

ABSTRACT
Thrombocytopenia is a hematological finding commonly encountered in daily clinical practice from asymptomatic clinic patients to critically ill intensive care unit patients. A broad spectrum of etiologies and variation in clinical presentation often present a diagnostic challenge. Furthermore, concomitant presence of thrombosis and thrombocytopenia, as in cases of thrombotic thrombocytopenia, complicates the management. In hospitalized patients, new-onset thrombocytopenia is an important reason for hematology consultation. Therefore, it is of utmost importance that the etiology is diagnosed accurately. In addition, a basic understanding of the pathophysiology and the differential diagnosis avoids delay in the diagnosis and leads to rapid initiation of treatment. This review will address causes of thrombocytopenia that arises in hospitalized patients with an emphasis on the pathophysiological basis of each disorder.
KEYWORDS: Thrombocytopenia, hospital acquired, pathophysiology, thrombopoiesis, thrombopoietin
1. Introduction
Normal adult platelet count ranges from 150–450 × 103 per µl. Thrombocytopenia is defined as a platelet count < 150 × 103 per µl [1]. Because normal values are traditionally determined as 2-standard deviations above and below the mean (approximately 95 percent), a proportion of normal population will have platelet count < 150 × 103 per µl. In addition, there is an annual and seasonal variation in the platelet count [2]. Therefore, a platelet count between 100–150 × 103 per µl may be normal and clinically insignificant under certain circumstances. Isolated thrombocytopenia is a common hematological finding in hospitalized patients. A broad spectrum of etiologies and variation in clinical presentation often present a diagnostic challenge. In this review, we will elucidate causes of isolated thrombocytopenia arising in hospitalized patients. We will develop a diagnostic approach and discuss each cause separately.
2. Incidence
Thrombocytopenia acquired during hospitalization is common [3]. Even though several studies have documented incidence of thrombocytopenia from 25 to 55% in intensive care units (ICU), no study has characterized its incidence in all hospitalized patients [4]. The incidence may be higher in surgical than medical ICUs. About 50% of all ICU patients have at least one platelet reading < 150 × 103 per µl [5]. Similarly, thrombocytopenia is seen in cardiac, obstetrics and gynecology, oncology, neurology, and general medical units. Up to 13% of patients with acute coronary syndrome may develop thrombocytopenia during their hospitalization [6].
3. Pathogenesis
Platelets are derived from fragmentation of megakaryocytes, hematopoietic cells residing in the bone marrow. A key regulator of thrombopoiesis is thrombobpoeitin (TPO), a hormone synthesized by the liver. It functions by promoting survival and proliferation of megakaryocytes [7]. Besides TPO, cytokines such as interleukin-3 (IL-3), interleukin-6 (IL-6), interleukin-11 (1L-11), and stem cell factor (SCF) have synergistic effects [8]. Platelets have a half-life of about eight days, after which they undergo intrinsic programmed apoptosis regulated by BAK/BCL-XL interaction [9]. Once senile, platelets are removed from the bloodstream by phagocytes in liver and spleen. Pathophysiologically, any process that disrupts the platelet life cycle could potentially cause thrombocytopenia. Primarily, there are two mechanisms involved; decreased platelet production and increased platelet destruction (Figure 1).
Figure 1.
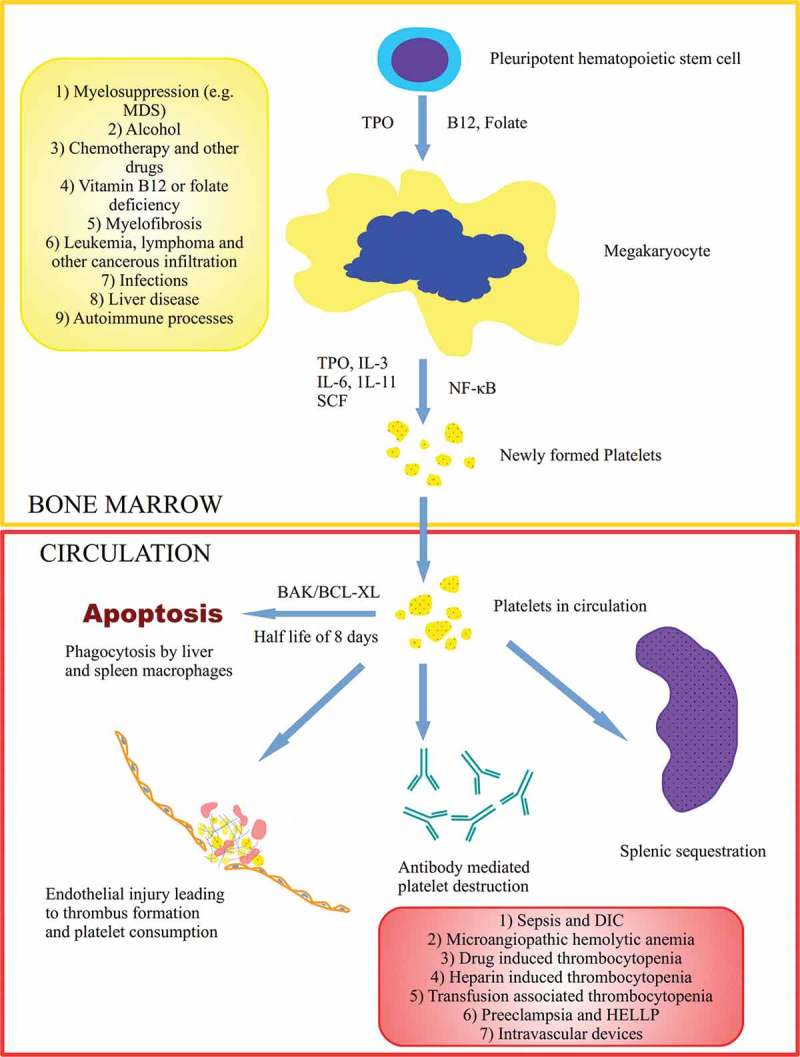
Thrombopoiesis begins in the bone marrow milieu with the differentiation of pleuripotent stem cell to megakaryocytes. Several key regulators (TPO, interleukins, B12, folate, NF-κB) are involved in platelet formation. Platelets have several destinations in the peripheral circulation including self-regulated apoptosis, consumption in response to injury, splenic sequestration, and platelet destruction. Causes of decreased platelet production (yellow box) and increased peripheral destruction (red box) are shown. Abbreviations: MDS (myelodysplastic syndrome), TPO (thrombopoietin), IL (interleukin), SCF (stem cell factor), NF-κB (nuclear factor kappa B), DIC (disseminated intravascular coagulation), HELLP (hemolysis, elevated liver enzymes and low platelets).
Several factors influence platelet formation. Reduction in the number of megakaryocytes due to destruction of the stem cells or megakaryocyte apoptosis leads to thrombocytopenia [10]. These processes are in part mediated by lack of TPO or auto-antibodies against it. Prevention of platelet budding from megakaryocytes by inhibition of NF-κB is another hypothesized mechanism [11]. Under certain circumstances, changes in megakaryocyte size and ploidy influence platelet production [12]. Ineffective megakaryopoiesis as a result of nutrient deficiency such as vitamin B-12 and folate is another cause. The second major mechanism is peripheral destruction as a result of humoral or complement mediated mechanisms. Auto-antibody mediated platelet destruction can be induced by drugs, infections, or autoimmune disorders. Other pathophysiologic mechanisms of thrombocytopenia include platelet sequestration, hemodilution, and consumption within thrombi. An algorithmic approach to thrombocytopenia in hospitalized patients is provided in Figure 2.
Figure 2.
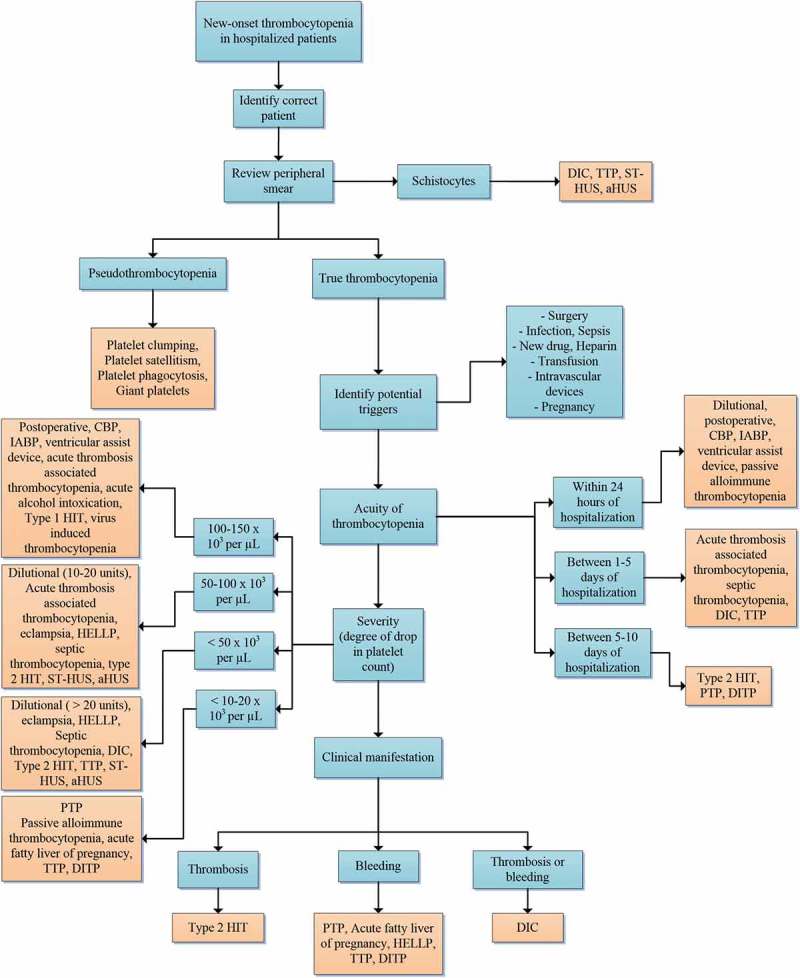
An algorithmic approach to hospital-induced thrombocytopenia. Abbreviations: DIC (disseminated intravascular coagulation), TTP (thrombotic thrombocytopenic purpura), ST-HUS (Shiga toxin-mediated hemolytic uremic syndrome), aHUS (atypical hemolytic uremic syndrome), CPB (cardiopulmonary bypass), IABP (intraaortic balloon pump), HIT (heparin-induced thrombocytopenia), DITP (drug-induced immune thrombocytopenia), HELLP ((hemolysis, elevated liver enzymes, and low platelets), PTP (post transfusion purpura).
4. Spurious thrombocytopenia
Technical errors in platelet counting can occasionally cause spurious thrombocytopenia or pseudothrombocytopenia (Figure 3). It is therefore important to recognize such conditions and avoid unnecessary testing or treatment delays. Platelet clumping is a laboratory artifact caused by ethylenediaminetetraacetic acid (EDTA) dependent auto-antibodies directed against glycoprotein IIb/IIIa receptor. In the presence of EDTA, these antibodies cause platelet aggregation and degranulation in vitro [13]. Hence, automated cell counters are unable to distinguish between individual platelets. Platelet satellitism is a rare phenomenon caused by EDTA-dependent IgG auto-antibodies directed against platelet glycoprotein IIb/IIIa receptor and neutrophil Fc gamma receptor III, perhaps due to cross-reacting epitopes [14]. This results in platelets adhering to the surface of polymorphonuclear leukocytes in a rosette-like fashion. Most commonly, it is seen as an artifact in normal individuals but conditions such as lupus, vasculitis, mantle cell lymphoma, and marginal zone lymphoma may be associated with it [14]. Platelet phagocytosis is a very rare EDTA-associated phenomenon characterized by phagocytosis of platelets by neutrophils or monocytes in vitro [15]. An accurate platelet count in these EDTA associated conditions can be determined by using EDTA-free, blood sample collecting tubes such as heparin-, citrate-, oxalate- or magnesium-containing tubes [13–15]. Lastly, giant platelets can be miscounted as leukocytes or erythrocytes by automated cell counters because of their size. Flow cytometry of immunologically stained platelets can be helpful in such cases for true platelet count [13–15].
Figure 3.
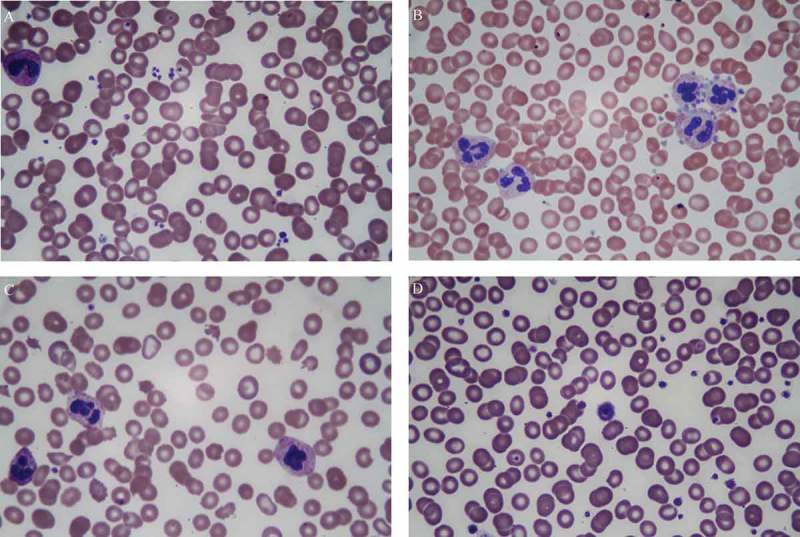
Peripheral smear examination demonstrating, (a) small scattered platelet clumps; (b) platelet satellitism where platelets are arranged in a rosette-like pattern around the neutrophils; (c) schistocytes, bite cells, and scarcity of platelets, (d) a giant platelet almost the size of a red blood cell.
5. Hemodilution
Large volume fluid resuscitation and massive blood transfusion are risk factors for dilutional thrombocytopenia. It is the most common hemostatic laboratory abnormality observed after massive transfusion (> 15 units) [16]. Primary mechanism of thrombocytopenia is replacement of lost blood by platelet deficient crystalloids or packed red blood cells [17]. Moreover, up to 75% of patients receiving > 20 units of packed red cells can develop thrombocytopenia with counts < 50 × 103 per µl [18].
A novel concept in treating trauma patients requiring massive blood transfusion is balanced ratio transfusion of RBC, platelets, and plasma (1:1:1). This has been considered as a paradigm shift in treating these patients as platelets and plasma are administered at the start of resuscitation along with packed red cells in contrast to traditional sequential administration of crystalloid, RBC, and platelets or plasma in that order [19]. This strategy has demonstrated higher platelet counts and better mortality outcomes [20]. Because of increased risk of bleeding and higher mortality associated thrombocytopenia of < 100 × 103 per µl in trauma patients, efforts should be made to keep the platelet count > 50–100 × 103 per µl.
6. Postoperative thrombocytopenia
Postoperative thrombocytopenia is commonly seen in cardiovascular, non-cardiac, and orthopedic surgeries [21]. Early-onset postoperative thrombocytopenia is physiological and inevitable. Surgery results in complement activation, neutrophil degranulation, and release of pro-inflammatory cytokines such as IL-6 and tumor necrosis factor-α (TNF-α), which cause endothelial cell dysfunction and release of tissue factor and von Willibrand (vWB) factor leading to platelet activation, aggregation, and consumption [22]. Physiologic postoperative thrombocytopenia is usually mild, not associated with clinical bleeding, and resolves spontaneously in three to four days [23]. If there is persistence of thrombocytopenia beyond five days or the onset is late (> 5 days), other causes should be considered.
7. Thrombocytopenia associated with cardiac bypass surgery
Cardiopulmonary bypass (CPB) for open heart surgery affects platelet count and function leading to quantitative and qualitative platelet dysfunctions, respectively. Thrombocytopenia develops because of hemodilution, filtration, and adhesion of platelet aggregates to synthetic surfaces. Platelet count starts to drop within five to ten minutes of initiation of CPB, usually stays above 100 × 103 per µl and recovers within five to seven days [24]. Qualitative platelet defects are caused by depletion of α-granules, impairment of platelet aggregation to adenosine diphosphate (ADP), and loss of platelet surface glycoprotein due to mechanical shear stress [25].
8. Thrombocytopenia associated with cardiac assist devices
Intraaortic balloon pumps (IABA) cause thrombocytopenia very frequently due to similar mechanisms discussed above. Platelet drop is usually mild and not associated with clinically significant bleeding. Patients on IABP frequently have concomitant heparin use for anticoagulation but the platelet drop is independent [26]. Continuous flow ventricular assist devices cause thrombocytopenia by causing platelet activation and aggregation due to non-pulsatile blood flow and mechanical shear stress [27].
9. Post transfusion purpura
Post transfusion purpura (PTP) is a rare and serious condition of acute thrombocytopenia developing after receiving platelet-containing blood products. It is caused by alloimmunization or sensitization to certain human platelet antigens (HPA-1a, HPA-1b, HPA-3a, HPA-4a, and HPA-5b) after pregnancy, transfusion, or bone marrow transplantation in patients who are HPA negative [28]. An anamnestic immunological response occurs after transfusion of HLA-1a positive donor products that not only causes destruction of donor platelets by anti-HLA-1a alloantibody but also paradoxically patient’s own platelets. There is a sudden drop in platelet count (sometimes < 10 × 103 per µl) usually five to ten days post transfusion. Clinical manifestations in PTP vary but can potentially cause life threatening bleeding and have 10–15% mortality. Diagnosis is based on the clinical picture as well as demonstration of anti-HLA alloantibodies in the patient’s serum [28]. Many cases will have spontaneous remission in a few weeks. But since clinical course cannot be predicted, aggressive treatment with intravenous immunoglobulin administration (IVIG) and/or steroids should be instituted early [29]. Platelet transfusion should generally be avoided even in HPA-1a negative platelets, and future transfusion should be from a platelet compatible donor [30].
10. Passive alloimmune thrombocytopenia
Preformed antiplatelet antibodies (anti-HPA-1a) in immunized antigen negative donor blood products, on rare occasions, can cause acute severe thrombocytopenia by passive transfer of these antibodies to the antigen positive recipient and causing platelet destruction. Onset is within hours of transfusion in contrast to PTP (where it is days) and platelet counts can fall < 10 × 103 per µl [28,31]. Spontaneous platelet count recovery can occur in about five days. There is no treatment guideline for this condition given its rarity, nevertheless, based on the prior case reports, IVIG, steroids, and platelet transfusion have been successful [31].
11. Acute thrombosis associated thrombocytopenia
Occasionally, platelet count will drop after an acute episode of venous thromboembolism (VTE). Since unfractionated heparin (UH) or low molecular weight heparin (LMWH) are the standard initial treatment for acute VTE, it is important to recognize this condition. Platelets have a propensity to adhere to the surfaces of fresh large clots likely due to exudation of thromboplastic substances. This process consumes circulating platelets and can cause acute thrombocytopenia. Platelet drop occurs as early as within 12 hours of thrombus formation and can continue to drop for 24–36 hours [32]. Up to 10% of patients with newly formed pulmonary emboli develop thrombocytopenia of < 150 × 103 per µl [33]. Platelet drop is usually mild but there are case reports where counts have dropped to 60 × 103 per µl. Recovery occurs usually in three days. Therefore, it is crucial to continue heparin as long as hemostasis remains intact [32].
12. Thrombocytopenia associated with liver disease
Thrombocytopenia is very common in liver disease and present in up to 70% of cirrhotic patients because of decreased megakaryopoiesis due to lack of TPO, splenic sequestration, interferon alpha and autoimmune mechanisms [34]. Thrombocytopenia due to chronic liver disease is rather chronic and present at baseline; however, acute worsening or new development of thrombocytopenia due to acute fulminant liver failure can happen in hospitalized patients. Thrombocytopenia in acetaminophen (APAP) hepatotoxicity was reported to be 3.4% in one study and correlated directly with the degree of hepatic damage [35]. APAP overdose not only impairs clotting factor synthesis but also induces coagulation cascade leading to consumption of clotting factors and platelets. Recently, platelets have been suggested to play a role in APAP-related hepatic injury at cellular and molecular level. Studies in mice have shown increased platelet concentration in the liver linking to APAP hepatotoxicity [36]. These results have yet to be studied or confirmed in humans.
13. Thrombcytopenia associated with alcohol abuse
Up to 80% of acutely ill, hospitalized alcoholic patients develop thrombocytopenia [37]. In general, thrombocytopenia is mild and self-limiting with resolution in five to seven days of abstinence. The mechanism by which acute alcohol abuse causes thrombocytopenia involves direct toxic effect on megakaryocytes preventing them from maturing as well as decreased life span of circulating platelets. Moreover, there is qualitative platelet dysfunction resulting in impaired platelet aggregation and prolongation of bleeding time [38]. Nutritional deficiencies (folate and vitamin B12) associated with alcoholism do not appear to be the cause in acute alcoholism-induced thrombocytopenia. A transient hematological response, rebound thrombocytosis, occurs in many alcoholics after an acute episode of thrombocytopenia where platelet count may increase up to 600–900 × 103 per µl [37].
14. Thrombocytopenia in pregnancy
Thrombocytopenia, usually an incidental finding on routine late pregnancy-related blood work, represents 70–80% of all thrombocytopenic pregnant women [39]. However, peripartum hospital acquired thrombocytopenia is more serious with maternal and fetal consequences. Preeclampsia, HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome and acute fatty liver of pregnancy are three pregnancy-specific causes closely associated and often difficult to differentiate from thrombotic microangiopathies [39]. Preeclampsia is characterized by hypertension and proteinuria after 20 weeks of pregnancy. Thrombocytopenia can be the initial manifestation and accounts for 21% of thrombocytopenia in pregnancy. Severe thrombocytopenia (< 50 × 103 per µL) with intravascular hemolysis can happen in < 5% of preeclapmtic patients [40]. HELLP syndrome, preceded by preeclampsia in 20% of the cases, causes severe hemolysis, elevation of liver enzymes (elevation of aspartate aminotransferase > 70 U/L is a diagnostic criterion), and thrombocytopenia [41]. Lastly, a rare life threatening condition called acute fatty liver of pregnancy presents with severe thrombocytopenia (less than 20 × 103 per µl) and markedly elevated liver enzymes [39]. Therapy is directed toward safe delivery of the fetus as early as possible as well as supportive treatment to keep platelet count greater than 50 × 103 per µl. There is evidence that steroids play some role particularly in maturation of the fetal pulmonary system [42].
15. Virus induced thrombocytopenia
Platelets have a far more extensive and complex interaction with viruses than was previously thought. As a result, mild thrombocytopenia is often seen with chronic viral infections. Some acute viral infections are associated with severe thrombocytopenia and life threatening hemorrhagic syndromes [43]. Several mechanisms are deployed by viruses including inappropriate platelet activation and consumption, suppression of hematopoietic stem cells and megakaryocytes, decreasing TPO synthesis by liver, direct interaction with platelets leading to their premature destruction, immune complex mediated removal of platelets, and splenic sequestration [43,44]. Direct interaction with viruses is mediated by several receptors on platelet cell membranes such as integrins, Toll-like receptors (TLR), and lectins [45]. The acuity and severity of thrombocytopenia depends on the type of virus involved. Treatment is usually supportive and of the underlying viral infection.
16. Septic thrombocytopenia and disseminated intravascular coagulation (DIC)
Moderate degree of thrombocytopenia (50–150 × 103 per µl) in septicemia without laboratory or clinical evidence of DIC is poorly understood and has been regarded as compensated or subclinical DIC [46]. Possible mechanisms involve bone marrow suppression, direct bacteria-platelet interaction and immune mediated destruction. Platelet associated IgG antibodies have been detected in a majority of patients with septicemia [47]. In addition, hemophagocytosis of platelets in response to infection results from elevated levels of macrophage colony stimulating factor leading to increased proliferation and activation of monocytes and macrophages. According to one study, hemophagocytosis is present in up to 64% of critically ill severely septic patients [48].
DIC is a dreaded complication of many disorders (Table 1). The morbidity and mortality associated with DIC depends on the underlying clinical disorder and the severity of coagulation defect [49]. In DIC, there is a widespread activation of platelets and coagulation cascade with thrombotic occlusion of small vessels in several organs. Several fold increase in fibrinogenesis is a result of tissue factor mediated activation of extrinsic pathway, impairment of anticoagulation system and inhibition of endogenous fibrinolytic pathways from plasminogen activator inhibitor 1 (PAI-1). Cytokines in particular IL-6 play an intermediary role [50]. Recently, further elucidation of the pathogenesis of DIC has identified NETosis as a crucial step. NETosis is a form for cell death, distinctive from apoptosis and necrosis, where cellular DNA, associated proteins (histones and high mobility group box protein 1) and cytoplasmic proteins are released into extracellular milieu forming web like structures called neutrophil extracellular traps (NETs). NETs provide scaffolding for factors XII and XI leading to activation of intrinsic coagulation pathway. In addition, cell free DNA and histones have strong procoagulant properties [51,52].
Table 1.
Various clinical conditions associated with DIC and their pathogenesis.
| Clinical condition | Pathogenetic trigger | |
|---|---|---|
| 1 | Sepsis and severe infections – gram positive and negative bacteria, viruses, fungi, parasites | Exotoxins and endotoxins (lipopolysaccharides) |
| 2 | Crush injuries and sever trauma | Phospholipids and fats from damaged tissues; fat embolism |
| 3 | Head injury | Phospholipids and fats from damaged tissues |
| 4 | Acute severe pancreatitis | Cytokines and severe inflammatory response |
| 5 | Solid tumors – pancreatic and prostate carcinoma | Cancer procoagulant |
| 6 | Hematological malignancies | Tissue factor; cytokines |
| 7 | Transfusion related hemolytic reactions – ABO and Rh incompatibility | Widespread endothelial damage |
| 8 | Obstetrical complications – placental abruption, amniotic fluid embolism, eclampsia, HELLP syndrome, RPOC, septic abortion, fetal demise | Leakage of procoagulant substances from placenta and amniotic fluid; widespread endothelial damage |
| 9 | Vascular malformation – giant hemangiomas, aneurysms | Localized activation of platelet aggregation and coagulation; can become widespread |
| 10 | Snake bites | Hemorrhagic metalloproteinases; thrombin-like enzymes; factor X and prothrombin activators [53] |
| 11 | Heat stroke, hyperthermia, and burns | Tissue factor; protein alteration with temperature changes |
| 12 | Acute fulminant hepatic failure | Accelerated platelet and coagulation activation; decreased pro- and anticoagulant synthesis complicates DIC |
| 13 | Drugs – allergic reactions and overdoses such as amphetamines | Severe inflammatory or immunological response; tissue necrosis and rhabdomyolysis in case of amphetamines |
| 14 | Purpura fulminans | Acute DIC and hemorrhagic cutaneous necrosis |
Abbreviations: HELLP (hemolysis, elevated liver enzymes, and low platelets) RPOC (retained products of contraception), DIC (disseminated intravascular coagulation).
DIC can be acute (overt) or chronic (non-overt), representing the inability or the ability of the liver to synthesize consumed factors respectively. Acute DIC most commonly presents with clinical bleeding, thrombocytopenia, low fibrinogen, increased D-dimers, elevated prothrombin (PT) time, and activated partial thromboplastin times (aPTT). On the contrary, in chronic DIC, these hemostatic parameters may be normal or mildly deranged and thromboembolic phenomenon may be prevalent [54]. The International Society on Thrombosis and Hemostasis (ISTH) developed a scoring system for identification of overt DIC [54]. A cutoff score of five or above provided very high sensitivity (91%) and specificity (97%) for overt DIC and the higher the score, the greater the mortality [55]. Treatment of DIC should be tailored towards treating the underlying condition. Supportive therapy in the form of fresh frozen plasma, cryoprecipitate and platelet transfusion may be necessary.
17. Thrombotic microangiopathies
Thrombotic microangiopathies (TMA) are a diverse group of disorders including thrombotic thrombocytopenic purpura (TTP), Shiga toxin-mediated hemolytic uremic syndrome (ST-HUS), and atypical hemolytic uremic syndrome (aHUS). These disorders can be genetic or acquired and present at any age. The most important diagnostic criteria are presence of nonimmune microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and varying degree of multiorgan injury [56]. Pathologically, there is a prothrombotic state due to various factors that leads to vascular endothelial injury, leukocyte recruitment, and platelet activation which, in turn, leads to microthrombi formation; the end result is organ dysfunction [55]. Recently, complement hyperactivation has been identified as a common factor in the pathogenesis of these three disorders [57]. Several secondary TMAs have been described in association with underlying disorders such as sepsis, autoimmune disorders and malignancies [56]. In general, thrombocytopenia is evident on presentation, nevertheless, TMAs can develop in hospitalized patients after surgery, sepsis, pancreatitis, and pregnancy related conditions. TTP triggered by cardiac surgery and acute pancreatitis has been well described [58,59]. Likewise, infections, drugs, and pregnancy have been common antecedents of aHUS episodes [60,61]. A summary of TTP, ST-HUS, and aHUS is given in Table 2.
Table 2.
Thrombotic microangiopathies.
| TTP | ST-HUS | aHUS | |
|---|---|---|---|
| Epidemiology | 4–11 cases per million | 6.1 cases per 100 000 children under five Overall 1–2 cases per 100 000 |
2 cases per million [62] |
| Age | Predominantly adults; rarely children | Predominantly children; less commonly adults | Predominantly children; 30% middle-aged; less common adults |
| Hereditary or acquired | Both | Acquired | Both |
| Pathogenesis | Severe deficiency of ADAMTS13 resulting in UL-vWF multimers leading to microvascular occlusive thrombi formation | Shiga toxin mediated injury to renal endothelial cells; Shiga toxin binds to Gb3 on the target cells and stimulates them leading to increased secretion of IL-1, IL-8, TNF-α, UL-vWF | Selective activation of alternative complement pathway due to congenital or rarely acquired defects in CFH, CHI, MCP, TM, and C3; result is complement mediated endothelial and platelet activation with thrombus formation |
| Potential triggers | Sporadic, surgery, acute pancreatitis, sepsis, pregnancy | Eschericia coli serotypes, Shigella dysentreae | Sporadic, infection, drugs, pregnancy |
| Clinical presentation | 50% patients with neurological problems, fever is uncommon, acute renal failure rare | Prodrome of abdominal pain and bloody diarrhea, acute renal failure | Acute kidney injury, hypertension, can have neurological symptoms |
| Laboratory features | MAHA, thrombocytopenia, < 5% ADAMTS13 activity (48–90% of patients) | MAHA, thrombocytopenia, marked derangements of renal function, Shiga toxin identification in stool | MAHA, thrombocytopenia, Decreased C3 but normal C4 level, specific hereditary defects |
| Acuity and degree of thrombocytopenia | Rapid onset of severe thrombocytopenia, direct correlation of degree of ADAMTS13 deficiency and thrombocytopenia | Generally slower than TTP, moderate to severe thrombocytopenia | Moderate to severe thrombocytopenia, Less commonly insidious onset with fluctuating thrombocytopenia for weeks to months |
| Treatment | Plasma exchange therapy, steroids, immunosuppressive therapy | Supportive, dialysis | Eculizumab, immunosuppressive therapy |
| Prognosis | Relapse is rare except when there is severe deficiency of ADAMTS13 (50%) | Good if survive the acute episode | Response to therapy, risk of relapse and progression to ESRD depend on specific mutations |
Abbreviations: TTP (thrombotic thrombocytopenic purpura), ST-HUS (Shiga toxin-mediated hemolytic uremic syndrome), aHUS (atypical hemolytic uremic syndrome), UL-vWF (ultra large von Willibrand Factor), IL (interleukin), TNF (tumor necrosis factor), CFH (complement factor H), CHI (complement factor I), MCP (membrane cofactor protein), TM (thrombomodulin), MAHA (microangiopathic hemolytic anemia), ESRD (end stage renal disease).
18. Heparin induced thrombocytopenia
Heparin induced thrombocytopenia (HIT), also known as type 2 HIT, is an important differential diagnosis of thrombocytopenia in hospitalized patients not only because of its thrombotic complications and mortality [63], but also because it is often over investigated requiring unnecessary interruption in heparin therapy. HIT is often confused with a transient heparin-associated thrombocytopenia, also referred to as type 1 HIT; a non-immune mediated platelet aggregation as a result of direct interaction between heparin and platelets. Type 1 HIT is much more common than true HIT with an incidence of about 10–20% as compared to 1–3% of heparin-exposed patients, respectively. Type 1 HIT is mild (platelet count rarely < 100 × 103 per µl), non-thrombotic, self-limiting, and occurs between one to four days of heparin exposure [64].
HIT is a clinicopathological entity of acquired paradoxical hypercoagulability in the setting of thrombocytopenia. This transient prothrombotic state is induced by heparins (UF and LMWH). Heparin, being a polyanion, binds to positively-charged platelet factor 4 (PF4) causing a conformational change in PF4 forming neoepitopes against which HIT autoantibodies (usually IgG) are generated [65]. IgG-heparin-PF4 immune complexes are attached to the surface of platelets, crosslinking them via Fc gamma receptors (FcγRIIa). These complexes also attach to and activate monocytes (via FcγRI) and endothelial cells. The end result is platelet activation and thrombin generation [66].
Diagnosis of HIT is often challenging due to confusion with type 1 HIT and lack of a single highly sensitive and specific diagnostic test. Nevertheless, diagnostic accuracy can be increased with the use of a stepwise approach. Pretest probability of HIT, commonly known as ‘4T’ or Warkentin score, should be determined first (Table 3) [67]. A low score (< 4) has a very high negative predictive value of 97–99% and essentially rules out HIT, whereas intermediate and high scores are not conclusive because of poor positive predictive values [67,68]. Therefore, detection of HIT IgG antibodies should follow intermediate or high probability scores. Furthermore, strength of reactivity on immunoassay should be taken into account since greater optical density (> 1.5) correlates strongly with HIT [69]. A negative test rules out HIT because of a very high negative predictive value of 99% [67]. A positive test should be followed by a functional assay such as serotonin release assays.
Table 3.
The “4-T” score.
| Variable | Score |
||
|---|---|---|---|
| 0 | 1 | 2 | |
| Thrombocytopenia | < 30% decrease or nadir ≤ 10 × 103 per µl | 30–50% decrease or nadir 10–19 × 103 per µl | > 50% decrease or nadir ≥ 20 × 103 per µl |
| Onset | < 4 days without any recent exposure to heparin | > 10 days, or < 1 day if exposure to heparin 30–100 days prior | 5–10 days, or < 1 day if exposure to heparin within 30 days |
| Thrombosis | No | Doubtful | Yes |
| Alternative cause | Yes | Doubtful | No |
Adapted from Lo et al. [67]. High score: 6–8, Intermediate score: 5–6, Low score: ≤ 4.
Treatment should promptly be initiated if clinical suspicion is high or there is evidence of thromboembolism. Generally, argatroban, a direct thrombin inhibitor, is the first choice. Danapiroid, fondaparinux, and bivalirudin are also utilized. Platelet transfusions should be avoided. It is important to realize that warfarin is contraindicated in HIT because of increased risk of gangrene until the platelet count has normalized. Refractory or high risk cases may require IVIG, but the data is very limited [65].
19. Drug induced immune thrombocytopenia
The term ‘drug induced immune thrombocytopenia’ (DITP) refers only to immune mediated destruction of platelets rather than myelosuppression [70]. DITP is an important yet under-recognized cause of thrombocytopenia in hospitalized patients and true incidence is unknown, predominantly because many cases are often misdiagnosed as ITP [71]. DITP is an idiosyncratic immune mediated reaction to a wide spectrum of drugs. Investigators have proposed several mechanisms for this reaction. In one model, antibodies only react with platelets in the presence of sensitizing drug. In the absence of drug, the binding affinity of these antibodies is too low to cause significant thrombocytopenia. When the drug is present, the antibody-drug-platelet interaction is enhanced culminating in severe thrombocytopenia [72,73]. Other pathogenetic models include induction of a conformational change in native platelet proteins such as GP IIb-IIIa or GP Ib-V-IX by the drug and hapten-dependent antibodies [73].
Typically, DITP occurs in one to weeks following the exposure to the drug. However, onset can occur within one day of exposure if the drug was previously taken, indicating that drug-induced antibodies can remain inside the body for many months. Nadir platelet count can drop < 20 × 103 per µl with the potential to cause severe bleeding and death [70]. Platelet count begins to rise in one to two days after stopping the drug. It is often challenging to identify the implicated drug as patients may be receiving multiple drugs with the potential to cause DITP. Some of the common drugs known to cause DITP are summarized in Table 4.
Table 4.
Drugs implicated in DITP on various medical floors.
| Drugs implicated in DITP on various medical floors | ||||||
|---|---|---|---|---|---|---|
| Cardiology | Neurology | Hematology & oncology | Infectious disease | General medicine | Psychiatry | Miscellaneous |
| Aspirin/Clopidogrel | Aspirin/Clopidogrel | Bortezomib | Acyclovir | acetaminophen | Clonazepam | Methyldopa |
| Gp IIb/IIIa blockers (abciximab, tirofiban, eptifibatide) | Butobarbital | Allopurinol/colchicine Deferoxamine |
Ampicillin/amoxicillin | Cimetidine/Famotidine/pantoprazole /ranitidine | Haloperidol | Gold |
| Atorvastatin/simvastatin | Carbamazepine | Ferrous sulfate and gluconate | Cephalosporins (Ceftriaxone, cefotetan, cefazolin) | Desmopressin | Amitryptyline | INF-alpha |
| Amlodipine | Diazepam | Filgastrim | Ciprofloxacin/levofloxacin | Diclofenac | Lithium | Danazol |
| Amiodarone | Lamotrigine | Oxaliplatin | Azithromycin/clarithromycin | Glucagon | Mirtazepine | Infliximab |
| Bumetanide/furosemide | Levetiracetam | Protamine sulfate | Fluconazole | Glyburide | Oxcarbezepine | Hepatitis B vaccine |
| Captopril/Enalapril | Phenytoin | Rituxumab | Isoniazid/rifampin/ethambutol | Ibuprofen/indomethacin/naproxen | Olanzapine | MMR vaccine |
| Carvedilol | Valproic acid | Transtuzomab | Linezolid | Lidocaine | Prednisone | |
| Chlorothiazide/Hydrochlorothiazide | Acetazolamide | Bleomycin | Penecillin | Morphine | Salfasalazine | |
| Digoxin/Cardizem | Bromocriptine | Fludarabine | Piperacillin-tazobactum | Ondansetron | ||
| Hydralazine | Quinidine/quinine | |||||
| Nadolol | Sulfamethoxazole-trimethoprim | |||||
| Procainamide | Vancomycin | |||||
Drugs in bold letters are more commonly implicated.
20. Idiopathic thrombocytopenic purpura
Idiopathic thrombocytopenic purpura (ITP) is a diagnosis of exclusion which is characterized by immune mediated destruction of platelets or inhibition of their production in the bone marrow. Most cases are idiopathic but some secondary conditions (for example infections such as Helicobacter pylori, cytomegalovirus, varicella zoster virus etc.) have been associated with it [74]. In most cases, ITP is managed in the outpatient setting. However, when the platelet count drops significantly, for example < 20 × 103 per μl, hospitalization and acute intervention are warranted. Typically, intravenous steroid is effective first line therapy, but in resistant cases, IVIG, immune suppression, and rituximab are needed. Detailed discussion on ITP is described elsewhere [74].
21. Conclusion
Thrombocytopenia is a commonly encountered hematological abnormality in hospitalized patients. It is important to recognize that the differential diagnosis of thrombocytopenia is distinctive from those encountered in the outpatient setting. Therefore, an understanding of the pathophysiology and a stepwise approach will avoid delays in accurate diagnosis and allow rapid initiation of treatment.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Buckley MF, James JW, Brown DE, et al. A novel approach to the assessment of variations in the human platelet count. Thromb Haemost. 2000;83(3):480–484. [PubMed] [Google Scholar]
- [2].Maes M, Scharpé S, Cooreman W, et al. Components of biological, including seasonal, variation in hematological measurements and plasma fibrinogen concentrations in normal humans. Experientia. 1995;51(2):141–149. [DOI] [PubMed] [Google Scholar]
- [3].McMahon CM, Cuker A.. Hospital-acquired thrombocytopenia. Hosp Pract. 2014;42(4):142–152. [DOI] [PubMed] [Google Scholar]
- [4].Akca S, Haji-Michael P, de Mendonça A, et al. Time course of platelet counts in critically ill patients. Crit Care Med. 2002;30(4):753–756. [DOI] [PubMed] [Google Scholar]
- [5].Crowther MA, Cook DJ, Meade MO, et al. Thrombocytopenia in medical-surgical critically ill patients: prevalence, incidence, and risk factors. J Crit Care. 2005;20(4):348–353. [DOI] [PubMed] [Google Scholar]
- [6].Wang TY, Ou F-S, Roe MT, et al. Incidence and prognostic significance of thrombocytopenia developed during acute coronary syndrome in contemporary clinical practice. Circulation. 2009;119(18):2454–2462. [DOI] [PubMed] [Google Scholar]
- [7].Kaushansky K. Thrombopoiesis. Semin Hematol. 2015;52(1):4–11. [DOI] [PubMed] [Google Scholar]
- [8].Broudy VC, Lin NL, Kaushansky K. Thrombopoietin (c-mpl ligand) acts synergistically with erythropoietin, stem cell factor, and interleukin-11 to enhance murine megakaryocyte colony growth and increases megakaryocyte ploidy in vitro. Blood. 1995;85(7):1719–1726. [PubMed] [Google Scholar]
- [9].Mason KD, Carpinelli MR, Fletcher JI, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–1186. [DOI] [PubMed] [Google Scholar]
- [10].Zeuner A, Signore M, Martinetti D, et al. Chemotherapy-induced thrombocytopenia derives from the selective death of megakaryocyte progenitors and can be rescued by stem cell factor. Cancer Res. 2007;67(10):4767–4773. [DOI] [PubMed] [Google Scholar]
- [11].Lonial S, Waller EK, Richardson PG, et al. Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. Blood. 2005;106(12):3777–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mattia G, Vulcano F, Milazzo L, et al. Different ploidy levels of megakaryocytes generated from peripheral or cord blood CD34+ cells are correlated with different levels of platelet release. Blood. 2002;99(3):888–897. [DOI] [PubMed] [Google Scholar]
- [13].Mant MJ, Doery JC, Gauldie J, et al. Pseudothrombocytopenia due to platelet aggregation and degranulation in blood collected in EDTA. Scand J Haematol. 1975;15(3):161–170. [DOI] [PubMed] [Google Scholar]
- [14].Bobba RK, Doll DC. Platelet satellitism as a cause of spurious thrombocytopenia. Blood. 2012;119(18):4100. [DOI] [PubMed] [Google Scholar]
- [15].Senzel L, Chang C. Platelet phagocytosis by neutrophils. Blood. 2013;122(9):1543. [DOI] [PubMed] [Google Scholar]
- [16].Mannucci PM, Federici AB, Sirchia G. Hemostasis testing during massive blood replacement. A study of 172 cases. Vox Sang. 1982;42(3):113–123. [DOI] [PubMed] [Google Scholar]
- [17].Sihler KC, Napolitano LM. Complications of massive transfusion. Chest. 2010;137(1):209–220. [DOI] [PubMed] [Google Scholar]
- [18].Leslie SD, Toy PT. Laboratory hemostatic abnormalities in massively transfused patients given red blood cells and crystalloid. Am J Clin Pathol. 1991;96(6):770–773. [DOI] [PubMed] [Google Scholar]
- [19].Johansson PI, Stensballe J, Oliveri R, et al. How I treat patients with massive hemorrhage. Blood. 2014;124(20):3052–3058. [DOI] [PubMed] [Google Scholar]
- [20].Borgman MA, Spinella PC, Perkins JG, et al. The ratio of blood products transfused affects mortality in patients receiving massive transfusions at a combat support hospital. J Trauma. 2007;63(4):805–813. [DOI] [PubMed] [Google Scholar]
- [21].Chang JC. Review: postoperative thrombocytopenia: with etiologic, diagnostic, and therapeutic consideration. Am J Med Sci. 1996;311(2):96–105. [DOI] [PubMed] [Google Scholar]
- [22].Ruel M, Khan TA, Voisine P, et al. Vasomotor dysfunction after cardiac surgery. Eur J Cardio-Thoracic Surg. 2004;26(5):1002–1014. [DOI] [PubMed] [Google Scholar]
- [23].Satchidanand RY, Nandhara GS, Chowdary PP, et al. Immediate postoperative thrombocytopenia following elective abdominal aortic anuerysm repair and aortic vascular surgery for occlusive disease. Int J Angiol. 2002;11(2):73–76. [Google Scholar]
- [24].Solis RT, Kennedy PS, Beall AC, et al. Cardiopulmonary bypass. Microembolization and platelet aggregation. Circulation. 1975;52(1):103–108. [DOI] [PubMed] [Google Scholar]
- [25].Harker LA, Malpass TW, Branson HE, et al. Mechanism of abnormal bleeding in patients undergoing cardiopulmonary bypass: acquired transient platelet dysfunction associated with selective alpha-granule release. Blood. 1980;56(5):824–834. [PubMed] [Google Scholar]
- [26].Roy SK, Howard EW, Panza JA, et al. Clinical implications of thrombocytopenia among patients undergoing intra-aortic balloon pump counterpulsation in the coronary care unit. Clin Cardiol. 2010;33(1):30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Eckman PM, John R. Bleeding and thrombosis in patients with continuous-flow ventricular assist devices. Circulation. 2012;125(24):3038–3047. [DOI] [PubMed] [Google Scholar]
- [28].Waters AH. Post-transfusion purpura. Blood Rev. 1989;3(2):83–87. [DOI] [PubMed] [Google Scholar]
- [29].Becker T, Panzer S, Maas D, et al. High-dose intravenous immunoglobulin for post-transfusion purpura. Br J Haematol. 1985;61(1):149–155. [DOI] [PubMed] [Google Scholar]
- [30].Allen DL, Samol J, Benjamin S, et al. Survey of the use and clinical effectiveness of HPA-1a/5b-negative platelet concentrates in proven or suspected platelet alloimmunization. Transfus Med. 2004;14(6):409–417. [DOI] [PubMed] [Google Scholar]
- [31].Pavenski K, Webert KE, Goldman M. Consequences of transfusion of platelet antibody: a case report and literature review. Transfusion. 2008;48(9):1981–1989. [DOI] [PubMed] [Google Scholar]
- [32].Kitchens CS. Thrombocytopenia due to acute venous thromboembolism and its role in expanding the differential diagnosis of heparin-induced thrombocytopenia. Am J Hematol. 2004;76(1):69–73. [DOI] [PubMed] [Google Scholar]
- [33].Sautter RD. The urokinase-pulmonary embolism trial. JAMA J Am Med Assoc. 1974;227(10):1168. [DOI] [PubMed] [Google Scholar]
- [34].Hancox SH, Smith BC. Liver disease as a cause of thrombocytopenia. QJM. 2013;106(5):425–431. [DOI] [PubMed] [Google Scholar]
- [35].Fischereder M, Jaffe JP. Thrombocytopenia following acute acetaminophen overdose. Am J Hematol. 1994;45(3):258–259. [DOI] [PubMed] [Google Scholar]
- [36].Miyakawa K, Joshi N, Sullivan BP, et al. Platelets and protease-activated receptor-4 contribute to acetaminophen-induced liver injury in mice. Blood. 2015;126(15):1835–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ballard HS. The hematological complications of alcoholism. Alcohol Health Res World. 1997;21(1):42–52. [PMC free article] [PubMed] [Google Scholar]
- [38].Torres Duarte AP, Dong QS, Young J, et al. Inhibition of platelet aggregation in whole blood by alcohol. Thromb Res. 1995;78(2):107–115. [DOI] [PubMed] [Google Scholar]
- [39].Gernsheimer T, James AH, Stasi R. How I treat thrombocytopenia in pregnancy. Blood. 2013;121(1):38–47. [DOI] [PubMed] [Google Scholar]
- [40].McCrae KR. Thrombocytopenia in pregnancy. Hematology. 2010;2010(1):397–402. [DOI] [PubMed] [Google Scholar]
- [41].Sibai BM, Ramadan MK, Usta I, et al. Maternal morbidity and mortality in 442 pregnancies with hemolysis, elevated liver enzymes, and low platelets (HELLP syndrome). Am J Obstet Gynecol. 1993;169(4):1000–1006. [DOI] [PubMed] [Google Scholar]
- [42].Lindheimer MD, Taler SJ, Cunningham FG. Hypertension in pregnancy. J Am Soc Hypertens. 2010;4(2):68–78. [DOI] [PubMed] [Google Scholar]
- [43].Assinger A. Platelets and infection - an emerging role of platelets in viral infection. Front Immunol. 2014;5:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chelucci C, Federico M, Guerriero R, et al. Productive human immunodeficiency virus-1 infection of purified megakaryocytic progenitors/precursors and maturing megakaryocytes. Blood. 1998;91(4):1225–1234. [PubMed] [Google Scholar]
- [45].Speth C, Löffler J, Krappmann S, et al. Platelets as immune cells in infectious diseases. Future Microbiol. 2013;8(11):1431–1451. [DOI] [PubMed] [Google Scholar]
- [46].Neame PB, Kelton JG, Walker IR, et al. Thrombocytopenia in septicemia: the role of disseminated intravascular coagulation. Blood. 1980;56(1):88–92. [PubMed] [Google Scholar]
- [47].Kelton JG, Neame PB, Gauldie J, et al. Elevated platelet-associated IgG in the thrombocytopenia of septicemia. N Engl J Med. 1979;300(14):760–764. [DOI] [PubMed] [Google Scholar]
- [48].François B, Trimoreau F, Vignon P, et al. Thrombocytopenia in the sepsis syndrome: role of hemophagocytosis and macrophage colony-stimulating factor. Am J Med. 1997;103(2):114–120. [DOI] [PubMed] [Google Scholar]
- [49].Gando S, Nakanishi Y, Tedo I. Cytokines and plasminogen activator inhibitor-1 in posttrauma disseminated intravascular coagulation: relationship to multiple organ dysfunction syndrome. Crit Care Med. 1995;23(11):1835–1842. [DOI] [PubMed] [Google Scholar]
- [50].Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341(8):586–592. [DOI] [PubMed] [Google Scholar]
- [51].Liaw PC, Ito T, Iba T, et al. DAMP and DIC: the role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev. 2016;30(4):257–261. [DOI] [PubMed] [Google Scholar]
- [52].Kazzaz NM, Sule G, Knight JS. Intercellular interactions as regulators of NETosis. Front Immunol. 2016;7:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].White J. Snake venoms and coagulopathy. Toxicon. 2005;45(8):951–967. [DOI] [PubMed] [Google Scholar]
- [54].Siegal T, Seligsohn U, Aghai E, et al. Clinical and laboratory aspects of disseminated intravascular coagulation (DIC): a study of 118 cases. Thromb Haemost. 1978;39(1):122–134. [PubMed] [Google Scholar]
- [55].Taylor FB, Toh CH, Hoots WK, et al. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001;86(5):1327–1330. [PubMed] [Google Scholar]
- [56].Bakhtiari K, Meijers JCM, de Jonge E, et al. Prospective validation of the International Society of Thrombosis and Haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med. 2004;32(12):2416–2421. [DOI] [PubMed] [Google Scholar]
- [57].George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654–666. [DOI] [PubMed] [Google Scholar]
- [58].Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8(11):622–633. [DOI] [PubMed] [Google Scholar]
- [59].McDonald V, Laffan M, Benjamin S, et al. Thrombotic thrombocytopenic purpura precipitated by acute pancreatitis: a report of seven cases from a regional UK TTP registry. Br J Haematol. 2009;144(3):430–433. [DOI] [PubMed] [Google Scholar]
- [60].Chang JC, Shipstone A, Llenado-Lee MA. Postoperative thrombotic thrombocytopenic purpura following cardiovascular surgeries. Am J Hematol. 1996;53(1):11–17. [DOI] [PubMed] [Google Scholar]
- [61].Neumann HPH, Salzmann M, Bohnert-Iwan B, et al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet. 2003;40(9):676–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Adamski J. Thrombotic microangiopathy and indications for therapeutic plasma exchange. Hematology. 2014;2014(1):444–449. [DOI] [PubMed] [Google Scholar]
- [63].Lubenow N, Eichler P, Lietz T, et al. Lepirudin in patients with heparin-induced thrombocytopenia - results of the third prospective study (HAT-3) and a combined analysis of HAT-1, HAT-2, and HAT-3. J Thromb Haemost. 2005;3(11):2428–2436. [DOI] [PubMed] [Google Scholar]
- [64].Brieger DB, Mak KH, Kottke-Marchant K, et al. Heparin-induced thrombocytopenia. J Am Coll Cardiol. 1998;31(7):1449–1459. [DOI] [PubMed] [Google Scholar]
- [65].Greinacher A. Heparin-induced thrombocytopenia. N Engl J Med. 2015;373(19):1882–1884. [DOI] [PubMed] [Google Scholar]
- [66].Warkentin TE. Heparin-induced thrombocytopenia: pathogenesis and management. Br J Haematol. 2003;121(4):535–555. [DOI] [PubMed] [Google Scholar]
- [67].Lo GK, Juhl D, Warkentin TE, et al. Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006;4(4):759–765. [DOI] [PubMed] [Google Scholar]
- [68].Cuker A, Arepally G, Crowther MA, et al. The HIT Expert Probability (HEP) score: a novel pre-test probability model for heparin-induced thrombocytopenia based on broad expert opinion. J Thromb Haemost. 2010;8(12):2642–2650. [DOI] [PubMed] [Google Scholar]
- [69].Warkentin TE, Sheppard JI, Moore JC, et al. Quantitative interpretation of optical density measurements using PF4-dependent enzyme-immunoassays. J Thromb Haemost. 2008;6(8):1304–1312. [DOI] [PubMed] [Google Scholar]
- [70].George JN, Aster RH. Drug-induced thrombocytopenia: pathogenesis, evaluation, and management. Hematology. 2009;2009(1):153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Neylon AJ, Saunders PWG, Howard MR, et al. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol. 2003;122(6):966–974. [DOI] [PubMed] [Google Scholar]
- [72].Bougie DW, Wilker PR, Aster RH. Patients with quinine-induced immune thrombocytopenia have both “drug-dependent” and “drug-specific” antibodies. Blood. 2006;108(3):922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med. 2007;357(6):580–587. [DOI] [PubMed] [Google Scholar]
- [74].Cines DB, Bussel JB, Liebman HA, et al. The ITP syndrome: pathogenic and clinical diversity. Blood. 2009;113(26):6511–6521. [DOI] [PMC free article] [PubMed] [Google Scholar]