

Abstract
Key points
Mechanical signals play a critical role in the regulation of muscle mass, but the molecules that sense mechanical signals and convert this stimulus into the biochemical events that regulate muscle mass remain ill‐defined.
Here we report a mass spectrometry‐based workflow to study the changes in protein phosphorylation that occur in mouse skeletal muscle 1 h after a bout of electrically evoked maximal‐intensity contractions (MICs).
Our dataset provides the first comprehensive map of the MIC‐regulated phosphoproteome.
Using unbiased bioinformatics approaches, we demonstrate that our dataset leads to the identification of many well‐known MIC‐regulated signalling pathways, as well as to a plethora of novel MIC‐regulated events.
We expect that our dataset will serve as a fundamentally important resource for muscle biologists, and help to lay the foundation for entirely new hypotheses in the field.
Abstract
The maintenance of skeletal muscle mass is essential for health and quality of life. It is well recognized that maximal‐intensity contractions, such as those which occur during resistance exercise, promote an increase in muscle mass. Yet, the molecules that sense the mechanical information and convert it into the signalling events (e.g. phosphorylation) that drive the increase in muscle mass remain undefined. Here we describe a phosphoproteomics workflow to examine the effects of electrically evoked maximal‐intensity contractions (MICs) on protein phosphorylation in mouse skeletal muscle. While a preliminary phosphoproteomics experiment successfully identified a number of MIC‐regulated phosphorylation events, a large proportion of these identifications were present on highly abundant myofibrillar proteins. We subsequently incorporated a centrifugation‐based fractionation step to deplete the highly abundant myofibrillar proteins and performed a second phosphoproteomics experiment. In total, we identified 5983 unique phosphorylation sites of which 663 were found to be regulated by MIC. GO term enrichment, phosphorylation motif analyses, and kinase‐substrate predictions indicated that the MIC‐regulated phosphorylation sites were chiefly modified by mTOR, as well as multiple isoforms of the MAPKs and CAMKs. Moreover, a high proportion of the regulated phosphorylation sites were found on proteins that are associated with the Z‐disc, with over 74% of the Z‐disc proteins experiencing robust changes in phosphorylation. Finally, our analyses revealed that the phosphorylation state of two Z‐disc kinases (striated muscle‐specific serine/threonine protein kinase and obscurin) was dramatically altered by MIC, and we propose ways these kinases could play a fundamental role in skeletal muscle mechanotransduction.
Keywords: exercise, hypertrophy, mechanotransduction, phosphoproteomics, protein synthesis, skeletal muscle
Key points
Mechanical signals play a critical role in the regulation of muscle mass, but the molecules that sense mechanical signals and convert this stimulus into the biochemical events that regulate muscle mass remain ill‐defined.
Here we report a mass spectrometry‐based workflow to study the changes in protein phosphorylation that occur in mouse skeletal muscle 1 h after a bout of electrically evoked maximal‐intensity contractions (MICs).
Our dataset provides the first comprehensive map of the MIC‐regulated phosphoproteome.
Using unbiased bioinformatics approaches, we demonstrate that our dataset leads to the identification of many well‐known MIC‐regulated signalling pathways, as well as to a plethora of novel MIC‐regulated events.
We expect that our dataset will serve as a fundamentally important resource for muscle biologists, and help to lay the foundation for entirely new hypotheses in the field.
Abbreviations
- CAMK
Ca2+/calmodulin‐dependent protein kinases
- HPLC
high performance liquid chromatography
- Ig
immunoglobulin
- LFQ
label free quantification by mass spectrometry
- MAPK
mitogen activated protein kinases
- MIC
maximal‐intensity contraction
- MS
mass spectrometry
- mTOR
mechanistic target of rapamycin
- PKC
protein kinase C
- RP
reverse phase
- SPEG
striated muscle‐specific serine/threonine protein kinase
- TMT
tandem mass tags
Introduction
Comprising approximately 45% of the body's mass, skeletal muscles not only are the motors that drive locomotion, but also play a critical role in breathing, whole body metabolism, and maintaining a high quality of life (Seguin & Nelson, 2003; Izumiya et al. 2008). Both sedentary and active adults will lose 30–40% of their muscle mass by the age of 80, and this reduction is associated with disability, loss of independence, increased risk of morbidity and mortality, along with an estimated $18.5 billion in annual healthcare costs in the USA alone (Pahor & Kritchevsky, 1998; Proctor et al. 1998; Seguin & Nelson, 2003; Janssen et al. 2004). Thus, the development of therapies that can restore, maintain, and/or increase muscle mass are of great clinical and fiscal significance. To develop such therapies, however, we must establish a comprehensive understanding of the molecular mechanisms that regulate skeletal muscle mass.
Skeletal muscle mass can be regulated by a variety of different stimuli including nutrients, growth factors, and mechanical signals (Adams & Bamman, 2012; Bodine, 2013; Piccirillo et al. 2014). For instance, maximal‐intensity contractions (MIC), such as those which occur during resistance exercise, promote an increase in muscle mass (Egan & Zierath, 2013; Schoenfeld et al. 2016). Yet, the molecules that sense the mechanical information and convert this stimulus into the signalling events (e.g. phosphorylation) that drive the increase in muscle mass remain undefined (Goodman et al. 2015).
Mass spectrometry (MS) has emerged as a powerful analytical methodology that could shed light on how mechanical stimuli regulate muscle mass. For instance, Hoffman et al. (2015) recently used MS to map the phosphoproteomic alterations that occur in response to aerobic exercise. Importantly, it is known that aerobic exercise induces an increase in the oxidative capacity of skeletal muscle (Rivera‐Brown & Frontera, 2012), and the work of Hoffman et al. provided a tremendous amount of novel insight into the signalling events that potentially drive this. However, aerobic exercise does not induce a substantial increase in skeletal muscle mass (Farup et al. 2012), and thus, the primary signalling events that control the mechanical regulation of muscle mass were likely not to have been identified in the work by Hoffman et al. Indeed, numerous studies have shown that mechanistic target of rapamycin (mTOR) signalling is robustly activated by resistance exercise, and the activation of mTOR signalling is considered to be one of the key events through which resistance exercise induces an increase in muscle mass (Watson & Baar, 2014). Yet, the results presented by Hoffman et al. indicate that mTOR signalling was actually inhibited by the aerobic exercise stimulus that was employed in their study. Thus, a map of the phosphoproteomic alterations that occur in response to resistance exercise would likely provide an abundance of novel and important information for the field.
To successfully detect low stoichiometric phosphorylation events, phosphoproteomic studies generally involve extensive enrichment procedures and offline pre‐fractionation by HPLC (Grimsrud et al. 2012; Zhou et al. 2013; Sharma et al. 2014; Mondal et al. 2015; Riley & Coon, 2015). Importantly, skeletal muscles are composed of abundant contractile proteins (e.g. myosin, titin, etc.), and the abundance of these proteins can limit the total phosphoproteomic coverage that can be obtained in a single MS experiment (Murgia et al. 2015). For instance, titin contributes to approximately 16% of the total protein mass in skeletal muscle (Deshmukh et al. 2015). Titin is also an atypically large protein of > 35,000 amino acid residues and having a total mass to > 3 MDa (Opitz et al. 2003). Upon digestion with trypsin, titin alone produces 6197 peptides in silico without taking into account tryptic missed cleavages. In such a challenging matrix, offline HPLC fractionation produces some gains in total identified peptides and proteins (Cao et al. 2012), but even with these measures the extreme size and abundance of contractile proteins still hampers the effectiveness of subsequent MS analysis.
Here we report the use of two bottom‐up proteomics workflows for examining the effect of electrically evoked maximal‐intensity contractions (MIC) on protein phosphorylation in mouse skeletal muscle. Specifically, these workflows used either (i) a traditional proteomics method incorporating trypsin digestion, tandem mass tag (TMT) labelling to multiplex sample throughput, and reverse phase (RP) fractionation to increase overall MS sampling depth, or (ii) a centrifugation‐based fractionation method prior to trypsin digestion to separate the muscle proteome into two major fractions prior to TMT labelling and RP fractionation. Using the phosphoproteomic results of both workflows, we identified a total of 663 different phosphorylation sites that experienced major MIC‐induced alterations (greater than ±1.5‐fold and Benjamini–Hochberg adjusted P‐value ≤ 0.05). As such, we have generated the first comprehensive map of the MIC‐regulated phosphoproteome. We then demonstrate that our dataset, in conjunction with various bioinformatics tools, leads to identification of many well‐known MIC‐regulated signalling pathways, as well as to a plethora of novel MIC‐regulated events.
Methods
Ethical approval
A total of six male C57BL6 mice at 8–10 weeks of age were randomly assigned to different experimental groups. After tissue extraction, the mice were killed by cervical dislocation. All animals were housed in a room maintained with a 12–12 h light–dark cycle (lights on at 06.00–18.00 h) and, unless otherwise indicated, all animals received food and water ad libitum. The Institutional Animal Care and Use Committee at the University of Wisconsin‐Madison, which operates under the guidelines of the animal welfare act and the public health service policy on the humane care and use of laboratory animals, approved all of the methods employed in this study.
Maximal‐intensity contractions
The model previously described by O'Neil et al. (2009) was used to induce MIC in the tibialis anterior muscle. Specifically, ad libitum fed mice were first anaesthetized with 5% isoflurane in O2 and then maintained at a surgical level of anaesthesia with 1–4% isoflurane in O2. Before initiating any procedures, a determination for the proper level of anaesthetic administration was performed by pinching the pad of the foot and ensuring that the animal showed no signs of reactivity. Once the animal was appropriately anaesthetized, electrodes were placed on the sciatic nerve of the right leg and contractions were elicited by stimulating the sciatic nerve with an SD9E Grass stimulator (Grass Instruments, Quincy, MA, USA) at 100 Hz, 4–8 V pulse, for 10 sets of six contractions. Each contraction lasted 3 s and was followed by a 10 s rest period, and a 1 min rest period was provided between each set. The left tibialis anterior muscle was not stimulated and was used as a control. Following the last set of contractions, the animal was allowed to recover from the anaesthesia in a cage that contained water but was devoid of food. Previous studies have shown that this model of MIC leads to a progressive increase in the activation of mTOR signalling, and that the increase in mTOR signalling peaks 1 h after the last set of contractions (O'Neil et al. 2009). This is an important point because the activation of mTOR signalling is considered to be one of the key events that drive mechanically induced changes in muscle mass (Watson & Baar, 2014). Therefore, to ensure that signalling events associated with the activation of mTOR signalling were encapsulated in the phosphoproteomic analyses, the animal was reanaesthetized so that both the left (control) and the right (MIC) muscles could be collected at 1 h after the last set of contractions. All of the aforementioned interventions were performed between 10.00 and 15.00 h.
Tissue lysis and centrifugation
For the urea‐based lysis procedure in the first TMT 6‐plex experiment, the skeletal muscles were homogenized in 1 ml of lysis buffer A, which consisted of 8 m urea, 50 mm Tris (pH 8.0), a PhosSTOP tablet (Roche, San Jose, CA, USA), and a Complete Mini EDTA‐Free Protease Inhibitor Cocktail Tablet (Roche). For the fractionation procedure in the second TMT 6‐plex experiment, muscles were homogenized in 1 ml of buffer B, which consisted of 40 mm Tris (pH 7.5), 1 mm EDTA, 5 mm EGTA, 0.5% Triton X‐100, a PhosSTOP tablet (Roche), and a Complete Mini EDTA‐Free Protease Inhibitor Cocktail Tablet (Roche). Samples were homogenized with a Polytron for 20 s and then centrifuged at 6000 g for 1 min to remove bubbles and confirm complete homogenization. The pellet portion of each sample was then resuspended and the whole homogenate was incubated on ice for 30 min. For the fractionation procedure, the volume of the whole homogenate was measured and then transferred to a microcentrifuge tube to be centrifuged at 2000 g for 5 min. The supernatant was carefully transferred to a new tube and the pellet (i.e. the portion enriched with myofibrillar proteins) was saved in the original tube. The pellet was resuspended in 1 ml of buffer B, pipette flushed, and centrifuged at 2000 g for 5 min. The supernatant was discarded, and the procedure was repeated with an additional wash. The pellet was then resuspended to the same volume as the original whole homogenate. The original supernatant sample was centrifuged at 2000 g for 5 min, and the resultant supernatant was removed and transferred to a new tube. This procedure was repeated one additional time to remove any remaining contaminant proteins. In all cases, a 100 μl aliquot of each was saved for Western blotting and the remainder of each sample was reserved for MS analysis.
Western blot analysis
Western blot analyses were performed as previously described (Frey et al. 2014). Briefly, samples were subjected to electrophoretic separation by SDS‐PAGE. Following electrophoretic separation, proteins were transferred to a polyvinylidene fluoride membrane, blocked with 5% powdered milk in Tris‐buffered saline containing 0.1% Tween 20 (TBST) for 1 h followed by an overnight incubation at 4°C with primary antibody dissolved in TBST containing 1% bovine serum albumin. All primary antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). After an overnight incubation with the primary antibody, the membranes were washed for 30 min in TBST and then probed with a peroxidase‐conjugated secondary antibody for 1 h at room temperature. Following 30 min of washing in TBST, the blots were developed on film or with a Chemi410 camera mounted to a UVP Autochemi system (UVP, Upland, CA, USA) using regular enhanced chemiluminescence (ECL) reagent (Pierce: Thermo Fisher Scientific, Waltham, MA, USA) or ECL Prime reagent (GE Healthcare: Amersham, Piscataway, NJ, USA). Once the appropriate image was captured, the membranes were stained with Coomassie Blue to verify equal loading in all lanes.
Protein precipitation, enzymatic digestion and peptide desalting
The total amount of protein was determined using a Pierce BCA Protein Assay Kit. Proteins were precipitated by bringing the original sample solution to a 90% concentration of methanol by volume and centrifuging the sample at 12,000 g for 5 min. The methanol solution was removed and the remaining protein precipitate was resuspended in 8 m urea, 50 mm Tris (pH 8.0), 10 mm tris(2‐carboxyethyl)phosphine (TCEP) and 40 mm chloroacetamide and incubated for 30 min with shaking to completely reduce and alkylate the proteins. The sample was diluted to a concentration of 1.5 m urea with 50 mm Tris (pH 8.0) and digested with trypsin (1:50 enzyme:protein ratio) at 37°C for 15 h. The enzymatic digestion was quenched by acidifying the sample to pH < 2 with 10% trifluoroacetic acid (TFA). Strata‐X desalting columns (Phenomenex, Torrance, CA, USA) were prepared by flowing 1 ml of 100% acetonitrile (ACN) over the column, followed by 1 ml of 0.1% TFA. Individual samples were spun down and the acidic supernatant was collected and gravity filtered through the Strata‐X columns. The bound peptides were washed with 1 ml 0.1% TFA, followed by elution into a fresh tube using 500 μl of 40% ACN and 0.1% TFA, and finally an additional elution of 300 μl of 80% ACN and 0.1% TFA. Eluted peptides were dried down by vacuum centrifugation. A Pierce Quantitative Colorimetric Peptide Assay (Thermo Fisher Scientific) was performed to determine peptide concentrations prior to TMT labelling.
TMT labelling
A total of 1 mg of peptides for each of the six samples was incubated with 6‐plex tandem mass tags (TMT) reagents according to instructions available in the commercially available kit (Thermo Fisher Scientific) for the quantification of relative abundances of tryptic peptides. Samples were incubated with shaking for 3 h at room temperature. Samples were quenched using 5% hydroxylamine and incubated at room temperature for 15 min with shaking. An aliquot of all six samples was mixed in a 1:1 ratio across all channels and analysed using an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) to ensure complete TMT peptide labelling and compare peptide ratios in this ‘test mix’. These preliminary mixing ratios were used as a guide to create a final sample mix, where the six muscle samples were mixed at a 1:1 ratio. The final pooled sample containing TMT labelled peptides from all six samples was desalted using a Strata‐X desalting column. The pooled sample was enriched using Immobilized Metal Affinity Chromatography (IMAC) Ni‐NTA magnetic agarose beads (Qiagen). The resultant phosphopeptide sample and flow through non‐phosphopeptide sample were each fractionated using a reverse phase HPLC to produce 12 total phosphopeptide fractions and 12 total non‐phosphopeptide fractions. Each fraction was dried using a vacuum centrifuge and resuspended in MS‐grade water with 0.2% formic acid for subsequent mass spectrometry analysis.
Nano‐liquid chromatography–tandem mass spectrometry methods
Each sample in the first TMT experiment (without homogenate centrifugation) was introduced to an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) during a 90 min nano‐liquid chromatography separation using a nanoAcquity UPLC (Waters, Milford, MA, USA). A Top 15 MS method was used to analyse eluting peptides, using a 60,000 resolving power survey scan followed by 15 tandem mass spectrometry (MS/MS) scans collected at 15,000 resolving power. Peptides were fragmented using higher energy collisional dissociation (HCD) at 35% normalized collision energy (Kocher et al. 2009). Non‐phosphopeptide fractions were analysed with automatic gain control (AGC) targets of 1 × 106 and 5 × 104 for MS and MS/MS scans, respectively. MS maximum injection times for these fractions were set at 100 ms for MS scans and 200 ms for MS/MS. Phosphopeptide fractions were analysed with automatic gain control (AGC) targets of 1 × 106 and 1 × 105 for MS and MS/MS scans, respectively. These phosphopeptide fractions were also analysed with MS maximum injection times set at 50 ms for MS scans and 250 ms for MS/MS. Only peptides with charge states from +2 to +8 were selected for MS/MS using an exclusion duration of 40 s. Each phosphopeptide fraction was run in duplicate. Each sample in the second TMT experiment (which incorporated homogenate centrifugations steps) was analysed using an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) during a 90 min nano‐liquid chromatography separation using a Dionex UltiMate 3000 RSLCnano system (Thermo Fisher Scientific). Samples were analysed using an MS1 AGC target of 1 × 106 and maximum injection times of 100 ms. MS cycle time was set to 2 s, and MS runs used an exclusion duration of 45 s, a quadrupole isolation window of 1.2 m/z, and HCD fragmentation using a collision energy of 35% and a stepped collision energy of 5%. The scan range for MS acquisition was 300–1500 m/z, and peptides with charge states of +2 to +8 were selected for fragmentation. Phosphopeptide fragment ions were analysed in the Orbitrap at 60,000 resolving power, with an AGC target of 1 × 105 ions and 120 ms maximum injection times. Non‐phosphopeptide fractions’ peptide fragments were analysed in the Oribtrap with 30,000 resolving power with an AGC target of 1 × 105 ions and 75 ms maximum injection times. Each phosphopeptide fraction was analysed in duplicate.
MS data analysis
Data were searched using both MaxQuant (version 1.5.3.51) with the Andromeda search algorithm (Cox et al. 2014) and the COMPASS software suite (Wenger et al. 2011b). Thermo RAW files were searched against a Mus musculus target‐decoy database (UniProt, downloaded 08/14/2015). Peptide and phosphopeptide datasets were searched using a 50 ppm precursor mass tolerance and 0.02 Da fragment tolerance for b and y ions produced by HCD fragmentation. All fractions were searched with static carbadimomethyl of cysteine residues, static TMT 6‐plex modifications of peptide N‐termini and lysines, and dynamic methionine oxidation. Phosphopeptide fractions were searched with additional dynamic phosphorylation modifications of serine, threonine and tyrosine residues. Resulting peptide identifications were filtered to 1% false discovery rate (FDR). MaxQuant and COMPASS were used to obtain the 6‐plex TMT protein and phosphopeptide quantification for all of the 12 peptide fractions and 12 phosphopeptide fractions. Peptides were mapped back to their parent proteins using both MaxQuant and COMPASS and filtered to a 1% FDR at the protein level. COMPASS utilized the PhosphoRS localization algorithm (Taus et al. 2011) to localize phosphorylation to amino acid residues with a fragment tolerance of 0.02 Da automatically considering neutral loss peaks for HCD and considering a maximum of 200 maximum position isoforms per phosphopeptide. 6‐Plex TMT reporter ions present for each identified peptide were mapped to their parent proteins.
Statistical analysis
The reporter intensities for localized phosphopeptides and quantified proteins were log2 transformed and mean normalized to obtain the relative phosphopeptide and protein quantification for each tissue and condition. The datasets for both TMT experiments (experiment 1 and experiment 2, which incorporated the optimized sample centrifugation steps) were aligned in Microsoft Excel so that comparisons could be made between the phosphopeptides and proteins identified in each dataset; specifically, the phosphopeptides and proteins identified in the first 6‐plex TMT experiment's dataset were compared to the phosphopeptides and proteins identified in the supernatant and pellet fractions in the second TMT experiment. Because the first 6‐plex TMT experiment produced an overlapping proteome and phosphoproteome dataset with the soluble and pellet fractions of the second 6‐plex experiment, each peptide and phosphopeptide was observed with n = 3 or n = 6. Significantly regulated phosphopeptides and proteins were determined using the moderated t test documented in the LIMMA package in R (Smyth, 2004; Hoffman et al. 2015), and FDR was controlled using the Benjamini–Hochberg method (Benjamini & Hochberg, 1995). Proteins and phosphopeptides were determined to have experienced a ‘major MIC‐induced alteration’ if they had a fold‐change of ±1.5‐fold in response to MIC, with Benjamini–Hochberg adjusted P‐values < 0.05.
Label‐free quantification of skeletal muscle proteins
Label‐free quantification (LFQ) was performed using peptide and protein intensity outputs from MaxQuant. A minimum LFQ ratio count of 2 and fast LFQ were utilized. Spectra were searched using a 20 ppm first search peptide tolerance and a 4.5 ppm main search peptide tolerance. MS/MS spectra were analysed with a 20 ppm fragment match tolerance. Protein quantification was defined using unique and razor peptides for quantification. Large LFQ values were stabilized and required MS/MS for LFQ comparisons. Proteins identified in each TMT experiment were aligned and their relative LFQ values were compared to identify proteins observed in experiment 1, experiment 2, or both experiments.
Reactome, DAVID and GoMiner analysis
The phosphopeptides with localized sites of phosphorylation were mapped back to their parent proteins, and proteins that contained major MIC‐induced alterations in phosphorylation (greater than ±1.5‐fold change, Benjamini–Hochberg adjusted P ≤ 0.05) were used to identify the pathways, molecular functions, biological processes, and intracellular components that were overrepresented in samples that had been subjected to MIC. Reactome pathways were identified with the default workflow employed by Reactome (http://www.reactome.org). The Database for Annotation, Visualization and Integrated Discovery (DAVID) was used to identify Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, molecular functions and biological processes. The analyses with DAVID were performed while using all of the phosphoproteins observed in experiments 1 and 2 as the background (Huang da et al. 2009). The high‐throughput version of GoMiner was used to identify cellular compartments and these analyses were performed with the following criteria: background = all phosphoproteins observed in experiment 1 and experiment 2, data source = MGI (M. musculus), evidence codes = all, lookup setting = cross reference and synonym, and number of randomizations = 100.
Motif‐X analysis
Motif‐X was used to identify overrepresented amino acid sequences that surrounded the MIC‐regulated sites of phosphorylation (Schwartz & Gygi, 2005). Specifically, the identified phosphopeptides with localized sites of phosphorylation were filtered for those that showed a greater than 1.5‐fold increase, or greater than 1.5‐fold decrease, in response to MIC (Benjamini–Hochberg adjusted P‐values ≤ 0.05). The list of phosphopeptides was then submitted to Motif‐X and analysed with the following criteria: width = 15 residues, occurrences ≥ 25, and P ≤ 0.0001. All of the phosphopeptides that were identified in experiments 1 and 2 were used as the background.
NetworKIN analysis
The individual FASTA sequences for proteins that contained major MIC‐induced alterations in phosphorylation (greater than ±1.5‐fold change, Benjamini–Hochberg adjusted P‐values ≤ 0.05) were uploaded into the NetworKIN browser and then the regulated sites of phosphorylation were manually selected for analysis. Kinase predictions were performed and then filtered with the following criteria: minimum score = 0.22, maximum score difference = 10, and maximum number of predictions = 10.
Results
Validation of MIC‐induced signalling events and centrifugation methods prior to 6‐plex TMT sample preparation
In an effort to identify MIC‐regulated phosphorylation events we first employed a traditional MS phosphoproteomics workflow. Specifically, as explained in the methods, mouse skeletal muscles were subjected to a bout of MIC or the control condition (in triplicate) and then collected after a 1 h recovery period. The muscles were extracted and homogenized in a urea lysis buffer, and then prior to MS, the samples were subjected to Western blot analysis to confirm that the bout of MIC had induced signalling through two pathways that are known to be highly sensitive to mechanical stimulation (mTOR [P‐p70S6K(389)] and p38 [P‐p38(180/2)], Fig. 1; Hornberger et al. 2004; O'Neil et al. 2009). The samples were then subjected to MS analysis using the workflow described in Fig. 2. In total, 3381 phosphopeptides were identified, and 479 of these exhibited a major MIC‐induced alteration in abundance (greater than ±1.5‐fold and Benjamini–Hochberg adjusted P‐value ≤ 0.05, see experiment 1 data in Supplemental Table S1 in online Supporting Information). Upon further examination of the results, it became apparent that a large proportion of the identified phosphopeptides originated from highly abundant myofibrillar proteins. For example, 13% of all the phosphopeptides identified in the experiment were mapped to isoforms of titin. Therefore, to help increase our total phosphoproteome coverage, we performed a second MS experiment in which a new set of samples were homogenized in a Triton X‐100 lysis buffer and then centrifuged to pellet the myofibrillar proteins (Solaro et al. 1971). Western blot analysis and staining of all proteins with Coomassie blue demonstrated that individual proteins and phosphoproteins could be effectively separated into the pellet and supernatant fractions. Preliminary single‐shot MS experiments also confirmed that myofibrillar proteins such as titin could be successfully removed from the supernatant fraction (Fig. 3).
Figure 1. Validation of maximal‐intensity contraction‐induced signalling events and centrifugation methods prior to 6‐plex TMT sample preparation.
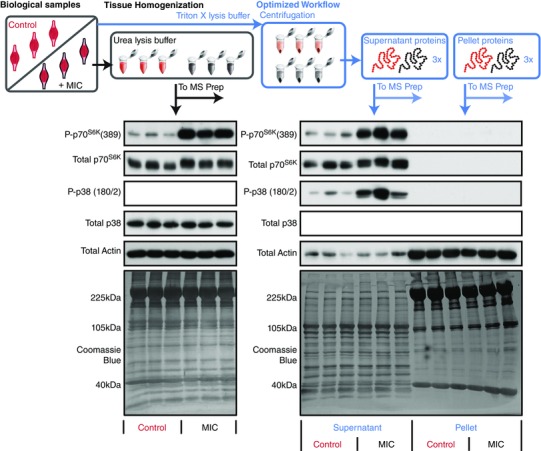
Tibialis anterior muscles were subjected to a bout of maximal‐intensity contractions (MIC) or the control condition. In the first workflow (black) the muscles were homogenized in a urea lysis buffer, while in the second workflow (blue) the muscles were homogenized in Triton X‐100 lysis buffer and then centrifuged to separate the proteome into pellet and supernatant fractions. Prior to MS analysis, all samples were subjected to Western blotting to confirm that the bout of MIC had induced the activation of signalling through mTOR [P‐p70S6K(389)] and p38 [P‐p38(180/2)]. Western blotting of actin, as well as Coomassie blue staining of all proteins, was also performed to illustrate the differences in the proteome of the pellet and supernatant fractions.
Figure 2. General experimental design for the proteomic and phosphoproteomic analyses.
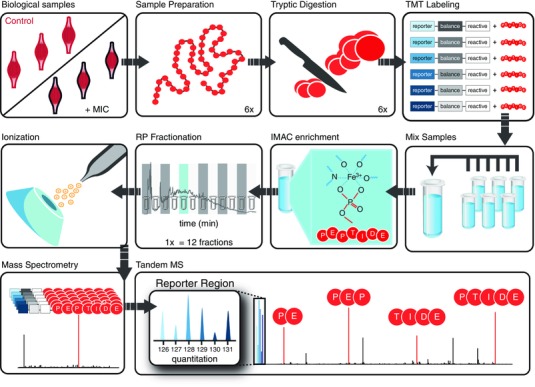
The right tibialis anterior muscles of mice were stimulated with a bout of maximal‐intensity contractions (MIC) while the left contralateral muscles were not stimulated and were used as controls. Tissues were homogenized, and their extracted proteins were denatured and tryptically digested. Each sample was labelled with one of six isobaric tandem mass tags (TMT) and mixed to produce a single pooled sample. Immobilized metal affinity chromatography (IMAC) was used to enrich the pooled sample for phosphopeptides. The enriched and unenriched samples were then fractionated offline by reverse phase (RP) chromatography. Twelve phosphopeptide fractions and 12 non‐phosphopeptide fractions were analysed by mass spectrometry. Peptides were isolated and fragmented during each instrument run, revealing peptide sequence information and generating reporter ions which revealed the relative abundance of each peptide across the six samples.
Figure 3. Assessment of titin and myosin abundance in pellet and supernatant fractions.
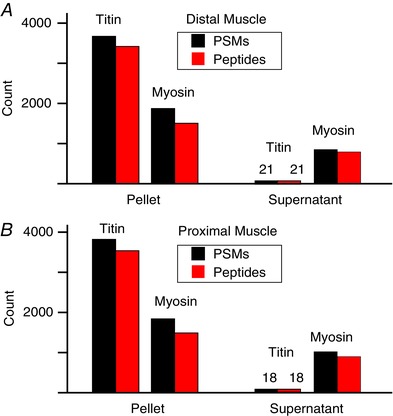
A single mouse plantar flexor muscle complex was split into distal and proximal halves. Each of the halves was centrifuged to separate the proteome into pellet and supernatant fractions and then prepared separately for MS analysis (without TMT labelling or phosphopeptide enrichment). The number of peptide spectral matches (PSMs) and unique peptide sequences identified for all titin and myosin isoforms were compared between the pellet and supernatant fractions. These PSM and peptide numbers were compared between the distal (A) and proximal (B) halves of the muscle tissue.
Centrifugation‐based fractionation adds to proteome and phosphoproteome characterization
Figure 4 A shows the overlap of phosphopeptides with localized phosphorylation sites identified in the supernatant and pellet fractions (denoted as experiment 2 supernatant or pellet fraction) compared to the phosphopeptides identified in the first experiment which lacked the fractionation step (experiment 1). In the first experiment, 3381 phosphopeptides were identified, while in the second experiment, the identifications increased to a total of 3836 phosphopeptides. Additionally, Fig. 4 B shows the overlap in total proteins that were quantified in the two 6‐plex TMT experiments, with > 80% overlap between the pelleted proteins and those identified in the first experiment. On the other hand, the supernatant fraction contained an additional 1490 proteins that were not detected in either experiment 1 or the pellet fraction of experiment 2.
Figure 4. Comparison of the phosphopeptides and proteins that were identified with the different sample preparation methods.
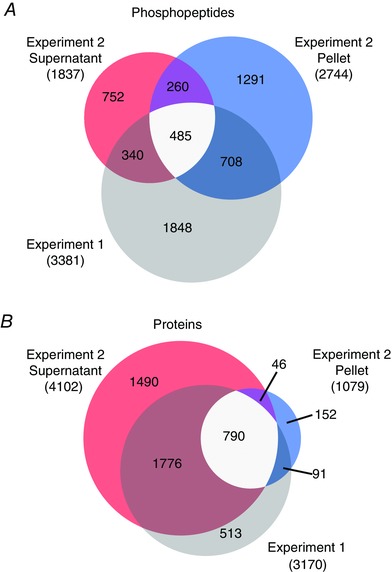
Mouse tibialis anterior muscles were subjected to a bout of maximal‐intensity contractions (MIC) or the control condition and then prepared for MS analysis. Samples in experiment 1 (n = 3 per group) were prepared via homogenization in a urea lysis buffer, while samples in experiment 2 (n = 3 per group) were prepared via homogenization in a Triton X‐100 buffer and then further separated by centrifugation into supernatant and pellet fractions. The differently prepared samples/fractions from each experiment were subjected to separate 6‐plex TMT MS analyses. Venn diagrams were generated to compare the unique phosphopeptide (A) and protein (B) populations that were quantified in experiment 1 (grey) and the supernatant (red) and pellet (blue) fractions in experiment 2. Intermediately shaded regions indicate the phosphopeptides or proteins that were observed in multiple experiments.
To further investigate our depth of proteome coverage, we used protein LFQ to estimate the abundance of proteins identified across both TMT experiments and coded the proteins according to whether they were observed in experiment 1, experiment 2, or both (Fig. 5 and Supplemental Table S2) (Cox et al. 2014). We then used DAVID to identify the biological processes and cellular components that were enriched in each quartile of the relative abundance curve (Huang da et al. 2009). The results indicated that the most abundant proteins were involved in biological processes such as actin–myosin filament sliding, muscle contraction, and glucose metabolism, and that these proteins were localized to muscular components such as the sarcomere, myosin complexes, and contractile fibres. Less abundant proteins were involved with biological processes such as protein translation, post‐transcriptional regulation and RNA splicing, while the least abundant proteins were principally involved with nuclear‐mediated processes and mRNA processing (primarily localized to chromatin and nuclear protein complexes). It is also worth noting that over 57% of the proteins identified in the lowest abundance quartile were only detected in experiment 2. In other words, the results indicate that the Triton X‐100–centrifugation workflow helped to increase the total coverage of low‐abundance proteins across the experiments.
Figure 5. Rank ordered protein abundance and GO term analysis.
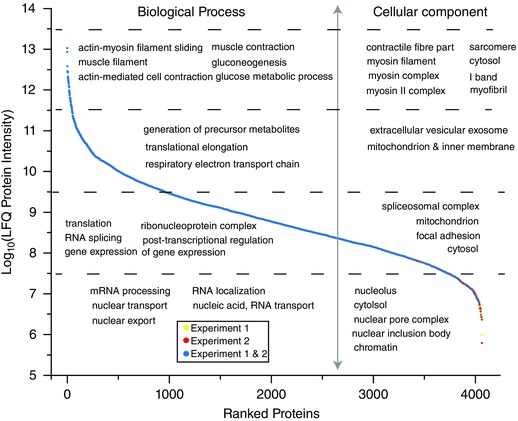
The proteins identified in the first (experiment 1) and second (experiment 2) workflows were analysed by MaxQuant for label‐free quantification (LFQ) of relative protein abundance. Proteins were rank ordered by their LFQ intensities and coloured according to whether they were observed in experiment 1 (yellow), experiment 2 (red), or both experiments (blue). Each quartile of proteins was also analysed for overrepresentation of gene ontology (GO) terms that corresponded to biological processes and cellular components. GO terms in each quartile were displayed if they were significantly overrepresented in the dataset.
MIC phosphoproteomics quantitation and GO term enrichment
The TMT reporter ions for each peptide were used to compare the relative fold‐change of that peptide's abundance between the MIC and control samples. The phosphopeptide abundances were normalized to protein levels and directly compared, while non‐phosphopeptides were mapped back to their parent proteins to assess MIC‐induced changes in the proteome. The results from the first TMT experiment were merged with the results of the second TMT experiment to generate a combined dataset that contained quantitative information for 4858 proteins and 5684 phosphopeptides that contained 5983 unique phosphorylation sites (n = 3–6 independent samples per group). Phosphopeptides and proteins with a greater than ±1.5‐fold change and Benjamini–Hochberg adjusted P‐values of ≤ 0.05 were considered to have experienced a major MIC‐induced alteration. Using these criteria, no proteins were concluded to have experienced a major MIC‐induced alteration in abundance (Supplemental Table S3). In stark contrast, 621 different phosphopeptides (corresponding to 313 different phosphorylated proteins) revealed major MIC‐induced alterations in phosphorylation (Supplemental Table S1). As shown in Fig. 6 A, the majority of these alterations (531 phosphopeptides) corresponded to increases in phosphorylation (blue region); however, a considerable number (90 phosphopeptides) also revealed decreases in phosphorylation (red region). Within the 621 phosphopeptides that changed in abundance, phosphorylation was successfully localized to 663 distinct sites. Through the use of Perseus (ww.perseus‐framework.org), it became apparent that the MIC‐regulated network of phosphorylation sites was quite unique. For instance, < 50% of the MIC‐regulated phosphorylation sites have been annotated in PhosphoSitePlus, and only 12 of the phosphorylation sites have known upstream kinases. Finally, Western blot analyses were performed in an effort to confirm that specific phosphorylation sites which were concluded to have experienced no change (threonine 287 on CAMK2β), a major increase (serine 86 on heat shock protein 27 (HSP27)), or a major decrease (serine 229 on glucocorticoid receptor) in phosphorylation were in fact modified as determined by MS (Fig. 6 B).
Figure 6. Overall analysis of the effect of maximal‐intensity contractions on phosphopeptide abundance.
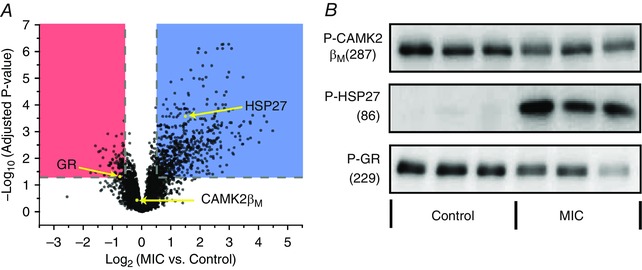
In two independent experiments, mouse tibialis anterior muscles were subjected to a bout of maximal‐intensity contractions (MIC) or the control condition and then prepared for MS analysis. In the first experiment (n = 3 per group) the samples were homogenized in a urea lysis buffer, while in the second experiment (n = 3 per group) the samples were homogenized in a Triton X‐100 lysis buffer and then centrifuged to separate the proteome into pellet and supernatant fractions. A, the results from the first and second experiment were merged and the fold‐changes (MIC/Control) of the phosphopeptides were log2 transformed and plotted versus their corresponding –log10 P‐values. Phosphopeptides that revealed major MIC‐induced alterations (> 1.5‐fold, P ≤ 0.05) are highlighted in blue (increased) or red (decreased). B, Western blot analysis was performed on specific phosphorylation sites which were concluded by MS to have experienced no change (threonine 287 on calmodulin kinase 2 (CAMK2β)), an increase (serine 86 on heat shock protein 27 (HSP27)), or a decrease (serine 229 on glucocorticoid receptor (GR)) in phosphorylation. Western blot analysis for actin was used to verify equal loading of protein in all lanes.
To examine which pathways were regulated by MIC we applied the Reactome and DAVID/KEGG algorithms to the list of proteins that experienced major MIC‐induced alterations in phosphorylation. The top 15 most highly overrepresented Reactome and KEGG pathways included signatures for the regulation of mTOR, mitogen activate protein kinase (MAPK), and insulin signalling, ubiquitin‐mediated protein degradation and various immune responses (Fig. 7 A and B). These results were mirrored by strong signatures for alterations in molecular functions such as kinase activity and ubiquitin–protein transferase activity, as well as biological processes such as the regulation of transcription, translation and proteolysis (Fig. 7 C and D). Finally, we also used GoMiner to identify which cellular compartments were overrepresented with proteins that experienced major MIC‐induced alterations in their phosphorylation state and then manually overlaid the results on a schematic representation that included many of the cellular compartments that have been implicated in mechanotransduction (Fig. 7 E) (Hornberger & Esser, 2004; Lyon et al. 2015). Unexpectedly, the results indicated that cellular compartments such as focal adhesions and the dystrophin‐associated glycoprotein complex were not overrepresented with proteins that experienced major MIC‐induced alterations in phosphorylation. On the other hand, myofibrillar proteins were found to be highly enriched with MIC‐induced changes in phosphorylation and, of particular interest, it was determined that > 74% of the identified Z‐disc proteins exhibited widespread MIC‐induced alterations in their phosphorylation state. Based on this observation, it appears that the Z‐disc is a particularly hot‐site for MIC‐induced signalling events.
Figure 7. Characteristics of the phosphoproteins that are regulated by maximal‐intensity contractions.
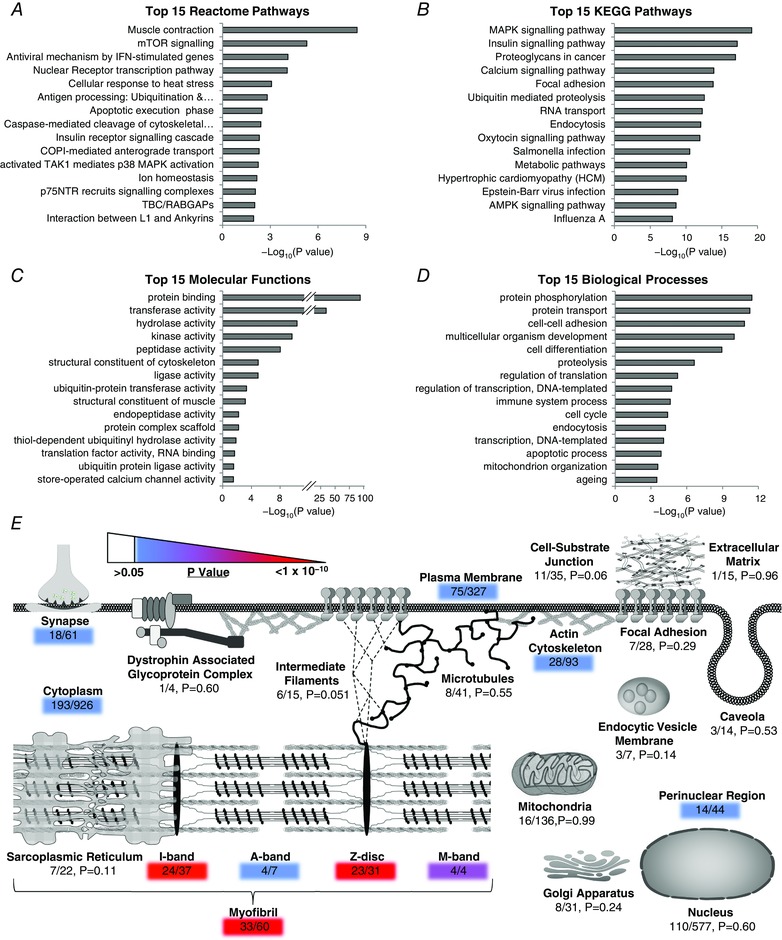
Phosphopeptides that revealed maximal‐intensity contraction (MIC)‐induced alterations (> 1.5‐fold, P ≤ 0.05) were mapped back to their parent proteins and the list of proteins was submitted to (A) Reactome for identification of the 15 most highly overrepresented pathways, or (B–D) DAVID for identification of the 15 most highly overrepresented KEGG pathways (B), molecular functions (C) and biological processes (D). The same list of proteins was submitted to GoMiner to identify which cellular compartments were overrepresented with proteins that experienced MIC‐induced alterations in phosphorylation. The results were manually overlaid on a schematic that includes many of the cellular compartments that have been implicated in skeletal muscle mechanotransduction (E). The colour of the shaded numbers highlights significantly overrepresented cellular compartments according to their associated P‐value. The ratio of proteins within each compartment that experienced MIC‐induced alterations in their phosphorylation state are also highlighted.
Substrate motif analysis
The list of sites that revealed major MIC‐induced alterations in phosphorylation was divided into sites that revealed an increase or a decrease in phosphorylation. These sites were then analysed with Motif‐X to identify overrepresented amino acid sequences that surrounded the site of phosphorylation. The results of these analyses revealed that four different sequence patterns were highly overrepresented in the dataset (Fig. 8 A). Three of the sequence patterns (PxSP – where x is any amino acid, SP and TP) conform to the recognition motif that is phosphorylated by proline directed kinases such as the MAPK and cyclin‐dependent kinase family members (Pearson et al. 2001; Rust & Thompson, 2011). The other sequence pattern (RxxS) conforms to the recognition motif that is phosphorylated by basophilic kinases such as protein kinase A, protein kinase C (PKC), and Ca2+/calmodulin‐dependent protein kinase 2 (CAMK2) family members (Pearson & Kemp, 1991; Rust & Thompson, 2011). Interestingly, this motif was overrepresented in both the list of sites that showed an increase and the list of sites that showed a decrease, in phosphorylation which suggests that they are regulated by distinct sets of kinases/phosphatases.
Figure 8. Phosphorylation sequence motif analysis and kinase–substrate predictions.
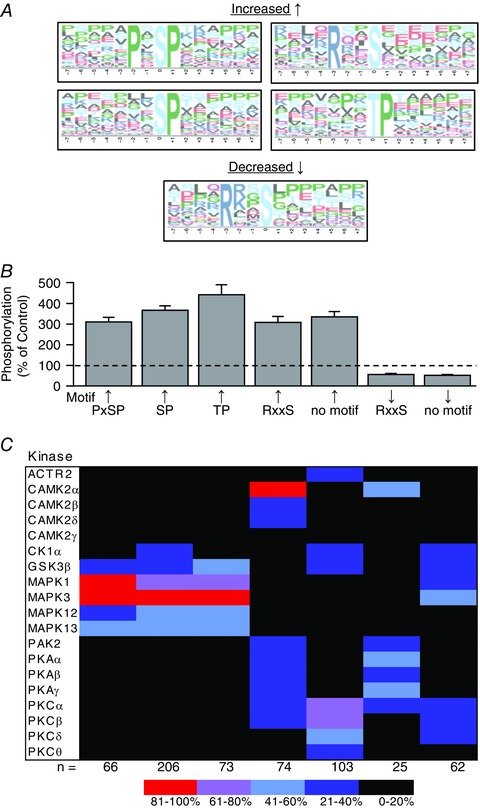
The phosphorylation sites that revealed maximal‐intensity contraction (MIC)‐induced alterations in phosphorylation (> 1.5‐fold, P ≤ 0.05) were grouped according to whether they showed an increase or decrease in phosphorylation. A, overrepresented sequence motifs within each of these groups were identified with Motif‐X (P ≤ 0.0001). B, the average change in phosphorylation for the sites within each motif cluster was plotted on a graph (values are the mean + SEM). C, NetworKIN was used to predict which kinases regulate the phosphorylation sites within each of the motif clusters and the results were displayed on a heat map. The colour of the boxes in the heat map represent the percentage of time that the kinase appeared in the top 10 list of kinases that were predicted to phosphorylate the sites within a given motif cluster. The values at the bottom of the heat map indicate the total number of sites within each motif cluster that were successfully analysed by NetworkKIN.
Prediction of regulated kinases
To investigate which kinases controlled the phosphorylation of the overrepresented motifs we first utilized NetworKIN, which combines protein‐associated network and preferred recognition motif information to predict the likelihood of kinase–substrate relationships (Horn et al. 2014). Specifically, in these analyses, we first clustered the different phosphorylation sites according to which, if any, overrepresented motif they contained, and then the average magnitude of change in phosphorylation for each cluster was determined (Fig. 8 B). Next, each substrate within a given cluster was individually analysed with NetworKIN and a maximum of the 10 most likely kinases were identified. Within each cluster, the number of times that a given kinase was identified was divided by the total number of substrates in that cluster. The results of these analyses were used to calculate the prediction frequency for each kinase within the different motif clusters, and this information was subsequently displayed as a motif heat map. Finally, the motif heat map was filtered to retain only the kinases that scored a > 20% prediction frequency and were identified by our MS analyses as being present in skeletal muscle (Fig. 8 C).
The results of the NetworKIN analyses revealed very high prediction frequencies (> 80%) for the activation of MAPK1 (ERK2), MAPK3 (ERK1) and CAMK2α, and moderately high prediction frequencies (61–80%) for the activation of PKCα and, PKCβ. Fairly strong prediction frequencies (41–60%) were also observed for alterations in the activity of GSK3β, MAPK12 (p38γ), MAPK13 (p38δ), PKAα, PKAγ and PKCδ. It's worth noting that many of these predictions are consistent with the results of previous studies. For instance, a large number of studies have shown that intense muscle contractions induce the activation of signalling through MAPK1, MAPK3, MAPK12 and MAPK13 (Kramer & Goodyear, 2007). A smaller body of evidence also indicates that intense muscle contractions can promote an increase in PKC and CAMK2 activity (Richter et al. 1987; Chin, 2005). On the other hand, we are not aware of any studies that have clearly addressed whether intense muscle contractions regulate the activity of protein kinase A.
The mouse genome encodes over 500 different kinases and the current version of NetworKIN (version 3.0) can perform predictive analyses on 213 of these (Caenepeel et al. 2004). In general, the kinases examined by NetworKIN are included because they have been highly studied, and their preferred recognition motifs have been firmly defined. However, not all highly studied kinases are included in the NetworKIN analyses. For instance, the preferred recognition motif of mTOR remains ill‐defined, and thus, mTOR is not incorporated in the NetworKIN analyses. These are important points because the limitations in kinase coverage, and the reliance on a priori knowledge, restricts the ability of programs like NetworKIN to identify less‐studied or novel signalling pathways. Therefore, in an effort to expand our prediction of regulated kinases, we identified which kinases experienced major MIC‐induced alterations in their phosphorylation state. The rationale for this approach was based on previous studies which have shown that the activity of individual kinases is often regulated by changes in their own phosphorylation state (Nolen et al. 2004). Therefore, to identify these kinases, we searched the list of proteins that experienced major MIC‐induced alterations in phosphorylation for members that possess protein kinase activity and/or function as part of a multi‐subunit protein kinase complex. In total, these analyses led to the identification of 23 different kinases (Fig. 9). For instance, we determined that the phosphorylation of the T320 residue on MAPK‐activated protein kinase 2 was significantly elevated in muscles that had been subjected to MIC. This result is worth highlighting because (i) the phosphorylation of the T320 residue is known to regulate MAPK‐activated protein kinase 2 activity (Ben‐Levy et al. 1995), and (ii) it demonstrates that assessing changes in the phosphorylation state of individual kinases can result in predictions that would have otherwise gone unnoticed with programs such as NetworKIN.
Figure 9. Maximal‐intensity contractions promote widespread changes in the phosphorylation of the CAMK family of protein kinases.
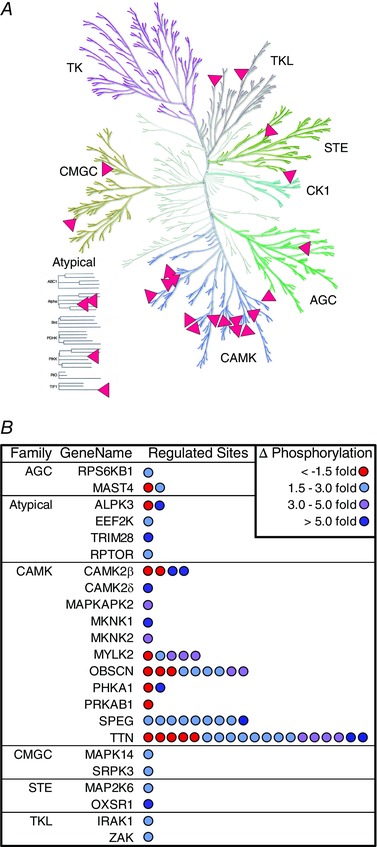
Phosphopeptides that revealed maximal‐intensity contraction (MIC)‐induced alterations (> 1.5‐fold, P ≤ 0.05) were mapped back to their parent proteins and then the list of proteins was interrogated for members that function as protein kinases or as part of a multi‐subunit protein kinase complex. A, Kinome Render (58) was used to plot the matches on a phylogenetic tree of the human kinome (illustration reproduced courtesy of Cell Signaling Technology, Inc.). B, the list of the matching proteins was grouped by protein kinase family and the number of regulated phosphorylation sites on each protein was represented with individual dots. The colour of the dots indicate the magnitude of change (∆) in phosphorylation that occurred on each site.
Next, we sought to gain further insight into the types of kinases that experienced major MIC‐induced alterations in phosphorylation, and thus, we plotted the position of the regulated kinases on a phylogenetic tree of the human kinome. As shown in Fig. 9 A, this visualization‐based approach revealed that a large proportion of regulated kinases were members of the CAMK family of protein kinases. Moreover, we found that the number of regulated phosphorylation sites on the CAMK family members was very high, with nearly 80% of the kinase phosphorylation events occurring on members of this family (Fig. 9 B). Amongst these family members were kinases which have been widely implicated in the regulation of contraction‐induced signalling events such as titin (Herzog, 2014), as well as less‐studied kinases that have never been associated with contraction‐induced signalling such as the striated muscle‐specific serine/threonine‐protein kinase (SPEG) and obscurin (Fig. 10). The potential connection of SPEG and obscurin with MIC‐induced signalling events was a particularly intriguing observation, and this point will be further addressed in Discussion.
Figure 10. Domain architecture of obscurin and SPEG and the regulation of their phosphorylation by maximal‐intensity contractions.
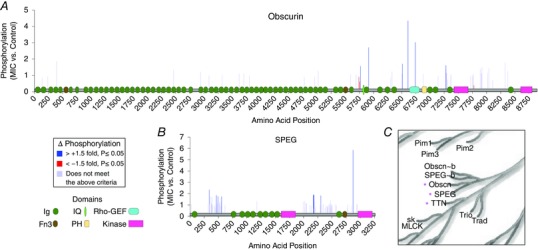
A and B, schematic illustration of the domains that are found on obscurin (A) and SPEG (B). The illustrations include: immunoglobulin I‐set (Ig); fibronectin type III (Fn3); plexstrin homology (PH); IQ calmodulin‐binding motif (IQ); RhoGEF; and protein kinase domains. The illustrations were overlaid on bar graphs which display the position and relative phosphorylation status of all sites that were identified in muscles that had been subjected to a bout of maximal‐intensity contractions (MIC). The sites that revealed a MIC‐induced increase (> +1.5‐fold, P ≤ 0.05) or decrease (> −1.5‐fold, P ≤ 0.05) in phosphorylation are indicated with blue and red bars, respectively. Sites which did not meet these criteria are shown in grey. C, a magnified region from the phylogenetic tree of the human kinome highlights the close relatedness of obscurin (Obscn), SPEG and titin (TTN). The illustration was reproduced courtesy of Cell Signaling Technology, Inc.
Discussion
While iTRAQ 4‐ or 8‐plex, and TMT 6‐ through 10‐plex tags differ in their structure, characteristic fragmentation patterns, and use of heavy isotopes, the increased throughput offered by these isobaric tags has been adopted by the proteomics community as a means for enhancing the reproducibility and precision of proteome and post translational modification quantification (Pichler et al. 2010, 2011; Ting et al. 2011; Wenger et al. 2011a; Erickson et al. 2017). Here we report a comparative analysis of sample preparation methods for skeletal muscle phosphoproteome using isobaric TMT chemical labels. Using a 6‐plex quantitative proteomics design we were able to measure changes in phosphopeptide and protein abundance in skeletal muscles that had been subjected to a bout of MIC. Our initial TMT experiment examined muscle tissue samples which were prepared using a traditional bottom‐up proteomics workflow without protein depletion steps. While we identified 479 phosphopeptides (containing 497 phosphorylation sites) that experienced major MIC‐induced alterations in phosphorylation, the majority of these sites mapped back to highly abundant structural and myofibrillar proteins such as titin and myosin. Therefore, to help increase our detection of phosphorylation events due to MIC, we performed a second MS experiment in which a new set of samples were homogenized in a Triton X‐100 lysis buffer and then centrifuged to pellet the myofibrillar proteins. This centrifugation procedure was inserted prior to enzymatic digestion, and it allowed us to split each sample into pellet and supernatant fractions, which were then prepared separately and analysed by MS. Preliminary studies demonstrated that a higher proportion of myofibrillar peptides were obtained from the pellet fractions. By incorporating this centrifugation‐based procedure into a new TMT 6‐plex experiment, we successfully identified an additional 1688 proteins. Importantly, with the addition of these proteins we were able to generate a rank ordered abundance list for over 4000 proteins in adult skeletal muscle (Supplemental Table S2), and we expect that this list will serve as a very valuable resource for muscle biologists.
By incorporating the centrifugation‐based procedure into a new TMT 6‐plex experiment, we also successfully identified an additional 2303 phosphopeptides (2492 phosphorylation sites) from skeletal muscle tissue, and 168 of the additional phosphopeptides (188 sites) were shown to have experienced a major MIC‐induced alteration in their phosphorylation state. The boost in identified phosphorylation sites with the second TMT experiment allowed us to piece together a more comprehensive map of the signalling events that are altered in response to MIC. We then used our dataset, and various bioinformatics tools, to develop a better understanding of the pathways that are regulated by MIC. Many of the results from the bioinformatics analyses pointed towards well‐known contraction regulated pathways such as the activation of MAPK and mTOR signalling (e.g. compare the results in Figs 1 and 7 A and B). The identification of the known signalling pathways lent confidence to the more novel outcomes of our analyses. For instance, one of the most striking observations was that myofibrillar proteins were highly enriched with major MIC‐induced alterations in phosphorylation. Specifically, we identified a total of 1607 different phosphorylated proteins, of which only 604 were annotated to the term ‘myofibril’. Yet, nearly 20% of the major MIC‐induced alterations in phosphorylation occurred on this small subset of proteins. Further analysis revealed that the majority (∼75%) of the ‘myofibril’ proteins that experienced major changes in phosphorylation were also annotated with the term ‘Z‐disc’. In other words, our results indicated that the Z‐disc is very heavily regulated by MIC‐induced phosphorylation events.
The basic contractile machinery of skeletal muscle is organized into structures called sarcomeres, and the outer boundary of individual sarcomeres is defined by the Z‐disc. The Z‐disc is directly linked to all of the major contractile proteins within the sarcomere (Pyle & Solaro, 2004; Frank et al. 2006). Through numerous protein–protein interactions, the Z‐disc facilitates the longitudinal transmission of forces from one sarcomere to the next, as well as the lateral transmission of forces from the sarcomere to costameres (functional analogues of focal adhesions) which lie at the periphery of the muscle fibre (Frank et al. 2006). With such a fundamental role in force transmission, it should not be surprising that the Z‐disc has been widely considered as an ideal site for the conversion of mechanical information into biochemical signals (i.e. mechanotransduction) (Pyle & Solaro, 2004; Frank et al. 2006; Frank & Frey, 2011). Our finding that the Z‐disc is heavily regulated by MIC‐induced phosphorylation events is consistent with this notion, and it raises questions about the kinase(s) that regulate these phosphorylation events.
One kinase that might regulate the MIC‐induced changes in Z‐disc phosphorylation is titin. Titin is an attractive candidate because one end of titin is anchored to the Z‐disc while the other end is anchored to the centre of the sarcomere. Titin also contains extensive repeats of immunoglobulin (Ig) domains which, via folding and unfolding, allow it to function as a molecular spring (Rief et al. 1997; Hsin et al. 2011). In addition, molecular dynamic simulations have shown that mechanical strain can induce a conformational change in the kinase domain of titin, and therefore, regulate its catalytic activity (Grater et al. 2005; Puchner et al. 2008). Based on these points, titin has been commonly labelled as a mechanosensitive kinase (Puchner et al. 2008). However, the kinase domain of titin is anchored near the centre of the sarcomere (∼1.1 μm away from the Z‐disc), and thus, it is very unlikely that titin directly contributes to the MIC‐induced changes in Z‐disc protein phosphorylation (Obermann et al. 1996).
Inspired by the above point, we set out to determine if our phosphoproteomics data could be used to expose other MIC‐regulated and Z‐disc‐associated kinases. To accomplish this, we first compiled a list of the kinases that were predicted to be regulated by MIC (Figs 8 and 9), and then searched this list for kinases that have been localized to the Z‐disc. Our analyses led to the identification of SPEG and obscurin as two very intriguing candidates. We were interested in these kinases for several reasons: (i) both are preferentially expressed in striated muscle (Sutter et al. 2004), (ii) both experienced major MIC‐induced alterations in phosphorylation at multiple sites (Fig. 10 A and B), (iii) the kinase domains of SPEG, and certain splice variants of obscurin, have been shown to colocalize with the Z‐disc (Bowman et al. 2007; Agrawal et al. 2014), and (iv) SPEG, obscurin and titin are immediate neighbours on the phylogenetic tree of the human kinome, which suggests that they could possess the same kind of mechanosensitive properties that have been ascribed to titin (Fig. 10 C). Indeed, both SPEG and obscurin are similar to titin in that they contain extensive repeats of Ig domains, which are known to endow titin with its molecular spring‐like properties (Fig. 10 A and B) (Hsin et al. 2011). Combined, these attributes vividly highlight the potential for SPEG and obscurin to function as mechanosensitive kinases at the Z‐disc.
Conclusions, limitations and future directions
In 2015, Hoffman et al. published a paper which described the phosphoproteomic alterations that occur in response to aerobic‐based exercise (Hoffman et al. 2015). It is well known that aerobic‐based exercise induces an increase in the oxidative capacity of skeletal muscle, and the work of Hoffman et al. provided a tremendous amount of novel insight into the signalling events that potentially drive this event. However, in contrast to aerobic‐based exercise, resistance‐based exercise induces a robust increase in skeletal muscle mass. Thus, the signalling events that are induced by aerobic‐ and resistance‐based exercise are expected to be, in many ways, very different. This point was readily highlighted in a follow‐up article that was published by Hawley and Krook in which it was stressed that ‘the networks induced by resistance‐based exercise urgently need to be determined’ (Hawley & Krook, 2016). In our study, we have addressed this need by performing a phosphoproteomic analysis on mouse skeletal muscles that had been subjected to a bout of MIC; a mouse model of human resistance exercise. Specifically, we have generated the first comprehensive map of the MIC‐regulated phosphoproteome and demonstrated that our dataset, in conjunction with various bioinformatics tools, leads to the identification of several well‐known MIC‐regulated events (e.g. activation of signalling through mTOR and p38). The identification of these known signalling events is important because it lends confidence to the novel outcomes of our analyses. For instance, our analyses indicate that the Z‐disc is a particularly hot site for MIC‐induced phosphorylation events, and that SPEG and obscurin might be mechanosensitive Z‐disc kinases that drive this response. Although provocative in nature, it is important to emphasize that these are purely data‐driven hypotheses, and follow‐up studies will be needed to both test and validate them.
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
G.K.P., J.J.C. and T.A.H. designed the experiments; G.K.P., A.S.H., M.S.W., J.S.Y., R.M.M., R.B. and T.A.H. acquired and analysed data; G.K.P., A.S.H., J.J.C. and T.A.H. wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number AR057347 to TAH. The research was also supported by the National Institutes of Health under Award Number R35 GM118110 to JJC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors declare no conflicts of interest.
Data availability
Raw mass spectrometry data (Thermo RAW files) for proteins and phosphopeptides identified in these experiments have been deposited online and will be made publicly available through the Chorus project (https://chorusproject.org/pages/index.html).
Supporting information
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Supplemental Table S1. Quantified mouse skeletal muscle phosphopeptides.
Supplemental Table S2. Rank ordered abundance of skeletal muscle proteins by LFQ.
Supplemental Table S3. Quantified mouse skeletal muscle proteins.
Linked articles This article is highlighted by a Perspective by Burniston. To read this Perspective, visit https://doi.org/10.1113/JP274672.
This is an Editor's Choice article from the 1 August 2017 issue.
References
- Adams GR & Bamman MM (2012). Characterization and regulation of mechanical loading‐induced compensatory muscle hypertrophy. Compr Physiol 2, 2829–2870. [DOI] [PubMed] [Google Scholar]
- Agrawal PB, Pierson CR, Joshi M, Liu X, Ravenscroft G, Moghadaszadeh B, Talabere T, Viola M, Swanson LC, Haliloglu G, Talim B, Yau KS, Allcock RJ, Laing NG, Perrella MA & Beggs AH (2014). SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am J Hum Genet 95, 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Levy R, Leighton IA, Doza YN, Attwood P, Morrice N, Marshall CJ & Cohen P (1995). Identification of novel phosphorylation sites required for activation of MAPKAP kinase‐2. EMBO J 14, 5920–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y & Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B 57, 289–300. [Google Scholar]
- Bodine SC (2013). Disuse‐induced muscle wasting. Int J Biochem Cell Biol 45, 2200–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman AL, Kontrogianni‐Konstantopoulos A, Hirsch SS, Geisler SB, Gonzalez‐Serratos H, Russell MW & Bloch RJ (2007). Different obscurin isoforms localize to distinct sites at sarcomeres. FEBS Lett 581, 1549–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caenepeel S, Charydczak G, Sudarsanam S, Hunter T & Manning G (2004). The mouse kinome: discovery and comparative genomics of all mouse protein kinases. Proc Natl Acad Sci USA 101, 11707–11712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Tang HY, Wang H, Liu Q & Speicher DW (2012). Systematic comparison of fractionation methods for in‐depth analysis of plasma proteomes. J Proteome Res 11, 3090–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin ER (2005). Role of Ca2+/calmodulin‐dependent kinases in skeletal muscle plasticity. J Appl Physiol (1985) 99, 414–423. [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N & Mann M (2014). Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh AS, Murgia M, Nagaraj N, Treebak JT, Cox J & Mann M (2015). Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol Cell Proteomics 14, 841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B & Zierath JR (2013). Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17, 162–184. [DOI] [PubMed] [Google Scholar]
- Erickson BK, Rose CM, Braun CR, Erickson AR, Knott J, McAlister GC, Wuhr M, Paulo JA, Everley RA & Gygi SP (2017). A strategy to combine sample multiplexing with targeted proteomics assays for high‐throughput protein signature characterization. Mol Cell 65, 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farup J, Kjolhede T, Sorensen H, Dalgas U, Moller AB, Vestergaard PF, Ringgaard S, Bojsen‐Moller J & Vissing K (2012). Muscle morphological and strength adaptations to endurance vs. resistance training. J Strength Cond Res 26, 398–407. [DOI] [PubMed] [Google Scholar]
- Frank D & Frey N (2011). Cardiac Z‐disc signaling network. J Biol Chem 286, 9897–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank D, Kuhn C, Katus HA & Frey N (2006). The sarcomeric Z‐disc: a nodal point in signalling and disease. J Mol Med (Berl) 84, 446–468. [DOI] [PubMed] [Google Scholar]
- Frey JW, Jacobs BL, Goodman CA & Hornberger TA (2014). A role for Raptor phosphorylation in the mechanical activation of mTOR signaling. Cell Signal 26, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CA, Hornberger TA & Robling AG (2015). Bone and skeletal muscle: Key players in mechanotransduction and potential overlapping mechanisms. Bone 80, 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grater F, Shen J, Jiang H, Gautel M & Grubmuller H (2005). Mechanically induced titin kinase activation studied by force‐probe molecular dynamics simulations. Biophys J 88, 790–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsrud PA, Carson JJ, Hebert AS, Hubler SL, Niemi NM, Bailey DJ, Jochem A, Stapleton DS, Keller MP, Westphall MS, Yandell BS, Attie AD, Coon JJ & Pagliarini DJ (2012). A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab 16, 672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley JA & Krook A (2016). Metabolism: One step forward for exercise. Nat Rev Endocrinol 12, 7–8. [DOI] [PubMed] [Google Scholar]
- Herzog W (2014). The role of titin in eccentric muscle contraction. J Exp Biol 217, 2825–2833. [DOI] [PubMed] [Google Scholar]
- Hoffman NJ, Parker BL, Chaudhuri R, Fisher‐Wellman KH, Kleinert M, Humphrey SJ, Yang P, Holliday M, Trefely S, Fazakerley DJ, Stockli J, Burchfield JG, Jensen TE, Jothi R, Kiens B, Wojtaszewski JF, Richter EA & James DE (2015). Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise‐regulated kinases and AMPK substrates. Cell Metab 22, 922–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn H, Schoof EM, Kim J, Robin X, Miller ML, Diella F, Palma A, Cesareni G, Jensen LJ & Linding R (2014). KinomeXplorer: an integrated platform for kinome biology studies. Nat Methods 11, 603–604. [DOI] [PubMed] [Google Scholar]
- Hornberger TA & Esser KA (2004). Mechanotransduction and the regulation of protein synthesis in skeletal muscle. Proc Nutr Soc 63, 331–335. [DOI] [PubMed] [Google Scholar]
- Hornberger TA, Stuppard R, Conley KE, Fedele MJ, Fiorotto ML, Chin ER & Esser KA (2004). Mechanical stimuli regulate rapamycin‐sensitive signalling by a phosphoinositide 3‐kinase‐, protein kinase B‐ and growth factor‐independent mechanism. Biochem J 380, 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin J, Strumpfer J, Lee EH & Schulten K (2011). Molecular origin of the hierarchical elasticity of titin: simulation, experiment, and theory. Annu Rev Biophys 40, 187–203. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT & Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, Hamilton JA, Ouchi N, LeBrasseur NK & Walsh K (2008). Fast/glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab 7, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen I, Shepard DS, Katzmarzyk PT & Roubenoff R (2004). The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc 52, 80–85. [DOI] [PubMed] [Google Scholar]
- Kocher T, Pichler P, Schutzbier M, Stingl C, Kaul A, Teucher N, Hasenfuss G, Penninger JM & Mechtler K (2009). High precision quantitative proteomics using iTRAQ on an LTQ Orbitrap: a new mass spectrometric method combining the benefits of all. J Proteome Res 8, 4743–4752. [DOI] [PubMed] [Google Scholar]
- Kramer HF & Goodyear LJ (2007). Exercise, MAPK, and NF‐kappaB signaling in skeletal muscle. J Appl Physiol (1985) 103, 388–395. [DOI] [PubMed] [Google Scholar]
- Lyon RC, Zanella F, Omens JH & Sheikh F (2015). Mechanotransduction in cardiac hypertrophy and failure. Circ Res 116, 1462–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal A, Potts GK, Dawson AR, Coon JJ & Mehle A (2015). Phosphorylation at the homotypic interface regulates nucleoprotein oligomerization and assembly of the influenza virus replication machinery. PLoS Pathog 11, e1004826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgia M, Nagaraj N, Deshmukh AS, Zeiler M, Cancellara P, Moretti I, Reggiani C, Schiaffino S & Mann M (2015). Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep 16, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen B, Taylor S & Ghosh G (2004). Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell 15, 661–675. [DOI] [PubMed] [Google Scholar]
- Obermann WM, Gautel M, Steiner F, van der Ven PF, Weber K & Furst DO (1996). The structure of the sarcomeric M band: localization of defined domains of myomesin, M‐protein, and the 250‐kD carboxy‐terminal region of titin by immunoelectron microscopy. J Cell Biol 134, 1441–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil TK, Duffy LR, Frey JW & Hornberger TA (2009). The role of phosphoinositide 3‐kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. J Physiol 587, 3691–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz CA, Kulke M, Leake MC, Neagoe C, Hinssen H, Hajjar RJ & Linke WA (2003). Damped elastic recoil of the titin spring in myofibrils of human myocardium. Proc Natl Acad Sci USA 100, 12688–12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahor M & Kritchevsky S (1998). Research hypotheses on muscle wasting, aging, loss of function and disability. J Nutr Health Aging 2, 97–100. [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K & Cobb MH (2001). Mitogen‐activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22, 153–183. [DOI] [PubMed] [Google Scholar]
- Pearson RB & Kemp BE (1991). Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol 200, 62–81. [DOI] [PubMed] [Google Scholar]
- Piccirillo R, Demontis F, Perrimon N & Goldberg AL (2014). Mechanisms of muscle growth and atrophy in mammals and Drosophila . Dev Dynam 243, 201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichler P, Kocher T, Holzmann J, Mazanek M, Taus T, Ammerer G & Mechtler K (2010). Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4‐plex compared to TMT 6‐plex and iTRAQ 8‐plex on LTQ Orbitrap. Anal Chem 82, 6549–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichler P, Kocher T, Holzmann J, Mohring T, Ammerer G & Mechtler K (2011). Improved precision of iTRAQ and TMT quantification by an axial extraction field in an Orbitrap HCD cell. Anal Chem 83, 1469–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor DN, Balagopal P & Nair KS (1998). Age‐related sarcopenia in humans is associated with reduced synthetic rates of specific muscle proteins. J Nutr 128, 351S–355S. [DOI] [PubMed] [Google Scholar]
- Puchner EM, Alexandrovich A, Kho AL, Hensen U, Schafer LV, Brandmeier B, Grater F, Grubmuller H, Gaub HE & Gautel M (2008). Mechanoenzymatics of titin kinase. Proc Natl Acad Sci USA 105, 13385–13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle WG & Solaro RJ (2004). At the crossroads of myocardial signaling: the role of Z‐discs in intracellular signaling and cardiac function. Circ Res 94, 296–305. [DOI] [PubMed] [Google Scholar]
- Richter EA, Cleland PJ, Rattigan S & Clark MG (1987). Contraction‐associated translocation of protein kinase C in rat skeletal muscle. FEBS Lett 217, 232–236. [DOI] [PubMed] [Google Scholar]
- Rief M, Gautel M, Oesterhelt F, Fernandez JM & Gaub HE (1997). Reversible unfolding of individual titin immunoglobulin domains by AFM. Science 276, 1109–1112. [DOI] [PubMed] [Google Scholar]
- Riley NM & Coon JJ (2015). Phosphoproteomics in the age of rapid and deep proteome profiling. Anal Chem 88, 74–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera‐Brown AM & Frontera WR (2012). Principles of exercise physiology: responses to acute exercise and long‐term adaptations to training. PM R 4, 797–804. [DOI] [PubMed] [Google Scholar]
- Rust HL & Thompson PR (2011). Kinase consensus sequences: a breeding ground for crosstalk. ACS Chem Biol 6, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfeld BJ, Ogborn D & Krieger JW (2016). Effects of resistance training frequency on measures of muscle hypertrophy: a systematic review and meta‐analysis. Sports Med 46, 1689. [DOI] [PubMed] [Google Scholar]
- Schwartz D & Gygi SP (2005). An iterative statistical approach to the identification of protein phosphorylation motifs from large‐scale data sets. Nat Biotechnol 23, 1391–1398. [DOI] [PubMed] [Google Scholar]
- Seguin R & Nelson ME (2003). The benefits of strength training for older adults. Am J Prev Med 25, 141–149. [DOI] [PubMed] [Google Scholar]
- Sharma K, D'Souza Rochelle CJ, Tyanova S, Schaab C, Wiśniewski Jacek R, Cox J & Mann M (2014). Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr‐based signaling. Cell Reports 8, 1583–1594. [DOI] [PubMed] [Google Scholar]
- Smyth GK (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3, Article3. [DOI] [PubMed] [Google Scholar]
- Solaro RJ, Pang DC & Briggs FN (1971). The purification of cardiac myofibrils with Triton X‐100. Biochim Biophys Acta 245, 259–262. [DOI] [PubMed] [Google Scholar]
- Sutter SB, Raeker MO, Borisov AB & Russell MW (2004). Orthologous relationship of obscurin and Unc‐89: phylogeny of a novel family of tandem myosin light chain kinases. Dev Genes Evol 214, 352–359. [DOI] [PubMed] [Google Scholar]
- Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C & Mechtler K (2011). Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res 10, 5354–5362. [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP & Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson K & Baar K (2014). mTOR and the health benefits of exercise. Semin Cell Dev Biol 36, 130–139. [DOI] [PubMed] [Google Scholar]
- Wenger CD, Lee MV, Hebert AS, McAlister GC, Phanstiel DH, Westphall MS & Coon JJ (2011a). Gas‐phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat Methods 8, 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger CD, Phanstiel DH, Lee MV, Bailey DJ & Coon JJ (2011b). COMPASS: A suite of pre‐ and post‐search proteomics software tools for OMSSA. Proteomics 11, 1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJR & Mohammed S (2013). Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res 12, 260–271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Supplemental Table S1. Quantified mouse skeletal muscle phosphopeptides.
Supplemental Table S2. Rank ordered abundance of skeletal muscle proteins by LFQ.
Supplemental Table S3. Quantified mouse skeletal muscle proteins.
Data Availability Statement
Raw mass spectrometry data (Thermo RAW files) for proteins and phosphopeptides identified in these experiments have been deposited online and will be made publicly available through the Chorus project (https://chorusproject.org/pages/index.html).