

Abstract
Placental insufficiency and intrauterine growth restriction (IUGR) of the fetus affects approximately 8% of all pregnancies and is associated with short‐ and long‐term disturbances in metabolism. In pregnant sheep, experimental models with a small, defective placenta that restricts delivery of nutrients and oxygen to the fetus result in IUGR. Low blood oxygen concentrations increase fetal plasma catecholamine concentrations, which lower fetal insulin concentrations. All of these observations in sheep models with placental insufficiency are consistent with cases of human IUGR. We propose that sustained high catecholamine concentrations observed in the IUGR fetus produce developmental adaptations in pancreatic β‐cells that impair fetal insulin secretion. Experimental evidence supporting this hypothesis shows that chronic elevation in circulating catecholamines in IUGR fetuses persistently inhibits insulin concentrations and secretion. Elevated catecholamines also allow for maintenance of a normal fetal basal metabolic rate despite low fetal insulin and glucose concentrations while suppressing fetal growth. Importantly, a compensatory augmentation in insulin secretion occurs following inhibition or cessation of catecholamine signalling in IUGR fetuses. This finding has been replicated in normally grown sheep fetuses following a 7‐day noradrenaline (norepinephrine) infusion. Together, these programmed effects will potentially create an imbalance between insulin secretion and insulin‐stimulated glucose utilization in the neonate which probably explains the transient hyperinsulinism and hypoglycaemia in some IUGR infants.
Keywords: β‐cell, developmental programming, epinephrine, intrauterine growth restriction, norepinephrine
Abbreviations
- AGA
appropriate‐for‐gestational‐age
- GPAIS
glucose‐potentiated arginine‐induced insulin secretion
- GSIS
glucose‐stimulated insulin secretion
- HG‐IUGR
hypoglycaemia‐induced intrauterine growth restriction
- IUGR
intrauterine growth restriction
- PI‐IUGR
placental insufficiency‐induced IUGR
- SGA
small‐for‐gestational age
Introduction
Placental insufficiency, defined as a failure of the placenta to deliver adequate oxygen and nutrients to maintain fetal growth rates at the genetically determined potential, results in intrauterine growth restriction (IUGR). This is associated with small‐for‐gestational age (SGA) infants and is estimated to affect 8% of all births (McCowan et al. 2010; Monteith et al. 2017). Considerable epidemiological evidence in humans and experimental evidence from animal models of IUGR demonstrate that fetal undernutrition, usually associated with placental insufficiency, produces developmental adaptations that predispose offspring to metabolic disorders of insulin secretion and glucose utilization (insulin sensitivity) at later stages of life (Barker et al. 1993; Fernandez‐Twinn & Ozanne, 2006; Harder et al. 2007; Owens et al. 2007b; Green et al. 2010; Zimmermann et al. 2015). SGA children develop glucose intolerance more frequently, and in adulthood exhibit impaired insulin secretion and a greater risk for developing Type 2 diabetes (Hales et al. 1991; Phipps et al. 1993; Yajnik et al. 1995; Mericq et al. 2005). However, glucose intolerance appears to be preceded in some formerly IUGR infants by a period of hyperinsulinism which transitions over the first few months to years of life (Bazaes et al. 2003; Soto et al. 2003; Milovanovic et al. 2014). Furthermore, evidence from human data is accumulating which shows that some formerly IUGR fetuses demonstrate neonatal hypoglycaemia associated with the transient form of hyperinsulinism, implying increased insulin concentrations and secretion relative to their insulin sensitivity (Miralles et al. 2002; Stanley et al. 2015). In severe cases this transient form of hyperinsulinism and hypoglycaemia may persist for several months (Hoe et al. 2006; Arya et al. 2013).
Observational data from humans indicate that the developmental adaptations of both β‐cell function and insulin sensitivity to IUGR result in a lifelong pathophysiological process which allows the fetus to maintain normal metabolic rates for survival to term at the expense of normal fetal growth but produces glucose intolerance and diabetes later in life (Fall et al. 1998; Jensen et al. 2002, 2007; Newsome et al. 2003). Similarly, in sheep, surgical removal of placental attachment sites, or caruncles, before pregnancy produces placental restriction and fetal IUGR that lead to deficiencies in insulin secretion in the fetus, month‐old lambs, and yearling sheep (De Blasio et al. 2007; Owens et al. 2007a; Gatford et al. 2008). The uterine carunclectomy model of placental restriction shows that fetal complications, such as low oxygen and glucose concentrations, lead to the development of glucose intolerance in adulthood, a finding consistent with several rodent models of fetal undernutrition as well (Robinson et al. 1979; Jones & Robinson, 1983; Green et al. 2010; Liu et al. 2015). While the development of diabetes in later life is well established in humans and animal models of IUGR, what has been less well studied are the mechanisms responsible for the period of increased insulin sensitivity and secretion immediately after birth and how these fit into the developmental pathophysiological processes which align IUGR and the later development of Type 2 diabetes. Identification of the mechanisms responsible for these developmental adaptations to IUGR will allow for targeted interventions to prevent both short and long term complications of placental insufficiency.
Factors contributing to IUGR and developmental programming of insulin secretion
In the literature, human studies generally compare outcomes in SGA neonates (usually defined as <10th percentile for birth weight) to appropriate‐for‐gestational‐age neonates (AGA) because birth weight is the most accessible proxy for fetal growth restriction. Although this statistical classification introduces heterogeneity by including newborns who are genetically predetermined to be small along with cases of IUGR where pathological fetal vascular patterns can be identified with Doppler assessments of the middle cerebral and umbilical artery, aorta and the ductus venosus, these studies provide important observations on insulin secretion and sensitivity which have been confirmed experimentally in animal models of placental insufficiency (Platz & Newman, 2008; Monteith et al. 2017). Such observations include those made by umbilical cord blood sampling in human SGA fetuses prior to delivery or the onset of labour. Human SGA fetuses develop hypoxaemia defined as low blood oxygen concentrations, hypoglycaemia and hypoinsulinaemia relative to AGA controls (Economides et al. 1989; Nicolini et al. 1990). Additionally, human SGA fetuses are characterized by increased plasma catecholamine concentrations (Greenough et al. 1990). In human fetuses, prior to the onset of labour or maternal anaesthesia, glucose‐stimulated insulin secretion (GSIS), measured by placing an umbilical vein catheter for fetal blood sampling and glucose infusion, is absent in IUGR fetuses. This is in contrast to a robust GSIS response in control fetuses (Nicolini et al. 1990). Clinical studies determining the mechanisms responsible for impaired GSIS in IUGR fetuses are limited to histological evaluation of the pancreas. One study found that fetuses with severe SGA (<5th percentile) have smaller islets and a lower fraction of β‐cells within the islets compared to AGA newborns at ∼34 weeks of gestation (Van Assche et al. 1977). In a different study with less severe SGA cases (<10th percentile), there was no difference in the pancreatic β‐cell area (Beringue et al. 2002). These data indicate that insulin secretion is blunted in human IUGR fetuses, yet reduced insulin secretion is not exclusively explained by less β‐cell mass. Therefore, impairments in insulin secretion are postulated to result not just from structural defects of the pancreatic islet and fewer β‐cells, but also from extrinsic factors such as hypoxaemia, hypoglycaemia, hypercatecholaminaemia, or the combination of these conditions.
Isolating IUGR complications that impair fetal insulin secretion
Due to ethical considerations, mechanistic studies in humans are limited. IUGR models in sheep offer a unique advantage to investigate the fetal β‐cell physiology both in vivo and in vitro because of the ability to catheterize sheep fetuses for experimental challenges and to study islets isolated from the same fetus. Using different experimental approaches in pregnant sheep, potential mechanisms responsible for the β‐cell dysfunction in placental insufficiency and IUGR can be tested (Green et al. 2010). For example, comparing β‐cell dysfunction in IUGR sheep fetuses created with hyperthermia‐induced placental insufficiency (PI‐IUGR) and maternal insulin infusion‐induced chronic hypoglycaemia (HG‐IUGR) is particularly informative (Bell et al. 1987; DiGiacomo & Hay, 1990). Fetal glucose concentrations are reduced to equivalent levels (∼50%) in these two sheep models of IUGR, but there are stark differences in duration of nutrient restriction, blood oxygen concentrations, and plasma catecholamine concentrations. Placental insufficiency restricts transfer of oxygen, glucose and amino acids to the fetus resulting in hypoglycaemia and hypoxaemia as early as 0.7 of gestation, prior to notable differences in fetal weight (Thureen et al. 1992; Ross et al. 1996; de Vrijer et al. 2004; Limesand et al. 2013). The fetal hypoglycaemia and hypoxaemia identified at 0.7 of gestation progressively worsen as the pregnancy gets closer to term and the IUGR becomes prominent (Limesand et al. 2007, 2013). In contrast, maternal hypoglycaemia experimentally restricts placental transport of glucose but does not affect placental oxygen and amino acid transfer to the fetus (DiGiacomo & Hay, 1990; Limesand et al. 2009). Therefore, while these fetuses are hypoglycaemic, they are not hypoxaemic and their degree of fetal IUGR is much less than in the PI‐IUGR model (DiGiacomo & Hay, 1990). Another difference is that PI‐IUGR sheep fetuses have higher circulating catecholamine concentrations compared to HG‐IUGR sheep fetuses, whereas cortisol concentrations tend to increase in both models (Narkewicz et al. 1993; Limesand & Hay, 2003; Limesand et al. 2006; Thorn et al. 2012).
By integrating findings from different sheep models of IUGR, we can identify mechanisms linking specific conditions of IUGR and β‐cell dysfunction (Fig. 1). While fetal GSIS is impaired in both models, glucose‐stimulated insulin concentrations are much lower in PI‐IUGR fetuses than in HG‐IUGR fetuses (Limesand & Hay, 2003; Limesand et al. 2006). Other important differences between the models can be identified with in vitro experiments using isolated islets from fetal sheep. In isolated islets from PI‐IUGR fetuses GSIS is reduced due to less insulin production by the β‐cell. However, the fraction of insulin present that is released in response to stimulatory glucose concentrations (secreted insulin/total islet insulin content) is actually greater in PI‐IUGR islets compared to controls. PI‐IUGR islets also have a deficiency in glucose metabolism. Compared to control isolated islets, PI‐IUGR islet oxidative glucose metabolism is significantly lower in hyperglycaemic conditions that stimulated insulin release (Limesand et al. 2006). In summary, PI‐IUGR islets have decreased insulin secretion due to reduced insulin biosynthesis and storage, which might be in response to impaired islet oxidative metabolism of glucose. However, in vitro PI‐IUGR islets compensate for these deficits to some extent by augmenting their capacity to release meager insulin stores. In this respect, the PI‐IUGR islets appear to be hyper‐responsive to glucose with regards to their ability to release the insulin that is produced and stored by the β‐cell. These findings contrast with the situation for islets isolated from HG‐IUGR fetuses (Rozance et al. 2006, 2007). In HG‐IUGR islets, GSIS also is reduced, but this is not due to reduced islet insulin content or glucose metabolism; instead the reduced GSIS results from a significantly lower fraction release of insulin present in the β‐cell in response to secretagogues. Essentially, the HG‐IUGR islets are hypo‐responsive to glucose. Therefore, PI‐IUGR islets have impaired in vitro insulin secretion due to decreased islet insulin production and storage, which is partly compensated for by their hyper‐responsiveness to glucose in terms of their greater capacity to release the insulin that is present. In contrast, HG‐IUGR in vitro islet insulin secretion is impaired due to an intrinsic inability of the β‐cell to release the insulin that it has available, or glucose hypo‐responsiveness. Comparing the in vivo phenotypes of these two different models leads to consideration of a variety of factors in the PI‐IUGR fetus, which may be responsible for differences in β‐cell dysfunction between the PI‐IUGR and HG‐IUGR models. Our focus is on factors that differ between the two models, such as elevated plasma catecholamines and lower blood oxygen concentrations.
Figure 1. Comparison of insulin secretion defects in sheep models of IUGR.
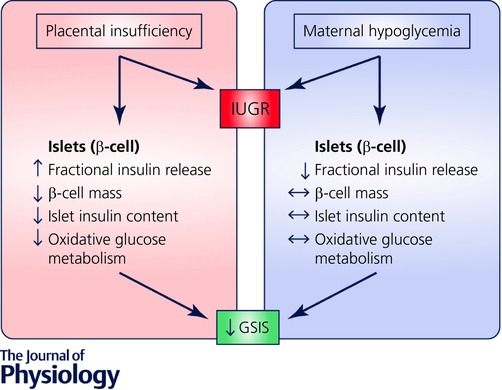
Two sheep models of IUGR, placental insufficiency and maternal insulin infusion‐induced chronic hypoglycaemia, exhibit reduced glucose‐stimulated insulin secretion (GSIS) in vivo. Isolated islets from these treatments distinguish alternative defects that are responsible for the β‐cell dysfunction.
Actions of hypoxaemia and hypercatecholaminaemia on insulin secretion in IUGR
In fetal sheep, hypoxaemia stimulates release of noradrenaline (norepinephrine) and adrenaline (epinephrine) from adrenal chromaffin cells, which elevates catecholamines in the circulation (Cohen et al. 1991; Adams & McMillen, 2000). In human cases of placental insufficiency, there is a sustained elevation of circulating catecholamines in response to chronic fetal hypoxaemia. This tight, negative association between blood oxygen content and plasma noradrenaline concentrations in PI‐IUGR sheep fetuses is observed at 0.7 and 0.9 of gestation (Limesand et al. 2006; Leos et al. 2010; Macko et al. 2013). In fact, a negative association between oxygen and noradrenaline is also observed in normally growing sheep fetuses and uterine carunclectomy IUGR fetuses (Cheung, 1990; Simonetta et al. 1997). As for acute hypoxaemia, chronic hypoxaemia increases noradrenaline in PI‐IUGR fetal sheep, which is postulated to lower oxidative metabolism and spare oxygen (Limesand et al. 2013; Milley, 1997). Evidence for this is twofold. First, an experimental infusion of noradrenaline into the fetal circulation increases fetal blood oxygen concentrations (Bassett & Hanson, 1998; Chen et al. 2014). Second, acute pharmacological inhibition of adrenergic receptors decreases blood oxygen content in both control and PI‐IUGR sheep fetuses (Leos et al. 2010; Macko et al. 2013). In human fetuses noradrenaline concentrations increase from 16 to 34 weeks of gestation, and similar to the experimental evidence in sheep, hypoxaemia elevates circulating noradrenaline in human fetuses (Greenough et al. 1990).
In fetal sheep, as in several other species, catecholamines inhibit insulin secretion from the pancreatic β‐cells (Yates et al. 2012). Adrenergic antagonists have shown that catecholamines mediate this effect via α2‐adrenergic receptors (Sperling et al. 1980; Jackson et al. 1993, 2000). Surgical ablation of the fetal chromaffin cells in the adrenal medulla prevents acute hypoxaemia‐induced noradrenaline and adrenaline secretion and inhibition of fetal GSIS (Yates et al. 2012). Furthermore, this experimental paradigm comparing GSIS under normoxaemic and hypoxaemic conditions indicates that catecholamines from the adrenal gland are responsible for the suppression of insulin secretion and not the fetal hypoxaemia. This is important because in this study the degree of hypoxaemia achieved is equivalent to the concentrations seen in PI‐IUGR fetuses, suggesting that hypoxaemia inhibits insulin secretion indirectly through adrenergic stimulation.
To test this, adrenergic receptor antagonists were administered to PI‐IUGR fetuses and GSIS was measured (Fig. 2). The results show that elevated catecholamines persistently inhibit insulin secretion between 0.7 and 0.9 of gestation (Leos et al. 2010; Macko et al. 2013). Importantly, during the pharmacological adrenergic blockade GSIS in PI‐IUGR fetuses is increased compared to control sheep fetuses. This is despite lower blood oxygen concentrations. The in vivo hyper‐responsiveness of the PI‐IUGR fetus to glucose stimulation occurs despite markedly lower pancreatic β‐cell mass and significantly less insulin per β‐cell (Limesand et al. 2006, 2013; Leos et al. 2010; Macko et al. 2013). The findings are consistent with the hyper‐responsiveness of the PI‐IUGR islet to glucose stimulation when tested in vitro without exposure to exogenous catecholamines. Another notable finding is that at 0.7 of gestation plasma concentrations of noradrenaline in the PI‐IUGR fetuses are substantially lower compared to the near‐term PI‐IUGR fetuses. The concentrations are actually similar to noradrenaline concentrations found in the near‐term control fetus. This shows that higher concentrations of catecholamines are effective throughout the final third of gestation, but progressively increase due to the maturation of the adrenal gland responsiveness and worsening hypoxaemia conditions in PI‐IUGR sheep fetuses. Taken together, these in vitro and in vivo findings across the latter third of gestation indicate that, compared to control sheep fetuses, fetal β‐cells in PI‐IUGR fetuses develop a hyper‐responsiveness for insulin secretion to stimulatory glucose concentration independent of low blood oxygen concentrations. However, increased GSIS in vivo is suppressed to levels lower than control fetuses by chronic adrenergic signalling.
Figure 2. Adrenergic programming of insulin secretion in PI‐IUGR fetuses.

The proposed actions and responses for placental insufficiency leading to decreased glucose‐stimulated insulin secretion (GSIS) are depicted. Experimental approaches used to disrupt the proposed actions of catecholamines are indicated along with the major conclusions for the induction of hyper‐responsiveness in insulin secretion.
Preventing high catecholamines partially restores GSIS in PI‐IUGR sheep fetuses
To fully elucidate the effects of catecholamines in PI‐IUGR fetuses, we surgically ablated the adrenal medullae before 0.7 of gestation to prevent the onset of chronically elevated catecholamines (Fig. 2; Macko et al. 2016). As expected, PI‐IUGR fetuses with the adrenal medullae ablation had lower circulating noradrenaline and adrenaline concentrations compared to intact PI‐IUGR fetuses near term despite being hypoxaemic and hypoglycaemic. At 0.9 of gestation GSIS is greater in PI‐IUGR fetuses which have the adrenal medullae ablation compared to intact PI‐IUGR fetuses. The maximum glucose‐stimulated insulin concentrations are similar to control fetuses with adrenal medullae ablation, though lower than intact control fetuses. Moreover, PI‐IUGR fetuses did not display hyper‐responsiveness to glucose stimulation following adrenal medullae ablation at 0.7 of gestation. Comparing GSIS in PI‐IUGR fetuses with chronically low circulating catecholamine concentrations with GSIS in the PI‐IUGR fetuses following acute pharmacological adrenergic blockade demonstrates a principal role for chronically elevated catecholamines in IUGR to induce hyper‐responsive insulin secretion that is independent of hypoxaemia, hypoglycaemia, and other fetal conditions associated with placental insufficiency. Therefore, adaptations to chronic adrenergic stimulation such as compensatory gain in β‐cell responsiveness were not present in PI‐IUGR fetuses that had undergone adrenal medullae ablation, indicating that chronic adrenergic stimulation produces this effect.
In addition to the profound effects of chronically high circulating catecholamines on GSIS in PI‐IUGR fetuses, elevated plasma catecholamines also promote growth restriction and asymmetric growth in PI‐IUGR fetuses, at least in tissues other than the endocrine pancreas (Davis et al. 2015). Approximately 50% of the growth restriction is associated with chronically elevated plasma catecholamines in PI‐IUGR fetuses because ablation of the adrenal medullae increases fetal weights by this amount despite hypoxaemia and hypoglycaemia. Sparing of brain and heart weights relative to fetal weight is a hallmark of asymmetric growth in IUGR and is present in intact PI‐IUGR fetuses. The asymmetric growth characteristics of these tissues are lost in PI‐IUGR fetuses with adrenal medullae ablation at 0.7 of gestation. However, the increase in fetal and pancreas weight does not translate to greater numbers of pancreatic endocrine cells because β‐cell mass is similar between intact and adrenal medulla‐ablated PI‐IUGR fetuses (Davis et al. 2015). This finding is the result of less immunopositive area for β‐cells in PI‐IUGR fetuses with adrenal medullae ablation. Furthermore, rates of β‐cell replication are not different between PI‐IUGR surgical treatments, which are both less than intact controls. These data indicate that high circulating catecholamines suppress somatic growth but do not constrain proliferation or neogenesis of pancreatic endocrine cells. Factors other than catecholamines are likely to be the cause of reduction in β‐cell mass. These other factors include growth factors, islet vascularity, amino acids, hypoglycaemia and hypoxaemia to name a few that have been investigated in PI‐IUGR fetuses (Chen et al. 2012; Lavezzi et al. 2013; Rozance et al. 2015; Brown et al. 2016; Hay et al. 2016; Macko et al. 2016; Benjamin et al. 2017).
Enhanced GSIS persists following sustained high noradrenaline
Integration of in vivo and in vitro data from studies on PI‐IUGR and HG‐IUGR fetuses leads to the hypothesis that chronic exposure to elevated catecholamines enhances β‐cell responsiveness to secretagogues after cessation of the adrenergic signalling. To study the specific effects of chronically elevated noradrenaline on β‐cell responsiveness, we continuously infused noradrenaline intravenously into normally grown sheep fetuses for 7 days near the end of gestation (Chen et al. 2014). During the infusion, noradrenaline concentrations are increased and insulin concentrations are decreased to levels similar to those in PI‐IUGR fetuses, but, unlike PI‐IUGR fetuses, glucose and oxygen concentrations are increased in noradrenaline‐infused fetuses. To simulate the acute adrenergic blockade performed in the PI‐IUGR fetuses, the noradrenaline infusion was terminated 3 h before measuring insulin secretion responsiveness. In the noradrenaline‐infused fetuses, GSIS and glucose‐potentiated arginine‐induced insulin secretion (GPAIS) are approximately fourfold greater than age‐matched control fetuses or pretreatment assessment of GSIS and GPAIS in the noradrenaline‐infused fetuses. The hyper‐responsiveness in insulin secretion is present despite the fact that plasma noradrenaline concentrations remain modestly elevated compared to controls. Lower mRNA expression of α2A‐adrenergic receptor (α2A‐AR), α2C‐adrenergic receptor (α2C‐AR) and G protein subunit alpha i2 (Gαi‐2), and adrenergic desensitization in islets from noradrenaline‐infused fetuses explains increased insulin secretion after termination of the infusion despite persistently elevated noradrenaline concentrations (Chen et al. 2014, 2017). Although no differences are seen in insulin, pancreatic and duodenal homeobox 1, and glucose transporter 2 mRNA concentrations, expression of uncoupling protein 2 is lower in islets from noradrenaline‐infused fetuses, which indicates improved insulin stimulus–secretion coupling (Zhang et al. 2001). It is also worth mentioning that uncoupling protein 2 expression is lower in PI‐IUGR islets as well, indicating a potential shared programming mechanism for both models with hypercatecholaminaemia (Kelly et al. 2017). The findings from the noradrenaline‐infused fetuses show that following acute removal from the chronically elevated adrenergic stimulation, fetal insulin secretion becomes hyper‐responsive to secretagogues. This is associated with adrenergic desensitization and greater potential of stimulus–secretion coupling in pancreatic islets.
In a follow‐up study, we tested the hypothesis that the compensatory increase in insulin secretion following chronic elevation of noradrenaline is independent of hyperglycaemia and persists in conjunction with adrenergic desensitization of islets (Chen et al. 2017). The noradrenaline infusion was performed as explained above, but in this cohort fetal euglycaemia was maintained with a maternal insulin infusion. The noradrenaline infusion was terminated at 7 days, 1 and 5 days prior to measuring GSIS and GPAIS respectively. In noradrenaline fetuses GSIS is approximately twofold greater than controls at both time points, indicating that the significant enhancement in insulin secretion persisted. The GPAIS is also significantly enhanced in noradrenaline‐infused fetuses. Adrenergic sensitivity was measured in pancreatic islets collected from these fetuses, and the half‐maximal inhibitory concentration for noradrenaline is greater in noradrenaline‐infused islets. The in vitro analysis also shows that the maximum GSIS in islets isolated from noradrenaline‐infused fetuses is significantly greater than controls, but islet insulin content and intracellular calcium signalling are not different between treatments. These findings show that chronic noradrenaline exposure and not hyperglycaemia produce persistent adaptations in pancreatic islets that augment β‐cell responsiveness in part through decreased adrenergic sensitivity.
Clinical implications to fetal catecholamine exposure
The actions for elevated catecholamines discussed above have direct relevance for the clinical problem of neonatal hypoglycaemia in formerly IUGR newborns. Some human IUGR newborns have evidence of increased insulin sensitivity for glucose (Bazaes et al. 2003), a finding supported by hyperinsulinaemic‐euglycaemic clamps in PI‐IUGR fetal sheep (Thorn et al. 2012). When insulin secretion and insulin concentrations are elevated inappropriately for the degree of insulin sensitivity, glucose supply to the newborn from milk feeding and/or intravenous dextrose infusions may not be enough to prevent a transient form of hypoglycaemia associated with hyperinsulinism (Miralles et al. 2002; Hoe et al. 2006; Arya et al. 2013). The developmental adaptations that result from chronically elevated catecholamines in pregnant sheep models explain how this disturbance might occur. Once sustained adrenergic signalling is alleviated, either experimentally in fetal sheep or by delivery and stabilization in humans, β‐cell hyper‐responsiveness will be unmasked and lead to inappropriately increased insulin secretion and concentrations (Fig. 3). While the PI‐IUGR fetal sheep is exposed to elevated catecholamine concentrations for much of the third trimester (Leos et al. 2010; Macko et al. 2013), like some human IUGR fetuses (Greenough et al. 1990), we have demonstrated that exposure to elevated noradrenaline for as short a time as 7 days is all that is required to establish β‐cell hyper‐responsiveness (Chen et al. 2014). We also have shown that the β‐cell hyper‐responsiveness persists at least 5 days in this experimental paradigm (Chen et al. 2017). It is important to point out that we have not defined the maximum duration that the persistent β‐cell hyper‐responsiveness will last following either the 7‐day noradrenaline infusion or experimental hyperthermia‐induced placental insufficiency. Thus, a longer duration of exposure in utero may lead to the prolonged courses of transient hyperinsulinism‐associated hypoglycaemia demonstrated in some formerly IUGR newborns (Hoe et al. 2006; Arya et al. 2013). Similarly, we have not defined the minimum period of exposure to elevated noradrenaline concentrations required to establish β‐cell hyper‐responsiveness. In this respect, it is worth noting that the pathophysiological process we have discussed for placental insufficiency and IUGR could potentially apply to any pregnancy complicated by low fetal oxygen concentrations depending on the degree and duration of this exposure, including chronic perinatal asphyxia, placental abruption, fetal anaemia, and even some pregnancies complicated by maternal diabetes (Madsen, 1986; Pschera et al. 1986; Hoe et al. 2006; Teramo, 2010; Stanley et al. 2015). Collectively, the hyperinsulinism‐associated hypoglycaemia present in these conditions and following IUGR has been termed perinatal stress hyperinsulinism and may share a common pathophysiological mechanism as described in the current review (Miralles et al. 2002; Hoe et al. 2006; Arya et al. 2013; Stanley et al. 2015; Thornton et al. 2015).
Figure 3. Translation of adrenergic programming in IUGR infants.
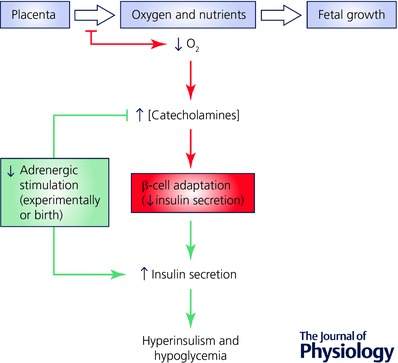
Normal placental transfer of oxygen and nutrients to the growing fetus is presented in black. Placental insufficiency, represented in red, reduces the delivery of oxygen and nutrients to the fetuses resulting in IUGR. Fetal hypoxaemia raises plasma catecholamine concentrations and subsequently lowers insulin secretion. The chronic elevation in plasma catecholamines causes β‐cell adaptations. Loss of the sustained adrenergic signalling following parturition exposes β‐cell hyper‐responsiveness, which is represented in green. It is hypothesized that hyperinsulinism and hypoglycaemia develop following delivery and stabilization of the neonate that experienced perinatal stress.
Conclusion
Synthesis of the findings from several well‐established models of IUGR in fetal sheep reveals that the consequence of nutrient restriction is β‐cell dysfunction, but the type of nutrient restriction causing IUGR also has different effects on the β‐cell phenotype. Placental insufficiency reduces nutrient transport globally causing a decrease in β‐cell mass, islet metabolism, and insulin production. In contrast, experimentally produced hypoglycaemia impaired distal components in the exocytosis of insulin. A major difference between these models is fetal hypoxaemia and hypercatecholaminaemia, and the latter inhibits insulin secretion and fetal growth to a greater extent than low glucose and insulin concentrations. In addition, chronically elevated noradrenaline makes β‐cells hyper‐responsive to secretagogues once the adrenergic stimulation subsides by the experimental procedures outlined, or as we propose delivery and stabilization of the IUGR fetus. Although the adaptations are expected to be transient, we have shown the hyper‐secretion persists for several days in the fetus following cessation of chronically elevated noradrenaline concentrations. The expectation is that IUGR infants will develop transient hyperinsulinism and hypoglycaemia, which may be asymptomatic but have the potential in severe cases to persist for months and may eventually transition to a state of increased insulin resistance, hyperglycaemia, and eventually Type 2 diabetes.
Additional information
Competing interests
None declared.
Funding
This work was supported by the National Institutes of Health grants R01DK084842 (S.W.L., Principal Investigator) and R01DK088139 (P.J.R., Principal Investigator). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Acknowledgements
We thank all our colleagues who have assisted in the primary experiments.
Biographies
Sean Limesand is a Professor of Endocrinology in the School of Animal and Comparative Biomedical Sciences at the University of Arizona. He obtained his PhD in Molecular Endocrinology from Colorado State University and was a Postdoctoral Fellow in Perinatal Biology at the University of Colorado, School of Medicine. His current research programmes use an integrative approach at the whole animal, isolated organ, cellular and molecular levels to determine developmental adaptations in pancreatic β‐cells and insulin sensitivity that result from early life risk factors such as intrauterine growth restriction and increase risk of glucose intolerance and diabetes in later life.

Paul Rozance is a board certified neonatologist and an Associate Professor in the Department of Pediatrics, Section of Neonatology at the University of Colorado, School of Medicine. He received his medical degree from Georgetown University School of Medicine, completed Pediatric Residency at Stanford University School of Medicine, and fellowship training in Neonatal‐Perinatal Medicine at the University of Colorado, School of Medicine. He remained on faculty at University of Colorado in order to pursue his research in β‐cell biology and intrauterine growth restriction. His overall goals are to understand how the fetus translates nutrient signals (supply from the placenta) into anabolic signals for growth (insulin secretion).
This review was presented at the symposium “Stress during pregnancy: Physiological consequences for intrauterine development”, which took place at Physiology 2016, Dublin, Ireland, 29–31 July 2016.
References
- Adams MB & McMillen IC (2000). Actions of hypoxia on catecholamine synthetic enzyme mRNA expression before and after development of adrenal innervation in the sheep fetus. J Physiol 529, 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya VB, Flanagan SE, Kumaran A, Shield JP, Ellard S, Hussain K & Kapoor RR (2013). Clinical and molecular characterisation of hyperinsulinaemic hypoglycaemia in infants born small‐for‐gestational age. Arch Dis Child Fetal Neonatal Ed 98, F356–F358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K & Clark PM (1993). Type 2 (non‐insulin‐dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 36, 62–67. [DOI] [PubMed] [Google Scholar]
- Bassett JM & Hanson C (1998). Catecholamines inhibit growth in fetal sheep in the absence of hypoxemia. Am J Physiol 274, R1536–R1545. [DOI] [PubMed] [Google Scholar]
- Bazaes RA, Salazar TE, Pittaluga E, Pena V, Alegria A, Iniguez G, Ong KK, Dunger DB & Mericq MV (2003). Glucose and lipid metabolism in small for gestational age infants at 48 hours of age. Pediatrics 111, 804–809. [DOI] [PubMed] [Google Scholar]
- Bell AW, Wilkening RB & Meschia G (1987). Some aspects of placental function in chronically heat‐stressed ewes. J Dev Physiol 9, 17–29. [PubMed] [Google Scholar]
- Benjamin JS, Culpepper CB, Brown LD, Wesolowski SR, Jonker SS, Davis MA, Limesand SW, Wilkening RB, Hay WW Jr & Rozance PJ (2017). Chronic anemic hypoxemia attenuates glucose‐stimulated insulin secretion in fetal sheep. Am J Physiol Regul Integr Comp Physiol 312, R492–R500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beringue F, Blondeau B, Castellotti MC, Breant B, Czernichow P & Polak M (2002). Endocrine pancreas development in growth‐retarded human fetuses. Diabetes 51, 385–391. [DOI] [PubMed] [Google Scholar]
- Brown LD, Davis M, Wai S, Wesolowski SR, Hay WW Jr, Limesand SW & Rozance PJ (2016). Chronically increased amino acids improve insulin secretion, pancreatic vascularity, and islet size in growth‐restricted fetal sheep. Endocrinology 157, 3788–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Green AS, Macko AR, Yates DT, Kelly AC & Limesand SW (2014). Enhanced insulin secretion responsiveness and islet adrenergic desensitization after chronic norepinephrine suppression is discontinued in fetal sheep. Am J Physiol Endocrinol Metab 306, E58–E64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Kelly AC, Yates DT, Macko AR, Lynch RM & Limesand SW (2017). Islet adaptations in fetal sheep persist following chronic exposure to high norepinephrine. J Endocrinol 232, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Rozance PJ, Hay WW Jr & Limesand SW (2012). Insulin‐like growth factor and fibroblast growth factor expression profiles in growth‐restricted fetal sheep pancreas. Exp Biol Med 237, 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung CY (1990). Fetal adrenal medulla catecholamine response to hypoxia‐direct and neural components. Am J Physiol 258, R1340–R1346. [DOI] [PubMed] [Google Scholar]
- Cohen WR, Piasecki GJ, Cohn HE, Susa JB & Jackson BT (1991). Sympathoadrenal responses during hypoglycemia, hyperinsulinemia, and hypoxemia in the ovine fetus. Am J Physiol 261, E95–E102. [DOI] [PubMed] [Google Scholar]
- Davis MA, Macko AR, Steyn LV, Anderson MJ & Limesand SW (2015). Fetal adrenal demedullation lowers circulating norepinephrine and attenuates growth restriction but not reduction of endocrine cell mass in an ovine model of intrauterine growth restriction. Nutrients 7, 500–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Blasio MJ, Gatford KL, McMillen IC, Robinson JS & Owens JA (2007). Placental restriction of fetal growth increases insulin action, growth, and adiposity in the young lamb. Endocrinology 148, 1350–1358. [DOI] [PubMed] [Google Scholar]
- de Vrijer B, Regnault TR, Wilkening RB, Meschia G & Battaglia FC (2004). Placental uptake and transport of ACP, a neutral nonmetabolizable amino acid, in an ovine model of fetal growth restriction. Am J Physiol Endocrinol Metab 287, E1114–E1124. [DOI] [PubMed] [Google Scholar]
- DiGiacomo JE & Hay WW Jr (1990). Effect of hypoinsulinemia and hyperglycemia on fetal glucose utilization. Am J Physiol 259, E506–E512. [DOI] [PubMed] [Google Scholar]
- Economides DL, Proudler A & Nicolaides KH (1989). Plasma insulin in appropriate‐ and small‐for‐gestational‐age fetuses. Am J Obstet Gynecol 160, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Fall CH, Stein CE, Kumaran K, Cox V, Osmond C, Barker DJ & Hales CN (1998). Size at birth, maternal weight, and type 2 diabetes in South India. Diabet Med 15, 220–227. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Twinn DS & Ozanne SE (2006). Mechanisms by which poor early growth programs type‐2 diabetes, obesity and the metabolic syndrome. Physiol Behav 88, 234–243. [DOI] [PubMed] [Google Scholar]
- Gatford KL, Mohammad SN, Harland ML, De Blasio MJ, Fowden AL, Robinson JS & Owens JA (2008). Impaired β‐cell function and inadequate compensatory increases in β‐cell mass after intrauterine growth restriction in sheep. Endocrinology 149, 5118–5127. [DOI] [PubMed] [Google Scholar]
- Green AS, Rozance PJ & Limesand SW (2010). Consequences of a compromised intrauterine environment on islet function. J Endocrinol 205, 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenough A, Nicolaides KH & Lagercrantz H (1990). Human fetal sympathoadrenal responsiveness. Early Hum Dev 23, 9–13. [DOI] [PubMed] [Google Scholar]
- Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C & Winter PD (1991). Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303, 1019–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder T, Rodekamp E, Schellong K, Dudenhausen JW & Plagemann A (2007). Birth weight and subsequent risk of type 2 diabetes: a meta‐analysis. Am J Epidemiol 165, 849–857. [DOI] [PubMed] [Google Scholar]
- Hay WW Jr, Brown LD, Rozance PJ, Wesolowski SR & Limesand SW (2016). Challenges in nourishing the IUGR foetus – Lessons learned from studies in the IUGR foetal sheep. Acta Paediatr 10, 881–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe FM, Thornton PS, Wanner LA, Steinkrauss L, Simmons RA & Stanley CA (2006). Clinical features and insulin regulation in infants with a syndrome of prolonged neonatal hyperinsulinism. J Pediatr 148, 207–212. [DOI] [PubMed] [Google Scholar]
- Jackson BT, Cohn HE, Morrison SH, Baker RM & Piasecki GJ (1993). Hypoxia‐induced sympathetic inhibition of the fetal plasma insulin response to hyperglycemia. Diabetes 42, 1621–1625. [DOI] [PubMed] [Google Scholar]
- Jackson BT, Piasecki GJ, Cohn HE & Cohen WR (2000). Control of fetal insulin secretion. Am J Physiol Regul Integr Comp Physiol 279, R2179–R2188. [DOI] [PubMed] [Google Scholar]
- Jensen CB, Storgaard H, Dela F, Holst JJ, Madsbad S & Vaag AA (2002). Early differential defects of insulin secretion and action in 19‐year‐old caucasian men who had low birth weight. Diabetes 51, 1271–1280. [DOI] [PubMed] [Google Scholar]
- Jensen CB, Storgaard H, Madsbad S, Richter EA & Vaag AA (2007). Altered skeletal muscle fiber composition and size precede whole‐body insulin resistance in young men with low birth weight. J Clin Endocrinol Metab 92, 1530–1534. [DOI] [PubMed] [Google Scholar]
- Jones CT & Robinson JS (1983). Studies on experimental growth retardation in sheep. Plasma catecholamines in fetuses with small placenta. J Dev Physiol 5, 77–87. [PubMed] [Google Scholar]
- Kelly AC, Bidwell CA, McCarthy FM, Taska DJ, Anderson MJ, Camacho LE & Limesand SW (2017) RNA sequencing exposes adaptive and immune responses to intrauterine growth restriction in fetal sheep islets. Endocrinology 158, 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavezzi JR, Thorn SR, O'Meara MC, LoTurco D, Brown LD, Hay WW Jr & Rozance PJ (2013). Increased fetal insulin concentrations for one week fail to improve insulin secretion or β‐cell mass in fetal sheep with chronically reduced glucose supply. Am J Physiol Regul Integr Comp Physiol 304, R50–R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leos RA, Anderson MJ, Chen X, Pugmire J, Anderson KA & Limesand SW (2010). Chronic exposure to elevated norepinephrine suppresses insulin secretion in fetal sheep with placental insufficiency and intrauterine growth restriction. Am J Physiol Endocrinol Metab 298, E770–E778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limesand SW & Hay WW Jr (2003). Adaptation of ovine fetal pancreatic insulin secretion to chronic hypoglycaemia and euglycaemic correction. J Physiol 547, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limesand SW, Rozance PJ, Brown LD & Hay WW Jr (2009). Effects of chronic hypoglycemia and euglycemic correction on lysine metabolism in fetal sheep. Am J Physiol Endocrinol Metab 296, E879–E887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limesand SW, Rozance PJ, Macko AR, Anderson MJ, Kelly AC & Hay WW Jr (2013). Reductions in insulin concentrations and β‐cell mass precede growth restriction in sheep fetuses with placental insufficiency. Am J Physiol Endocrinol Metab 304, E516–E523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limesand SW, Rozance PJ, Smith D & Hay WW Jr (2007). Increased insulin sensitivity and maintenance of glucose utilization rates in fetal sheep with placental insufficiency and intrauterine growth restriction. Am J Physiol Endocrinol Metab 293, E1716–E1725. [DOI] [PubMed] [Google Scholar]
- Limesand SW, Rozance PJ, Zerbe GO, Hutton JC & Hay WW Jr (2006). Attenuated insulin release and storage in fetal sheep pancreatic islets with intrauterine growth restriction. Endocrinology 147, 1488–1497. [DOI] [PubMed] [Google Scholar]
- Liu H, Schultz CG, De Blasio MJ, Peura AM, Heinemann GK, Harryanto H, Hunter DS, Wooldridge AL, Kind KL, Giles LC, Simmons RA, Owens JA & Gatford KL (2015). Effect of placental restriction and neonatal exendin‐4 treatment on postnatal growth, adult body composition, and in vivo glucose metabolism in the sheep. Am J Physiol Endocrinol Metab 309, E589–E600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCowan LM, Roberts CT, Dekker GA, Taylor RS, Chan EH, Kenny LC, Baker PN, Moss‐Morris R, Chappell LC & North RA (2010). Risk factors for small‐for‐gestational‐age infants by customised birthweight centiles: data from an international prospective cohort study. BJOG 117, 1599–1607. [DOI] [PubMed] [Google Scholar]
- Macko AR, Yates DT, Chen X, Green AS, Kelly AC, Brown LD & Limesand SW (2013). Elevated plasma norepinephrine inhibits insulin secretion, but adrenergic blockade reveals enhanced β‐cell responsiveness in an ovine model of placental insufficiency at 0.7 of gestation. J Dev Orig Health Dis 4, 402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macko AR, Yates DT, Chen X, Shelton LA, Kelly AC, Davis MA, Camacho LE, Anderson MJ & Limesand SW (2016). Adrenal demedullation and oxygen supplementation independently increase glucose‐stimulated insulin concentrations in fetal sheep with intrauterine growth restriction. Endocrinology 157, 2104–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen H (1986). Fetal oxygenation in diabetic pregnancy. With special reference to maternal blood oxygen affinity and its effectors. Dan Med Bull 33, 64–74. [PubMed] [Google Scholar]
- Mericq V, Ong KK, Bazaes R, Pena V, Avila A, Salazar T, Soto N, Iniguez G & Dunger DB (2005). Longitudinal changes in insulin sensitivity and secretion from birth to age three years in small‐ and appropriate‐for‐gestational‐age children. Diabetologia 48, 2609–2614. [DOI] [PubMed] [Google Scholar]
- Milley JR (1997). Ovine fetal metabolism during norepinephrine infusion. Am J Physiol 273, E336–E347. [DOI] [PubMed] [Google Scholar]
- Milovanovic I, Njuieyon F, Deghmoun S, Chevenne D, Levy‐Marchal C & Beltrand J (2014). SGA children with moderate catch‐up growth are showing the impaired insulin secretion at the age of 4. PLoS One 9, e100337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles RE, Lodha A, Perlman M & Moore AM (2002). Experience with intravenous glucagon infusions as a treatment for resistant neonatal hypoglycemia. Arch Pediatr Adolesc Med 156, 999–1004. [DOI] [PubMed] [Google Scholar]
- Monteith C, Flood K, Mullers S, Unterscheider J, Breathnach F, Daly S, Geary MP, Kennelly MM, McAuliffe FM, O'Donoghue K, Hunter A, Morrison JJ, Burke G, Dicker P, Tully EC & Malone FD (2017). Evaluation of normalization of cerebro‐placental ratio as a potential predictor for adverse outcome in SGA fetuses. Am J Obstet Gynecol 216, 285.e1–6. [DOI] [PubMed] [Google Scholar]
- Narkewicz MR, Carver TD & Hay WW Jr (1993). Induction of cytosolic phosphoenolpyruvate carboxykinase in the ovine fetal liver by chronic fetal hypoglycemia and hypoinsulinemia. Pediatr Res 33, 493–496. [DOI] [PubMed] [Google Scholar]
- Newsome CA, Shiell AW, Fall CH, Phillips DI, Shier R & Law CM (2003). Is birth weight related to later glucose and insulin metabolism? – A systematic review. Diabet Med 20, 339–348. [DOI] [PubMed] [Google Scholar]
- Nicolini U, Hubinont C, Santolaya J, Fisk NM & Rodeck CH (1990). Effects of fetal intravenous glucose challenge in normal and growth retarded fetuses. Horm Metab Res 22, 426–430. [DOI] [PubMed] [Google Scholar]
- Owens JA, Gatford KL, De Blasio MJ, Edwards LJ, McMillen IC & Fowden AL (2007a). Restriction of placental growth in sheep impairs insulin secretion but not sensitivity before birth. J Physiol 584, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens JA, Thavaneswaran P, De Blasio MJ, McMillen IC, Robinson JS & Gatford KL (2007b). Sex‐specific effects of placental restriction on components of the metabolic syndrome in young adult sheep. Am J Physiol Endocrinol Metab 292, E1879–E1889. [DOI] [PubMed] [Google Scholar]
- Phipps K, Barker DJ, Hales CN, Fall CH, Osmond C & Clark PM (1993). Fetal growth and impaired glucose tolerance in men and women. Diabetologia 36, 225–228. [DOI] [PubMed] [Google Scholar]
- Platz E & Newman R (2008). Diagnosis of IUGR: traditional biometry. Semin Perinatol 32, 140–147. [DOI] [PubMed] [Google Scholar]
- Pschera H, Persson B & Lunell NO (1986). Interrelationship between amniotic fluid C‐peptide and catecholamines in the last trimester of diabetic pregnancy. Am J Obstet Gynecol 154, 48–52. [DOI] [PubMed] [Google Scholar]
- Robinson JS, Kingston EJ, Jones CT & Thorburn GD (1979). Studies on experimental growth retardation in sheep. The effect of removal of a endometrial caruncles on fetal size and metabolism. J Dev Physiol 1, 379–398. [PubMed] [Google Scholar]
- Ross JC, Fennessey PV, Wilkening RB, Battaglia FC & Meschia G (1996). Placental transport and fetal utilization of leucine in a model of fetal growth retardation. Am J Physiol 270, E491–E503. [DOI] [PubMed] [Google Scholar]
- Rozance PJ, Anderson M, Martinez M, Fahy A, Macko AR, Kailey J, Seedorf GJ, Abman SH, Hay WW Jr & Limesand SW (2015). Placental insufficiency decreases pancreatic vascularity and disrupts hepatocyte growth factor signaling in the pancreatic islet endothelial cell in fetal sheep. Diabetes 64, 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozance PJ, Limesand SW & Hay WW Jr (2006). Decreased nutrient stimulated insulin secretion in chronically hypoglycemic late gestation fetal sheep is due to an intrinsic islet defect. Am J Physiol Endocrinol Metab 291, E404–E411. [DOI] [PubMed] [Google Scholar]
- Rozance PJ, Limesand SW, Zerbe GO & Hay WW Jr (2007). Chronic fetal hypoglycemia inhibits the later steps of stimulus‐secretion coupling in pancreatic β‐cells. Am J Physiol Endocrinol Metab 292, E1256–E1264. [DOI] [PubMed] [Google Scholar]
- Simonetta G, Rourke AK, Owens JA, Robinson JS & McMillen IC (1997). Impact of placental restriction on the development of the sympathoadrenal system. Pediatr Res 42, 805–811. [DOI] [PubMed] [Google Scholar]
- Soto N, Bazaes RA, Pena V, Salazar T, Avila A, Iniguez G, Ong KK, Dunger DB & Mericq MV (2003). Insulin sensitivity and secretion are related to catch‐up growth in small‐for‐gestational‐age infants at age 1 year: results from a prospective cohort. J Clin Endocrinol Metab 88, 3645–3650. [DOI] [PubMed] [Google Scholar]
- Sperling MA, Christensen RA, Ganguli S & Anand R (1980). Adrenergic modulation of pancreatic hormone secretion in utero: studies in fetal sheep. Pediatr Res 14, 203–208. [DOI] [PubMed] [Google Scholar]
- Stanley CA, Rozance PJ, Thornton PS, De Leon DD, Harris D, Haymond MW, Hussain K, Levitsky LL, Murad MH, Simmons RA, Sperling MA, Weinstein DA, White NH & Wolfsdorf JI (2015). Re‐evaluating ‘transitional neonatal hypoglycemia’: mechanism and implications for management. J Pediatr 166, 1520–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teramo KA (2010). Obstetric problems in diabetic pregnancy ‐ The role of fetal hypoxia. Best Pract Res Clin Endocrinol Metab 24, 663–671. [DOI] [PubMed] [Google Scholar]
- Thorn SR, Brown LD, Rozance PJ, Hay WW Jr & Friedman JE (2012). Increased hepatic glucose production in fetal sheep with intrauterine growth restriction is not suppressed by insulin. Diabetes 62, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton PS, Stanley CA, De Leon DD, Harris D, Haymond MW, Hussain K, Levitsky LL, Murad MH, Rozance PJ, Simmons RA, Sperling MA, Weinstein DA, White NH & Wolfsdorf JI (2015). Recommendations from the pediatric endocrine society for evaluation and management of persistent hypoglycemia in neonates, infants, and children. J Pediatr 167, 238–245. [DOI] [PubMed] [Google Scholar]
- Thureen PJ, Trembler KA, Meschia G, Makowski EL & Wilkening RB (1992). Placental glucose transport in heat‐induced fetal growth retardation. Am J Physiol 263, R578–R585. [DOI] [PubMed] [Google Scholar]
- Van Assche FA, De Prins F, Aerts L & Verjans M (1977). The endocrine pancreas in small‐for‐dates infants. Br J Obstet Gynaecol 84, 751–753. [DOI] [PubMed] [Google Scholar]
- Yajnik CS, Fall CH, Vaidya U, Pandit AN, Bavdekar A, Bhat DS, Osmond C, Hales CN & Barker DJ (1995). Fetal growth and glucose and insulin metabolism in four‐year‐old Indian children. Diabet Med 12, 330–336. [DOI] [PubMed] [Google Scholar]
- Yates DT, Macko AR, Chen X, Green AS, Kelly AC, Anderson MJ, Fowden AL & Limesand SW (2012). Hypoxemia‐induced catecholamine secretion from adrenal chromaffin cells inhibits glucose‐stimulated hyperinsulinemia in fetal sheep. J Physiol 590, 5439–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang L & Liu L (2001). The study of levels of norepinephrine and dopamine‐beta‐hydroxylase in patients with pregnancy‐induced hypertension. Zhonghua Yi Xue Za Zhi (Taipei) 64, 351–356. [PubMed] [Google Scholar]
- Zimmermann E, Gamborg M, Sorensen TI & Baker JL (2015). Sex differences in the association between birth weight and adult type 2 diabetes. Diabetes 64, 4220–4225. [DOI] [PubMed] [Google Scholar]