

SUMMARY
Intratumor genetic heterogeneity underlies the ability of tumors to evolve and adapt to different environmental conditions. Using CRISPR/Cas9 technology and specific DNA barcodes, we devised a strategy to recapitulate and trace the emergence of subpopulations of cancer cells containing a mutation of interest. We used this approach to model different mechanisms of lung cancer cell resistance to EGFR inhibitors and to assess effects of combined drug therapies. By overcoming intrinsic limitations of current approaches, CRISPR-barcoding also enables investigation of most types of genetic modifications, including repair of oncogenic driver mutations. Finally, we used highly complex barcodes inserted at a specific genome location as a means of simultaneously tracing the fates of many thousands of genetically labeled cancer cells. CRISPR-barcoding is a straightforward and highly flexible method that should greatly facilitate the functional investigation of specific mutations, in a context that closely mimics the complexity of cancer.
In Brief
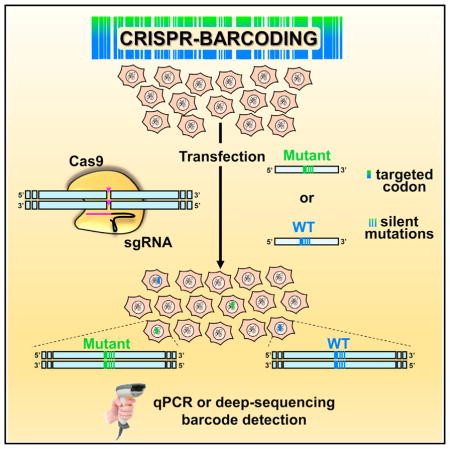
Guernet et al. used CRISPR/Cas9 technology to genetically label and trace tumor cells within a mass population through insertion of a series of point mutations at a specific genomic location. The effects of a particular experimental condition on the fraction of barcoded cells can be assessed by qPCR or deep sequencing.
INTRODUCTION
Staggering advances in sequencing technologies have provided a detailed overview of the multiple genetic aberrations in cancer and also demonstrated that, within an individual tumor, such mutations are often distributed in intricate patterns of multiple and heterogeneous subclonal populations (Gerlinger et al., 2012; McGranahan and Swanton, 2015). This complex genetic reservoir fuels the capacity of tumor cells to adapt to various environmental conditions, with major clinical implications for cancer progression and resistance to therapeutic intervention (Gillies et al., 2012; Greaves and Maley, 2012; McGranahan and Swanton, 2015). Although the ability of tumors to adapt and evolve through the emergence of clonal subpopulations containing de novo mutations has been known for decades (Nowell, 1976), tumors are generally treated as genetically homogeneous entities (Clevers, 2011). Computational modeling based on analysis of deep-sequencing data sets, including single-cell sequencing, now provides the means to dissect the clonal architecture of a tumor through the identification of its different cell subpopulations (Ding et al., 2014; McGranahan and Swanton, 2015). However, although these new powerful technologies can capture a picture of the genetic complexity and diversity of a given tumor, they cannot experimentally recapitulate cancer evolution through the emergence of new mutations.
CRISPR (clustered regularly interspaced short palindromic repeats) is a new DNA editing technology based on the Cas9 nuclease from S. pyogenes and a single-guide RNA (sgRNA), which as a complex can specifically recognize and cleave a genomic sequence of interest (Doudna and Charpentier, 2014; Hsu et al., 2014). The double-strand DNA break induced by CRISPR/Cas9 can trigger two distinct cellular mechanisms for DNA repair: error-prone nonhomologous end-joining (NHEJ) and high-fidelity homology-directed repair (HDR). While NHEJ-induced indels can be exploited to inactivate a gene of interest, HDR enables precise DNA editing (Doudna and Charpentier, 2014; Hsu et al., 2014). Despite its tremendous potential, widespread use of HDR has been curbed by intrinsic limitations, mostly related to its low efficiency and the necessity to derive clones.
By turning the low efficiency of HDR-mediated DNA editing to our advantage, we devised a strategy to recapitulate and trace intratumor heterogeneity and the emergence of genetically distinct cancer subpopulations on the basis of silent DNA barcodes coupled to a desired mutation in the sequence of a gene of interest. These genetic labels can then be “read” by real-time qPCR from genomic DNA (gDNA) to measure the relative proportion of the modified cells within an unmodified mass population. We used this CRISPR-barcoding strategy to model different mechanisms of non-small-cell lung cancer (NSCLC) resistance to epidermal growth factor receptor (EGFR) inhibitors, and we established a multiplex system to evaluate the efficacy of combined drug therapies aimed at preventing or delaying the emergence of resistant cells. Through a similar approach, we assessed for the first time the effects of repairing oncogenic driver mutations in addicted cancer cells directly at the genome level. Finally, we used a highly complex set of CRISPR-barcodes as a means to simultaneously label several thousand different breast and lung cancer cells, and we compared their relative fitness to grow in vivo after inoculation in immunodeficient mice or in vitro upon treatment with a targeted therapeutic agent.
DESIGN
Compared with other DNA editing tools, including zinc finger nucleases and transcription activator-like effector nucleases, which are based on protein-DNA recognition, the CRISPR/Cas9 technology is remarkably more flexible and easy to use (Boettcher and McManus, 2015). However, despite the undeniable potential of this new technology, major intrinsic limitations need to be considered when applying CRISPR/Cas9 for HDR genome manipulation of cultured cells. First, in certain contexts this system can tolerate a few mismatches between the sgRNA and its target sequence, which can result in off-target DNA cleavage (Doudna and Charpentier, 2014; Fu et al., 2013; Hsu et al., 2014). Although tools have been designed to identify in silico potential off-target sequences, occasional minor shifts in the secondary structure of the RNA-DNA duplex that could make such predictions more problematic have been recently reported (Lin et al., 2014). Another limitation is related to the fact that the efficiency of HDR-mediated DNA editing can be extremely low, depending on the cell model, the sgRNA sequence, and the targeted DNA. Thus, in a given experiment, only a fraction of cells within a population will contain the desired genetic modification. Hence, the derivation and the subsequent analysis of a certain number of individual clones constitute an almost inevitable step (Ran et al., 2013). Besides the potential issues related to clonal variability, this approach requires a reasonably high efficiency of CRISPR-mediated editing, and it is obviously not compatible with modifications that have a negative impact on cell growth.
To overcome these drawbacks, we have devised CRISPR-barcoding, a new strategy in which a potentially functional modification in the sequence of a gene of interest is coupled with a series of silent point mutations, serving as a genetic label for cell tracing. In parallel, a second barcode consisting of distinct silent mutations is inserted in the same cell population and used as a control for possible CRISPR off-target effects. gDNA from the resulting mixture of CRISPR-modified and unmodified cells is then probed using qPCR to assess the relative proportion of each barcode. By exposing the cells to a given selective condition, this approach can be used to functionally characterize the effects of different types of mutations of a particular gene of interest. We applied this strategy to manipulate the endogenous sequence of different oncogenes and tumor suppressors, including EGFR, KRAS, anaplastic lymphoma kinase (ALK), TP53, and adenomatous polyposis coli (APC) in various cancer cell models. We then assessed the effects of such modifications on signaling pathway activation, cell growth and invasion, or resistance to chemotherapy, both in vitro and in vivo. Through a similar approach, we also labeled breast cancer cells using a degenerate barcode inserted at a specific location of their genome, to investigate the contribution of cell heterogeneity to tumor formation. By overcoming the limitations associated with the low efficiency and potential off-target effects of DNA editing, our work demonstrates that CRISPR-barcoding can be easily implemented in functional studies to investigate the effects of a specific genetic modification.
RESULTS
Modeling Drug Resistance in NSCLC Cells
A major limitation of cancer-targeted therapies is the almost inevitable development of acquired resistance, as a result of the emergence of a subpopulation of cancer cells that become insensitive to the treatment, generally through particular genetic aberrations. Although such mutations could conceivable originate de novo during the treatment, recent evidence from different types of tumors indicates that resistant clones are often already present before the onset of the therapy (Bhang et al., 2015; Diaz et al., 2012; Misale et al., 2012; Turke et al., 2010).
As a typical example of drug resistance, gefitinib is a small-molecule ATP-competitive reversible EGFR inhibitor used in the clinic for the treatment of advanced NSCLC harboring EGFR-activating mutations. After an initial response, the tumors invariably relapse, in half of the cases because of the appearance of the T790M “gatekeeper” secondary mutation in the catalytic domain of this receptor, resulting in an increased affinity for ATP (Chong and Jänne, 2013). To generate a new model of NSCLC resistance to EGFR inhibitors, we designed a specific sgRNA and a donor single-stranded DNA oligonucleotide (ssODN) containing the T790M mutation, as well as a few additional silent mutations to serve as a genetic barcode. As a control for potential CRISPR/Cas9 off-target cleavage, we generated a ssODN (EGFR-T790T) containing a distinct set of silent mutations (Figure 1A). To prevent the potential incorporation of the two DNA sequences into different alleles within the same cell, EGFR inhibitor-sensitive PC9 cells were transfected with the CRISPR/Cas9 plasmid, encoding Cas9 and the sgRNA, together with one of the two ssODNs. Immediately after transfection, the cells were pooled to form a mixed population of unmodified, EGFR-T790M, and EGFR-T790T cells, and PCR primers were designed to specifically recognize each barcode (Figure 1B).
Figure 1. CRISPR-Barcoding to Recapitulate NSCLC Resistance to EGFR Inhibitors.

(A) Schematic representation of the incorporation of EGFR-T790M and EGFR-T790T (control) barcodes in NSCLC cells.
(B) The specificity of the qPCR primers for EGFR barcodes was assessed using gDNA from 293T cells transfected with a control GFP plasmid or the CRISPR/Cas9 vector with either the EGFR-T790T or the EGFR-T790M ssODN. The control (Ctrl) primers allowed amplification of a different, unmodified region of the EGFR gene.
(C) Nine days after transfection for EGFR CRISPR-barcoding, PC9 cells were treated in the presence or the absence of gefitinib (10 nM, arrow). qPCR was performed from gDNA extracted at the indicated time points. The results correspond to the mean values of the EGFR-T790M to EGFR-T790T barcode ratio (±SEM; n = 3) of one representative of three independent experiments.
(D) PC9 cells containing the EGFR-T790M and EGFR-T790T barcodes were treated for 4 days with the indicated concentrations of gefitinib. Mean ± SEM; n = 4 of one representative of three independent experiments.
(E) Effects of 4 days of gefitinib (0.1 μM) and/or WZ4002 (0.5 μM) on the EGFR-T790M to EGFR-T790T ratio. Mean ± SEM; n = 4 of one representative of three independent experiments.
(F) Effects of gefitinib (1 μM, 5 days) on the KRAS-G12D to KRAS-G12G ratio in PC9 cells using two distinct sgRNAs (A or B). Mean ± SEM; n = 5 of one representative of four independent experiments. *p < 0.05 and **p < 0.01 compared with control (Mann-Whitney test).
(G) Effects of WZ4002 (0.5 μM, 4 days) on the KRAS-G12D to KRAS-G12G ratio. Mean ± SEM; n = 4 of one representative of three independent experiments.
(H) Strategy for PC9 cell invasion studies using CRISPR-barcoding.
(I) qPCR analysis of EGFR-T790T and EGFR-T790M barcodes from PC9 cells outside (OUT) or inside (IN) the Boyden chamber, or seeded in parallel in a regular well (WELL), as illustrated in (H). The mean values (±SEM; n = 3) of one representative of three independent experiments were normalized to the total amount of gDNA using EGFR_Ctrl primers (B). ***p < 0.001 compared with the cells inside the Boyden chamber (Student’s t test).
See also Figure S1.
As shown in Figures 1C and S1A, gefitinib treatment provoked a gradual increase in the EGFR-T790M to EGFR-T790T barcode ratio, consistent with acquired resistance to the inhibitor conferred by the mutation. Similar results were obtained using a different sgRNA (Figure S1B). Because of the EGFR-T790T internal control, the CRISPR-barcoding approach allows specificity over a wide range of inhibitor concentrations. Indeed, an enrichment of EGFR-T790M containing cells was readily detectable after 4 days of gefitinib treatment at a concentration of 10 nM, while a stronger effect was observed at higher doses (Figure 1D).
Recent clinical trials have shown the efficacy of third-generation EGFR irreversible inhibitors against NSCLCs that developed resistance to gefitinib and erlotinib through the T790M mutation (Jänne et al., 2015). We tested the effects of one of these compounds, WZ4002 (Zhou et al., 2009), in our CRISPR-barcoding model for drug resistance. Figure 1E shows that WZ4002 completely abolished the enrichment of the EGFR-T790M bar-code in the presence of gefitinib, consistent with the high affinity of this compound for the receptor containing the gatekeeper mutation. To model a different type of genetic aberration leading to resistance to EGFR inhibitors in NSCLCs, we used a similar approach to generate a subpopulation of PC9 cells containing a mutation of the oncogene KRAS (Figure S1C), a well-known negative predictor for primary responsiveness of NSCLCs to EGFR inhibitors (Pao and Chmielecki, 2010). As shown in Figures 1F and S1D, the KRAS mutant barcode was significantly enriched in the presence of gefitinib, consistent with a downstream activation of EGFR signaling in these cells. Of note, a similar enrichment was observed in the presence of WZ4002 (Figure 1G), indicating that therapies based on receptor inhibition could result in the selection of clonal subpopulations containing alternative mechanisms of resistance.
EGFR can stimulate cell migration and invasion through different mechanisms, including Src and STAT5 activation, or by promoting epithelial to mesenchymal transition (Avraham and Yarden, 2011; Huveneers and Danen, 2009). However, these effects are often masked by the overwhelming impact of this receptor on cell proliferation and survival. To investigate the specific contribution of EGFR inhibition in restraining NSCLC invasiveness, independently of its effects on cell growth, EGFR-T790-barcoded PC9 cells were pre-treated for 2 days with gefitinib, then transferred into Boyden chambers containing Matrigel. Twenty-four hours later, the cells on both sides of the polycarbonate membrane were collected, and gDNA was extracted for qPCR analysis (Figure 1H). As shown in Figure 1I, the relative proportion of the EGFR-T790M barcode was dramatically increased in cells that migrated through the Matrigel-coated membrane, indicating that, in addition to the effect on cell growth, EGFR inhibition has also a profound impact on the invasive properties of NSCLC cells. As a control, in the absence of gefitinib the proportion of the two EGFR barcodes remained stable in each compartment (Figure S1E). By measuring the fraction of barcoded cells on both sides of the chamber, our bar-coding strategy allows specific and quantitative analysis of cell invasion, regardless of any potential effect on cell proliferation and/or survival.
Multiplex CRISPR-Barcoding for In Vitro and In Vivo Studies
Identification of chromosomal rearrangements involving the gene encoding ALK receptor tyrosine kinase in a significant fraction of metastatic NSCLCs (Soda et al., 2007) led to the approval of specific ALK inhibitors for the treatment of this type of cancer (Solomon et al., 2014). The most frequent of such rearrangements corresponds to an inversion on chromosome 2, resulting in the expression of a constitutively active fusion protein, composed of the echinoderm microtubule-associated protein-like 4 (EML4) and ALK (Hallberg and Palmer, 2013). As recently described in other models (Choi and Meyerson, 2014; Maddalo et al., 2014), we used CRISPR/Cas9 technology to generate the EML4-ALK inversion in PC9 cells. To increase the efficiency of the chromosomal rearrangement, the cells were co-transfected with a ssODN encompassing the EML4-ALK fusion sequence, and qPCR primers were designed to specifically recognize the inverted locus (Figures S2A–S2C), providing a genetic barcode for cells expressing the oncogenic EML4-ALK protein. As shown in Figure 2A, gefitinib treatment increased the fraction of PC9 cells containing EML4-ALK, indicating that this chromosomal rearrangement could represent a new mechanism of resistance to EGFR inhibition.
Figure 2. Multiplex Model for NSCLC Resistance to EGFR Inhibition.

(A) EML4-ALK chromosomal inversion was induced in PC9 cells using CRISPR/Cas9, and the fraction of cells containing the EML4-ALK barcode was assessed in the presence or the absence of gefitinib (1 μM, 5 days). The mean values (±SEM; n = 4) of one representative of three independent experiments were normalized to the total amount of gDNA using EGFR_Ctrl primers.
(B) Multiplexed CRISPR-barcoding in PC9 cells for in vitro or in vivo studies.
(C) Effects of gefitinib (1 μM, 5 days) in PC9-EKE cells. The proportion of each barcode was normalized using EGFR_Ctrl primers. Mean (±SEM; n = 4) of one representative of three independent experiments.
(D) Effects of combinations of gefitinib (0.5 μM, 5 days) with WZ4002 (0.5 μM) and TAE684 (0.5 μM) in PC9-EKE cells. Mean (±SEM; n = 4) of one representative of three independent experiments.
(E) Effects of gefitinib (0.5 μM, 5 days) with or without trametinib (50 nM) in EKE cells. Mean (±SEM; n = 5) of one representative of three independent experiments. *p < 0.05 and **p < 0.01 compared with Ctrl or gefitinib-treated cells (Mann-Whitney test).
See also Figure S2.
The fact that different mechanisms of resistance can coexist within the same tumor or in metastases from the same patient suggests that multiple resistant clones may be present before the onset of therapy (Chong and Jänne, 2013). To recapitulate this type of genetic heterogeneity, we co-introduced the EGFR, KRAS, and EML4-ALK mutations described above in different subpopulations of the same PC9 cell culture, hereafter designed as PC9-EKE (Figure 2B). Consistent with the data obtained by mutating individual genes, treatment of this heterogeneous cell population with gefitinib increased the proportion of cells containing the EGFR-T790M, KRAS-G12D, and EML4-ALK barcodes (Figure 2C). This type of multiplex model is particularly suited to evaluate the efficacy of combined therapies aimed at preventing or delaying the emergence of resistant cells. As shown in Figure 2D, although WZ4002 and the ALK inhibitor TAE684 could efficiently prevent gefitinib resistance conferred by the T790M mutation or the EML4-ALK fusion, respectively, these compounds had no effect on the other resistant populations, indicating that strategies based on targeting single mechanisms of resistance likely have limited effectiveness in the long term.
It has recently been proposed that targeting the MAPK pathway can be an effective strategy to inhibit the growth of NSCLCs addicted to different oncogenes (Tricker et al., 2015). Using our multiplex model of NSCLC resistance to gefitinib, we tested the effects of trametinib, the first MEK inhibitor approved for cancer treatment. Figure 2E shows that a clinically relevant concentration of trametinib blocked the growth of all gefitinib-resistant populations, indicating that combination therapy including downstream inhibitors of EGFR signaling may prevent or delay NSCLC resistance.
To illustrate the potential applications of our strategy for in vivo studies, PC9-EKE cells were subcutaneously and bilaterally injected in immunocompromised mice. In the presence of gefitinib, the growth of tumors was initially arrested and, in certain cases, reversed, but it eventually resumed after a few weeks (Figure 3A), mimicking the pattern typically observed in the clinic for NSCLC patients treated with this class of inhibitors. To assess whether PC9 tumor relapse resulted from amplification of the small fraction of cells made resistant through CRISPR/Cas9, barcode frequency was measured by qPCR in gDNA from each tumor sample and normalized to the levels observed in the same batch of cells prior to mouse injection. Figure 3B shows that the proportion of all three mutations was markedly increased in the tumors from gefitinib treated mice, indicating that all three mechanisms of resistance were selected in vivo. Of note, the relative enrichment of the three barcodes was different in each tumor; for instance, in mouse number 8, the right-flank tumor showed extremely high levels of EGFR-T790M, while the KRAS-G12D barcode was predominant in the left-flank tumor (Figures 3B and S2D). This profile is reminiscent of the genetic heterogeneity that can be observed in tumors and metastases from the same patients and suggests that the selection of a particular mechanism of resistance may be guided by competition among different clonal subpopulations, as well as by stochastic events.
Figure 3. NSCLC In Vivo Resistance to Gefitinib.

(A) Multiplex CRISPR-barcoded PC9 cells were subcutaneously injected in the left (L) and right (R) flanks of SCID mice, and the volume of the tumors was measured by caliper. At day 16, mice 6–10 were treated with gefitinib (25 mg/kg/day). The mice were sacrificed when at least one of the tumors reached 550 mm3.
(B) The fraction of the EML4-ALK, KRAS-G12D, and EGFR-T790M barcodes in each tumor was assessed by qPCR and normalized to the total amount of gDNA. The proportions of the three barcodes in the cells prior to mouse injection were analyzed in parallel and arbitrarily set to 1.
See also Figure S2.
CRISPR-Barcoding to Investigate the Effects of Gene Inactivation
Analysis of the distribution of the most common driver mutations within individual tumors has uncovered variable patterns of clonal frequencies (Shah et al., 2012). The coexistence of cancer cells containing or not a particular oncogenic mutation can reflect not only a different stage of tumor evolution but also an improved ability of the tumor to adapt to different environmental conditions. We used our CRISPR-barcoding strategy to model the presence of a subclonal population lacking the tumor suppressor TP53, a major inducer of growth arrest and apoptosis in response to a variety of cellular stressors (Muller and Vousden, 2014). In diploid cells, CRISPR/Cas9 editing can affect one or both alleles, resulting in heterozygous or homozygous mutations, but it is well established that upon CRISPR/Cas9 cleavage, the efficiency of DNA repair through NHEJ is considerably higher compared with HDR (Doudna and Charpentier, 2014; Hsu et al., 2014). If in a particular cell the CRISPR/Cas9 activity is sufficient for the insertion of a barcode on one allele, we reasoned that in most cases it should also provoke inactivation of the second allele through NHEJ, thus resulting in a dominant effect of the barcode. We designed a ssODN (TP53-STOP) to introduce a STOP codon to replace Phe109, located at the beginning of the TP53 DNA-binding domain (Figures S3A and S3B), as well as a control ssODN (TP53-WT) containing only silent mutations. MCF7 (breast) and HCT-116 (colon) cancer cells, which contain a wild-type (WT) TP53 gene, were transfected for CRISPR-bar-coding and treated or not with Nutlin 3, an inhibitor of MDM2-mediated degradation of TP53 (Vassilev et al., 2004). As shown in Figures 4A, 4B, S3C, and S3D, Nutlin 3 significantly increased the fraction of MCF7 and HCT-116 cells containing the TP53-STOP barcode, consistent with a selective advantage for cells lacking TP53 activity. We used a similar barcoding approach to model TP53 “DNA contact” mutation R273H (Figure S3E), a frequent genetic aberration in human tumors resulting in a dominant-negative form of TP53 (Muller and Vousden, 2014). Nutlin 3 treatment significantly increased the fraction of HCT-116 cells containing the TP53-R273H barcode (Figures S3F and S3G), indicating that this mutation exerts an effect similar to TP53 loss in our model.
Figure 4. Inactivation of the Tumor Suppressor TP53 through CRISPR-Barcoding.

(A) Effects of Nutlin-3 (N3; 10 μM, 7 days) on the TP53-STOP to TP53-WT ratio in MCF7 cells using two distinct sgRNAs (A or B). Mean ± SEM; n = 4 of one representative of three independent experiments. *p < 0.05 (Mann-Whitney test).
(B) Effects of N3 (10 μM; 7 days) or doxorubicin (Dox) on the TP53-STOP to TP53-WT ratio in HCT-116 cells. Mean ± SEM; n = 4 of one representative of three independent experiments.
(C) The cells in (B) were treated for 7 days with KU-55933 (10 μM) and/or Dox (50 nM), and the TP53-STOP to TP53-WT ratio was assessed by qPCR. Mean ± SEM of seven independent experiments.
(D) Effects of KU-55933 (10 μM) and/or N3 (10 μM, 7 days) on the TP53-STOP to TP53-WT ratio in HCT-116 cells. Mean ± SEM; n = 4 of one representative of three independent experiments.
See also Figure S3.
Despite exerting similar or stronger effects than Nutlin 3 on TP53 induction and cell growth inhibition (Figure S3H and data not shown), the genotoxic agent doxorubicin did not affect the relative proportion of the two barcodes (Figures 4B and S3D), supporting the notion that TP53 is not strictly required for the activation of DNA damage checkpoints (Jackson and Bartek, 2009). It has been shown that inhibition of the ataxia telangiectasia mutated (ATM) kinase, a major regulator of the response to DNA damage, can specifically sensitize TP53 null cells to genotoxic stress by promoting mitotic catastrophe and subsequent cell death (Jiang et al., 2009). Consistent with this model, co-treatment with doxorubicin and the ATM inhibitor KU-55933 reduced the fraction of HCT-116 cells containing the TP53-STOP barcode (Figure 4C). In a seemingly paradoxical manner, a recent genome-wide small hairpin RNA (shRNA) screen revealed that ATM inhibition exerts an opposite effect upon nongenotoxic activation of TP53, in that it promoted apoptosis of TP53 WT cells, possibly through the inhibition of an ATM-dependent prosurvival pathway (Sullivan et al., 2012). Indeed, Figure 4D shows that Nutlin 3 and KU-55933 synergistically increased the fraction of HCT-116 cells containing the TP53-STOP barcode, thus indicating that different combination strategies targeting the same oncogenic lesion can potentially lead to opposite therapeutic outcomes.
Repair of a Mutated Tumor Suppressor Gene through CRISPR-Barcoding
Unlike other CRISPR/Cas9 strategies, our barcoding approach enables tracing of the mutated cells immediately after DNA editing without the need to derive clones, thus providing a unique means to investigate the effects of HDR-mediated modifications, regardless of their potential impact on cell growth. The APC tumor suppressor gene encodes a major component of the destruction complex that promotes the degradation of free β-catenin, and it is mutated in more than 80% of colorectal cancers (Clevers and Nusse, 2012; Polakis, 2012). We used CRISPR-barcoding to investigate the effects of repairing the homozygous frameshift mutation at position c.4248 of this gene in DLD-1 colon cancer cells (Figure S4A). We designed specific sgRNA constructs and two donor DNAs for APC HDR, containing either the repaired (APC-WT) or a STOP codon, as well as a two series of silent mutations to serve as specific barcodes (Figure S4B).
To assess the effects of APC restoration on Wnt signaling activation, we used a lentiviral reporter containing a destabilized form of GFP, driven by a minimal promoter containing 14 Wnt-responsive elements (Figures S4C and S4D). Five days after CRISPR-barcoding transfection, DLD-1 cells stably containing the Wnt reporter were sorted using fluorescence-activated cell sorting (FACS) to isolate the 10% of cells displaying the highest (GFPhi) or lowest (GFPlo) levels of GFP (Figure 5A). As shown in Figures 5B and S4E, qPCR analysis from gDNA revealed that the APC-WT to APC-STOP ratio was strongly increased in GFPlo compared with GFPhi or unsorted cells, consistent with an enrichment of the repaired APC allele in cells displaying low levels of Wnt reporter activity.
Figure 5. Repair of the Mutated APC Gene in Colon Cancer Cells.

(A) FACS sorting of DLD-1 cells containing the Wnt-responsive GFP reporter and transfected for APC CRISPR-barcoding.
(B) APC-WT to APC-STOP ratio in the cells FACS sorted in (A). Mean ± SEM; n = 3 of one representative of three independent experiments. **p < 0.01 (Student’s t test).
(C) APC-WT to APC-STOP ratio at different time points after transfection of DLD-1 cells. Mean of one representative of three independent experiments.
(D) Deep sequencing analysis of representative gDNA samples used in (C). The percentage of reads containing the APC-WT (WT) and APC-STOP (STOP) barcodes compared with the endogenous sequence is shown.
See also Figures S4 and S5.
Using inducible shRNAs, it has recently been shown that restoration of APC expression can promote regression of small intestine and colon tumors (Dow et al., 2015). To investigate the effects on colon cancer cell growth of directly repairing the mutated APC gene, DLD-1 cells were trypsinized 3 days after CRISPR-barcoding transfection (Tref), replated into four different flasks, and propagated for about 4 weeks. At each cell passage, gDNA was extracted for qPCR analysis (Figure S5A). We found that the fraction of DLD-1 cells containing a repaired APC gene strongly decreased over time, reaching a plateau at 2 weeks after transfection (Figures 5C and S5B–S5D), indicating that aberrant activation of Wnt signaling is strictly required for the growth of these colon cancer cells. We confirmed these data by deep sequencing the samples from Figure 5C and found that the proportion of APC-WT reads dramatically decreased over time compared with the reads containing the APC-STOP barcode (Figure 5D). Of note, a small fraction of APC-WT reads carried additional frameshift mutations, likely originating from errors during the HDR process or the synthesis of the ssODN. Such mutations, which are expected to prevent restoration of a full-length APC protein, showed a 6-fold increase at day 27 compared to the Tref and most likely accounted for the persistence of a small but stable population of cells retaining the APC-WT barcode at longer time points (Figures 5C, 5D, and S5B–S5D).
Repair of a Mutated Oncogene in Neuroblastoma Cells
Although different strategies can conceivably be used to investigate the oncogenic properties of a putative gain-of-function mutation, the definitive demonstration of its role as a driver force in a particular cancer cell would require the restoration of a WT sequence, which is not possible with current DNA editing approaches (see above). We chose the receptor tyrosine kinase ALK, mutated in 6%–10% of neuroblastomas (Cheung and Dyer, 2013; Hallberg and Palmer, 2013), and we used CRISPR-barcoding to correct the ALK-F1174L activating mutation in Kelly neuroblastoma cells (Figures S6A and S6B) by designing three distinct ssODNs: (1) ALK-F1174F, to repair the oncogenic mutation; (2) ALK-F1174L, as an internal control for the mutated receptor; and (3) ALK-STOP, to generate a truncated protein lacking most of the catalytic domain. Because the parental cells contain a WT and a F1174L allele, incorporation of each barcode could result in four possible combinations; for three of them, including the likely most frequent barcode/indel, the phenotypically dominant sequence expressed in the cell is expected to be the one encoded by the barcode (Figure S6C). As shown in Figures 6A and 6B, ALK-F1174F and ALK-STOP barcodes similarly decreased over time, while the fraction of ALK-F1174L cells remained stable. The same results were obtained using a distinct sgRNA (Figures S6D and S6E), indicating that these neuroblastoma cells are addicted to the F1174L mutation of ALK.
Figure 6. Repair of the Oncogenic ALK-F1174L Mutation in Neuroblastoma Cells.

(A) Relative proportions of the three ALK barcodes in Kelly cells at 3 (Tref) 12 or 17 days after transfection, normalized using ALK_Ctrl primers. Mean ± SEM; n = 4 of one representative of three independent experiments. *p < 0.05 compared with the F1174L control barcode (Mann-Whitney test).
(B) The data in (A) were normalized to the ALK-F1174L barcode.
(C) The silent mutations on the F1174F and F1174L barcodes were swapped (SWP), and an experiment was performed as in (A) and (B). Mean ± SEM; n = 4 of one representative of three independent experiments.
(D) Effects of TAE684 (0.2 μM, 7 days) on normalized ALK-F1174F and ALK-STOP barcodes in Kelly cells. gDNA derived from the same cells at time zero was used as the reference (Tref). Mean ± SEM; n = 4 of one representative of three independent experiments. *p < 0.05 compared with untreated cells (Mann-Whitney test).
See also Figure S6.
The silent mutations that compose a CRISPR-barcode may conceivably have an impact on the stability of the corresponding transcript. Although irrelevant for a STOP barcode, variations in the expression levels of other modified sequences could potentially result in false-positive or false-negative effects. With the possible exception of TP53-STOP, the relative proportions of the different APC, TP53, and ALK barcodes were similar in gDNA versus cDNA extracted in parallel from the same cells (Figure S6F). As an additional control, we repeated the ALK barcoding experiment by switching the silent mutations between ALK-F1174F and ALK-F1174L. The “swapped” barcodes confirmed a similar growth disadvantage of cells containing the WT or the STOP codon (Figures 6C and S6G), thus excluding an effect of the silent mutations on ALK mRNA stability.
To further demonstrate the specificity of our approach, Figure 6D shows that the fractions of ALK-F1174F and ALK-STOP cells decreased significantly less in the presence of a small-molecule ALK inhibitor, indicating that the selective pressure that favors the expression of a mutant receptor is abolished upon inhibition of ALK catalytic activity.
Highly Complex CRISPR-Barcodes to Probe Intratumor Heterogeneity
Randomly integrated lentiviral libraries have been used to tag and trace the fate of the different cells comprising a tumor mass population (Bhang et al., 2015; Nguyen et al., 2014). As an alternative approach, we adapted CRISPR-barcoding to label distinct subsets of cancer cells through insertion of a degenerate sequence at a specific genomic location. We chose the adeno-associated virus integration site 1 (AAVS1) locus on chromosome 19, a genomic “safe harbor” widely used for transgene insertion (DeKelver et al., 2010), and used BT474 and PC9 cells, derived from breast cancer and NSCLC, respectively (Figure 7A). The cells were sequentially transfected for CRISPR-barcoding with two distinct sgRNAs and a partially degenerate ssODN containing a SalI restriction site for restriction fragment length polymorphism analysis. As shown by surveyor assay, the AAVS1 locus was edited in the vast majority of the transfected cells (Figure S7A and data not shown), while HDR efficiency was about 22% and 8% for BT474 and PC9, respectively (Figure S7B, Table S1, and data not shown).
Figure 7. Probing Intratumor Heterogeneity Using Complex CRISPR-Barcodes.

(A) Insertion of a degenerate sequence in the AAVS1 locus of BT474 or PC9 cells.
(B) Barcode distribution in BT474 cells maintained in culture for 21, 28, or 35 days. Linear regression was calculated and the coefficient of determination (r2) is shown.
(C) Barcode profile in tumors versus the corresponding cell cultures (35 days after inoculation).
(D) Barcode distribution in different tumors from the same (up) or different (low) mice.
(E) Distribution of enriched AAVS1 barcodes in PC9 and PC9-EKE cells treated with or without gefitinib (1 μM, 14 days). Each dot represents a unique barcode upregulated at least 5-fold compared with the control cells in each of the four gefitinib replicates.
(F) The barcodes in (E) are represented according to their mean fold enrichment in the presence of gefitinib.
See also Figure S7.
After barcoding, BT474 cells were either maintained in culture or injected bilaterally in the fat pads of immunocompromised mice (Figures 7A and S7C). gDNA was derived from both tumors and cells in culture at 21, 28, and 35 days after inoculation, and the barcoded AAVS1 locus was deep-sequenced. In each sample, we detected several thousands of distinct sequences, among which the most frequent corresponded to the parental sequence and to small indels at the two CRISPR/Cas9 cleavage sites induced by NHEJ (Figure S7D, corresponding to the dots in the top right section of the plots shown in Figures 7B–7D and S7E). While the distribution of the barcodes was similar in the three tissue culture samples (Figure 7B), comparison between each tumor and its matched cell culture sample resulted in more scattered profiles (Figures 7C and S7E). Of note, high correlation was found among the different tumors, originated either in the same or in different animals (Figures 7D and S7E), indicating that although the great majority of BT474 cells could contribute to tumor formation, certain subpopulations of cells showed an intrinsically different fitness for in vivo growth.
We used a similar approach to compare the effects of EGFR inhibition in PC9 and PC9-EKE cells (Figure 7A). Deep-sequencing analysis revealed that in the presence of gefitinib (1 μM, 14 days) a few hundred barcodes were consistently enriched compared with the control in all four replicates of both cell lines (Figure 7E), indicating that certain subpopulations are constitutively less sensitive to the inhibitor. Of note, although the number of barcodes upregulated between 10- and 50-fold was similar in both lines, PC9-EKE cells showed a higher fraction of barcodes upregulated more than 50-fold (Figure 7F). Consistent with a recent study using lentiviral barcoded cells (Hata et al., 2016), our data suggest that before the beginning of the treatment, the mass population already contains not only fully resistant clones, probably resulting from preexisting additional genetic aberrations, such as those we artificially inserted through CRISPR/Cas9 in PC9-EKE cells, but also slower growing, drug-tolerant cells, which were similarly represented in both parental and EKE cells and could constitute a sort of reservoir for new mutations potentially leading to acquired resistance.
DISCUSSION
DNA barcoding is a taxonomic method to identify known or new species through sequencing of standardized genomic regions, which is widely used in ecology to assess and monitor environmental biodiversity (Valentini et al., 2009). On the basis of the idea that cancer can be viewed as an evolutionary and ecological process (Merlo et al., 2006), we devised a new type of genetic barcoding to label and follow the fate of a cluster of tumor cells exposed to genetic drift and selective pressure. By coupling the barcode to a potentially functional mutation, this approach provides a unique means to model intratumor heterogeneity and cancer progression.
The extraordinary capacity of cancer cells to evade therapy largely depends on the complex genetic diversity within individual tumors. Compared with current approaches, our barcoding strategy not only provides strong evidence that a certain mutation confers resistance, but also allows this conclusion to be made very rapidly (Figures 1C and 1D). Of note, a recent study demonstrated that therapeutic inhibition of oncogenic driver mutations can induce a complex array of paracrine signals that promote the emergence, expansion, and metastatic spread of resistant clones (Obenauf et al., 2015). Compared with other approaches, our system reproduces the potential crosstalk between genetically distinct cancer cells both in the presence and in the absence of therapy, thus providing a model that more closely recapitulates the complexity of the response to treatment.
One of the major advantages of CRISPR-barcoding is that it can be easily adapted to multiplexing, which allows comparison of the effects of different mutations within the same heterogeneous cell population. As an example, we used this type of approach to assess the efficacy of different combinations of inhibitors in lung cancer cells containing various resistance mutations. Of note, whereas in our model we could not discriminate between homo- or heterozygous mutations, new strategies have been recently developed to specifically target either one or both alleles (Paquet et al., 2016). By integrating known mechanisms used by a particular type of tumor to escape therapy, multiplex models of resistance can be applied to design and validate combinations of compounds with the best potential of achieving tumor remission or long-term stabilization of the disease.
Besides making it possible to recapitulate and model intratumor genetic heterogeneity, CRISPR-barcoding can also be used to investigate the effects of a particular mutation, regardless of its potential effects on cell growth and/or survival. Indeed, although the CRISPR revolution has greatly simplified DNA editing and genome manipulation, the low efficiency of HDR imposes strong limitations as to the types of modifications and cell models that can be used. From a cancer perspective, with current approaches it is extremely difficult, if not impossible, to repair mutations that drive the growth of cancer cells. By enabling monitoring of the cell phenotype immediately after DNA editing, we demonstrated how our strategy could be used to establish and trace any kind of modification in most cancer types, including reversal of powerful oncogenic events, such as APC or ALK mutations, thus providing ultimate proof of the selective growth advantages such mutations confer. Finally, we describe a strategy to separately label at a specific genomic location thousands of different cancer subpopulations, as an alternative to the time-consuming generation and optimization of complex viral libraries.
Although the proofs of concept reported here are related to cancer models, this technology can be implemented in different fields of biological research. Through the concurrent use of a control barcode, consisting of silent mutations, our approach directly tackles the potential non-specific effects that can derive from off-target DNA cleavage induced by the sgRNA-Cas9 complex (Doudna and Charpentier, 2014; Fu et al., 2013; Hsu et al., 2014).
Despite recent attempts to increase HDR efficiency through small molecules (Yu et al., 2015; Chu et al., 2015; Maruyama et al., 2015) or improved design of the donor DNA (Richardson et al., 2016), the fact that only a fraction of the transfected cells contains the desired mutation constitutes a major limitation for CRISPR/Cas9 technology. By combining the extreme sensitivity of barcode detection through qPCR or deep sequencing, without the need to select and amplify individual clones, CRISPR-bar-coding does not require particularly high levels of DNA editing efficiency (Table S1). Hence, our approach can be used in most cell types to generate different kinds of genetic manipulations, including missense point mutations, gene inactivation through insertion of a STOP codon, and chromosomal rearrangements, regardless of their potential effects on cell growth and/or survival.
In conclusion, CRISPR-barcoding is a fast and highly flexible means to investigate the effects of different kinds of genomic modifications in a broad range of functional assays. On the basis of a simple idea, this approach enables a series of novel applications, which would be otherwise extremely difficult, if not impossible, with other methods. Indeed, we demonstrated how this strategy can be shaped to recapitulate and trace intratumor genetic heterogeneity, through generation of novel cancer models for a better understanding of specific oncogenic mechanisms, as well as to develop and validate new therapeutic protocols. Given its adaptability to multiplexing, as well as the possibility to use different methods to “read” the barcodes, including deep sequencing (Figures 5D and 7), TaqMan genotyping probes (data not shown) and Droplet Digital PCR, this approach could also serve as a customizable platform for high-throughput screening of compounds targeting specific genetic alterations, such as those underlying resistance to cancer therapy.
Limitations
Although a limitation of this approach could conceivably derive from the fact that the silent mutations forming the barcode might have an effect on mRNA stability, we demonstrated how this potential issue can be addressed by simply swapping the silent mutations between barcodes. Another potential drawback is the need for some sort of selective pressure in order to establish a positive or negative effect on the relative fraction of cells containing the desired DNA modification. Although CRISPR-barcoding cannot be adapted to all kinds of assays, we provided several proofs of concept to illustrate how it can be easily implemented to investigate effects on cell growth and survival, invasion, transcriptional activity, and resistance and sensitivity to a particular agent in culture and in vivo.
Despite being considerably faster and easier to implement compared with viral libraries, the use of highly complex CRISPR-barcodes to trace cellular heterogeneity requires good levels of HDR efficiency, which could limit its potential applications. However, considering the new approaches for improved HDR efficiency (Yu et al., 2015; Chu et al., 2015; Maruyama et al., 2015; Richardson et al., 2016) and the fact that indels generated by NHEJ can also act as barcodes (Figure S7D), this strategy can be probably used in a relatively broad array of cell models. Finally, depending on the targeted locus, one cell can conceivably contain distinct barcodes on different alleles, which should be taken into account during the analysis of the data, for example through the identification of barcodes displaying identical patterns. To overcome this issue, a haploid region of the genome could be chosen as a target in certain models, such as a locus on the X or Y chromosomes in cells derived from a male individual.
EXPERIMENTAL PROCEDURES
Cell Culture, Transfection, and Inhibitors
293T (human embryonic kidney), DLD-1, HCT-116 (colorectal carcinoma), MCF7, and BT474 (breast cancer) cells were obtained from ATCC; PC9 cells (NSCLC) were obtained from ECACC-Sigma-Aldrich; and Kelly cells (neuroblastoma) were a kind gift from Dr. C. Einvik. HCT-116 and MCF7 cells are WT for TP53 (http://p53.iarc.fr/CellLines.aspx). All cells were grown in DMEM (Life Technologies), except Kelly and PC9 cells, grown in RPMI medium (Life Technologies), both supplemented with 10% fetal bovine serum (Life Technologies) and 0.6% penicillin/streptomycin (Life Technologies). Cells were transfected with a Nucleofector II device (Lonza) using the Amaxa Nucleofector kit (Lonza) and electroporation program recommended by the manufacturer. 293T cells were transfected using polyethylenimine (Polysciences). The efficiency of each transfection was assessed in parallel using a GFP-containing plasmid.
Nutlin 3 was purchased from SelleckChem, and doxorubicin was purchased from Abcam. Gefitinib, WZ4002, TAE684, trametinib, and KU-55933 were purchased from Santa Cruz Biotechnology.
CRISPR-Barcoding
sgRNA target sequences (Table S2) were designed using the CRISPR Design tool hosted by the Massachusetts Institute of Technology (http://crispr.mit.edu) to minimize potential off-target effects. Oligos encoding the targeting sequence were then annealed and ligated into the pSpCas9(BB)-2A-Puro (Ran et al., 2013) vector digested with BbsI (New England Biolabs). The sequence of the ssODNs (Integrated DNA Technologies) used for CRISPR/Cas9-mediated HDR, containing one missense/nonsense mutation coupled to different silent mutations, are provided in Table S2. The set of silent mutations is designed to enable PCR specificity and to avoid recognition by the corresponding sgRNA used to cleave the endogenous sequence. For each targeted locus, cells were co-transfected with 2 μg of the CRISPR/Cas9 plasmid and 2 μl of either the control or the sense/nonsense ssODN (50 μM) to prevent the potential incorporation of the two donor DNA sequences into different alleles within the same cell. Immediately after transfection, the cells were pooled in the same flask. For AAVS1 barcoding, BT474 cells were subjected to two rounds of transfection with a 2-week interval using CRISPR/Cas9 plasmids encoding two distinct sgRNAs, together with a ssODN containing nine degenerate nucleotides and a SalI restriction site.
qPCR
gDNA was extracted using the NucleoSpin Tissue kit (Macherey-Nagel). The sequence of the different PCR primers, designed using Primer-BLAST (National Center for Biotechnology Information), is provided in Table S3. To avoid potential amplification from ssODN molecules not integrated in the correct genomic locus, one of the two primers was designed to target the endogenous genomic sequence flanking the region sharing homology with the ssODNs. Primer specificity for each particular barcode was assessed. qPCR was performed from 100 ng of gDNA using SYBR Green (Life Technologies) on a 7900 HT Fast-Real-Time, a Q-PCR ABI PRISM 7500, or a QuantStudio Flex PCR System (Life Technologies). qPCR analysis was performed using the standard curve or the Pfaffl methods (Pfaffl, 2001).
Statistical Analysis
Statistical analysis was performed with GraphPad Prism software using the Mann-Whitney test or Student’s two-tailed t test. Linear regression of the AAVS1 barcode normalized frequencies between different samples was calculated using Excel (Microsoft).
Supplementary Material
Highlights.
CRISPR-barcoding is a versatile method to trace clonal dynamics in tumor populations
Sensitive barcode detection allows DNA editing even when HDR efficiency is low
A silent barcode is used as a control for potential Cas9 off-target cleavage
Highly complex barcodes are used to label thousands of cells within a mass population
Acknowledgments
We thank G. Liu, J.Y. Lee, J.M. Flaman, D. Vaudry, and members of INSERM U982 for helpful discussion. Some of the experiments were performed at the Cell Imaging Platform of Normandie (PRIMACEN). We thank G. Riou (IRIB Flow Cytometry Facility) and M. Di Giovanni (PRIMACEN) for technical assistance. This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), the Université de Rouen Normandie, the Ligue Contre le Cancer de Haute-Normandie, the National Cancer Institute (P01CA080058), and the Breast Cancer Research Foundation. A.G. is recipient of a doctoral fellowship from the Normandie Region. L.G. was supported by a Chair of Excellence program from INSERM and the Université de Rouen Normandie. A patent application related to this work has been submitted by INSERM and the Université de Rouen Normandie.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2016.06.017.
AUTHOR CONTRIBUTIONS
L.G. conceived the original idea and designed and supervised the study. A.G., S.K.M., D.C., D.A., S.A.A., Y.A., and L.G. designed and/or analyzed experiments. A.G., S.K.M., D.C., S.Y., H.A., and L.G. performed experiments. R.S., A.J., and G.R. performed deep sequencing analysis. M.V., F.C., S.C., and I.T. performed deep sequencing analysis for the APC gene. S.A. and O.B. performed cell sorting and FACS analysis. L.G. wrote the manuscript, with input and editing from the other authors.
References
- Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–117. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V, Caushi JX, Zhao R, Hims MM, Singh AP, Kao I, Rakiec D, Shaw P, et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med. 2015;21:440–448. doi: 10.1038/nm.3841. [DOI] [PubMed] [Google Scholar]
- Boettcher M, McManus MT. Choosing the right tool for the job: RNAi, TALEN, or CRISPR. Mol Cell. 2015;58:575–585. doi: 10.1016/j.molcel.2015.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi PS, Meyerson M. Targeted genomic rearrangements using CRISPR/Cas technology. Nat Commun. 2014;5:3728. doi: 10.1038/ncomms4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- DeKelver RC, Choi VM, Moehle EA, Paschon DE, Hockemeyer D, Meijsing SH, Sancak Y, Cui X, Steine EJ, Miller JC, et al. Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010;20:1133–1142. doi: 10.1101/gr.106773.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA, Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Wendl MC, McMichael JF, Raphael BJ. Expanding the computational toolbox for mining cancer genomes. Nat Rev Genet. 2014;15:556–570. doi: 10.1038/nrg3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Dow LE, O’Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, Lowe SW. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell. 2015;161:1539–1552. doi: 10.1016/j.cell.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12:487–493. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013;13:685–700. doi: 10.1038/nrc3580. [DOI] [PubMed] [Google Scholar]
- Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22:262–269. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveneers S, Danen EH. Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci. 2009;122:1059–1069. doi: 10.1242/jcs.039446. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372:1689–1699. doi: 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;23:1895–1909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P, Sarode N, Wile BM, Vertino PM, Stewart FJ, Bao G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42:7473–7485. doi: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N, de Stanchina E, de Stanchina E, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–427. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–317. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LV, Cox CL, Eirew P, Knapp DJ, Pellacani D, Kannan N, Carles A, Moksa M, Balani S, Shah S, et al. DNA barcoding reveals diverse growth kinetics of human breast tumour subclones in serially passaged xenografts. Nat Commun. 2014;5:5871. doi: 10.1038/ncomms6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368–372. doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4:a008052. doi: 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34:339–344. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Solomon B, Wilner KD, Shaw AT. Current status of targeted therapy for anaplastic lymphoma kinase-rearranged non-small cell lung cancer. Clin Pharmacol Ther. 2014;95:15–23. doi: 10.1038/clpt.2013.200. [DOI] [PubMed] [Google Scholar]
- Sullivan KD, Padilla-Just N, Henry RE, Porter CC, Kim J, Tentler JJ, Eckhardt SG, Tan AC, DeGregori J, Espinosa JM. ATM and MET kinases are synthetic lethal with nongenotoxic activation of p53. Nat Chem Biol. 2012;8:646–654. doi: 10.1038/nchembio.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, Pratilas CA, Rosen N, Gray NS, Wong KK, Jänne PA. Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR-mutant lung cancer. Cancer Discov. 2015;5:960–971. doi: 10.1158/2159-8290.CD-15-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentini A, Pompanon F, Taberlet P. DNA barcoding for ecologists. Trends Ecol Evol. 2009;24:110–117. doi: 10.1016/j.tree.2008.09.011. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Yu C, Liu Y, Ma T, Liu K, Xu S, Zhang Y, Liu H, La Russa M, Xie M, Ding S, Qi LS. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell. 2015;16:142–147. doi: 10.1016/j.stem.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.