

SUMMARY
Repair of interstrand crosslinks by the Fanconi anemia (FA) pathway requires both the monoubiquitination and de-ubiquitination of the FANCI/FANCD2 (FANCI/D2) complex. In the standing model, the phosphorylation of six sites in the FANCI S/TQ cluster domain occurs upstream of, and promotes, FANCI/D2 monoubiquitination. We generated phospho-specific antibodies against three different S/TQ cluster sites (Serines 556, 559, and 565) on human FANCI and found that, in contrast to the standing model, distinct FANCI sites were phosphorylated either predominantly upstream (ubiquitination-independent; Serine 556) or downstream (ubiquitination-linked; Serines 559 and 565) of FANCI/D2 monoubiquitination. Ubiquitination-linked FANCI phosphorylation inhibited FANCD2 de-ubiquitination and bypassed the need to de-ubiquitinate FANCD2 to achieve effective interstrand crosslink repair. USP1 depletion suppressed ubiquitination-linked FANCI phosphorylation despite increasing FANCI/D2 monoubiquitination, providing an explanation of why FANCD2 de-ubiquitination is important for function of the FA pathway. Our work results in a refined model of how FANCI phosphorylation activates the FANCI/D2 complex.
eTOC Blurb
How the Fanconi anemia pathway is activated to repair interstrand crosslinks is incompletely understood. Cheung et al. use FANCI phosphorylation-specific antibodies to identify ubiquitination-linked FANCI phosphorylation. Ubiquitination-linked FANCI phosphorylation fully activates the FANCI/D2 complex such that it can function without needing to undergo de-ubiquitination.
INTRODUCTION
Fanconi anemia (FA) is a genetic disorder characterized by bone marrow failure, cancer predisposition, and cellular sensitivity to interstrand crosslinks (ICLs). Patients with FA carry inherited mutations in one of 21 FA genes, which encode proteins that interact in a DNA repair pathway called the FA pathway (Ceccaldi et al., 2016). The function of this pathway is dependent on the monoubiquitination of FANCD2, which is catalyzed by the FA core complex that is comprised of eight FA proteins (FANCA, B, C, E, F, G, L and M) (Garcia-Higuera et al., 2001; Huang et al., 2014; Taniguchi et al., 2002b). This modification permits FANCD2 to localize to sites of DNA damage (foci formation) and orchestrate the downstream repair of ICLs, thereby conferring cellular resistance to ICL-inducing agents such as mitomycin C (MMC).
While FANCD2 monoubiquitination is necessary for ICL repair, it is not sufficient. FANCD2 de-ubiquitination by the ubiquitin peptidase, USP1, is also required for the FA pathway to function properly. Studies using genetic and chemical disruption of USP1 have demonstrated the importance of FANCD2 de-ubiquitination for FANCD2 foci formation and ICL repair (Liang et al., 2014; Murai et al., 2011; Oestergaard et al., 2007). Usp1-deficient mice have an FA-like phenotype (Kim et al., 2009). The FA pathway therefore depends on USP1 to maintain a proper equilibrium between monoubiquitinated and non-ubiquitinated FANCD2. FANCD2 monoubiquitination by itself is an insufficient indicator of effective ICL repair.
FANCD2 forms a heterodimer (the FANCI/D2 complex) with its binding partner, FANCI. FANCI is also monoubiquitinated in response to ICLs, and the monoubiquitinations of both FANCI and FANCD2 are reciprocally dependent (Smogorzewska et al., 2007). Previous studies have suggested that ATR-mediated phosphorylation of six sites on FANCI, located within a domain known as the S/TQ cluster, is critical for monoubiquitination of FANCI/D2 (Ishiai et al., 2008; Shigechi et al., 2012; Tomida et al., 2013). These studies have largely relied on an electrophoretic mobility-based method to detect the collective phosphorylation of the chicken FANCI (chFANCI) S/TQ cluster in DT40 cells. Using this approach, phosphorylation of the monoubiquitination-deficient chFANCI K525R mutant could be detected by gel shift in response to ICLs. This finding led to a standing model where FANCI phosphorylation occurs without prior FANCI/D2 monoubiquitination (Boisvert and Howlett, 2014; Chen et al., 2015; Gibbs-Seymour et al., 2015; Ishiai et al., 2008; Shigechi et al., 2012; Tomida et al., 2013). Because FANCI phosphorylation is an upstream event in this model, it makes little consideration of whether this phosphorylation plays a role in the FA pathway beyond promoting FANCD2 monoubiquitination. Furthermore, little is known about which of the six FANCI S/TQ cluster sites are actually phosphorylated in response to DNA damage.
The inability to analyze the phosphorylation of individual sites within the FANCI S/TQ cluster has resulted in an incomplete understanding of a post-translational modification that is critical for activating the FA pathway. In this study, we generated phospho-specific antibodies to three sites in the human FANCI (hFANCI) S/TQ cluster (Serine 556 (S556), Serine 559 (S559), and Serine 565 (S565)) and found that, in contrast to the standing model, the phosphorylation of certain FANCI sites (S559 and S565) were linked to the monoubiquitinated form of FANCI. Interestingly, we found that an S559D-S565D FANCI mutant carrying phosphomimetic mutations at these ubiquitination-linked sites strongly inhibited FANCD2 de-ubiquitination, yet increased cellular resistance to MMC, when compared to wild-type FANCI. Our findings suggest that ubiquitination-linked FANCI phosphorylation is able to bypass the need to de-ubiquitinate FANCD2 for efficient function of the FA pathway. This study establishes ubiquitination-linked FANCI phosphorylation as a post-translational modification that acts downstream of FANCI/D2 monoubiquitination, to sustain FANCD2 monoubiquitination in a functionally meaningful manner.
RESULTS
Characterization of Phospho-Specific Antibodies Against Human FANCI S/TQ Cluster Sites
There is limited information available regarding which sites within the hFANCI S/TQ cluster are actually phosphorylated. Therefore, we performed mass spectrometry to establish sites of actual phosphorylation within the S/TQ cluster of GFP-hFANCI that was immunoprecipitated from irradiated 293T cells (as described in Supplemental Experimental Procedures). We found that two of these sites, S556 and S565, were phosphorylated under the conditions of this experiment (Figures 1A, S1A and S1B). We designed immunogenic phospho-peptides (Figure 1B) that were injected into rabbits to generate antibodies that could specifically detect the phosphorylation of these two sites. Based on a prior structural study of mouse Fanci/d2 implicating T558 (S559 of hFANCI) phosphorylation in stabilizing the interaction between the two proteins (Joo et al., 2011), we also generated a phospho-specific antibody to S559, although its phosphorylation was not established by our mass spectrometry.
Figure 1. Characterization of Phospho-Specific Antibodies Against Human FANCI S/TQ Cluster Sites.

(A) Schematic of human FANCI (hFANCI), indicating the six putative phosphorylation sites within the S/TQ cluster domain. Sites whose phosphorylation was confirmed by mass spectrometry in this study, and sites against which phospho-specific antibodies were generated, are indicated in the table. The FANCI monoubiquitination site (K523) is also shown.
(B) Schematic of the immunogenic phospho-peptides used to generate hFANCI phospho-specific antibodies.
(C) Immunoblot with hFANCI phospho-S556, phospho-S559 and phospho-S565 antibodies of U2OS cells that were either untreated (Utx), or treated with 60 ng/mL MMC for the indicated times. An ATR inhibitor (VE821) was used to test the ATR-dependence of the FANCI phosphorylation sites.
(D) Immunoblot of FANCI-deficient (F010191) cells complemented with either MYC-tagged wild-type FANCI (FANCIwt), or MYC-tagged FANCI mutants that are alanine-substituted at the relevant sites. Cells were either untreated, or treated with 60 ng/mL MMC for 24 hours.
Ub refers to the monoubiquitinated form of FANCI, and non-Ub refers to the non-ubiquitinated form. The asterisk (*) indicates a non-specific band.
See also Figure S1.
The antibodies were tested by ELISA for phospho-specificity (Figures S1C–S1E). By necessity, the phospho-peptide used to generate the phospho-S559 antibody included an S556 residue that was not phosphorylated. This antibody could detect a phospho-S559 peptide provided that S556 is non-phosphorylated, and could not detect a phospho-peptide that is doubly phosphorylated at S556 and S559 (Figure S1D). The phospho-S556 and phospho-S565 antibodies were generated using phospho-peptides containing only the single relevant phosphorylation site.
The antibodies could detect the ATR-dependent phosphorylation of each of the three FANCI sites in U2OS cells, in response to MMC (Figure 1C). Using FANCI-deficient (F010191) cells that were complemented with MYC-tagged wild-type, or mutant, FANCI constructs, we confirmed that phospho-disruptive mutations at S556, S559 or S565 led to predictable losses of signal, indicating that each of the phospho-antibodies were site specific (Figure 1D). Our data establishes that hFANCI is phosphorylated in response to ICLs, in an ATR-dependent manner, on at least three sites within the hFANCI S/TQ cluster.
FANCI Phosphorylation is Co-Regulated with FANCI/D2 Monoubiquitination During Cell Cycle Progression
We next analyzed FANCI phosphorylation in U2OS cells at various time points after release from double thymidine (Figure 2A) or nocodazole (Figure 2B) blocks. Cell cycle analysis by flow cytometry was performed at each time point to track cell cycle progression. Consistent with the reported role of FANCI phosphorylation in regulating FANCD2 monoubiquitination, all three sites were phosphorylated during S-phase, when FANCD2 monoubiquitination is known to occur (Figure S2; Taniguchi et al., 2002a). This finding suggests a role of FANCI phosphorylation not only in response to exogenous DNA damage, but during normal cell cycle progression as well.
Figure 2. FANCI Phosphorylation Occurs During S-Phase of the Cell Cycle and Occurs Predominantly on Chromatin.

(A and B) Immunoblot of U2OS cells that were synchronized by, and released from, double thymidine block (A) or nocodazole block (B). Cell cycle phases at each time point were determined by flow cytometry of DNA content. Asynchronous cells were included for comparison.
(C) Immunoblot of soluble and pellet (chromatin-containing) fractions prepared from U2OS cells that were either untreated (Utx), or treated with 60 ng/mL MMC for 24 hours.
Ub refers to the monoubiquitinated forms of FANCI and FANCD2, and non-Ub refers to the non-ubiquitinated forms. The asterisk (*) indicates a non-specific band.
(D) Indirect immunofluorescence with FANCI phospho-S565 and FLAG antibodies of U2OS 3xFLAG-FANCD2 cells that were either untreated (Utx), or treated with 60 ng/mL MMC for 24 hours. Cells siRNA-depleted of FANCI, or λ phosphatase-treated on coverslips prior to staining, were included as negative controls for phospho-S565 FANCI antibody staining.
See also Figure S2.
Ubiquitination-Independent and Ubiquitination-Linked Phosphorylation of the FANCI S/TQ Cluster
Where FANCI phosphorylation occurs in relation to FANCI/D2 chromatin loading is unknown. In a cellular fractionation experiment (Figure 2C), we found that, relative to the soluble fraction, the phosphorylation of S556, S559 and S565 increased dramatically in the pellet (chromatin-containing) fraction of U2OS cells after MMC treatment. This suggested that the majority of FANCI phosphorylation occurs on chromatin-bound FANCI. We were able to validate one of the antibodies (to phospho-S565) for immunofluorescence staining. In U2OS cells expressing a FLAG-tagged FANCD2 (3xFLAG-FANCD2), phospho-S565 FANCI formed foci in response to MMC treatment, which colocalized with FANCD2 foci (Figure 2D). This suggests that S565 phosphorylation, at least, persists at sites of DNA damage.
Until this point, we observed that the phosphorylation of S556, S559 and S565 were differentially distributed between the monoubiquitinated and non-ubiquitinated forms of FANCI (Figures 1C and 2A–2C). Taking total FANCI levels into account, S556 phosphorylation was detected predominantly on non-ubiquitinated FANCI. In contrast, S559 and S565 phosphorylation was detected mostly on monoubiquitinated FANCI. We also observed this differential phosphorylation of S556 and S565 in primary human foreskin fibroblasts (Figure S3). This was an unexpected finding, as previous studies had suggested that FANCI phosphorylation does not require prior monoubiquitination, and had assumed that different FANCI sites are similarly phosphorylated (Ishiai et al., 2008; Shigechi et al., 2012; Tomida et al., 2013).
To investigate further, we examined the phosphorylation of endogenous FANCI S556, S559 and S565 in cell lines that are deficient in FA core complex proteins (Figure 3A; FANCA: GM6914; FANCF: TOV21G; FANCG: 326SV). MMC-induced S556 phosphorylation of non-ubiquitinated FANCI could occur in the absence of the core complex proteins. However, phosphorylation of S559/S565 on non-ubiquitinated FANCI was either partially or completely impaired. Only the restoration of FANCI monoubiquitination by complementation with the appropriate core complex protein permitted optimal phosphorylation of S559/S565 on monoubiquitinated FANCI.
Figure 3. Ubiquitination-Independent and Ubiquitination-Linked FANCI Phosphorylation.

(A) Immunoblot of FANCA-deficient (GM6914), FANCF-deficient (TOV21G) and FANCG-deficient (326SV) cells, and their complemented counterparts, that were either untreated (Utx), or treated with 60 ng/mL MMC for 24 hours.
(B) Immunoblot of FANCD2-deficient (PD20F) cells, complemented with either wild-type FANCD2 (FANCD2wt) or a monoubiquitination-deficient (K561R) mutant, that were either untreated, or treated with 60 ng/mL MMC for 24 hours.
(C) Immunoblot of pellet (chromatin containing) fractions prepared from FANCI-deficient (F010191) cells complemented with either wild-type FANCI (FANCIwt) or a monoubiquitination-deficient (K523R) mutant, that were either untreated, or treated with 60 ng/mL MMC for 24 hours. Irrelevant lanes have been removed from these blots.
(D) Immunoblot of FANCI-deficient (HCT116 FANCI−/−) cells complemented with an empty vector, wild-type FANCI, or the indicated mutants. Cells were then either untreated, or treated with 60 ng/mL MMC for 24 hours. Ratios of monoubiquitinated to non-ubiquitinated FANCD2 (D2 Ub:non-Ub) are indicated beneath the corresponding lanes.
Ub refers to the monoubiquitinated forms of FANCI and FANCD2, and non-Ub refers to the non-ubiquitinated forms. The asterisk (*) indicates a non-specific band.
See also Figures S3 and S4.
In FANCD2-deficient cells (Figure 3B; PD20F), a monoubiquitination-deficient mutant of FANCD2 (K561R) was unable to rescue S559 phosphorylation, and was able to only partially rescue S565 phosphorylation on non-ubiquitinated FANCI. Only restoration of wild-type FANCD2 expression was able to fully restore the phosphorylation of S559/S565 on monoubiquitinated FANCI. S556 phosphorylation of non-ubiquitinated FANCI was, again, relatively unaffected in the K561R cells. A similar result was obtained with FANCI-deficient (F010191) cells complemented with either wild-type FANCI, or a FANCI K523R mutant where the monoubiquitination site is disrupted. In FANCI-complemented F010191 cells, where FANCI is expressed at relatively high levels, immunoblot of whole cell lysates resulted in poor detection of total monoubiquitinated FANCI (Figure S4A), making it difficult to appreciate any preferential phosphorylation of this isoform. We found that by performing cellular fractionation of the complemented cells and analyzing the pellet (chromatin-containing) fraction, we could detect both non-ubiquitinated and monoubiquitinated forms of FANCI (Figure S4B). Consistent with our previous data, FANCI K523R was largely intact in S556 phosphorylation, but suppressed in S559 and S565 phosphorylation (Figure 3C). The mild phosphorylation of S565 on non-ubiquitinated FANCI that is observed in PD20F K561R cells (Figure 3B) and F010191 K523R cells (Figure 3C) indicates that the phosphorylation of this site is not absolutely dependent on monoubiquitination. However, S565 phosphorylation was clearly higher on monoubiquitinated FANCI. Taken together, our data suggests that optimal phosphorylation of FANCI S559/S565 occurs only when FANCI/D2 are monoubiquitinated.
We next tested the contribution of each of these sites to FANCD2 monoubiquitination in response to ICLs in FANCI-deficient (HCT116 FANCI−/−) cells complemented with wild-type FANCI, or phospho-disruptive (A)/phosphomimetic (D) mutants of the relevant sites (Figure 3D). All three sites positively regulated FANCD2 monoubiquitination in response to MMC. Phospho-disruptive mutations of each site impaired FANCD2 monoubiquitination, and combinations of these mutations had additive effects. Phosphomimetic mutations at each site increased FANCD2 monoubiquitination, especially in MMC-untreated conditions.
Our findings demonstrate that ubiquitination-independent (S556) and ubiquitination-linked (S559/S565) FANCI S/TQ phosphorylation sites positively regulate FANCD2 monoubiquitination.
USP1 is Required for Optimal Ubiquitination-Linked FANCI Phosphorylation
We next investigated whether simply increasing FANCI/D2 monoubiquitination would increase the phosphorylation of ubiquitination-linked FANCI sites in response to ICLs. As expected, depletion of USP1 resulted in increased monoubiquitination of FANCI and FANCD2 in U2OS cells, even without MMC treatment (Figure 4A, lanes 4 and 7). However, the monoubiquitinated FANCI that accumulated at baseline was not phosphorylated at ubiquitination-linked sites. Furthermore, in response to MMC, there was reduced induction of ubiquitination-linked FANCI phosphorylation in the USP1-depleted cells, compared to control cells (Figure 4A). A similar result was obtained when using a chemical inhibitor of USP1, ML323 (Figure 4B), suggesting that the de-ubiquitinating activity of USP1 is required for optimal phosphorylation of these sites. Interestingly, the ubiquitination-independent phosphorylation of S556 in response MMC was relatively unaffected by USP1 disruption, highlighting another biochemical distinction between S556 phosphorylation and S559/S565 phosphorylation. Our data demonstrates that increasing FANCI/D2 monoubiquitination by impairing USP1 is associated with the suppressed phosphorylation of ubiquitination-linked FANCI sites.
Figure 4. USP1 is Required for Optimal Ubiquitination-Linked FANCI Phosphorylation.

(A) Immunoblot of U2OS cells transfected with the indicated siRNAs and either untreated (Utx), or treated with 60 ng/mL MMC for the indicated times.
(B) Immunoblot of U2OS cells that were either untreated, or treated with 60 ng/mL MMC for the indicated times, either in the presence or absence of 30 μM ML323.
Ub refers to the monoubiquitinated forms of FANCI and FANCD2, and non-Ub refers to the non-ubiquitinated forms. The asterisk (*) indicates a non-specific band.
Ubiquitination-Linked Phosphomimetic FANCI Bypasses the Need to De-Ubiquitinate FANCD2 for Efficient Function of the FA Pathway
While ubiquitination-linked phosphorylation of S559/S565 was predominantly present on monoubiquitinated FANCI, both sites positively regulated FANCD2 monoubiquitination. This suggested that S559/S565 phosphorylation plays a role in inhibiting FANCD2 de-ubiquitination. We therefore devised an experiment to test whether de-phosphorylation of FANCI S559/S565 was associated with FANCD2 de-ubiquitination (Figure 5A). Cells were treated with MMC for 24 hours before withdrawing the MMC into the presence or absence of an ATR inhibitor (VE821), or an ATR inhibitor combined with a USP1 inhibitor (ML323). In U2OS cells, we again observed the induction of FANCI phosphorylation and FANCI/D2 monoubiquitination after MMC treatment for 24 hours (Figure 5B, lanes 1–2). The withdrawal of MMC alone was insufficient to trigger significant FANCD2 de-ubiquitination or FANCI de-phosphorylation (Figure 5B, lane 3). However, when ATR inhibitor was added, both FANCI and FANCD2 were rapidly de-ubiquitinated. This de-ubiquitination was associated with the progressive de-phosphorylation of S559 and S565 (Figure 5B, lanes 4–8). While S556 phosphorylation also dropped initially, it persisted on non-ubiquitinated FANCI at low levels after MMC withdrawal into ATR inhibitor. The addition of the USP1 inhibitor impaired FANCI/D2 de-ubiquitination in this experiment, most notably during the first four hours after MMC withdrawal. However, this only slightly delayed, and did not prevent, the de-phosphorylation of S559/S565 (Figure 5B, lanes 9–11). Our data suggests that FANCI phosphorylation sites, and S559/S565 in particular, are regulated by a phosphatase whose action is unmasked by inhibition of ATR.
Figure 5. Phosphomimetic Mutations at Ubiquitination-Linked FANCI Sites Bypass the Need to De-Ubiquitinate FANCD2 for ICL Repair.

(A) Outline of experiment to determine the relationship between FANCI de-phosphorylation and FANCI/D2 de-ubiquitination. Whole cell lysates for immunoblot were prepared at the various points during the experiment (triangles).
(B) Immunoblots of U2OS cells subjected to the experiment described in Figure 5A. Ratios of monoubiquitinated to non-ubiquitinated FANCD2 (D2 Ub:non-Ub) are indicated beneath the corresponding lanes.
(C) Immunoblot of HCT116 FANCI −/− cells complemented with either wild-type FANCI (FANCIwt) or a ubiquitination-linked phosphomimetic FANCI mutant (S559D-S565D), and subjected to the VE821-only component of the experiment described in Figure 5A. Ratios of monoubiquitinated to non-ubiquitinated FANCD2 (D2 Ub:non-Ub) are indicated beneath the corresponding lanes.
Ub refers to the monoubiquitinated forms of FANCI and FANCD2, and non-Ub refers to the non-ubiquitinated forms. The asterisk (*) indicates a non-specific band.
(D) Quantification of FANCD2 foci formation in HCT116 FANCI−/− cells complemented with either wild-type FANCI (FANCIwt) or a ubiquitination-linked phosphomimetic FANCI mutant (S559D-S565D), and either untreated, or treated with 60 ng/mL MMC for 24 hours.
(E) MMC cell survival assay of HCT116 FANCI−/− cells complemented with the indicated FANCI constructs, or with an empty vector.
(F) MMC cell survival assay of HCT116 FANCI−/− cells complemented with wild-type FANCI, either in the presence or absence of 10 μM ML323. Cells complemented with monoubiquitination-deficient FANCI (K523R) were included in this experiment for comparison.
(G) Quantification of fold decrease in cell survival caused by USP1 inhibition with 10 μM ML323 in HCT116 FANCI−/− cells complemented with either wild-type FANCI or S559D-S565D FANCI, and treated with 2 different concentrations of MMC.
All error bars represent S.E.M in experiments that were performed at least in triplicate, n.s. = not statistically significant.
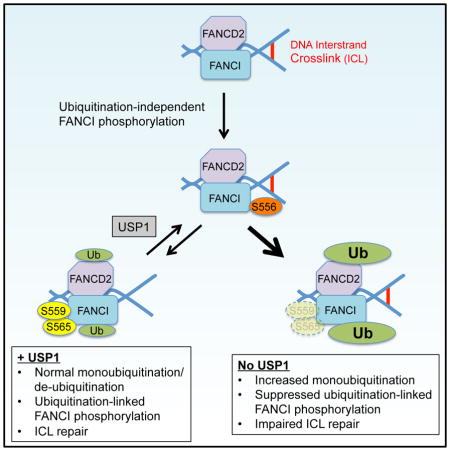
(H) Model of how ubiquitination-independent and ubiquitination-linked phosphorylation regulates FANCD2 monoubiquitination and downstream function of the FA pathway.
(I) Increased FANCI/D2 monoubiquitination in the context of USP1 deficiency leads to hypophosphorylation of ubiquitination-linked FANCI sites and inefficient ICL repair.
The distinct behaviors of S559 and S565 suggested that FANCI phosphorylation may function downstream of FANCI/D2 monoubiquitination. To specifically address the role of FANCI phosphorylation that is linked to monoubiquitinated FANCI, we created a FANCI construct carrying phosphomimetic substitutions at the two sites that we identified as ubiquitination-linked in their phosphorylation (S559D-S565D FANCI). Phosphomimicry of S556 was excluded from this construct, due to the likelihood that, relative to S559/S565, this site is hypophosphorylated on monoubiquitinated FANCI. This construct avoids the potentially excessive mutagenesis associated with the FANCI Dx6 mutant (which contains phosphomimetic mutations at all six S/TQ cluster sites) that is typically used in FANCI phosphorylation studies. The Dx6 mutant has been previously shown to disrupt, either partially or completely, both FANCD2 foci formation and cellular resistance to MMC (Castella et al., 2015; Chen et al., 2015; Ishiai et al., 2008). The combination of mutations used in the S559D-S565D construct, which mimics constitutive ubiquitination-linked FANCI phosphorylation, has not been investigated before.
Compared to wild-type FANCI, FANCI-deficient HCT116 FANCI−/− cells complemented with S559D-S565D FANCI exhibited significantly increased FANCD2 monoubiquitination, both at baseline and after MMC treatment (Figure 5C, lanes 1–2; lanes 8–9). Therefore, the effect of S559D-S565D FANCI on FANCD2 monoubiquitination is similar to that seen with USP1 depletion or inhibition. Furthermore, relative to wild-type FANCI, S559D-S565D FANCI strongly delayed FANCD2 de-ubiquitination after MMC withdrawal into an ATR inhibitor (Figure 5C, lanes 3–7; lanes 10–14).
USP1 is essential for FANCD2 foci formation and ICL repair. Epistasis studies have demonstrated that in the context of USP1 deficiency or inhibition, impaired FANCD2 de-ubiquitination is the cause of a poorly functioning FA pathway (Liang et al., 2014; Murai et al., 2011; Oestergaard et al., 2007). In addition, a FANCD2-ubiquitin fusion protein, which cannot be de-ubiquitinated, is impaired in its ability to repair ICLs (Matsushita et al., 2005). Based on these previous studies, we expected that cells expressing S559D-S565D FANCI, which behave similarly to USP1-deficient cells in that they are impaired in FANCD2 de-ubiquitination, would also have suppressed FA pathway function.
Surprisingly, we found that the relatively impaired FANCD2 de-ubiquitination observed in HCT116 FANCI −/− cells expressing S559D-S565D FANCI (Figure 5C) had no effect on MMC-induced FANCD2 foci formation when compared to wild-type FANCI (Figures 5D and S5). FANCI-deficient cells complemented with S559D-S565D FANCI exhibited intact, and even slightly increased, MMC resistance, when compared to wild-type FANCI (Figure 5E).
Consistent with previous studies (Dexheimer et al., 2010; Liang et al., 2014), treatment of FANCI-deficient cells complemented with wild-type FANCI with a USP1 inhibitor (ML323) resulted in sensitization to MMC (Figure 5F). At MMC concentrations that induced moderate cell death (2.5 ng/mL and 5 ng/mL), S559D-S565D FANCI conferred a mild, but statistically significant, protective effect against ML323-induced sensitization to MMC, when compared to wild-type FANCI (Figure 5G). While previous studies have indicated a role of FANCD2 de-ubiquitination in ICL repair, it is possible that an additional mechanism (for example, another USP1 substrate) contributes to the deficient ICL repair that we observe with USP1 impairment. Taken together, our results suggest that sustained phosphorylation of ubiquitination-linked FANCI sites is able to bypass the need to de-ubiquitinate FANCD2 for efficient FA pathway function, and temper the sensitization to MMC that occurs with USP1 inhibition.
DISCUSSION
In this study, we demonstrate that different FANCI phosphorylation sites regulate FANCD2 monoubiquitination from both sides of the equilibrium. Ubiquitination-independent phosphorylation of FANCI S556 occurs upstream of, and promotes, FANCD2 monoubiquitination. Ubiquitination-linked phosphorylation of S559/S565 occurs predominantly downstream of monoubiquitination, and acts to inhibit FANCD2 de-ubiquitination by USP1. Our findings provide evidence that FANCI de-phosphorylation is required for FANCI/D2 de-ubiquitination.
Between FANCI phosphorylation and monoubiquitination of the FANCI/D2 complex, monoubiquitination has been considered the more downstream modification (Boisvert and Howlett, 2014). This standing model was derived from prior studies that used electrophoretic mobility to detect the phosphorylation of multiple FANCI sites collectively, in a non-quantitative manner (Ishiai et al., 2008; Shigechi et al., 2012; Tomida et al., 2013). This method did not allow previous investigators to identify certain sites (S559 and S565) as being predominantly phosphorylated on monoubiquitinated FANCI. We present a refined model involving interplay between phosphorylation and monoubiquitination of the FANCI/D2 complex that regulates FANCD2 recycling between its monoubiquitinated and non-ubiquitinated states. In this model, ubiquitination-linked FANCI phosphorylation is supported by monoubiquitination, and is able to mediate ICL repair despite impairing FANCD2 de-ubiquitination (Figure 5H).
Our data suggest that ubiquitination-linked phosphorylation of FANCI, while dependent on FANCI/D2 monoubiquitination, is also suppressed by the excess monoubiquitination caused by USP1 impairment. In a prior study (Guervilly et al., 2011), USP1 depletion suppressed both Chk1 phosphorylation and steady state levels in an FA pathway-dependent manner, suggesting that FANCD2 monoubiquitination acts to switch off Chk1 through a negative feedback mechanism. It is possible that the effect of USP1 impairment on FANCI S559/S565 phosphorylation is mediated through a similar mechanism. Alternatively, USP1 deficiency may cause the accumulation of monoubiquitinated FANCI at genomic sites away from DNA damage, where it cannot undergo S559/S565 phosphorylation by ATR. In the absence of USP1 activity, excess FANCI/D2 monoubiquitination is less able to facilitate ICL repair without the appropriate level of ubiquitination-linked FANCI phosphorylation (Figure 5I).
Different mechanisms of increasing FANCD2 monoubiquitination have contrasting effects on the activity of the monoubiquitinated FANCI/D2 complex. In the case of USP1 disruption, impaired FANCD2 de-ubiquitination suppresses both ubiquitination-linked FANCI phosphorylation and ICL repair. However, if ubiquitination-linked FANCI phosphorylation is sustained, the resulting impairment of FANCD2 de-ubiquitination results in intact ICL repair. In other words, a FANCI/D2 complex that is both monoubiquitinated and phosphorylated at ubiquitination-linked sites is activated to the point that it does not require de-ubiquitination to function. While the S559D-S565D FANCI mutant has a mildly increased ability to confer MMC resistance, this construct omits phosphomimetic mutations of other possible ubiquitination-linked FANCI sites that were not identified by our study, but could potentially make further contributions to ICL repair. Ultimately, we have found that ubiquitination-linked FANCI phosphorylation is an effective marker of an efficient FA pathway (Figures 5H and 5I; Table S1).
This study re-defines the activated FANCI/D2 complex as being not only monoubiquitinated, but also phosphorylated in a ubiquitination-linked manner. Our work suggests that modulating FANCI phosphorylation can mediate an increase in FANCD2 monoubiquitination that is functionally, and perhaps therapeutically, meaningful.
EXPERIMENTAL PROCEDURES
Immunoblots/Antibodies
Rabbit polyclonal antibodies against phospho-S556, phospho-S559, and phospho-S565 of hFANCI were generated by PhosphoSolutions. Anti-FANCI (Santa Cruz, sc-271316), anti-FANCD2 (Abcam, ab2187), anti-MYC tag 9E10 (Abcam, ab32), anti-USP1 (C-term; gift from T. Huang), anti-FLAG M2 (Sigma, F1804), anti-Histone H3 (Cell Signaling, 9715) and anti-Vinculin (Sigma, V9131) were used. Whole cell lysates were prepared by lysing cells in 0.05 M Tris-HCl (pH 6.8), 2% SDS, 6%-mercaptoethanol and boiling for 5 minutes. Lysates were subjected to SDS-PAGE using NuPAGE 3–8% Tris-Acetate or 4–12% Tris-Glycine gels before transferring to nitrocellulose membranes. Horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies (Amersham) were used, and blots were developed using ECL reagent with image acquisition on ImageQuant LAS4000 (GE Healthcare).
Cell Fractionations
Cell fractionations were performed as described (Kim et al., 2008).
Cell Culture
U2OS, U2OS 3xFLAG-FANCD2 (Castella et al., 2015), 293T, GM6914, 326SV and TOV21G cells were cultured in DMEM + 10% FBS supplemented with L-glutamine. F010191 cells (gift from T. Huang) and primary human foreskin fibroblasts (gift from D. Galloway) were cultured in DMEM + 15% FBS supplemented with L-glutamine. HCT116 FANCI−/− cells (gift from E. Hendrickson) (Castella et al., 2015) were cultured in McCoy’s 5A media + 10% FBS supplemented with L-glutamine. The following enzyme inhibitors were used: ML323 (USP1 inhibitor; gift from Z. Zhuang), VE821 (ATR inhibitor; Axon Medchem, 1893).
Immunofluorescence
Cells on coverslips were permeabilized and fixed for 20 minutes with a solution containing 2% paraformaldehyde/0.5% Triton X-100 in PBS. Cells were then washed in PBS before blocking in PBS containing 3% BSA + 0.1% Tween 20. Cells were incubated in primary antibodies in blocking buffer at 4 °C overnight before washing in PBS + 0.1% Tween 20 and incubating in secondary antibodies (AlexaFluor 488 Goat Anti-Rabbit IgG and AlexaFluor 594 Goat Anti-Mouse IgG; Molecular Probes) in blocking buffer for 1 hour. The coverslips were then stained with 1 μg/mL DAPI (4,6-Diamidino-2-phenylindole) for 5 minutes, washed in PBS + 0.1% Tween 20 and mounted onto glass slides using Vectashield mounting media. Images were acquired with a TE2000 Nikon microscope equipped with a 40x immersion objective and a CCD camera (CoolSNAP ES, Photometrics). MetaVue software was used for image acquisition. For phosphatase treatment of fixed cells, coverslips were incubated in λ phosphatase (NEB) in the presence of MnCl2 at 30 °C for 2 hours prior to antibody staining.
Lentiviral Infections
Lentivirus was generated using a pLENTIX1-puro vector carrying a MYC-tagged human FANCI under a Ubiquitin C promoter (Castella et al., 2015). Mutagenesis of the FANCI coding sequence was achieved through an overlapping extension PCR method. Lentivirus was produced as described (Wang et al., 2011). Infections of F010191 and HCT116 FANCI−/− cells were performed by adding viral supernatant containing polybrene. Cells were selected with 2 μg/mL puromycin prior to their use for experiments.
Cell Synchronization and Cell Cycle Analysis
Synchronization of U2OS cells in G1/S and M phases using double thymidine and nocodazole blocks, respectively, and cell cycle analyses, were performed as described (Taniguchi et al., 2002a).
MMC Cell Survival Assay
Cell survivals over a range of MMC concentrations were measured using a crystal violet absorbance-based assay as described (Taniguchi et al., 2002a; Taniguchi et al., 2002b; Wang et al., 2011). Fold decreases in cell survival due to ML323 treatment were determined by using the formula: Fold decrease = (% survival of ML323 untreated cells)/(% survival of ML323 treated cells).
SiRNA Transfection
The following siRNA sequences were used: si-USP1_1: 5′ UCGGCAAUACUUGCUAUCUUA 3′, si-USP1_2: 5′ CGACAGCUAUGGAUUAUUU 3′, si-FANCI: 5′ CACGGGCAUCUGGGAGAUAUA 3′. siRNAs were transfected at 20nM concentration using Lipofectamine RNAiMAX reagent (Invitrogen), following the manufacturer’s protocol. Experiments involving gene knockdown were initiated 48 hours after siRNA transfection.
Statistics
Statistical analyses were performed using a Student’s t-test. A p-value <0.05 was considered statistically significant.
Supplementary Material
Highlights.
FANCI phosphorylation may be either independent of, or linked to, ubiquitination
Ubiquitination-linked FANCI phosphorylation inhibits FANCD2 de-ubiquitination
Ubiquitination-linked FANCI phosphorylation fully activates the FANCI/D2 complex
Impairing USP1 suppresses ubiquitination-linked FANCI phosphorylation
Acknowledgments
We thank Rebecca Reeves for technical assistance. We thank Drs. Tony Huang (New York University School of Medicine), Eric Hendrickson (University of Minnesota), Denise Galloway (FHCRC) and Zhihao Zhuang (University of Delaware) for reagents, and Drs. Tony Huang, Celine Jacquemont (FHCRC) and Alex Sobeck (University of Minnesota) for critical reading of the manuscript. This work was supported by Howard Hughes Medical Institute, Fanconi Anemia Research Fund, NIH/NCI R01 CA125636 and JSPS KAKENHI 15K21771 (to T.T.), NIH/Ruth L. Kirschstein National Research Service Award T32 CA009351 and NIH/NICHD University of Washington Child Health Research Center K12 HD043376 (to R.S.C.) and NSF Graduate Research Fellowship under Grant No. DGE-0718124 and Fred Hutch CMCTG T32 CA009657 (to A.A.). The Fred Hutch Proteomics Facility is partially funded by Cancer Center Support Grant P30 CA015704 from the NIH.
Footnotes
AUTHOR CONTRIBUTIONS
R.S.C designed and performed experiments, collected and analyzed data, and wrote the manuscript. M.C. provided the FANCI expression vector, designed experiments and reviewed the manuscript. A.A. and N.T. performed experiments. P.R.G. performed mass spectrometry. T.T. designed experiments, supervised the study, and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boisvert RA, Howlett NG. The Fanconi anemia ID2 complex: dueling saxes at the crossroads. Cell Cycle. 2014;13:2999–3015. doi: 10.4161/15384101.2014.956475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castella M, Jacquemont C, Thompson EL, Yeo JE, Cheung RS, Huang JW, Sobeck A, Hendrickson EA, Taniguchi T. FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2. PLoS genetics. 2015;11:e1005563. doi: 10.1371/journal.pgen.1005563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nature reviews Molecular cell biology. 2016;17:337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ, 3rd, Rothenberg E, Jallepalli PV, Huang TT. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Molecular cell. 2015;58:323–338. doi: 10.1016/j.molcel.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexheimer TS, Rosenthal AS, Liang Q, Chen J, Villamil MA, Kerns EH, Simeonov A, Jadhav A, Zhuang Z, Maloney DJ. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): 2010. Discovery of ML323 as a Novel Inhibitor of the USP1/UAF1 Deubiquitinase Complex. [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Gibbs-Seymour I, Oka Y, Rajendra E, Weinert BT, Passmore LA, Patel KJ, Olsen JV, Choudhary C, Bekker-Jensen S, Mailand N. Ubiquitin-SUMO circuitry controls activated fanconi anemia ID complex dosage in response to DNA damage. Molecular cell. 2015;57:150–164. doi: 10.1016/j.molcel.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guervilly JH, Renaud E, Takata M, Rosselli F. USP1 deubiquitinase maintains phosphorylated CHK1 by limiting its DDB1-dependent degradation. Human molecular genetics. 2011;20:2171–2181. doi: 10.1093/hmg/ddr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, Huong D, Takata M, Chen J, Li L. Modularized functions of the Fanconi anemia core complex. Cell Rep. 2014;7:1849–1857. doi: 10.1016/j.celrep.2014.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nature structural & molecular biology. 2008;15:1138–1146. doi: 10.1038/nsmb.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo W, Xu G, Persky NS, Smogorzewska A, Rudge DG, Buzovetsky O, Elledge SJ, Pavletich NP. Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science. 2011;333:312–316. doi: 10.1126/science.1205805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Kee Y, Gurtan A, D’Andrea AD. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111:5215–5222. doi: 10.1182/blood-2007-09-113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D’Andrea AD. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Developmental cell. 2009;16:314–320. doi: 10.1016/j.devcel.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Dexheimer TS, Zhang P, Rosenthal AS, Villamil MA, You C, Zhang Q, Chen J, Ott CA, Sun H, et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol. 2014;10:298–304. doi: 10.1038/nchembio.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita N, Kitao H, Ishiai M, Nagashima N, Hirano S, Okawa K, Ohta T, Yu DS, McHugh PJ, Hickson ID, et al. A FancD2-monoubiquitin fusion reveals hidden functions of Fanconi anemia core complex in DNA repair. Molecular cell. 2005;19:841–847. doi: 10.1016/j.molcel.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Murai J, Yang K, Dejsuphong D, Hirota K, Takeda S, D’Andrea AD. The USP1/UAF1 complex promotes double-strand break repair through homologous recombination. Molecular and cellular biology. 2011;31:2462–2469. doi: 10.1128/MCB.05058-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. Deubiquitination of FANCD2 is required for DNA crosslink repair. Molecular cell. 2007;28:798–809. doi: 10.1016/j.molcel.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigechi T, Tomida J, Sato K, Kobayashi M, Eykelenboom JK, Pessina F, Zhang Y, Uchida E, Ishiai M, Lowndes NF, et al. ATR-ATRIP kinase complex triggers activation of the Fanconi anemia DNA repair pathway. Cancer research. 2012;72:1149–1156. doi: 10.1158/0008-5472.CAN-11-2904. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D’Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002a;100:2414–2420. doi: 10.1182/blood-2002-01-0278. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D’Andrea AD. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002b;109:459–472. doi: 10.1016/s0092-8674(02)00747-x. [DOI] [PubMed] [Google Scholar]
- Tomida J, Itaya A, Shigechi T, Unno J, Uchida E, Ikura M, Masuda Y, Matsuda S, Adachi J, Kobayashi M, et al. A novel interplay between the Fanconi anemia core complex and ATR-ATRIP kinase during DNA cross-link repair. Nucleic acids research. 2013;41:6930–6941. doi: 10.1093/nar/gkt467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang JW, Li M, Cavenee WK, Mitchell PS, Zhou X, Tewari M, Furnari FB, Taniguchi T. MicroRNA-138 modulates DNA damage response by repressing histone H2AX expression. Molecular cancer research: MCR. 2011;9:1100–1111. doi: 10.1158/1541-7786.MCR-11-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.