

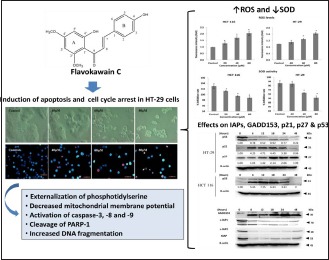
Abstract
Chalcones have been shown to exhibit anti-cancer properties by targeting multiple molecular pathways. It was, therefore, of interest to investigate flavokawain C (FKC), a naturally occurring chalcone, which can be isolated from Kava (Piper methysticum Forst) root extract. The aim of this study was to investigate the inhibitory effect of FKC on the growth of HT-29 cells and its underlying mechanism of action. Cell viability of HT-29 cells was assessed by Sulforhodamine B assay after FKC treatment. Induction of apoptosis was examined by established morphological and biochemical assays. ROS generation was determined by dichlorofluorescein fluorescence staining, and superoxide dismutase activity was measured using the spectrophotometric method. Western blotting was used to examine the changes in the protein levels. FKC markedly decreased the cell viability of HT-29 cells and the cells showed dramatic changes in cellular and nuclear morphologies with typical apoptotic features. The induction of apoptosis correlated well with the externalization of phosphatidylserine, DNA fragmentation, decreased mitochondrial membrane potential, activation of caspases, and PARP cleavage. This was associated with an increase in reactive oxygen species and a decrease in SOD activity. The protein levels of XIAP, c-IAP1, and c-IAP2 were downregulated, whereas the GADD153 was upregulated after FKC treatment. FKC induced cell cycle arrest at the G1 and G2/M phases via upregulation of p21 and p27 in a p53-independent manner. Our results provide evidence that FKC has the potential to be developed into chemotherapeutic drug for the treatment of colon adenocarcinoma.
SUMMARY:
Flavokawain C inhibited the growth of HT-29 human colon adenocarcinoma cells
Flavokawain C induced apoptosis in HT-29 cells, associated with an increase in reactive oxygen species and a decrease in SOD activity
Flavokawain C induced cell cycle arrest at the G1 and G2/M phases via upregulation of p21 and p27 in HT-29 cells
HT-29 cells treated with flavokawain C caused downregulation of XIAP, c-IAP1, and c-IAP2, and upregulation of GADD153.
Abbreviations used: FKC: Flavokawain C; SRB: Sulforhodamine B; ROS: Reactive oxygen species; SOD: Superoxide dismutase; PARP: Poly(ADP-ribose) polymerase; ER: Endoplasmic reticulum; IAPs: Inhibitor of apoptosis proteins; TUNEL: Transferase dUTP nick end labeling; Annexin V-FITC: Annexin V conjugated with fluorescein isothicyanate
Keywords: Apoptosis, cell cycle, colon adenocarcinoma, flavokawain C, HT-29, ROS
INTRODUCTION
Colorectal cancer (CRC) is the third leading cause of cancer-related death worldwide.[1] Although the highest incidence rates have been observed mostly in western countries, there is a rising trend in Asian countries, and this is mostly due to changes in diet and lifestyles.[2,3] Surgical resection is the main curative treatment that usually gives a long-term survival rate for the CRC patient. However, a great majority of the patients eventually relapse following surgery and develop resistance to the initial therapy.[4,5] Therefore, there is an urgent need to find new preventive and therapeutic agents for CRC.
Uncontrolled cell proliferation and evasion of apoptosis are the hallmark of cancer, and the induction of apoptosis has been one of the promising chemotherapeutic strategies.[6,7] Apoptosis or programmed cell death occurs through two major pathways, namely mitochondria and death receptor-mediated pathways. In addition, prolonged ER stress has been implicated in apoptotic execution.[8]
It is well-known that a moderate increase in reactive oxygen species (ROS) can stimulate cell growth and proliferation. However, excessive ROS accumulation causes cellular injury such as damage to DNA, protein, and lipid membrane.[9] Due to its harmful effect, ROS generation can be counterbalanced by the action of antioxidant enzymes.[10] The imbalance between the level of ROS and the endogenous antioxidants can result in oxidative stress that can provoke apoptosis in cells.[11]
Chalcones have been shown to demonstrate anticancer activities by targeting multiple molecular pathways such as apoptosis, cell cycle, p53 pathway, NF-kappa B, tubulin polymerization, ubiquitin-proteasome pathway, β-catenin/Wnt, and others.[12,13] In addition, chalcones have been shown to possess low toxicity against normal cells. Due to these properties, chalcones have emerged as the potential candidate for the development as an anticancer agent or for cancer prevention. In our earlier study, flavokawain C (FKC), a naturally occurring chalcone that can be isolated from the Kava-kava (Piper methysticum Forst) root extract, has been shown to inhibit the growth of colon carcinoma HCT 116 cells via apoptosis and cell cycle arrest.[14,15] Therefore, this has led us to focus our investigation in the present study on the cytotoxic and apoptotic activities of FKC (structure is shown [Figure 1]) in human adenocarcinoma HT-29 cells.
Figure 1.
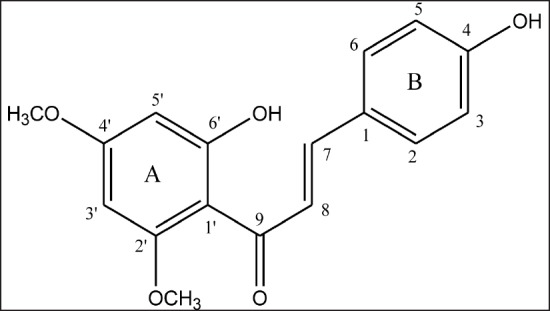
Structure of flavokawain C.
SUBJECTS AND METHODS
Cell culture, chemicals, and reagents
FKC was obtained from the Extrasynthase (Genay, France) and dissolved in Dimethyl sulfoxide (DMSO, Sigma). Human colon adenocarcinoma HT-29 and human carcinoma HCT 116 cells were purchased from the American Type Culture Collection (ATCC, USA). HT-29 cells were cultured in RPMI (Nacalai Tesque, Japan) and McCoy's 5A (Sigma) media, respectively. The media were supplemented with 10% heat-inactivated fetal bovine serum (Sigma), 1% penicillin/streptomycin (PAA Laboratories), 1% amphoteric B (PAA Laboratories).
The primary antibodies for cleaved PARP-1 (cleaved p25) and p53 were purchased from GeneTex; XIAP, c-IAP1, c-IAP2, GADD153/CHOP, p21WAF1/Cip1, and β-actin were purchased from Thermo Scientific; p27Kip1 was purchased from Cell Signaling. The horseradish peroxidase (HRP)-labeled antimouse and antirabbit secondary antibodies were purchased from Thermo Scientific.
Assessment of cell viability
Cytotoxicity activity was detected by the SRB assay as previously described.[15] Briefly, cells were seeded in 96-well plates with 150 μL per well at a concentration of cell suspension of 4.0×105 cells/mL, and incubated for overnight at 37°C in a humidified atmosphere with 5% CO2. The cells were treated with 40, 60, and 80 μM of FKC for 6, 12, 24, 48, and 72 h in the CO2 incubator at 37°C. The cells were fixed in ice-cold trichloroacetic acid (10%w/v) for 1 h. The cells were washed and stained 0.4% SRB dye for 30 min at room temperature. The excess SRB dye was removed and 100 μL of 10 mM Tris buffer (pH 10.5) was added. The absorbance value of each well was measured at 492 nm using a microplate reader (BioTek). All experiments were performed at least three times. The cell viability was calculated as follows:
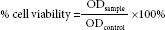
Morphological assessment
Cells were seeded in a 6-well plate at a concentration of 2.7×105 cells/well, and incubated for overnight at 37°C in a humidified atmosphere with 5% CO2, and then were treated with FKC at various concentrations of 40, 60, and 80 μM for 48 h. After incubating, cell morphology was observed with a phase contrast inverted microscope (Zeiss Axio Vert A1). For nuclear morphological analysis, the cells were stained with Hoechst 33342 (10 μg/mL) followed by propidium iodide (2.5 μg/mL). The cells were then mounted onto glass microscope slides and observed under the fluorescence microscope (Leica DFC 310 FX).
Annexin V-FITC and PI assay
Apoptosis of cells were quantified using the Annexin V-FITC Apoptosis Detection Kit (BD Pharmingen). Briefly, cells were seeded at a density of 2.7×105 cells in 6-well plates for overnight and then treated with FKC at the concentration of 40, 60, and 80 μM for 24 and 48 h. Following the treatment, cells were harvested and stained with Annexin V-FITC and PI according to the instruction of the kit. A total of 10,000 cells were analyzed by flow cytometry (Accuri C6, BD Biosciences, USA) using the BD CFlow software. The percentages of cells (viable cells, early apoptotic cells, late apoptotic cells, and dead cells/primary necrotic cells) were quantified from the mean fluorescence intensity in each of the four quadrants. The negative control was the cells in 0.5% DMSO without FKC.
Detection of DNA fragmentation
DNA fragmentation was detected using Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay kit (APO-BrdU; Invitrogen). Cells were seeded at a density of 1×106 cells in 60 mm petri dishes for overnight and then treated with FKC at the concentrations of 40, 60, and 80 μM for 48 h. Following the treatment, cells were harvested and fixed with 4% formaldehyde and permeabilized using 70% ethanol overnight. The cells were washed and DNA labeling steps were performed according to manufacturer's instructions. The cells were then analyzed using flow cytometry. Untreated cells in 0.5% DMSO served as the control.
Measurement of mitochondrial membrane potential (Δ Ψm)
The loss of Δ Ψm was assessed using the BD MitoScreen kit according to manufacturer's instructions. The cells were seeded at a density of 2.7×105 cells in 6-well plates for overnight and then treated with FKC at the concentrations of 40, 60, and 80 μM for 24 and 48 h. Following treatment, the cells were harvested and washed twice with PBS. The cells were then suspended in 500 μL of JC-1 working solution and incubated at 37°C for 30 min. A total of 10,000 events were recorded by flow cytometry and analyzed by the BD CFlow software.
Measurement of intracellular ROS level
Intracellular levels of ROS in cancer cells were determined using DCFH-DA. HT-29 cells were seeded at a density of 7500 cells per well in 150 μL of media in 96-well plates. After 24 h, cells were treated with 80 μM of FKC and incubated for 4 h. After treatment with FKC, the cells were washed thrice with PBS and added with HBSS containing DCFH-DA (20 μM). Then, cells were incubated at 37°C for 30 min in the dark. DCF fluorescence intensity was measured by the fluorescence microplate reader (BioTek) with an excitation source at 480 nm and emission at 530 nm.
SOD (superoxide dismutase) inhibition activity
The SOD activity was measured using the SOD assay Kit-WST (Sigma-Aldrich, USA). HT-29 and HCT 116 cells were seeded at a density of 7500 cells per well in 96-well plates and allowed to adhere overnight. Cells were then treated with 80 μM of FKC for 4 h. Cells were then harvested by centrifugation and lysed. The supernatants were collected, and protein concentration was determined by the Bradford assay. Cell lysates (35 μg) was used for determination of SOD activity according to the manufacturer's instructions.
Measurement of activated caspase-3, -8, and-9
Cells at a density of 1×106 were cultured in a 60-mm petri dish and then treated with FKC at the concentrations of 40, 60, and 80 μM for 48 h. After incubation, the cells were harvested and incubated in situ marker (FITC-DEVD-FMK for caspase-3, FITC-IETD-FMK for caspase-8, and FITC-LEHD-FMK for caspase-9; CaspILLUME, Genetex) for 20 min in 5% CO2 at 37°C before being analyzed by flow cytometry.
Cell cycle analysis
Cells were seeded in petri dishes at a concentration of 1.0×106 cells and allowed to adhere overnight. The cells were treated with FKC at the concentrations of 40, 60, and 80 μM. Next, the cells were harvested, washed with cold PBS, and fixed overnight with ice-cold 70% ethanol at -20°C. The cells were washed with PBS twice and stained with propidium iodide solution for 30 min in the dark before subjected to flow cytometry by using 15,000 cells per each sample. Cell cycle profiles were analyzed with the ModFit software (Verity Software House, Topsham, ME, USA).
Western blot
After treatment with FKC (80 μM) for the indicated time points (6, 12, 18, 24, and 48 h), the cells were extracted using the mammalian cell extraction kit (BioVision). The cell lysates (50 μg) were denatured at 100 °C for 5 min. Proteins were separated by SDS-PAGE and then electrophoretically transferred to the nitrocellulose membrane. The membrane was blocked for 1 h and followed by incubation with primary antibody at 4°C overnight. The membranes were incubated for 1 h with horseradish peroxidase-conjugated secondary antibody (1:10,000 dilution). After three washes in PBST, band signals were visualized using the Supersignal West Pico chemiluminescence substrate (Thermo Scientific), and the images were captured with the Fusion FX7 image system (Vilber Lourmat). β-actin served as the protein loading control. Densitometric quantification of the bands was performed using the ImageJ software, and the results were expressed as fold change relative to the control after normalization with β-actin.
Statistical analysis
All data were expressed as mean ± standard deviation of triplicates. Statistical analysis of data was performed using SPSS Statistics 17.0 and differences with a P < 0.05 were considered significant.
RESULTS
FKC inhibited the growth and induced G2/M arrest in HT-29 cells
The effect of FKC on the cell growth in HT-29 cells was first examined by the SRB assay, which evaluates cytotoxicity based on the total protein content which was proportional to the number of viable cells. The concentration of FKC used in this study was selected based on the IC50 value (39.00 ± 0.37 μM) found in our earlier study, and thus, the concentration used is equivalent to, or two and three times higher than, the IC50 value. FKC treatment (40, 60, and 80 μM) resulted in a decrease in cell viability for increasing time-point (6, 12, 24, 48, and 72 h). As shown in Figure 2A, the cell viability was reduced from 644.51% in control to 185.17, 111.81, and 104.94% after the cells at the concentration of 40, 60, and 80 μM, respectively, for 72 h. A comparable observation was noticed in the growth arrest between doses of 60 and 80 μM of FKC.
Figure 2.
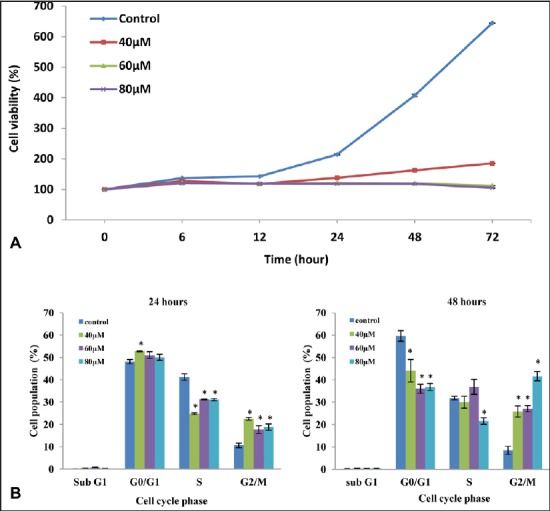
(A) The inhibitory effect of FKC on cell viability of HT-29 cells. In comparison to the control, the inhibition of cell proliferation was significantly increased in dose- and time-dependent manner. (B) The effect of FKC on the cell cycle in HT-29 cells. Data given are expressed as mean ± standard deviation of triplicates obtained from three independent experiments. *P<0.05 compared to the control.
To investigate the inhibition of cell growth as associated with cell cycle arrest, HT-29 cells was stained with propidium iodide (PI) and cell cycle distribution was analyzed by flow cytometry. At 24 h, FKC treatment caused a high accumulation of cells in the G2/M phase, although there was a small accumulation of cells in the G1 phase at the expense of a decrease in S-phase population in a dose-dependent manner over the control [Figure 2B]. After treatment of over 48 h, there was a marked increase in the G2/M phase population in a dose-dependent manner and the highest increase was observed at 80 μM of FKC [Figure 2B]. Based on the cell cycle analysis, it can be suggested that the decrease in cell viability was associated with G2/M arrest and an 80 μM dose of FKC was selected for further mechanistic studies.
FKC induced cell death through apoptosis
To investigate whether the inhibition of cell growth by FKC is associated with induction of apoptosis, the changes in cellular and nuclear morphologies of HT-29 cells were examined. Results from phase contrast microscopic analysis [Figure 3A] showed that the morphology of HT-29 cells underwent drastic changes after FKC treatment in comparison to the control. The cells showed typical apoptotic morphological changes such as cell shrinkage, formation of numerous blebs on the cell surface, and vacuoles in the cytoplasm.
Figure 3.
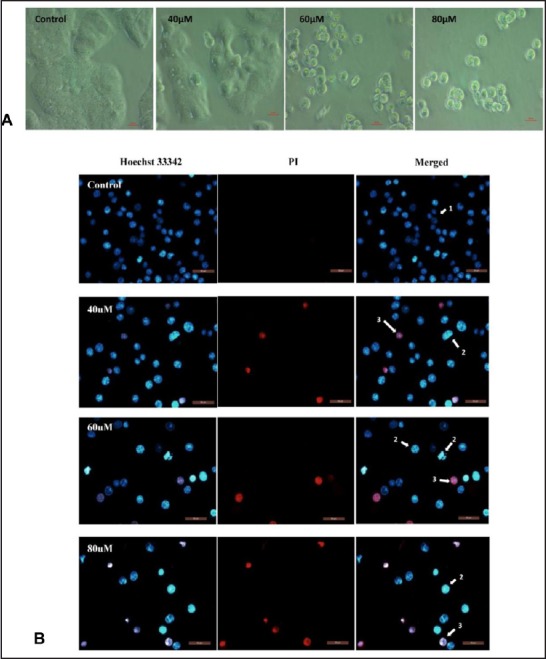
(A) Changes in morphology of HT-29 after FKC treatment for 48 h were detected by inverted phase contrast microscopy (400×). Cells became irregular, shrunken, and began to detach from the culture plate in a dose-dependent manner. (B) Changes in nuclear morphology of HT-29 cells were detected by the fluorescence microscope (400×). The arrows indicate the formation of chromatin fragmentation and apoptotic body.
Results from Hoechst 33342/PI staining [Figure 3B] showed that apoptosis occurred in HT-29 cells after FKC treatment for 48 h. FKC-treated HT-29 cells showed changes in nuclear morphology with typical apoptotic features such as chromatin condensation and/or fragmentation as shown in bright blue compared to dull blue in the control. In addition, a population of cells with pink fluorescence was also observed, indicating the presence of late apoptotic cells.
To further validate the apoptotic effect of FKC, Annexin V-FITC/PI double staining and TUNEL assay were performed. The Annexin V-FITC and PI analysis [Figure 4A] showed that FKC treatment caused a gradual increase in the population of Annexin V-FITC-positive cells (apoptotic cells) in the lower right quadrants of flow cytometry graphs in a dose- and time-dependent manner. Results from the TUNEL assay [Figure 4C] showed a concentration-dependent increase in the amount of apoptotic cells with fragmented DNA (P < 0.05); however, an obvious increase in TUNEL-positive cells (11.6 ± 0.1%) was observed at 80 μM following FKC treatment for 48 h in comparison to the control. Taken together, the results clearly showed that FKC caused induction of apoptosis in HT-29 cells.
Figure 4.
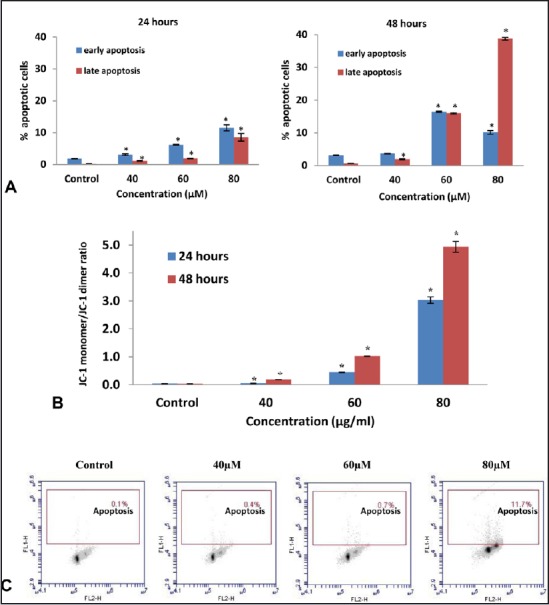
(A) The number of early and late apoptotic cells (from total 10,000 cells) of HT-29 cells was increased following FKC treatment for 24 and 48 h. *P<0.05 compared to the control. (B) The effect of FKC on the mitochondrial membrane potential (Δ Ψm) of HT-29 cells. Δ Ψm of HT-29 cells was significantly reduced by FKC in a dose- and time-dependent manner. *P<0.05 compared to the control. (C) FKC induces DNA fragmentation in HT-29 cells. Percentages of HT-29 cells that showed positive DNA fragmentation are represented in the upper quadrant from a dot plot.
FKC increased permeability of the outer mitochondrial membrane
The loss of mitochondrial membrane potential (Δ Ψm) due to the opening of the mitochondrial permeability transition pore has played a central role in the intrinsic apoptotic pathway.[16] The changes in Δ Ψm can be determined by JC-1, a cationic fluorescent probe and analyzed by flow cytometry. The flow cytometry analysis showed that FKC treatment resulted in an increase of green-fluorescent JC-1 monomers (lower quadrant) and a decrease red-fluorescent J-aggregates (upper quadrant). The quantitative data of the ratio of J-aggregate/JC-1 monomer [Figure 4B] showed a significant increase in the ratio in dose- and time-dependent manner, indicating that FKC induced Δ Ψm loss.
FKC induces apoptosis via activation of caspase-3, -8, and-9
Induction of apoptosis requires the activation of the caspase cascade in most cases, which may have played essential roles as executors in apoptosis.[17] Thus, the activated caspase-3, -8, and -9 were examined by flow cytometry in HT-29 cells after 48 h of FKC treatment. The results showed that the activities of caspase-3, -8, and -9 were significantly elevated by FKC in a dose-dependent manner [Figure 5A]. The highest increment in activated caspases was observed at 80 μM of FKC, and activation of caspase-8 was found to be more prominent than caspase-9. The activation of caspase-3 was further confirmed by the cleavage of PARP-1 via western blot. Moreover, the level of cleaved PAPR-1 (25kDa) was gradually increased over 48 h after FKC treatment in a time-dependent manner as shown in Figure 5B The results indicated that FKC treatment resulted in caspase-dependent apoptosis in HT-29 cells.
Figure 5.
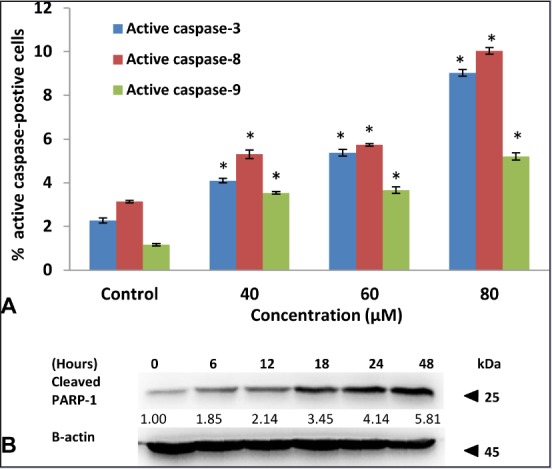
(A) FKC treatment increased the activity of caspase-3, -8, and -9 in HT-29 cells. Values given are expressed as mean ± standard deviation of triplicates obtained from three independent experiments. * P<0.05 compared to the control. (B) FKC caused the cleavage of PARP-1 in HT-29 cells.
Figure 6.
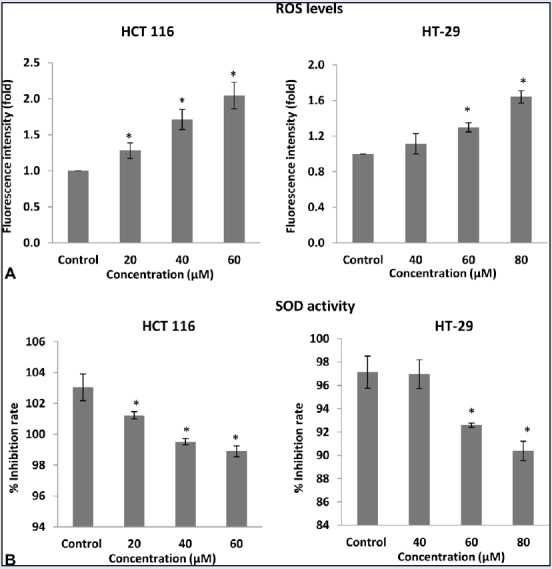
Concentration-dependent effect of FKC on ROS generation (A) and SOD activity (B) in HCT 116 and HT-29 cells. The data are presented as the mean ± standard deviation for three independent experiments. * P<0.05 compared to control.
FKC increased the ROS generation and reduced the SOD activity
ROS generation plays an important role in the pro-apoptotic activities. To examine whether oxidative stress damage was involved in FKC-induced apoptosis in HT-29 and HCT 116 cells, cells treated with FKC were stained with DCF-DA dye and also examined for SOD activity. As shown in Figure 6A, the intracellular ROS level of HT-29 and HCT 116 cells was significantly increased by FKC treatment in a dose-dependent manner. Next, the activity of superoxide dismutase (SOD), an enzymatic antioxidant in HT-29 and HCT 116 cells, was evaluated. The SOD activity was significantly decreased by FKC in a dose-dependent manner as shown in Figure 6B. The results suggested the involvement of the changes in redox status in FKC-induced apoptosis.
FKC triggered ER stress-mediated apoptosis and inactivated inhibitor of apoptosis proteins (IAPs)
The accumulation of unfolded proteins resulted in endoplasmic reticulum (ER) stress and the prolonged stress can lead to apoptosis. ER stress-dependent apoptosis is mainly mediated by the transcription factor GADD153.[18] Thus, western blot was carried out to determine the level of GADD153 in HT-29 cells treated with 80 μM of FKC. Treatment of FKC on HT-29 cells resulted in a marked increase in the GADD153 level at the early time point of 6 h and continue to increase up to 48 h [Figure 7B], and GADD153 was undetected in the untreated cells. This finding suggested that ER stress was involved in the FKC-induced apoptosis of HT-29 cells.
Figure 7.
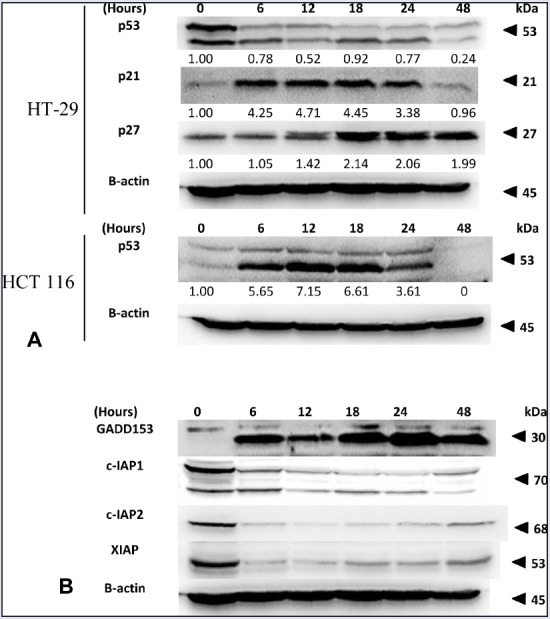
(A) Effect of FKC on the protein levels of p53, p21, and p27 in HT-29 and HCT 116 cells. (B) The changes in the protein levels of GADD153, c-IAP1, c-IAP2, and XIAP in HT-29 cells treated with FKC were analyzed by western blot.
IAPs have been implicated to play a key role in inhibiting apoptosis by inactivating caspases in cancer. The effect of FKC on these proteins was investigated by western blot. As shown in [Figure 7B], treatment with FKC caused a dramatic decrease in the level of XIAP, c-IAP1, and c-IAP2 at the early time of 6 h in HT-29 cells. Thus, the results showed that FKC inhibited the activation of IAPs, leading to activation of caspases.
FKC upregulated the cyclin kinase inhibitor proteins (p21 and p27) independently of p53
To identify the underlying mechanism of FKC-mediated apoptosis and cell cycle arrest, western blot was used to examine the changes in the levels of cell cycle modulator proteins (p53, p21, and p27) over 48 h. Following FKC treatment, the cells showed a dramatic increase (up to 4-fold) in the level of p21 within 6 h and sustained up to 18 h before the level started to decline to the control level at 48 h in HT-29 cells [Figure 7A]. In the case of p27, its level gradually increased after treatment, and a strong increase was observed at 18 h and remained at that level up to 24 h in HT-29 cells [Figure 7A].
Next, in order to determine whether the upregulation of p21 and p27 by FKC is p53 dependent, we examine the level of p53 in HT-29 cells and compared this with the p53 level in HCT 116 cells (colon carcinoma) following FKC treatment because HT-29 cells harbor mutant p53, whereas those in HCT 116 cells are of the wild-type. The western blot results showed that there was a high level of p53 in the untreated HT-29 cells that started to decrease upon treatment [Figure 7A]. As for HCT 116 cells, p53 was barely detected in the untreated cells and it started to increase dramatically upon treatment up to 18 h before the level declined to the basal level [Figure 7A]. In our early studies of FKC-treated HCT 116 cells, the levels of p21 and p27 were also found to increase in a similar pattern. Based on the results, it can be suggested that the activities of p21 and p27 in colon cancers are probably p53-independent.
DISCUSSION
In general, cancer is resistant to cell death by loss of apoptosis and accelerated cell proliferation. Manipulation of the apoptosis and cell cycle pathways represent an important chemotherapeutic strategy for the treatment of cancer. Therefore, a compound that can overcome this resistance may have the potential to be used in the treatment of cancer. In the present study, we demonstrated that FKC induced reduction in HT-29 cell proliferation, and the growth was arrested at concentrations of 60 and 80 μM FKC. Furthermore, the growth arrest was associated with G2/M phase arrest as shown in the cell cycle analysis.
It is well known that mitochondria play a major role in apoptosis by releasing the pro-apoptotic proteins such as cytochrome c, Smac/DIABLO, and AIF. In the present study, we also found that there was an increasing depolarization of outer mitochondrial membrane, suggesting that increased permeability of the mitochondrial membrane. In response to proapoptotic signals, a caspase cascade is initiated and resulted in morphological and biochemical changes. In our study, we observe that FKC could notably elevate activity of caspase-3, -8, and -9, and cleavage of PARP-1. The induction of apoptosis was evidenced by dramatic morphological changes, nuclear fragmentation, DNA fragmentation, and externalizaton of phosphatidylserine.
IAPs are well-known inhibitors of apoptosis through inactivation of caspases. These proteins contain one or more Baculovirus IAP Repeat (BIR) domains that are required for suppression of apoptosis. XIAP, c-IAP1, and c-IAP2 can directly or indirectly inhibit the activity of caspase-3, -8, and -9. In addition, c-IAP1 and c-IAP2 involve in the activation of tumor necrosis factor α (TNFα)-induced NF-κB via binding to TNFR-associated factor 2 (TRAF2), and this resulted in the inactivation of caspase-8.[19] In this study, XIAP, c-IAP1, and c-IAP2 were downregulated dramatically at the early apoptotic event-induced FKC treatment. In our earlier study, we also showed the downregulation of XIAP, c-IAP1, and surviving in HCT 116 cells after FKC treatment. Thus, the increased sensitivity of HT-29 cells to apoptosis could be due to the inactivation of IAPs by FKC.
ER stress has been shown to modulate both the mitochondrial and/or the death receptor apoptotic pathways.[20] The ER stress induces the expression of the GADD153 gene, which is known to promote apoptosis. Overexpression of GADD153 has been shown to downregulate the antiapoptotic protein Bcl-2 and proapoptotic proteins (such as DR5, GADD34, Trb3, Bim, PUMA).[20,21,22] The present study showed that FKC induced an early expression of GADD153, and this is similar to our previous finding on HCT 116 cells. Thus, this could suggest the possibility that FKC initiate the apoptotic signaling via ER stress.
Increased oxidative stress by increasing ROS generation could be a potential therapeutic strategy.[23] Reactive oxygen species act in multiple signaling cascades involving the development of cancer such as proliferation, survival, angiogenesis, and metastasis.[11] However, high levels of ROS have been reported to inhibiting cell proliferation by inducing cell cycle arrest and apoptosis. The level of ROS is tightly regulated by intracellular antioxidants for maintaining redox homeostasis.[24] Excessive ROS can induce apoptosis by damaging the mitochondrial membrane integrity that leads to the release of proapoptotic proteins.[25,26] ROS have been shown to induce the activation both caspase-8 and caspase-9.[27] In this study, apart from enhancing the ROS generation in both HT-29 and HCT 116 cells, FKC was also found to inhibit the activity of SOD in both HT-29 and HCT 116 cells. It has been proposed that a relatively high Mn-SOD content could stimulate the growth of the tumor cells by protecting them against high concentrations of ROS that can be produced by several anticancer therapies such as radiotherapy, chemotherapy, and photodynamic therapy.[28] Thus, the present study suggested that FKC-induced apoptosis could be associated with elevation of ROS and depletion of SOD activity. This may explain from our results on the disruption of the mitochondrial membrane potential by FKC, resulting in the activation of caspases. More study is needed to explore the underlying mechanisms by which FKC induces ROS.
p53, p21, and p27 play a significant role in preventing the onset of cancer and are involved in the elimination of damaged cells through induction of cell cycle arrest, apoptosis, DNA repair, and senescence.[29] In this study, a comparison of the p53 status between HT-29 and HCT 116 cells treated with FKC was examined. It is well known that p53 in HT-29 cells has a mutation in codon 273, whereas those in HCT 116 cells are of the wild type.[30] Thus, it is interesting to investigate the changes in the levels of mutated and wild-type p53 in colon cancer upon FKC treatment. In HCT 116 cells, the p53 level was upregulated after FKC treatment and was barely detected in untreated cells but the level decreased after 12 h of treatment. One possible explanation for the reduction is due to the complex biphasic nature of p53 alteration in which its activity is regulated by post-translational modifications on multiple sites such as phosphorylation, ubiquitination, acetylation, or methylation.[31] These post-translational modifications occur in response to typical cellular stresses such as DNA damage and oncogene activation.[32] Unlike in HCT 116 cells, the high level of p53 was found in untreated HT-29 cells but decreased after treated with increasing concentrations of FKC. The high level of p53 in HT-29 cells could be due to the mutated p53 gene that gives rise to a stable mutant protein in human cancers, which results in inhibition of MDM2-mediated p53 ubiquitination.[32] There is accumulating evidence supporting the view of p53 mutants are able to actively promote tumor development by several other means such as increased proliferation, evasion of apoptosis, and chemoresistance. Thus, the reduction of the p53 mutant level in HT-29 cells by FKC may contribute to the growth arrest; however, it remained unclear as to how FKC caused the reduction in mutant p53 by FKC.
It is known that cyclin-Cdk complexes in the cell cycle often bind to the endogenous inhibitor proteins (CKIs), p21WAF1/CIP1 and p27Kip1, which inhibit their kinase activities and prevent cell cycle progression. In the present study, western blot analysis showed that p21 and p27 were upregulated in HT-29 cells upon FKC treatment. The results are consistent with our earlier study on HCT 116 cells treated with FKC. Based on western blot analysis on the p53 status in both cell lines, the upregulation of p21 and p27 could occur in a p53-independent manner. Unlike p53, the somatic mutations in the Cip/Kip genes are uncommon and their expressions are frequently found relatively low in human tumor; thus, they have become important therapeutic targets.[33] Although p21 was initially identified to be the downstream mediator of p53 in cell cycle control and apoptosis in response to DNA damage; however, previous studies have shown that p21 can also be induced by other factors such as Chk2 kinase and p73.[34,35] p21 can also be induced by HuR, a RNA-binding protein by enhancing p21 mRNA stability.[36] p27 can be upregulated via activation of its translation by an internal ribosome entry site element (IRES) in its 5’untraslated region.[37] Thus, it can be proposed that the upregulation of p21 and p27 could be responsible for the inhibitory effect of FKC. Further studies need to be done to elucidate the molecular events of this pathway.
CONCLUSION
In summary, we have shown that FKC exhibits anticancer properties against colon adenocarcinoma HT-29 cells by inducing cell apoptosis and cell-cycle arrest. These effects are mediated by disruption of mitochondrial membrane, inactivation of the IAPs, and endoplasmic reticulum stress pathway. The induction of apoptosis was associated with the increased ROS generation and upregulation of p21 and p27. Although more studies are needed to elucidate the molecular mechanism or additional pharmacological studies, this study has proven its potential to be developed as anticancer drugs or used in combination with other approaches on the treatment of colon adenocarcinoma.
Financial source of support
This research is supported by High Impact Research MoE Grant UM.C/625/1/HIR/MoE/SC/02 from the Ministry of Education Malaysia, and University of Malaya PPP grant (PG121-2015B).
Conflicts of interest
There are no conflicts of interest
REFERENCES
- 1.Tarraga Lopez PJ, Albero JS, Rodriguez-Montes JA. Primary and secondary prevention of colorectal cancer. Clin Med Insights Gastroenterol. 2014;7:33–46. doi: 10.4137/CGast.S14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pourhoseingholi MA. Increased burden of colorectal cancer in Asia. World J Gastrointest Oncol. 2012;4:68–70. doi: 10.4251/wjgo.v4.i4.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. 2009;22:191–7. doi: 10.1055/s-0029-1242458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheer Az, Auer RA. Surveillance after curative resection of colorectal cancer. Clin Colon Rectal Surg. 2009;22:242–50. doi: 10.1055/s-0029-1242464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abhari BA, Cristofanon S, Kappler R, von Schweinitz D, Humphreys R, Fulda S. RIP1 is required for IAP inhibitor-mediated sensitization for TRAIL-induced apoptosis via a RIP1/FADD/caspase-8 cell death complex. Oncogene. 2013;32:3263–73. doi: 10.1038/onc.2012.337. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Sak K. Cytotoxicity of dietary flavonoids on different human cancer types. Pharmacogn Rev. 2014;8:122–46. doi: 10.4103/0973-7847.134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608–18. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Rosen DG, Zhou Y, Feng L, Yang G, Liu J, et al. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J Biol Chem. 2005;280:39485–92. doi: 10.1074/jbc.M503296200. [DOI] [PubMed] [Google Scholar]
- 10.Lobo V, Patil A, Phatak A, Chandra N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn Rev. 2010;4:118–26. doi: 10.4103/0973-7847.70902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther. 2008;7:1875–84. doi: 10.4161/cbt.7.12.7067. [DOI] [PubMed] [Google Scholar]
- 12.Jandial DD, Blair CA, Zhang S, Krill LS, Zhang YB, Zi X. Molecular targeted approaches to cancer therapy and prevention using chalcones. Curr Cancer Drug Targets. 2014;14:181–200. doi: 10.2174/1568009614666140122160515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yong WK, Ho YF, Malek SNA. Xanthohumol induces apoptosis and S phase cell cycle arrest in A549 non-small cell lung cancer cells. Pharmacogn Mag. 2015;11:S275–S83. doi: 10.4103/0973-1296.166069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dharmaratne HR, Nanayakkara NP, Khan IA. Kavalactones from Piper methysticum, and their 13C NMR spectroscopic analyses. Phytochemistry. 2002;59:429–33. doi: 10.1016/s0031-9422(01)00443-5. [DOI] [PubMed] [Google Scholar]
- 15.Phang CW, Karsani SA, Sethi G, Abd Malek SN. Flavokawain C inhibits cell cycle and promotes apoptosis, associated with endoplasmic reticulum stress and regulation of MAPKs and Akt signaling pathways in HCT 116 human colon carcinoma cells. PLoS One. 2016;11:e0148775. doi: 10.1371/journal.pone.0148775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis : an update. Apoptosis. 2003;8:115–28. doi: 10.1023/a:1022945107762. [DOI] [PubMed] [Google Scholar]
- 17.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5:a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. 2008;283:24295–9. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kadowaki H, Nishitoh H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes (Basel) 2013;4:306–33. doi: 10.3390/genes4030306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng WP, Hung HF, Wang BW, Shyu KG. The molecular regulation of GADD153 in apoptosis of cultured vascular smooth muscle cells by cyclic mechanical stretch. Cardiovasc Res. 2008;77:551–9. doi: 10.1093/cvr/cvm057. [DOI] [PubMed] [Google Scholar]
- 22.Wan Nor Hafiza WAG, Yazan LS, Tor YS, Foo JB, Armania N, Rahman HS. Endoplasmic reticulum stress-induced apoptotic pathway and mitochondrial dysregulation in HeLa cells treated with dichloromethane extract of Dillenia suffruticosa. Pharmacogn Mag. 2016;12:S86–95. doi: 10.4103/0973-1296.176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–91. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 24.Kovacic P, Jacintho JD. Mechanisms of carcinogenesis: focus on oxidative stress and electron transfer. Curr Med Chem. 2001;8:773–96. doi: 10.2174/0929867013373084. [DOI] [PubMed] [Google Scholar]
- 25.Gao J, Gao L, Zhang L, Yao W, Cao Y, Bao B, et al. 3-O-(2’E,4’Z-decadienoyl)-20-O-acetylingenol induces apoptosis in intestinal epithelial cells of rats via mitochondrial pathway. J Ethnopharmacol. 2015;174:331–8. doi: 10.1016/j.jep.2015.08.036. [DOI] [PubMed] [Google Scholar]
- 26.Mohanty S, Cock IE. The chemotherapeutic potential of Terminalia ferdinandiana: Phytochemistry and bioactivity. Pharmacogn Rev. 2012;6:29–36. doi: 10.4103/0973-7847.95855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Bowman L, Magaye R, Leonard SS, Castranova V, Ding M. Apoptosis induced by tungsten carbide-cobalt nanoparticles in JB6 cells involves ROS generation through both extrinsic and intrinsic apoptosis pathways. Int J Oncol. 2013;42:1349–59. doi: 10.3892/ijo.2013.1828. [DOI] [PubMed] [Google Scholar]
- 28.Miranda A, Janssen L, Bosman CB, van Duijn W, Oostendorp-van de Ruit MM, Kubben FJGM, et al. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clin Cancer Res. 2000;6:3183. [PubMed] [Google Scholar]
- 29.Jette L, Bissoon-Haqqani S, Le Francois B, Maroun JA, Birnboim HC. Resistance of colorectal cancer cells to 5-FUdR and 5-FU caused by Mycoplasma infection. Anticancer Res. 2008;28:2175–80. [PubMed] [Google Scholar]
- 30.Fatima N, Yi M, Ajaz S, Stephens RM, Stauffer S, Greenwald P, et al. Altered gene expression profiles define pathways in colorectal cancer cell lines affected by celecoxib. Cancer Epidemiol Biomarkers Prev. 2008;17:3051–61. doi: 10.1158/1055-9965.EPI-08-0261. [DOI] [PubMed] [Google Scholar]
- 31.Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–44. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res. 2003;1:1001–8. [PubMed] [Google Scholar]
- 33.Pruneri G, Pignataro L, Carboni N, Buffa R, Di Finizio D, Cesana BM, et al. Clinical relevance of expression of the CIP/KIP cell-cycle inhibitors p21 and p27 in laryngeal cancer. J Clin Oncol. 1999;17:3150–9. doi: 10.1200/JCO.1999.17.10.3150. [DOI] [PubMed] [Google Scholar]
- 34.Aliouat-Denis C-M, Dendouga N, Van den Wyngaert I, Goehlmann H, Steller U, van de Weyer I, et al. p53-Independent Regulation of p21 and lt;sup and gt;Waf1/Cip1 and lt;/sup and gt; Expression and Senescence by Chk2. Mol Cancer Res. 2005;3:627. doi: 10.1158/1541-7786.MCR-05-0121. [DOI] [PubMed] [Google Scholar]
- 35.Schmelz K, Wagner M, Dörken B, Tamm I. 5-Aza-2’-deoxycytidine induces p21WAF expression by demethylation of p73 leading to p53-independent apoptosis in myeloid leukemia. Int J Cancer. 2005;114:683–95. doi: 10.1002/ijc.20797. [DOI] [PubMed] [Google Scholar]
- 36.Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y, et al. HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol. 2000;20:760–9. doi: 10.1128/mcb.20.3.760-769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng Y, Miskimins WK. CUG-binding protein represses translation of p27Kip1 mRNA through its internal ribosomal entry site. RNA Biol. 2011;8:365–71. doi: 10.4161/rna.8.3.14804. [DOI] [PMC free article] [PubMed] [Google Scholar]