

Key Clinical Message
Clinical geneticists, neurologists, psychiatrists, and other healthcare providers can learn from this case report that patients with a behavioral phenotype that includes a mild learning disability may also require a thorough examination, including brain MRI and whole‐exome sequencing.
Keywords: Behavioral phenotype of genetic syndromes, corpus callosum hypogenesis, L1CAM mutation, X‐linked mental retardation
Introduction
Mutations in the gene encoding L1 cell adhesion molecule (L1CAM) are phenotypically characterized by X‐linked hydrocephalus (OMIM #307000) or—in milder cases—corpus callosum agenesis or hypogenesis (OMIM #304100) 1. The variability in clinical presentation reflects different functions of L1CAM. Hydrocephalus is a very complex disorder. Mutations in L1 as a cell adhesion molecule play a role in the pathogenesis of hydrocephalus. The corpus callosum hypogenesis reflects dysfunction as a neural recognition molecule through disorders in axonal growth and guidance 2.
The L1CAM gene is located on the X chromosome at Xq28 and is expressed primarily in the nervous system, where it plays important roles in neuronal development, including the guidance of neurite outgrowth, neuronal cell migration, axon bundling, synaptogenesis, myelination, neuronal cell survival, and long‐term potentiation 3.
The patients with hydrocephalus have a worse prognosis 4. This is in agreement with recent evidence that some forms of hydrocephalus and the inborn neurological impairment are inseparable phenomena. This is because both fetal onset hydrocephalus and abnormal neurogenesis have a common origin: a primary pathology of the embryonic ventricular zone, involving neural stem cells and multiciliated ependymal cells. Junctional failure and loss of neural stem cells underpin the etiology of neuronal migration defects at specific regions of brain. Thus, neonates are born with a neurodevelopmental deficit that derivative surgery cannot solve 5, 6.
Here, we report a patient with a generally mild physical presentation but with a more distinct behavioral phenotype.
Clinical Description
The proband (referred to here as “Patient A”) is a 56‐year‐old man born in a family with two brothers from nonconsanguineous healthy parents. One of his two brothers has a learning disability of unknown severity; the other does not. Their mother had 14 siblings; three of her brothers had a learning disability. Patient A's parents died around the age of 75. Patient A's daughter developed epilepsy at the age of 6; she initially experienced absence seizures, and later developed grand mal seizures. For the past 2 years, she has taken valproate and has been seizure‐free. Patient A's son is healthy.
The proband sought help from professional caregivers due to anxiety and depression following several major life events. He was fired 10 years ago and was unable to find a new job until he found employment at a sheltered workplace as a postal employee. In addition, he experienced the deaths of his parents and his parents‐in‐law. Patient A also experienced difficulty remembering essential information during work and at home. He lives with his wife and two adult children in the center of a large city in the Netherlands, where he was born. The patient requested a psychological and neurological evaluation because he experienced a subdural hematoma with cerebral contusion following an unexpected fall when he was 28 years of age. Although the subdural hematoma was treated, he wondered whether his recent complaints were related to this earlier injury.
A thorough patient history revealed a mild learning disability that was present since his earliest memories. At 10 years of age, he set fire to his family's house, after which he was removed from his home by child services. He also switched to a school for children with mild learning disabilities and behavioral difficulties. At 16 years of age, he experienced his first epileptic seizure and was treated with antiepileptic drugs. It is unclear whether the accident at 28 years of age, which led to the subdural hematoma, was caused by an epileptic seizure, as he frequently experienced unexpected falls.
The results of a psychological examination revealed overall difficulties with cognitive functioning, including impaired memory, attention, and executive functions. The patient's clinical history and test results revealed that psychological problems appeared at an early age, leading to social, emotional, and behavioral problems. Therefore, combined with his intellectual challenges, the patient lacked sufficient opportunities to learn the appropriate skills needed to cope during complex, stressful situations. These developmental issues, limited coping skills, and limited opportunities to express himself emotionally likely predisposed the patient to develop depression, particularly when experiencing stress due to a lack of structure in his daily life, thereby having a strongly disrupting effect on daily functioning and activities (Table 1).
Table 1.
Psychological test results of Patient A (measured April, 2013)
| Domain | Results | Test(s) |
|---|---|---|
| Intellectual functioning | Verbal IQ: 68; Performance IQ: 70; Full Scale IQ: 69; Verbal Comprehension Index 66; Perceptual Organization Index: 63; Working Memory Index: 88; Processing Speed Index: 76 | Wechsler Adult Intelligence Scale—Third Edition, Dutch version (WAIS‐III‐NL) 12 |
| Attention and working memory | Below the mean, but not significantly impaired compared with cognitive functioning. | Trail Making Test (TMT) 13; Test of Everyday Attention for Children (TEA‐Ch) 14, 15; Stroop Color Word Test (STROOP) 16; D2 Test of Attention (D2) 17, 18 |
| Executive functioning | Significantly decreased in all domains, including initiative, motivation, planning, and control/evaluation of behavior, flexibility and problem solving. The subject made perseverative mistakes. | Behavioral Assessment of the Dysexecutive Syndrome (BADS‐NL) 19; Word Fluency Test (WFT); Figure Fluency Test (FFT) 20 |
| Visual and visuo‐motor ability | Decreased; indicative of neurological syndrome. | WAIS‐III‐NL, RCFT 21; Hooper Visual Organization Test (VOT) 22 |
| Gross and fine motor function | Gross motor function was normal; fine motor function was decreased slightly; coordination tasks and switching of functions were decreased considerably. | Test of manual dexterity and bimanual coordination: Purdue Pegboard 23, 24 |
| Personality and social functioning | Characterized by emotional disturbances which were not overtly evident due to introvert coping. This led to depression, tense feelings, and feelings of inferiority and hopelessness. Social functioning was characterized by shyness, reduced self‐confidence, reduced confidence in others, and impaired communication. | Clinical interview with the proband and his wife; Dutch Personality Inventory (Nederlandse Persoonlijkheid‐Vragenlijst (NPV‐2) 25). |
Physical examination revealed a healthy adult male with the following parameters: height, 177.5 cm (−1 SD); weight, 77.6 kg (+1.3 SD); occipito‐frontal circumference, 57.3 cm (−0.33 SD); left ear length, 7.4 cm (75th–97th percentile); left hand length, 21.1 cm (above the 97th percentile); and mildly adducted thumbs. The patient presented with no gait abnormalities, and a neurological examination was unremarkable.
In addition, an MRI examination of the brain revealed hypogenesis of the corpus callosum, particularly in the splenium (Fig. 1), as well as signs of prior traumatic injuries to the right frontal brain parenchyma and mild, slightly asymmetric dilatation of the lateral ventricles due to underdevelopment and/or loss of white matter (Fig. 2); the loss of white matter in the right hemisphere may have been due in part to the prior traumatic brain injury.
Figure 1.
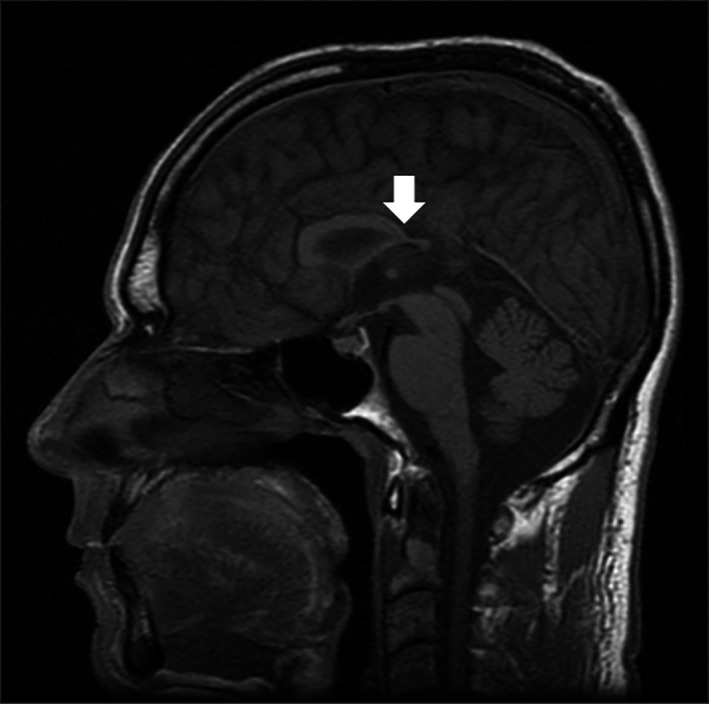
Midsagittal T1 SE image of the brain of Patient A illustrating the hypogenesis of the corpus callosum, in particular the splenium of the corpus callosum (arrow).
Figure 2.
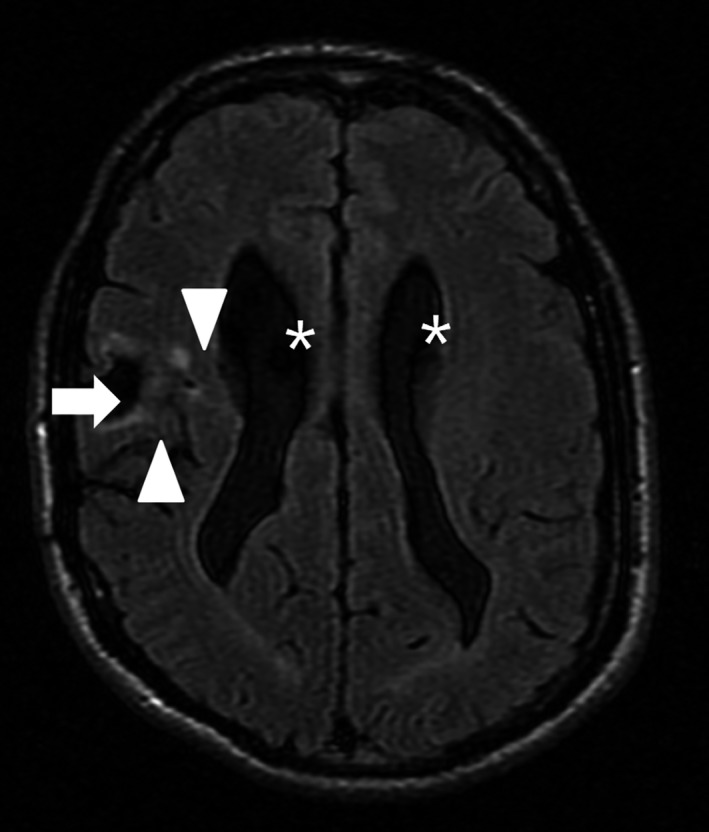
Axial T2 Flair image of the brain of Patient A shows the post‐traumatic loss of tissue in the right frontal lobe (arrow) with surrounding gliosis (scar tissue, arrowheads). There is a mild and slightly asymmetric dilatation of the lateral ventricles (asterisks).
A genetic examination was also performed. SNP array analysis revealed a normal male karyotype. Whole‐exome sequencing (WES) was performed essentially as reported previously 7. Exome capture was performed using the Agilent SureSelect Human All Exon v4 enrichment kit (Agilent Technologies, Santa Clara, CA), and WES was performed using the Illumina HiSeq platform (BGI, Copenhagen, Denmark). Data were analyzed using the BWA (read alignment) and GATK (variant calling) software packages. Variants were annotated using an in‐house‐developed pipeline and prioritized using an in‐house‐designed “variant interface” with manual curation. Putative causative variants were confirmed by Sanger sequencing. WES revealed the missense mutation c.998C>T (p.Pro333Leu) in the proband's L1CAM gene (NM_000425.3). Segregation analysis revealed that the same mutation was present in the proband's brother with a learning disability; in contrast, the proband's other brother—who did not have developmental abnormalities—did not carry this variant. It was not possible to obtain genetic material for testing the proband's deceased mother —who would have been an obligate carrier of the variant—or her brothers with mental disability.
This missense variant causes the substitution of a highly conserved nucleotide (phyloP: 5.29) and amino acid residue in the immunoglobulin‐like domain of the L1CAM gene and protein, respectively. This mutation has not been described previously in any SNP database, including dbSNP and the EXAC database (http://exac.broadinstitute.org/); moreover, several mutation prediction tools predict that this variant is likely to be pathogenic.
Discussion
In this clinical report, we present a male patient with a novel, putative pathogenic variant in the L1CAM gene. This variant affects the structure of the fourth immunoglobulin‐like domain in the protein, as Pro333 is believed to be one of the key residues required for proper folding of this domain 8. Therefore, this variant likely disrupts interactions between both L1‐L1 proteins and L1‐non‐L1 proteins, interactions that are important for protein function. Interestingly, a different variant in the same amino acid (p.Pro333Arg) was described in a patient with X‐linked hydrocephalus; this patient died before the age of 1 4. As we mentioned before, some forms of hydrocephalus are not just ventriculomegaly, but the tip of the iceberg of a complex neurodevelopmental disorder 5, 6.
In the family reported here, the c.998C>T variant was present in both the proband and his affected brother, but not in the proband's unaffected brother. Both the patient and his affected brother presented with a rather mild clinical course. In most families with L1 syndrome, the affected boys die before, during, or soon after birth; however, both interfamily variability and intrafamily variability have been reported 9. Basel‐Vanagaite et al. described two half‐brothers who had a mutation in the L1CAM gene and a hypoplastic corpus callosum, but no other characteristics of L1 syndrome 1. Epilepsy has also been reported in a minority of patients with L1 syndrome 10.
In our study, the proband presented with a hypoplastic corpus callosum and some minor clinical features of L1 syndrome. In this case, the patient's behavioral phenotype appeared to be more severe and distinct than his physical phenotype. Because social delays and behavioral abnormalities have been described in patients with corpus callosum abnormalities, an integrative examination of these functions should be included in the diagnostic protocol 11.
The value to this family from obtaining this diagnosis should not be underestimated. For example, several family members are now comforted by the fact that they—and their children—do not have an increased risk of recurrence. In addition, other family members—particularly the proband's adolescent daughter—now have the opportunity to seek clinical genetic advice when deciding whether to have children. Moreover, this family has learned that they should not expect the proband to achieve a higher level of functioning, and they have learned not to overestimate his abilities as either a husband or a father. After receiving this genetic diagnosis, his primary clinical goal—namely to help resolve his depressive symptoms—has waned.
Based on these findings, clinicians should be aware of the value of performing neuroimaging in persons with developmental disabilities, and they should offer their patients the opportunity to seek an etiological diagnosis, including genetic testing. Psychiatrists—particularly those who work in the field of intellectual disabilities/learning disabilities—should be made aware of the options available regarding diagnostic procedures based on clinical genetics and neuroimaging. These procedures can provide evidence‐based assistance to patients and their families, including preventive management, thereby improving quality of life.
Authorship
MO: main author, was responsible for writing and reviewing the manuscript, literature review, and provided direct neuropsychiatric care to the patient. MW: participated in study design, coordination, and data analysis, especially concerning clinical genetic aspects, and provided direct clinical genetic care to the patient and his family. MP: performed the psychological assessment and wrote Table 1. RP: performed the WES and analyzed the molecular data and co‐authored the description of the molecular details in the manuscript. YV: analyzed the molecular data and co‐authored the description of the molecular details in the manuscript. RJN: reviewed the MRI of the brain and provided the figures. CS: involved in overall supervision of the writing of the manuscript.
Conflict of Interest
The authors declare to have no conflict of interest.
Acknowledgement for publication
This study makes use of data shared through the PhenomeCentral repository. Funding for PhenomeCentral was provided by Genome Canada and Canadian Institute of Health Research (CIHR).
References
- 1. Basel‐Vanagaite, L. , Straussberg R., Friez M. J., Inbar D., Korenreich L., Shohat M., et al. 2006. Expanding the phenotypic spectrum of L1CAM‐associated disease. Clin. Genet. 69:414–419. [DOI] [PubMed] [Google Scholar]
- 2. Kenwrick, S. , Watkins A., and De Angelis E.. 2000. Neural cell recognition molecule L1: relating biological complexity to human disease mutations. Hum. Mol. Genet. 9:879–886. [DOI] [PubMed] [Google Scholar]
- 3. Stumpel, C. , Vos Y. J.. L1 Syndrome 2004. Apr 28 [Updated 2015 Mar 5]. In: Pagon R. A., Adam M. P., Ardinger H. H., et al., editors. GeneReviews® [Internet]. University of Washington, Seattle, Seattle (WA). 1993–2015. [PubMed] [Google Scholar]
- 4. De Angelis, E. , MacFarlane J., Du J. S., Yeo G., Hicks R., Rathjen F. G., et al. 1999. Pathological missense mutations of neural cell adhesion molecule L1 affect homophilic and heterophilic binding activities. EMBO J. 18:4744–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jimenez, A. J. , Dominguez‐Pinos M. D., Guerra M. M., Fernandez‐Llebrez P., and Perez‐Figares J. M.. 2014. Structure and function of the ependymal barrier and diseases associated with ependyma disruption. Tissue Barriers. 2:e28426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodriguez, E. M. , Guerra M. M., Vio K., Gonzalez C., Ortloff A., Batiz L. F., et al. 2012. A cell junction pathology of neural stem cells leads to abnormal neurogenesis and hydrocephalus. Biol. Res. 45:231–242. [DOI] [PubMed] [Google Scholar]
- 7. Neveling, K. , Feenstra I., Gilissen C., Hoefsloot L. H., Kamsteeg E. J., Mensenkamp A. R., et al. 2013. A post‐hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum. Mutat. 34:1721–1726. [DOI] [PubMed] [Google Scholar]
- 8. Bateman, A. , Jouet M., MacFarlane J., Du J. S., Kenwrick S., and Chothia C.. 1996. Outline structure of the human L1 cell adhesion molecule and the sites where mutations cause neurological disorders. EMBO J. 15:6050–6059. [PMC free article] [PubMed] [Google Scholar]
- 9. Schrander‐Stumpel, C. , Howeler C., Jones M., Sommer A., Stevens C., Tinschert S., et al. 1995. Spectrum of X‐linked hydrocephalus (HSAS), MASA syndrome, and complicated spastic paraplegia (SPG1): Clinical review with six additional families. Am. J. Med. Genet. 57:107–116. [DOI] [PubMed] [Google Scholar]
- 10. Ortega, E. , Munoz R. I., Luza N., Guerra F., Guerra M., Vio K., et al. 2016. The value of early and comprehensive diagnoses in a human fetus with hydrocephalus and progressive obliteration of the aqueduct of Sylvius: Case Report. BMC Neurol. 16:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Al‐Hashim, A. H. , Blaser S., Raybaud C., and MacGregor D.. 2016. Corpus callosum abnormalities: neuroradiological and clinical correlations. Dev. Med. Child Neurol. 58:475–484. [DOI] [PubMed] [Google Scholar]
- 12. Wechsler, D. , Van der Steene G., and Vertommen H.. 2005. WAIS III‐NL: Wechsler Adult Intelligence Scale III‐NL. Harcourt Test Publishers, Amsterdam. [Google Scholar]
- 13. Strauss, E. , Sherman E. M. S., and Spreen O.. 2006. A compendium of neuropsychological tests: administration, norms, and commentary, 3rd rev. ed Oxford University Press, Oxford. [Google Scholar]
- 14. Manly, T. , Robertson I. H., Anderson V., and Nimmo‐Smith I.. 1999. TEA‐Ch. Test of everyday attention for children. Thames Valley Test Company Limited, Bury St. Edmunds, UK. [Google Scholar]
- 15. Schittekatte, M. , Groenvynck H., Fontaine J. R. J., and Dekker P. H.. 2007. Aanvullend psychometrisch onderzoek met de Test of Everyday Attention for Children (TEACH): In Nederland en Vlaanderen aangepaste normen en nieuwe validiteits‐ en betrouwbaarheidsgegevens Harcourt, Amsterdam. [Google Scholar]
- 16. Hammes, J. G. W. 1978. Stroop Kleur‐Woord Test. Pearson Testpublisher, Amsterdam. [Google Scholar]
- 17. Brickenkamp, R. , and Oosterveld P.. 2002. d2 Aandachts‐ en concentratietest. Hogrefe, Amsterdam. [Google Scholar]
- 18. Ross, R. M. 2005. The D2 Test of Attention: An Examination of Age, Gender, and Cross‐cultural Indices: Argosy University.
- 19. Wilson, B. A. , Alderman N., Burgess P. W., Emslie H., Evans J. J., Krabbendam L., et al. 1997. BADS‐NL; behavioural assessment of the dysexecutive syndrome. Pearson Assessment and Information B.V, Amsterdam. [Google Scholar]
- 20. Mulder, J. , Dekker P., and Dekker R.. 2006. WFT & FFT Woord‐ en figuur‐fluencytest; handleiding. Hogrefe, Amsterdam. [Google Scholar]
- 21. Meyers, J. E. , and Meyers K. R.. 1995. Rey Complex Figure Test and Recognition Trial: RCFT: Psychological Assessment Resources.
- 22. Hooper, H. E. 1958. Manual: Hooper visual organization test. Western Psychological Services (WPS), Los Angeles, CA. [Google Scholar]
- 23. Lindstrom‐Hazel, D. K. , and VanderVlies Veenstra N.. 2015. Examining the Purdue pegboard test for occupational therapy practice. Open J. Occup. Ther. 3:5. [Google Scholar]
- 24. Tiffin, J. 1948. The Purdue Pegboard: norms and studies of reliability and validity. J. Appl. Psychol. 32:234–247. [DOI] [PubMed] [Google Scholar]
- 25. Barelds, D. P. H. , Luteijn F., van Dijk H., and Starren J.. 2007. Nederlandse Persoonlijkheids Vragenlijst‐2 Handleiding. Harcourt Test Publishers, Amsterdam. [Google Scholar]