsu K.; Martin C.; Britton W. J.; Triccas J. A. Protective immunity afforded by attenuated, PhoP-deficient Mycobacterium tuberculosis is associated with sustained generation of CD4+ T-cell memory. Eur. J. Immunol. 2012, 42, 385–392. 10.1002/eji.201141903. [DOI] [PubMed] [Google Scholar]
- Aporta A.; Arbues A.; Aguilo J. I.; Monzon M.; Badiola J. J.; de Martino A.; Ferrer N.; Marinova D.; Anel A.; Martin C.; Pardo J. Attenuated Mycobacterium tuberculosis SO2 vaccine candidate is unable to induce cell death. PLoS One 2012, 7, e45213 10.1371/journal.pone.0045213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo-Asensio J.; Soto C. Y.; Arbues A.; Sancho J.; Menendez M. d. C.; Garcia M. J.; Gicquel B.; Martin C. The Mycobacterium tuberculosis phoPR operon is positively autoregulated in the virulent strain H37Rv. J. Bacteriol. 2008, 190, 7068–7078. 10.1128/jb.00712-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S. T.; Brosch R.; Parkhill J.; Garnier T.; Churcher C.; Harris D.; Gordon S. V.; Eiglmeier K.; Gas S.; Barry C. E. 3rd; Tekaia F.; Badcock K.; Basham D.; Brown D.; Chillingworth T.; Connor R.; Davies R.; Devlin K.; Feltwell T.; Gentles S.; Hamlin N.; Holroyd S.; Hornsby T.; Jagels K.; Barrell B. G.; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Sinha A.; Sarkar D. Transcriptional autoregulation by Mycobacterium tuberculosis PhoP involves recognition of novel direct repeat sequences in the regulatory region of the promoter. FEBS Lett. 2006, 580, 5328–5338. 10.1016/j.febslet.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Wang S.Bacterial Two-Component Systems: Structures and Signaling Mechanisms. In Protein Phosphorylation in Human Health; Huang C., Ed.; InTech: Rijeka, Croatia, 2012; pp 439–466. [Google Scholar]
- Tanaka T.; Saha S. K.; Tomomori C.; Ishima R.; Liu D.; Tong K. I.; Park H.; Dutta R.; Qin L.; Swindells M. B.; Yamazaki T.; Ono A. M.; Kainosho M.; Inouye M.; Ikura M. NMR structure of the histidine kinase domain of the E. coli osmosensor EnvZ. Nature 1998, 396, 88–92. 10.1038/23968. [DOI] [PubMed] [Google Scholar]
- Albanesi D.; Martin M.; Trajtenberg F.; Mansilla M. C.; Haouz A.; Alzari P. M.; de Mendoza D.; Buschiazzo A. Structural plasticity and catalysis regulation of a thermosensor histidine kinase. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16185–16190. 10.1073/pnas.0906699106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casino P.; Rubio V.; Marina A. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 2009, 139, 325–336. 10.1016/j.cell.2009.08.032. [DOI] [PubMed] [Google Scholar]
- Marina A.; Waldburger C. D.; Hendrickson W. A. Structure of the entire cytoplasmic portion of a sensor histidine-kinase protein. EMBO J. 2005, 24, 4247–4259. 10.1038/sj.emboj.7600886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechaly A. E.; Sassoon N.; Betton J.-M.; Alzari P. M. Segmental helical motions and dynamical asymmetry modulate histidine kinase autophosphorylation. PLoS Biol. 2014, 12, e1001776 10.1371/journal.pbio.1001776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi J.; Arrar M.; Sycz G.; Cerutti M. L.; Berguer P. M.; Paris G.; Estrín D. A.; Martí M. A.; Klinke S.; Goldbaum F. A. Structural Insights into the HWE Histidine Kinase Family: The Brucella Blue Light-Activated Histidine Kinase Domain. J. Mol. Biol. 2016, 428, 1165–1179. 10.1016/j.jmb.2016.01.026. [DOI] [PubMed] [Google Scholar]
- Casino P.; Miguel-Romero L.; Marina A. Visualizing autophosphorylation in histidine kinases. Nat. Commun. 2014, 5, 3258. 10.1038/ncomms4258. [DOI] [PubMed] [Google Scholar]
- Marina A.; Mott C.; Auyzenberg A.; Hendrickson W. A.; Waldburger C. D. Structural and mutational analysis of the PhoQ histidine kinase catalytic domain. Insight into the reaction mechanism. J. Biol. Chem. 2001, 276, 41182–41190. 10.1074/jbc.m106080200. [DOI] [PubMed] [Google Scholar]
- Ashenberg O.; Keating A. E.; Laub M. T. Helix bundle loops determine whether histidine kinases autophosphorylate in cis or in trans. J. Mol. Biol. 2013, 425, 1198–1209. 10.1016/j.jmb.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomomori C.; Tanaka T.; Dutta R.; Park H.; Saha S. K.; Zhu Y.; Ishima R.; Liu D.; Tong K. I.; Kurokawa H.; Qian H.; Inouye M.; Ikura M. Solution structure of the homodimeric core domain of Escherichia coli histidine kinase EnvZ. Nat. Struct. Biol. 1999, 6, 729–734. 10.1038/11495. [DOI] [PubMed] [Google Scholar]
- Wang C.; Sang J.; Wang J.; Su M.; Downey J. S.; Wu Q.; Wang S.; Cai Y.; Xu X.; Wu J.; Senadheera D. B.; Cvitkovitch D. G.; Chen L.; Goodman S. D.; Han A. Mechanistic insights revealed by the crystal structure of a histidine kinase with signal transducer and sensor domains. PLoS Biol. 2013, 11, e1001493 10.1371/journal.pbio.1001493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulko M.; Berndt F.; Gruber M.; Linder J. U.; Truffault V.; Schultz A.; Martin J.; Schultz J. E.; Lupas A. N.; Coles M. The HAMP domain structure implies helix rotation in transmembrane signaling. Cell 2006, 126, 929–940. 10.1016/j.cell.2006.06.058. [DOI] [PubMed] [Google Scholar]
- Airola M. V.; Watts K. J.; Bilwes A. M.; Crane B. R. Structure of concatenated HAMP domains provides a mechanism for signal transduction. Structure 2010, 18, 436–448. 10.1016/j.str.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris H. U.; Dunin-Horkawicz S.; Hornig N.; Hulko M.; Martin J.; Schultz J. E.; Zeth K.; Lupas A. N.; Coles M. Mechanism of regulation of receptor histidine kinases. Structure 2012, 20, 56–66. 10.1016/j.str.2011.11.014. [DOI] [PubMed] [Google Scholar]
- Podgornaia A. I.; Casino P.; Marina A.; Laub M. T. Structural basis of a rationally rewired protein-protein interface critical to bacterial signaling. Structure 2013, 21, 1636–1647. 10.1016/j.str.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover C. K.; de la Cruz V. F.; Fuerst T. R.; Burlein J. E.; Benson L. A.; Bennett L. T.; Bansal G. P.; Young J. F.; Lee M. H.; Hatfull G. F.; et al. New use of BCG for recombinant vaccines. Nature 1991, 351, 456–460. 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- Lee M. H.; Hatfull G. F. Mycobacteriophage L5 integrase-mediated site-specific integration in vitro. J. Bacteriol. 1993, 175, 6836–6841. 10.1128/jb.175.21.6836-6841.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. H.; Pascopella L.; Jacobs W. R. Jr.; Hatfull G. F. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guerin. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 3111–3115. 10.1073/pnas.88.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira A. L.; Tsenova L.; Murray P. J.; Freeman S.; Bergtold A.; Chiriboga L.; Kaplan G. Aerosol infection of mice with recombinant BCG secreting murine IFN-γ partially reconstitutes local protective immunity. Microb. Pathog. 2000, 29, 175–185. 10.1006/mpat.2000.0382. [DOI] [PubMed] [Google Scholar]
- Manganelli R.; Voskuil M. I.; Schoolnik G. K.; Smith I. The Mycobacterium tuberculosis ECF sigma factor σE: role in global gene expression and survival in macrophages. Mol. Microbiol. 2001, 41, 423–437. 10.1046/j.1365-2958.2001.02525.x. [DOI] [PubMed] [Google Scholar]
- Menon S.; Wang S. Structure of the response regulator PhoP from Mycobacterium tuberculosis reveals a dimer through the receiver domain. Biochemistry 2011, 50, 5948–5957. 10.1021/bi2005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyne G. D.; Standaert R. F.; Karplus P. A.; Schreiber S. L.; Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 1993, 229, 105–124. 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 479–485. 10.1107/s0907444909038360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G.; Cohen S. X.; Lamzin V. S.; Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/s0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007, 63, 32–41. 10.1107/s0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.