

Abstract
Objective
Copy number variations (CNVs), a major cause of genetic hearing loss, most frequently involve the STRC gene, located on chr15q15.3 and causally related to autosomal recessive non-syndromic hearing loss (ARNSHL) at the DFNB16 locus. The interpretation of STRC sequence data can be challenging due to the existence of a virtually identical pseudogene, pSTRC that promotes complex genomic rearrangements in this genomic region. Targeted genomic enrichment with massively parallel sequencing (TGE+MPS) has emerged as the preferred method by which to provide comprehensive genetic testing for hearing loss. We aimed to identify CNVs in the STRC region using established and validated bioinformatics methods.
Methods
We used TGE+MPS to identify the genetic cause of hearing loss. CNV results were confirmed with customized array comparative genomic hybridization (array CGH).
Results
Three probands with progressive mild-to-moderate hearing loss were found among 40 subjects with ARNSHL to segregate homozygous STRC deletions and gene to pseudogene conversion. Array CGH showed that the deletions/conversions span multiple genes outside of the exons captured by TGE+MPS.
Conclusion
These data further validate the necessity to integrate the detection of both simple variant changes and complex genomic rearrangements in the clinical diagnosis of genetic hearing loss.
Keywords: Hearing loss, copy number variations, STRC, massively parallel sequencing, array CHG
Introduction
Complex genomic alterations are an important cause of disease. While attention is most frequently focused on single nucleotide variations (SNVs), copy number variations (CNVs), traditionally considered the alteration through deletion, insertion and/or duplication of more than ~1 kbp, are implicated in many different diseases and neurodevelopmental disorders1, 2. Included in this list is sensorineural hearing loss (SNHL), a disease phenotype in which CNVs play a major role. Using comprehensive genetic testing platforms, CNVs have been identified in about 20% of all deafness-causing genes and can be found in about 15% of patients who undergo genetic testing3.
STRC is the deafness-causing gene most frequently associated with CNV events, with the CNV event most often leading to complete deletion of this gene3. These deletions have an estimated prevalence of 1% in deaf populations4. Defining the extent of a STRC CNV is challenging because the genomic region includes four genes, three of which have highly homologous pseudogenes. For example, the pseudo-STRC gene (pSTRC) has 98% homology to the functional STRC gene. Accurately assessing the rearrangement size in this region takes on added importance because when the neighboring CATSPER2 gene is included in the CNV and is also deleted, the phenotype becomes complex5. In addition to hearing loss, males have infertility secondary to altered sperm motility and therefore carry the diagnosis of deafness infertility syndrome (DIS)6, 7. Female fertility is not affected.
In this study, we compare the results of TGE+MPS and high-resolution array CGH to characterize CNVs involving the STRC gene in three Japanese families segregating deafness at the DFNB16 locus.
Subjects and Methods
Families screened in this study were identified as part of a comprehensive genetic screen of 194 Japanese subjects (114 females) from unrelated and non-consanguineous families identified in 33 otolaryngology clinics in 28 prefectures across Japan. The complete details of this cohort have been previously reported 8. For each proband, informed consent was obtained to participate in this study, which was approved by the human subjects ethical committee associated with each clinic. Clinical information and blood samples were obtained for each proband and for all consenting affected and unaffected relatives.
Targeted Genomic Enrichment and Massively Parallel Sequencing
We sequenced all exons of all known non-syndromic hearing loss genes and non-syndromic hearing loss mimic genes as described previously. TGE and library preparation were completed using a modified solution-phase protocol based on the Agilent SureSelect target enrichment system. Prior to library preparation, genomic DNA quality was assessed by gel electrophoresis and Nanodrop 1000 spectrophotometer (Thermo Scientific, Inc) (260/280 ration = 1.8–2.2) and quantitated using a Qubit 2.0 fluorometer (Life Technologies, CA, USA). Three μg of gDNA was randomly fragmented to an average size of 250 bp by sonication with a Covaris acoustic solubilizer (Covaris Inc., Woburn, MA, USA), fragment ends were repaired, A-tails were added and sequencing adaptors were ligated prior to the first amplification. Solid phase reverse immobilization purification was performed between each enzymatic reaction. Hybridization and capture with RNA baits was followed by a second amplification before pooling for sequencing. In all cases, the smallest number of amplifications possible was used: typically eight cycles for the pre-hybridization PCR (range 8–10 cycles using NEB Phusion HF Master Mix) and 14 cycles for the post-hybridization PCR (range 12–16 cycles using Agilent Herculase II Fusion DNA Polymerase). All samples were barcoded and multiplexed before sequencing. We performed sequencing using the Illumina HiSeq system in pools of 48 samples per lane using 100-bp paired-end reads.
Copy number analysis on MPS data
We completed read mapping with BWA and re-alignment with GATK, using the indexed BAM file to call variants and generate a pileup using samtools, retaining only the position and depth information. CNV calling was completed using a custom R script, a previously published tool9. This method normalizes read-depth data by sample batch using a sliding-window approach. We used the 2011-07-01 version of the tool with default settings. All CNV calls were curated through manual inspection.
Array-CGH
We designed a custom aCGH platform using the Agilent SureDesign website to select for inclusion of 86 known deaf genes, with probes covering these genomic regions at 150–200 bp intervals on the Agilent 8x60K platform (Agilent Technologies, Santa Clara, CA). aCGH was completed using the same gDNA samples as for TGE+MPS. 10ul of gDNA solution (0.5ug of DNA) was fragmented, labeled with cyanine-3 (reference DNA) or cyanine-5 (subjects), and hybridized. Scanning of the array was completed following the manufacturer’s recommended protocols; scanned aCGH data were analyzed using CytoGenomics software v3.0.6.6 (Agilent Technologies).
Results
Case Details
Case 1: Patient # 167
This 12-year-old male was identified with hearing loss at a school check-up. His brother was also having hearing loss. Newborn hearing screening data were not available so the exact age-of-onset was not known. He did not have any complaints of vertigo or tinnitus. Both parents had normal hearing (Figure 1).
Figure 1.
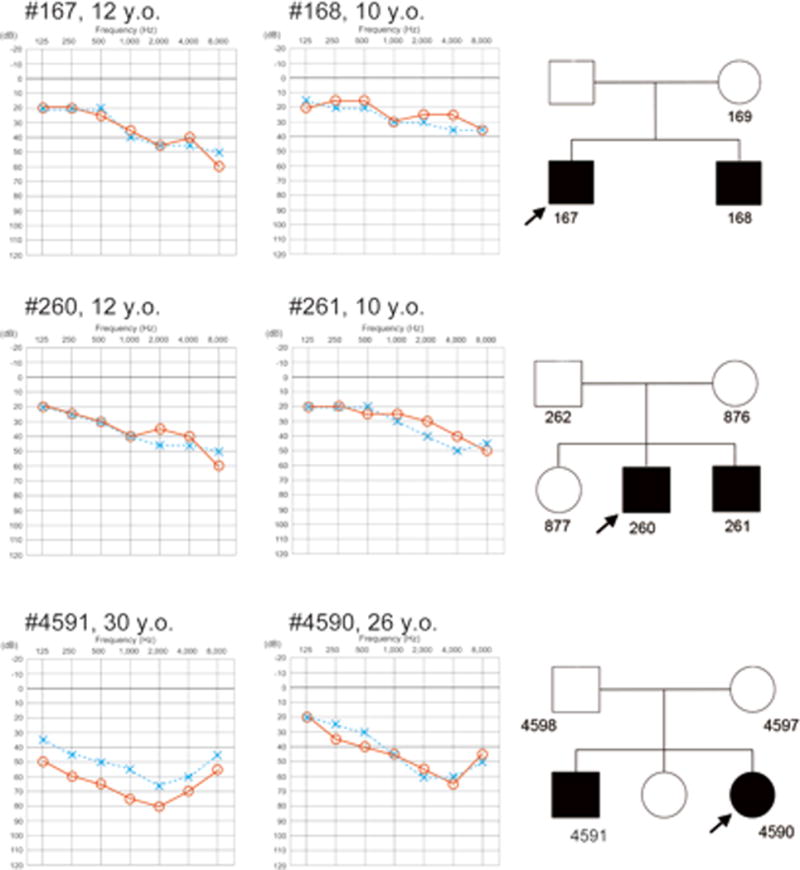
Audiograms of the proband and siblings with homozygous STRC deletions. Audiograms show mild-to-moderate hearing loss and mild age-associated progression of hearing loss. Each number in the pedigree is the assigned patient number.
Case 2: Patient # 260
This 12-year-old male was first noted to have hearing loss at a school check up. Although he was diagnosed with mild hearing loss, he was not aware of any difficulty hearing. His brother (patient #261) had a similar mild sloping hearing loss (Figure 1).
Case 3: Patient # 4590
This 26-year-old female noticed hearing loss in early childhood. Serial audiological assessments showed a slow deterioration in hearing. She had no episodes of vertigo however she did complain of tinnitus. Her elder brother (patient #4591) had more severely progressive hearing loss (Figure 1).
CNVs in the STRC genomic region
Two probands were homozygous for a deletion of STRC (prevalence amongst deaf subjects in this study, 7.5%). We also identified one subject who was heterozygous for a STRC gene deletion and one with a heterozygous gene duplication of STRC.
CNV analysis derived from TGE+MPS identified a homozygous deletion encompassing STRC and CATSPER2 genes in two probands (#167 and #4590) and a biallelic partial STRC gene to pseudogene conversion in one proband (#260) (Figure 2).
Figure 2.
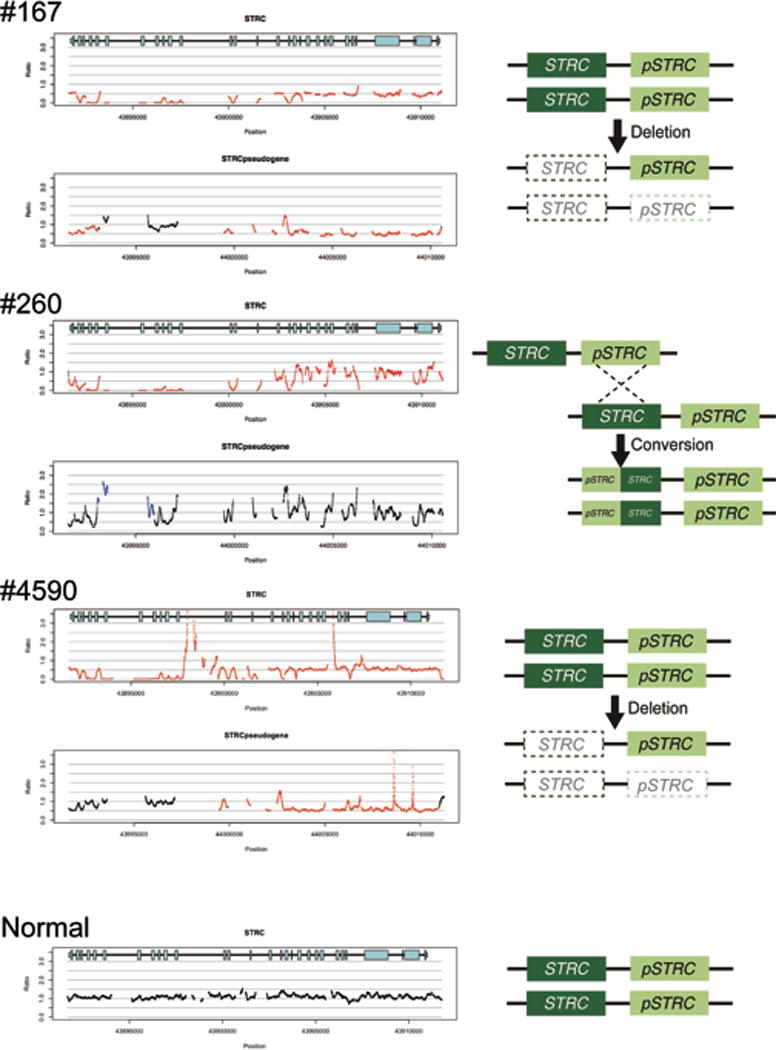
CNV identification by a CNV-calling script implemented with TGE+MPS. The horizontal axis indicates chromosomal position, and the vertical axis indicates the number of gene copies shown as a ratio; 1.0 indicates the normal 2 copies (black lines), and 0.5 and 0.0 indicates 1 copy (heterozygous deletion) or 0 copy (homozygous deletion), respectively indicated by red lines. Likewise, a figure above 1.0 shows copy gain (heterozygous or homozygous gene duplication) as indicated by blue lines.
A homozygous deletion in the STRC region and a heterozygous deletion in the pseudo-STRC gene (pSTRC) is seen in patients #167 and #4590. A homozygous deletion of the last 11 exons in STRC (left side in the gene diagram) and homozygous duplication of the corresponding region of pSTRC is seen in patient #260. The hypothesized mechanisms for these events are shown to the right.
As shown in Figure 3, array CGH confirmed a homozygous deletion of STRC through CATSPER2 in Case 1 and 3. Segregation analysis confirmed a heterozygous deletion in the parental samples of Case 3, with no obvious changes in the pSTRC region. Case 2 also carried a homozygous deletion of the STRC to CATSPER2 region, with at least two or more copies of the pSTRC region predicted (indicated by blue dots). Parental samples of Case 2 showed only heterozygous deletion of the STRC to CATSPER2 region.
Figure 3.
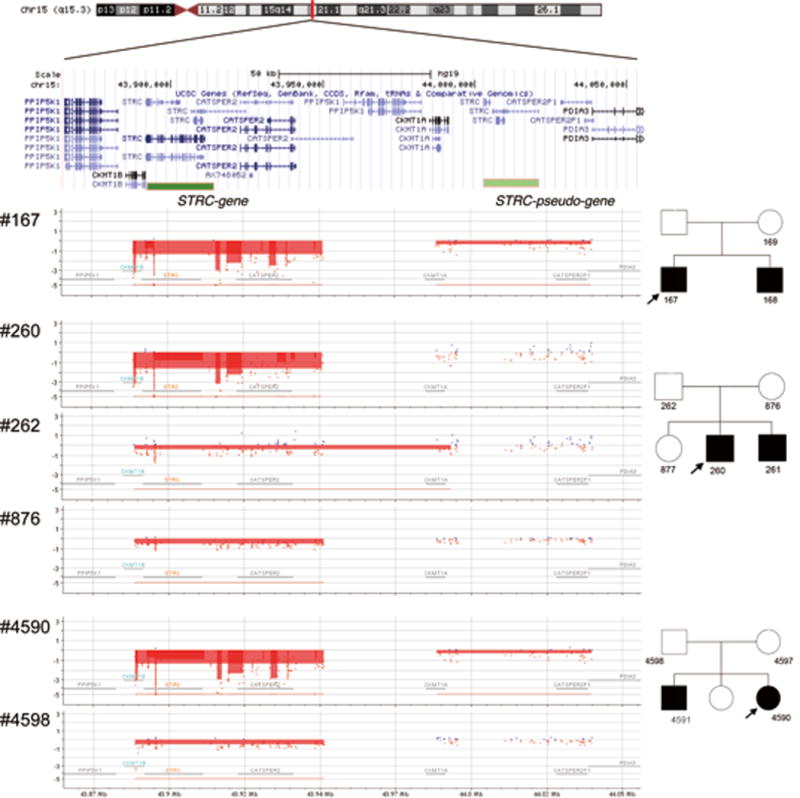
Results of array CGH for patients and parent(s). The upper part of the figure shows the genomic location across multiple genes. The results show deletion of both copies of STRC and hemizygous deletion of pSTRC in patients #167 and #4590 by CytoGnomics software, with parental sample #4598 showing hemizygous deletion of STRC but no changes in the pSTRC region. For the family of the proband #260, deletions in only STRC to CATSPER2 region were observed.
DISCUSSION
We identified three patients with ARNSHL caused by CNVs in STRC in a Japanese hearing loss population for an estimated prevalence of 7.5% (3 /40 ARNSHL probands). This figure is similar to that reported by Vona et al. who found six of 94 SNHL subjects (6.4%) with homozygous or compound heterozygous STRC deletions as a cause of hearing loss using whole-genome aCGH10. The carrier frequency of STRC deletions has been estimated at 1.1 to 2.5% 4, 11. We analyzed only a limited number of 40 ARNSHL samples, thus further study is necessary to clarify the prevalence of CNV in the STRC.
CNVs are found in 13% of the human genome and may be underestimated as a cause of SNHL to difficulties and limitations in their detection.13 Although several methods including multiplex ligation-dependent probe amplification (MLPA), SNP arrays, and aCGH can be used to identify CNVs, these analyses are not routinely included in genetic testing for SNHL. Fortunately, with the implementation of TGE+MPS in the comprehensive evaluation of SNHL, detection of CNVs can be integrated into the bioinformatics analysis. Furthermore, targeted genomic regions can be expanded with additional probes to improve CNV identification when the gene locations or positions in which CNVs commonly occurred were well established.
Using a customized aCGH pipeline, we confirm the validity of this approach for the detection of STRC-associated CNVs. There was high concordance between the MPS CNV analysis and the aCGH results provided more data throughout the STRC genomic region. This result is important as CNVs in STRC are a common cause of mild-to-moderate ARNSHL3, 14, 15. In summary, we propose that genomic alterations including CNVs should be considered as a major cause of SNHL and that genetic testing must integrate SNV and CNV identification.
Acknowledgments
This work was supported by grants-in-aid from the NIDCD (RO1s DC003544, DC002842 and DC012049 to RJHS), the Ministry of Health, Labour and Welfare, Japan (a Health and Labour Sciences Research Grant for Research on Rare and Intractable Diseases and Comprehensive Research on Disability Health and Welfare to SU), the Japan Agency for Medical Research and Development (a Practical Research Project for Rare/Intractable Disease Grant to SU), and the Ministry of Education, Culture, Sports, Science and Technology, Japan (a Grant-in-Aid for Scientific Research to SU and HM).
Footnotes
Declaration of conflicting interests
All authors have declared no conflicts of interest.
References
- 1.Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conrad DF, Pinto D, Redon R, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464(7289):704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shearer AE, Kolbe DL, Azaiez H, et al. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014;6(5):37. doi: 10.1186/gm554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoppman N, Aypar U, Brodersen P, Brown N, Wilson J, Babovic-Vuksanovic D. Genetic testing for hearing loss in the United States should include deletion/duplication analysis for the deafness/infertility locus at 15q15.3. Mol Cytogenet. 2013;6(1):19. doi: 10.1186/1755-8166-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knijnenburg J, Oberstein SA, Frei K, et al. A homozygous deletion of a normal variation locus in a patient with hearing loss from non-consanguineous parents. J. Med. Genet. 2009;46(6):412–417. doi: 10.1136/jmg.2008.063685. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Malekpour M, Al-Madani N, et al. Sensorineural deafness and male infertility: a contiguous gene deletion syndrome. J. Med. Genet. 2007;44(4):233–240. doi: 10.1136/jmg.2006.045765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaiswal D, Singh V, Dwivedi US, Trivedi S, Singh K. Chromosome microarray analysis: a case report of infertile brothers with CATSPER gene deletion. Gene. 2014;542(2):263–265. doi: 10.1016/j.gene.2014.03.055. [DOI] [PubMed] [Google Scholar]
- 8.Moteki H, Azaiez H, Booth KT, et al. Comprehensive genetic testing with ethnic-specific filtering by allele frequency in a Japanese hearing-loss population. Clin. Genet. 2015 doi: 10.1111/cge.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics. 2011;12:184. doi: 10.1186/1471-2164-12-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vona B, Hofrichter MA, Neuner C, et al. DFNB16 is a frequent cause of congenital hearing impairment: implementation of STRC mutation analysis in routine diagnostics. Clin. Genet. 2015;87(1):49–55. doi: 10.1111/cge.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Francey LJ, Conlin LK, Kadesch HE, et al. Genome-wide SNP genotyping identifies the Stereocilin (STRC) gene as a major contributor to pediatric bilateral sensorineural hearing impairment. Am J Med Genet A. 2012;158A(2):298–308. doi: 10.1002/ajmg.a.34391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girirajan S, Rosenfeld JA, Coe BP, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012;367(14):1321–1331. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1(1):4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shearer AE, Black-Ziegelbein EA, Hildebrand MS, et al. Advancing genetic testing for deafness with genomic technology. J. Med. Genet. 2013;50(9):627–634. doi: 10.1136/jmedgenet-2013-101749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sloan-Heggen CM, Bierer AO, Shearer AE, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016;135(4):441–450. doi: 10.1007/s00439-016-1648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]