

Abstract
Key points
Neuronal nicotinic acetylcholine receptors (nAChRs) play a fundamental role in the attentional circuitry throughout the mammalian CNS.
In the present study, we report a novel finding that ageing negatively impacts nAChR efficacy in auditory thalamus, and this is probably the result of a loss of nAChR density (B max) and changes in the subunit composition of nAChRs.
Our data support the hypothesis that age‐related maladaptive changes involving nAChRs within thalamocortical circuits partially underpin the difficulty that elderly adults experience with respect to attending to speech and other salient acoustic signals.
Abstract
The flow of auditory information through the medial geniculate body (MGB) is regulated, in part, by cholinergic projections from the pontomesencephalic tegmentum. The functional significance of these projections is not fully established, although they have been strongly implicated in the allocation of auditory attention. Using in vitro slice recordings, we have analysed postsynaptic function and pharmacology of neuronal nicotinic ACh receptors (nAChRs) in young adult and the aged rat MGB. We find that ACh produces significant excitatory postsynaptic actions on young MGB neurons, probably mediated by β2‐containing heteromeric nAChRs. Radioligand binding studies show a significant age‐related loss of heteromeric nAChR receptor number, which supports patch clamp data showing an age‐related loss in ACh efficacy in evoking postsynaptic responses. Use of the β2‐selective nAChR antagonist, dihydro‐β‐erythroidine, suggests that loss of cholinergic efficacy may also be the result of an age‐related subunit switch from high affinity β2‐containing nAChRs to low affinity β4‐containing nAChRs, in addition to the loss of total nAChR number. This age‐related nAChR dysfunction may partially underpin the attentional deficits that contribute to the loss of speech understanding in the elderly.
Keywords: aging, auditory system, cholinergic neural pathways, nicotinic receptor, thalamus
Key points
Neuronal nicotinic acetylcholine receptors (nAChRs) play a fundamental role in the attentional circuitry throughout the mammalian CNS.
In the present study, we report a novel finding that ageing negatively impacts nAChR efficacy in auditory thalamus, and this is probably the result of a loss of nAChR density (B max) and changes in the subunit composition of nAChRs.
Our data support the hypothesis that age‐related maladaptive changes involving nAChRs within thalamocortical circuits partially underpin the difficulty that elderly adults experience with respect to attending to speech and other salient acoustic signals.
Abbreviations
- ACh
acetylcholine
- aCSF
artificial cerebrospinal fluid
- DHβE
dihydro‐β‐erythroidine
- FBN
Fischer Brown Norway
- MGB
medial geniculate body
- mAChR
muscarinic ACh receptor
- MGd
dorsal subdivision of MGB
- MGv
ventral subdivision of MGB
- nAChR
nicotinic ACh receptor
- PMT
pontomesencephalic tegmentum
- RT
room temperature
Introduction
Age‐related hearing loss effects ∼30–35% of the population between 65 and 75 years of age and at least 50% of people aged older than 75 years of age (NIDCD Report, 2006). The decline in hearing sensitivity is most commonly attributed to changes in the auditory periphery, including the middle ear, hair cells, and acoustic nerve fibres. Although this may reflect a primary cause of the initial pathology of presbycusis, central auditory processing dysfunction (i.e. central presbycusis) is increasingly viewed as an important factor in the loss of speech understanding (Humes et al. 2012; Ouda et al. 2015; Harris & Dubno, 2017).
To compensate for the age‐related loss of auditory signal quality, additional cortical resources may play a role in knowledge‐based optimization of input. Convergence of the functions mediated by top‐down and bottom‐up processes is crucial to maintaining performance in tasks requiring continuous attention not only in the auditory system, but also in all sensory modalities (Sarter et al. 2001; Lesicko & Llano, 2017). Perception of sensorimotor signals is dynamically regulated by acetylcholine (ACh) in a behavioural state‐dependent manner at the level of the sensory thalamus (Mooney et al. 2003). The pontomesencephalic tegmentum (PMT) is a collection of brainstem nuclei that serve as the main source of cholinergic input to many structures in the auditory pathway including the medial geniculate body (MGB) (Schofield et al. 2011). Activation of cholinergic systems at the level of the auditory thalamus probably plays a major role in event detection, sensory gating, auditory conditioning, and various forms of associative learning and memory (Schofield et al. 2011).
Central cholinergic neurotransmission, mediated by neuronal nicotinic (nAChR) and muscarinic (mAChR) receptors, acts to affect the excitability of neuronal networks, altering the release of a wide variety of neurotransmitters, including GABA, glutamate, norepinephrine, serotonin, and dopamine (McGehee et al. 1995; Wonnacott, 1997). nAChR activation can coordinate the response patterns of groups of individual neurons (Wonnacott, 1997; Kawai et al. 2007; Changeux, 2010). In addition to presynaptic sites on terminal endings, nAChRs and mAChRs also exist postsynaptically on somatic and dendritic sites (Picciotto et al. 2012). Postsynaptic nAChRs are known to depolarize neurons, increasing their firing rates, and possibly contribute to increased levels of attention and long‐term potentiation (Radcliffe and Dani, 1998; Kawai et al. 2007). At the level of thalamus, it is unclear whether cholinergic afferents form synapses onto thalamocortical neurons or whether ACh is released by volume transmission (Hallanger et al. 1990; Dani & Bertrand, 2007). In the present study, we refer to nAChRs located on the somata of MGB neurons as postsynaptic.
In the mammalian CNS, heteromeric nAChRs comprise α and β subunit combinations, including α2‐α6 and β2‐β4, whereas homomeric nAChRs comprise only α subunits, using α7, α9, and α10 subunits. The most common combinations found in brain are α4β2* and α7 (Dani & Bertrand, 2007; Gotti et al. 2007). nAChRs in MGB are predominately heteromers, with almost no α‐bungarotoxin binding detected, which indicates a lack of homomeric nAChRs (Clarke et al. 1985; Breese et al. 1997; Spurden et al. 1997; Court et al. 2000). In whole thalamus of young rats, ∼90% of the nAChRs comprise α4 and β2 subunits, although the α5 subunit is also found in ∼15% of the receptors and the α3 and β4 subunits are seen in ∼17% and ∼12% of the nAChRs, respectively (Mao et al. 2008). The presence of various α and β nAChR subunits in a single receptor contributes to agonist affinity, as well as the rate of desensitization and calcium permeability (Quick & Lester, 2002), with β2‐containing receptors having higher affinity for nicotinic agonists than β4‐containing nAChRs (Luetje & Patrick, 1991; Parker et al. 1998; Xiao & Kellar, 2004). Also, the relative calcium permeability of α4β2 receptors is greater than that of α3β4 receptors (Haghighi & Cooper, 2000). The diversity of subunit combinations, as well as the presynaptic, axonal, and postsynaptic locations of nAChRs, indicate the diverse roles that they play in the CNS.
Ageing negatively impacts nAChRs in a number of brain structures. In adult rats, studies of whole thalamus show some of the highest levels of mRNA for the α4 and β2 nAChR subunits in young samples, as well as the largest age‐related decreases compared to other brain regions. Age‐related decreases of nAChR mRNA in whole thalamus were between 20% and 30% at 29 months of age and up to 50% by 32 months of age (Ferrari et al. 1999). Similarly, a postmortem human study found that whole thalamus underwent a 32% decline in β2‐containing nAChRs over the lifespan, a rate of ∼4.8% per decade (Mitsis et al. 2009). However, little is known about the impact of ageing on the individual sensory thalamic nuclei such as MGB.
The present study examines the impact of ageing on the presence, subunit composition, and physiological profile of postsynaptic nAChRs. We consider that age‐related maladaptive changes involving nAChRs within thalamocortical circuits partially underpin the difficulty experienced by elderly adults with respect to attending to speech and other salient acoustic signals. We report neurochemical and electrophysiological evidence of age‐related changes in normal adult cholinergic function.
Methods
Animals
Young adult (4–6 months old) or aged (28–32 months old) Fischer Brown Norway (FBN) rats were obtained from the National Institute of Aging rodent resource colonies, where they were individually housed under a reversed 12:12 h light/dark cycle with access to food and water available ad libitum. The FBN rat was chosen because it is an extensively studied rat model of ageing that exhibits presbycusic properties and a long life span with few other specific age‐related pathologies. Auditory brainstem responses of aged FBN rats exhibit a significant mean threshold shift of 23 dB upward for all frequencies studied compared to young FBN rats (Turner & Caspary, 2005; Caspary et al. 2008). For dose–response experiments (Figs 1 and 2), seven young and 10 aged animals were used. For experiments involving dihydro‐β‐erythroidine (DHβE) (Figs 3 and 4), six young and seven aged animals were used. For the autoradiography experiments, four young and four aged animals were used. One coronal slice containing both left and right MGB from each animal was taken for analysis. Procedures were performed in accordance with guidelines and protocols approved by the Southern Illinois University School of Medicine Animal Care and Use Committee.
Figure 1. Postsynaptic effects of varying concentrations of ACh puffed onto patch‐clamped neurons in the MGB slice preparation.

A, traces from one young (red) and one aged (blue) MGB neuron showing the postsynaptic response to local ACh application when voltage clamped at −70 mV. All experiments were carried out in the presence of atropine (20 μm) to block mAChRs. B, age‐related reduction of the postsynaptic nAChR response to local ACh application onto young (red) and aged (blue) MGB neurons. For 0.1 and 0.5 mm: young, n = 24; aged, n = 30. For 1 mm, young, n = 28; aged, n = 28. C, dose–response curves to compare response to local application of ACh in young and aged MGB. 0.1, 0.5, 1, 5, 10 mm ACh in aCSF was pressure ejected (100 ms, 10 p.s.i.) 10–20 μm away from patched MGB neurons. Peak amplitude in the 2 s immediately following the ACh application was measured. Red, young MGB; blue, aged MGB. For 0.1, 0.5, 1 mm, young, n = 28; aged, n = 28 aged. For 5 mm, young, n = 7, aged, n = 8. For 10 mm, young, n = 7; aged, n = 9. * P < 0.05, ** P < 0.005, *** P < 0.0005, **** P < 0.0001.
Figure 2. Differences in nAChR activation in dorsal and ventral MGB subdivisions.

Labelled cells with representative morphology characteristics of those in MGv (A) and MGd (B). C, response of young (red) vs. aged (blue) neurons in MGv and MGd to 0.1, 0.5 and 1 mm ACh application. * P < 0.05, **** P < 0.0001.
Figure 3. Postsynaptic response to ACh application using the β2‐selective antagonist DHβE to separate receptor subtype in young and aged MGB neurons.
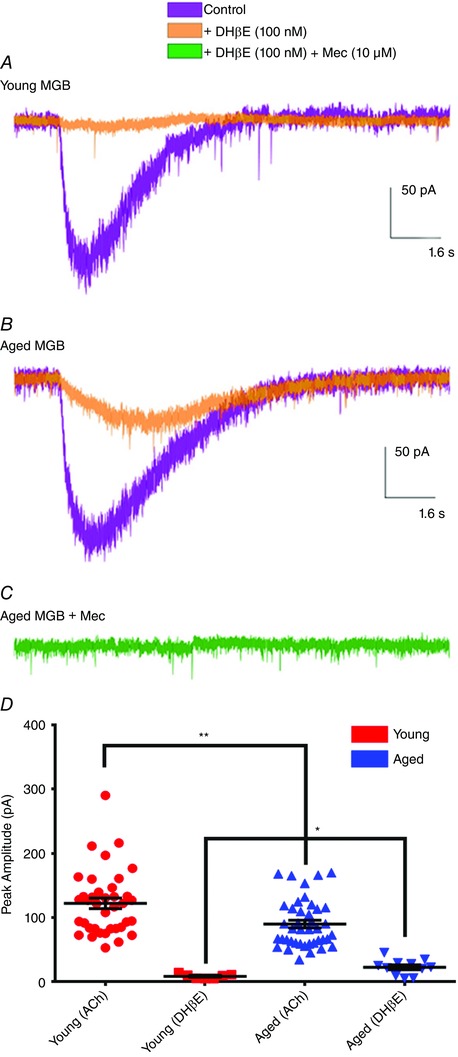
Postsynaptic effects of 1 mm ACh puffed onto patch‐clamped neurons in young (A) and aged (B and C) MGB with subunit selective antagonists. All experiments were carried out in the presence of atropine (20 μm) to block mAChRs. Total nAChR response is shown in purple. The β2‐specific antagonist DHβE was added and the remaining nAChR current is shown in orange. The non‐specific nAChR antagonist mecamylamine (10 μm) was added and blocked any remaining nAChR current in aged neurons (green). D, total nAChR responsiveness is significantly decreased with the condition of ageing (77.3% ± 10.7% of young nAChR amplitude). When 100 nm DHβE was added to the bath solution, it blocked only the high affinity β2‐containing nAChRs. After the addition of DHβE, maximal response to ACh application was 7.27% ± 1.14% of the initial nAChR response in young (red) compared to 30.37% ± 5.55% in aged (blue). nAChR: young, n = 38; aged, n = 41. DHβE: young, n = 7; aged, n = 11. * P < 0.05. ** P < 0.005.
Figure 4. Saturation binding of [3H]epibatidine using autoradiography in young and aged MGBs.

Scatchard plot (A) and saturation binding curves (B) show a significant age‐related loss of nAChRs density (B max). B max in young MGB (red) = 145 ± 4.2 fmol/mg protein and B max in aged MGB (blue) = 118.6 ± 3.8 fmol/mg protein (*** P < 0.0005). There was no change in the affinity (K d) of [3H]epibatidine with age (P = 0.634). K d in young MGB = 0.036 ± 0.08 nm and K d in aged MGB = 0.042 ± 0.009 nm (n = 4 animals/age group). C, autoradiographic images of a coronal section in which MGB is present with 375 pM of [3H]epibatidine.
Slice preparation
Similar to Richardson et al. (2013), young and aged FBN rats were anaesthetized using isoflurane (3%) and decapitated. Brains were quickly removed and submerged in cold (1–2°C) artificial cerebrospinal fluid (aCSF) maintained at pH 7.4 by bubbling with carbogen (95% O2–5% CO2). The aCSF composition was (in mm): 125 NaCl, 3 KCl, 1 MgCl2, 1.23 NaH2PO4, 2 CaCl2, 26 NaHCO3 and 10 glucose.
Horizontal slices (250–300 μm) were made using a Vibratome (Pelco, St Louis, MO, USA) and immediately incubated for 15 min at physiological temperature (37°C), followed by 45 min at room temperature (RT) (20–22°C) in carbogen‐bubbled aCSF. Individual slices were transferred to an immersion recording chamber (2 ml) placed on an upright microscope (BX50WI; Olympus, Tokyo, Japan) equipped with differential interference contrast image devices and viewed with a 40× water‐immersion objective. Slices were perfused at a rate of 2–3 ml min−1 with aCSF bubbled with carbogen at RT.
Whole cell recordings
Patch clamp recordings were performed in the whole‐cell configuration using 4–6 MΩ fire‐polished pipettes pulled from borosilicate glass (inner diameter 1.1 mm, outer diameter 1.7 mm; Garner Glass, Claremont, CA, USA). The internal patch pipette solution contained (in mm): 140.0 potassium gluconate, 1 NaCl, 2 MgCl2, 10 Hepes, 2 Mg‐ATP, 0.3 Na‐GTP and 6.88 KOH (osmolarity: 300 mosmol l−1), pH 7.3 (adjusted with KOH). Pipettes were connected to an Axoclamp 2B amplifier (Molecular Devices, Sunnyvale, CA, USA) and cells were recorded in the continuous current or voltage clamp modes. Under voltage clamp conditions, cells were held at −70 mV unless otherwise specified. The patch pipette was positioned in the MGB and neurons were viewed with a microscope (Olympus). MGB neurons to be patched were chosen based on thickness and health of slice area. Giga‐ohm seal intracellular recordings were achieved with series resistances ranging from 5 to 18 MΩ. Whole cell capacitance, input resistance and series resistance were determined by application of a 5 mV square pulse. Exclusion criteria included: (i) a resting membrane potential more depolarized than −60 mV; (ii) series resistance greater than 20 MΩ; and (iii) a resting input resistance less than 100 MΩ. Generated transistor‐transistor logic pulses, voltage commands, acquisition and display of the recorded signals were achieved using the Clampex program (Molecular Devices).
Drug application
All experiments were carried out in the presence of atropine (20 μm) (Sigma, St Louis, MO, USA) added to the aCSF bath solution. Using an on‐screen scale bar, a patch pipette (∼1 μm) containing varying concentrations of ACh was positioned 15–20 μm from the patched cell under study and pressure was ejected at 10 p.s.i using a Picospritzer (General Valve Corp., Fairfield, NJ, USA) every 60 s for 3 min.
Receptor autoradiography
FBN rats were decapitated and brains quickly removed, washed briefly in 0.1 m PBS (pH 7.4) slush, frozen in dry ice, and stored in −80°C. Frozen coronal sections (16 μm) through MGB (Bregma −6.04 mm to Bregma −4.80 mm) (Paxinos & Watson, 1998) were cut on a cryostat (model CM1850; Leica Microsystems, Buffalo Grove, IL, USA), collected on slides and stored at −20°C for less than 48 h. On the day of the binding experiment, after 10 min fixation in 4% paraformaldehyde (Sigma), slides were prewashed with 50 mm Tris‐HCl buffer containing 120 mm NaCl, 5 mm KCl, 2.5 mm CaCl2 and 1 mm MgCl2 (pH 7.4) at room temperature (RT) by quick dips. Residue of prewash buffer was blotted off and sections were incubated at RT for 60 min in the same buffer above with 0, 0.1, 0.25, 0.5, 0.75, 1 and 1.5 nm of [3H]epibatidine (Perkin Elmer, Waltham, MA, USA; specific activity = 62.2 Ci mmol−1). Non‐specific binding was determined by incubating adjacent sections with 300 μm (–)‐nicotine (Sigma) in addition to the various concentrations of [3H]epibatidine. Incubation was stopped by washing the slides twice with ice cold buffer for 5 min each, followed by dipping them quickly in ice‐cold dH2O. Slides were air dried overnight. Dried slides were opposed to [3H]‐hypersensitive phosphor screens (Perkin Elmer, Waltham, MA, USA) for 2 days at RT. The phosphor screens were scanned using a Cyclone Storage Phosphor System (Perkin Elmer). The MGB was outlined and analysed using OptiQuant Image Analysis software (Canberra Packard, Meriden, CT, USA), which provided tools for greyscale quantification in digital light units. Digital light units were then converted to nCi/mg protein using a standard curve generated from co‐exposed [3H]‐embedded plastic standards (American Radiolabeled Chemicals, St Louis, MO, USA) and further converted to fmol/mg protein. Values from the left and right MGB were combined. The present receptor binding protocol is similar to previous receptor binding autoradiographic protocols used by our laboratory with [3H]gaboxadol (Milbrandt & Caspary, 1995; Milbrandt et al. 1996, 1997, 2000; Bauer et al. 2000; Wang et al. 2009a, b). The present saturation [3H]epibatidine autoradiographic protocol for nAChRs was validated against studies reported by Tribollet et al. (2004) and Perry and Kellar (1995). Transverse sections from Tribollet et al. (2004) containing MGB show levels of receptor binding similar to sections (Bregma −5.2) used in the present study. Although Perry and Kellar (1995) did not examine sensory thalamus, we compared their nAChR binding values obtained from the Sprague Dawley rat parietal cortex with our values obtained from a similar transverse section (Bregma −5.2) from FBN rat parietal cortex. Perry and Kellar (1995) used a single [3H]epibatidine concentration at 0.46 nm and our nearest concentration was 0.58 nm. Specific binding (fmol/mg protein) was comparable across parietal cortex. A somewhat higher value obtained in the present study is probably the result of the lower concentration of [3H]epibatidine used by Perry and Kellar (1995) (0.46 vs. 0.58 nm), as well as differences between film vs. trans‐screen autoradiography with resultant densitometic measurement differences. One coronal slice including both left and right MGB from each of the four young and aged animals was analysed. The measurements were assessed objectively using Bioquant software (Heidelberg, Germany).
Statistical analysis
Electrophysiology data were recorded using Clampex (Molecular Devices). Responses to ACh were analysed offline using Clampfit, version 10.6 (Molecular Devices) or Mini Analysis (Synaptosoft, Fort Lee, NJ, USA). To determine the peak amplitude following application of ACh, the mean current before drug application (20 s) was taken as baseline and then, for a period of 2 s immediately after the addition of ACh, the maximum amplitude deflection from baseline mean was recorded. Two‐way ANOVA was used to analyse the dose response experiments (Fig. 1) and differences between MGB subdivisions (Fig. 2), as well as those separating nAChR subtype (Fig. 3). All statistical analysis was carried out using Prism (GraphPad Software Inc., La Jolla, CA, USA). GraphPad Prism 7 was also used for the analysis of autoradiography data to obtain B max and K d. P < 0.05 was considered statistically significant. All values are expressed as the mean ± SEM.
Results
Electrophysiological evidence of an age‐related loss of nAChR efficacy
All experiments were carried out using either young adult (4–6 months old) or aged (28–32 months old) FBN rats. nAChR responses were analysed after successful patching of MGB neurons in the presence of atropine (20 μm) to block mAChRs. There were no significant differences in resting membrane potential or input resistance between young and aged MGB neurons. When ACh was applied by puff into the immediate vicinity of a patched MGB neuron, there was a rapid increase in inward current seen in exemplars from whole cell recordings from one young and one aged MGB neuron (Fig. 1 A). Puff application of aCSF alone evoked no inward current in young or aged MGB neurons. A distinctly smaller inward current was seen in response to puffed ACh in the aged MGB neuron (Fig. 1 A). Group data showed significant age‐related decreases in nAChR efficacy when ACh was applied in the vicinity of aged MGB neurons compared to young MGB neurons (Fig. 1 B). Dose response curves compared responses to local ACh application onto young and aged MGB neurons (Fig. 1 C). A two‐way ANOVA using Tukey's post hoc test showed significant age‐related decreases in four out of the five concentrations tested in response to ACh application, with larger age‐related decreases seen for the higher ACh concentrations (Fig. 1 C). Mean and SEM for dose response curve are shown in Table 1.
Table 1.
Maximal nAChR‐mediated response elicited from young‐adult and aged MGB neurons
| [ACh] (m) | Young adult | Aged |
|---|---|---|
| 0.0001 | 60.07 ± 6.58 | 42.01 ± 2.54 |
| 0.0005 | 100.26 ± 9.46 | 69.59 ± 4.80 |
| 0.001 | 132.50 ± 9.80 | 87.70 ± 5.82 |
| 0.005 | 186.87 ± 17.83 | 111.86 ± 24.16 |
| 0.01 | 271.31 ± 25.97 | 149.49 ± 27.48 |
Data presented as the mean ± SEM (n = 28 neurons for both young adult and aged groups at 0.1, 0.5 and 1 mm; n = 7 at 5 mm; and n = 8 and 9 for 10 mm).
Neurons from dorsal (MGd) and ventral (MGv) subdivisions of MGB exhibit distinctive morphologies (Fig. 3; Clerici & Coleman, 1990; Winer 1992; Bartlett & Smith, 1999). MGv neurons are described as being ‘bitufted’ showing dendrites that polarize along the long axis of the cell, giving rise to numerous close proximity branches (Fig. 2 A). Neurons in MGd show a ‘stellate’ morphology as a result of their radially projecting, lower‐order dendrites, typically generating two higher‐order dendrites that diverge to form a star‐like configuration (Fig. 2 B). Many MGd neurons also display bitufted morphology but generally do not display oriented dendritic trees (Clerici & Coleman, 1990; Winer 1992). MGd neurons tended to show slightly larger responses to ACh application compared to neurons in MGv, although these differences are not statistically significant. The age‐related decrease in nAChR efficacy to applied ACh was greater in MGd compared to MGv (Fig. 2 C).
Electrophysiological and pharmacological evidence of an age‐related nAChR subunit switch
Antagonists differing in nAChR subunit selectivity were used to examine the postsynaptic actions of 1 mm ACh puffed onto young and aged MGB neurons. As above, all of the experiments were carried out in the presence of atropine (20 μm) to block mAChRs. We compared the effects of the non‐selective nAChR antagonist mecamylamine (10 μm) against DHβE, a β2‐selective nAChR competitive antagonist (100 nm) (Harvey & Luetje, 1996; Chavez‐Noriega et al. 1997; Xiao & Kellar, 2004) (Fig. 3). Although bath application of 100 nm DHβE almost eliminated responses to ACh application in patched recordings from young MGB neurons (Fig. 3 A), aged MGB neurons showed residual ACh evoked inward currents in the presence of 100 nm DHβE (Fig. 3 B), which were eliminated by the non‐specific nAChR antagonist mecamylamine (Fig. 3 C). Group data compare the nAChR responses to ACh from young and aged MGB neurons using DHβE to separate receptor subtypes (Fig. 3 D). A significant age‐related decrease in total nAChR current in response to ACh application shows that peak amplitude recorded from aged MGB neurons was ∼77.3% ± 10.7% of that seen in young MGB neurons (Fig. 3 D). After the addition of 100 nm DHβE, the remaining nAChR current was ∼7.27% ± 1.14% of the total nAChR response in young MGB neurons compared to 30.37% ± 5.55% in aged neurons. Collectively, these findings suggest that a small number of the nAChR subtypes located on young MGB neurons do not contain the β2 subunit but, instead possess the lower affinity β4 subunit, and this population of β4‐containing nAChRs increases with age.
Autoradiographic evidence of an age‐related loss of nAChRs
To further investigate the reduced actions of ACh application onto aged MGB neurons, saturation binding using autoradiography was carried out to determine number of heteromeric nAChR binding sites. [3H]epibatidine has high affinity for all heteromeric nAChR subtypes (Houghtling et al. 1995; Parker et al. 1998; Xiao & Kellar, 2004) and saturation binding allows the detection of the total number of nAChRs (B max), as well as the affinity (K d) of the radioligand used. A significant decrease in B max was seen in the aged MGB compared to young MGB (Fig. 4), although there was no significant difference in the K d of [3H]epibatidine (Fig. 4).
Discussion
The present study has addressed the postsynaptic function and pharmacology of nAChRs in the MGB using radioligand binding and whole‐cell recordings from MGB neurons in young adult and aged brain slices. These data show that (i) locally applied ACh evokes a significant nAChR‐mediated excitatory postsynaptic inward current in neurons from the two major subdivisions of MGB of both young and aged animals; (ii) MGB neurons show an age‐related deficit in the efficacy of ACh to evoke nAChR mediated postsynaptic currents; and (iii) age‐related changes are probably the result of an age‐related loss of nAChR density (B max) and changes in the subunit composition of nAChRs. Although observations indicating that nAChRs mediate an excitatory postsynaptic inward current in MGB neurons have been established previously (McCormick & Prince, 1987), to our knowledge, this is the first study to examine the impact of ageing on nAChR responses in sensory thalamic neurons.
Significant deficits in cholinergic systems are a common hallmark of ageing in the mammalian central nervous system (Bartus et al. 1982; Schliebs & Arendt, 2011). Nicotinic receptors have been implicated in a wide range of neurological dysfunctions associated with ageing, such as Alzheimer's disease and Parkinson's disease (Rinne et al. 1991; Newhouse et al. 1997; Terry & Buccafusco, 2003). Previous studies have noted a marked loss of high affinity nAChRs in cortical tissue isolated from patients with Alzheimer's disease (Coyle et al. 1983; Nordberg et al. 1992), as well as significant reductions in β2‐containing nAChR density in the whole thalamus (∼32% over human lifespan) (Mitsis et al. 2009). A lack of high affinity nAChRs has been shown to accelerate the development of structural, as well as cognitive, deficits associated with ageing, supporting the hypothesis that a loss of high affinity nAChRs during ageing can contribute to the development of neuronal degeneration and disruption of normal neural circuitry (Zoli et al. 1999). In spiral ganglion neurons that connect the cochlea to brainstem nuclei, it has been shown that the expression level of the β2 subunit is downregulated during the development of presbycusis in C57BL/6J mice. In addition, the expression level of the β2 subunit is increased in a C57BL/6J congenic strain resistant to presbycusis (Bao et al. 2005). These studies implicate the β2 nAChR subunit as being important for the neuronal survival not only of spiral ganglion neurons, but also neurons in central auditory structures as well.
We see a larger age‐related decrease of nAChR‐mediated current at higher ACh concentration. This is probably because, at lower concentrations, ACh is activating mainly β2‐containing receptors in both young and aged MGB, whereas higher concentrations activate not only β2‐containing receptors, but also β4‐containing nAChRs. ACh has a K i of 33–44 nm for α4β2 receptors, whereas the K i is ∼620–850 nm for α3β4 nAChRs (Xiao & Kellar, 2004). There are more β4‐containing receptors in the aged animals (Fig. 3) and, because these receptors are generally less calcium permeable, desensitize faster, and have lower affinity, we see a greater age‐related decrease of ACh at higher ACh concentrations.
The age‐related changes in the whole cell electrophysiology and pharmacology data reported in the present study are consistent with previous studies showing a loss of high affinity nAChRs in animal and human studies (Ferrari et al. 1999; Mitsis et al. 2009). Although the present saturation binding studies using autoradiography show a significant age‐related loss in the total number of nAChRs (B max), no age‐related change in nAChR affinity (K d) was observed (Fig. 4). Meanwhile, the β2‐selective antagonist DHβE was able to block ACh evoked currents from young adult MGB neurons, although it only partially blocked ACh evoked currents from aged MGB neurons indicating an age‐related change in nAChR subunit composition, and thus affinity (Fig. 3). The discordance between patch‐clamp slice recording pharmacology studies and autoradiography regarding receptor affinity is primarily a result of the inability of [3H]epibatidine used in the binding studies to discriminate between β subunits in nAChRs because of small affinity differences between β2‐containing (0.025–0.061 nm) and β4‐containing (0.095–0.97 nm) nAChRs (Whiteaker et al.1998; Xiao et al. 1998; Xiao & Kellar, 2004). On the other hand, DHβE has a ∼100‐fold affinity difference for β2‐containing nAChRs (600–6200 nm) compared to β4‐containing nAChRs (17–190 μm) (Xiao & Kellar, 2004). It is also possible that changes occur in the α subunit of MGB nAChRs as well, although we have not investigated these because there are no simple pharmacological means to carry this out using our patch clamp paradigm.
In addition to the alterations to nAChRs reported in the present study, age‐related changes in central auditory structures are also evident in other members of the cys‐loop ligand‐gated ion channel superfamily such as GABAA and glycine receptors (Caspary et al. 2008). The subunit makeup of GABAA receptor constructs is altered in the ageing thalamus and has been shown to result in altered response properties consistent with a net down‐regulation of functional inhibition (Richardson et al. 2011). This loss of neural inhibition combined with a loss of cholinergic efficacy probably has a negative impact on auditory processing and attentional gating.
Functionally, postsynaptic nAChRs play a strong role in mediating cognitive functions by rapidly changing the excitability of neuronal networks and thus altering synaptic efficacy. These postsynaptic nAChRs, at the level of MGB, potentially facilitate long‐term changes required for the establishment of neuronal networks, functions associated with learning and memory, and the associations of auditory stimuli with importance, reward, and other emotional components (Jones et al. 1999; Dajas‐Bailador & Wonnacott, 2004). Selective attention, mediated through cholinergic circuitry, results in an increased gain of sensory neurons representing the attended stimuli. The responsiveness of the cholinergic system is altered during normal ageing. ACh synthesis and stimulation‐induced release of ACh are diminished in aged animals (Decker, 1987). This cholinergic dysfunction, in addition to the loss of nAChR receptor number and loss of affinity for ACh itself caused by the subunit switch, produces a maladaptive change in these attentional circuits which probably underpins a mechanism contributing to loss of speech understanding seen in presbycusis.
Taken together, these results suggest that certain nAChR subunits found in aged MGB may not be normally translated or assembled in MGB thalamocortical neurons of aged animals. It is our hope that further investigations of the changes in the dynamic properties of cholinergic input to sensory thalamus during ageing may provide clarification of the relationship between cholinergic dysfunction and age‐related decline in auditory processing and may also provide novel insight to approach the treatment of presbycusis.
Additional information
Competing interests
The authors declare that they have no competing financial interests.
Author contributions
S.Y.S. (electrophysiology) and L.L. (receptor binding) performed the experiments and primary data analysis. S.Y.S., L.L, B.C.C., and D.M.C. were involved in designing the experiments, interpretation of data, preparation of manuscript and approval of the final version of the manuscript. All authors have read and approved the final submission. All authors approve of the final version of the manuscript, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved, and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by the National Institute on Deafness and Other Communication Disorders (DC000151‐34). Southern Illinois University School of Medicine Research Imaging facility equipment was supported by Award Number S10RR027716 from the National Centre for Research Resources‐Health.
Acknowledgements
The authors would like to thank Dr Kenneth Kellar and Dr Daniel Llano for their thoughtful guidance and support through the early phases of these studies and Dr Thomas Brozoski for his consultation regarding the statistical analysis of the data.
References
- Bao J, Lei D, Du Y, Ohlemiller KK, Beaudet AL & Role LW (2005). Requirement of nicotinic acetylcholine receptor subunit β2 in the maintenance of spiral ganglion neurons during aging. J Neurosci 25, 3041–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett EL & Smith PH (1999). Anatomic, intrinsic, and synaptic properties of dorsal and ventral division neurons in rat medial geniculate body. J Neurophysiol 81, 1999–2016. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Dean R, Beer B & Lippa AS (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science 217, 408–414. [DOI] [PubMed] [Google Scholar]
- Bauer CA, Brozoski TJ, Holder TM & Caspary DM (2000). Effects of chronic salicylate on GABAergic activity in rat inferior colliculus. Hear Res 147, 175–182. [DOI] [PubMed] [Google Scholar]
- Breese CR, Adams C, Logel J, Drebing C, Rollins Y, Barnhart M, Sullivan B, Demasters BK, Freedman R, Leonard S (1997). Comparison of the regional expression of nicotinic acetylcholine receptor alpha7 mRNA and [125I]‐alpha‐bungarotoxin binding in human postmortem brain. J Comp Neurol 387, 385–398. [DOI] [PubMed] [Google Scholar]
- Caspary DM, Ling L, Turner JG & Hughes LF (2008). Inhibitory neurotransmission, plasticity and aging in the mammalian central auditory system. J Exp Biol 211, 1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux JP (2010). Allosteric receptors: from electric organ to cognition. Ann Rev Pharmacol Toxicol 50, 1–38. [DOI] [PubMed] [Google Scholar]
- Chavez‐Noriega LE, Crona JH, Washburn MS, Urrutia A, Elliott KJ, Johnson EC (1997) Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors hα2β2, hα2β4, hα3β2, hα3β4, hα4β2, hα4β4 and hα7 expressed in Xenopus oocytes. J Pharmacol Exp Ther 280, 346–356. [PubMed] [Google Scholar]
- Clarke PB, Schwartz RD, Paul SM, Pert CB & Pert A (1985). Nicotinic binding in rat brain: autoradiographic comparison of [3H] acetylcholine,[3H] nicotine, and [125I]‐alpha‐bungarotoxin. J Neurosci 5, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici WJ & Coleman JR (1990). Anatomy of the rat medial geniculate body: I. Cytoarchitecture, myeloarchitecture, and neocortical connectivity. J Comp Neurol 297, 14–31. [DOI] [PubMed] [Google Scholar]
- Court JA, Martin‐Ruiz C, Graham A & Perry E (2000). Nicotinic receptors in human brain: topography and pathology. J Chem Neuroanat 20, 281–298. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Price DL & Delong MR (1983). Alzheimer's disease: a disorder of cortical cholinergic innervation. Science 219, 1184–1190. [DOI] [PubMed] [Google Scholar]
- Dajas‐Bailador F & Wonnacott S (2004). Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci, 25, 317–324. [DOI] [PubMed] [Google Scholar]
- Dani J & Bertrand D (2007). Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol Annu Rev Pharmacol Toxicol, 47, 699–729. [DOI] [PubMed] [Google Scholar]
- Decker MW (1987). The effects of aging on hippocampal and cortical projections of the forebrain cholinergic system. Brain Res Rev, 12, 423–438. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Pedrazzi P, Algeri S, Agnati LF & Zoli M (1999). Subunit and region‐specific decreases in nicotinic acetylcholine receptor mRNA in the aged rat brain. Neurobiol Aging 20, 37–46. [DOI] [PubMed] [Google Scholar]
- Gotti C, Moretti M, Gaimarri A, Zanardi A, Clementi F, Zoli M (2007). Heterogeneity and complexity of native brain nicotinic receptors. Biochem Pharmacol 74, 1102–1111. [DOI] [PubMed] [Google Scholar]
- Haghighi AP & Cooper E (2000). A molecular link between inward rectification and calcium permeability of neuronal nicotinic acetylcholine α3β4 and α4β2 receptors. J Neurosci 20, 529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallanger AE, Price SD, Lee HJ, Steininger TL & Wainer BH (1990). Ultrastructure of cholinergic synaptic terminals in the thalamic anteroventral, ventroposterior, and dorsal lateral geniculate nuclei of the rat. J Comp Neurol 299, 482–492. [DOI] [PubMed] [Google Scholar]
- Harris KC & Dubno JR (2017). Age‐related deficits in auditory temporal processing: unique contributions of neural dyssynchrony and slowed neuronal processing. Neurobiol Aging 53, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, Luetje CW (1996) Determinants of competitive antagonist sensitivity on neuronal nicotinic receptor β subunits. J Neurosci 16, 3798–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghtling RA, Davila‐Garcia MI & Kellar KJ (1995). Characterization of (+/‐)(‐)[3H] epibatidine binding to nicotinic cholinergic receptors in rat and human brain. Mol Pharmacol 48, 280–287. [PubMed] [Google Scholar]
- Humes LE, Dubno JR, Gordon‐Salant S, Lister JJ, Cacace AT, Cruickshanks KJ, Gates GA, Wilson RH & Wingfield A (2012). Central presbycusis: a review and evaluation of the evidence. J Am Acad Audiol 23, 635–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Sudweeks S & Yakel JL (1999). Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci 22, 555–561. [DOI] [PubMed] [Google Scholar]
- Kawai H, Lazar R, Metherate R (2007). Nicotinic control of axon excitability regulates thalamocortical transmission. Nature Neurosci 10, 1168–1175. [DOI] [PubMed] [Google Scholar]
- Lesicko AM & Llano DA (2017). Impact of peripheral hearing loss on top‐down auditory processing. Hear Res 343, 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luetje CW & Patrick J (1991). Both alpha‐ and beta‐subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J Neurosci 11, 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao D, Perry DC, Yasuda RP, Wolfe BB & Kellar KJ (2008). The α4β2α5 nicotinic cholinergic receptor in rat brain is resistant to up‐regulation by nicotine in vivo. J Neurochem 104, 446–456. [DOI] [PubMed] [Google Scholar]
- McCormick DA & Prince DA (1987). Actions of acetylcholine in the guinea‐pig and cat medial and lateral geniculate nuclei, in vitro. J Physiol 392, 147–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW (1995). Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269, 1692–1696. [DOI] [PubMed] [Google Scholar]
- Milbrandt JC & Caspary DM (1995). Age‐related reduction of [3H] strychnine binding sites in the cochlear nucleus of the Fischer 344 rat. Neuroscience 67, 713–719. [DOI] [PubMed] [Google Scholar]
- Milbrandt JC, Albin RL, Turgeon SM & Caspary DM (1996). GABAA receptor binding in the aging rat inferior colliculus. Neuroscience 73, 449–458. [DOI] [PubMed] [Google Scholar]
- Milbrandt JC, Holder TM, Wilson MC, Salvi RJ & Caspary DM (2000). GAD levels and muscimol binding in rat inferior colliculus following acoustic trauma. Hear Res 147, 251–260. [DOI] [PubMed] [Google Scholar]
- Mitsis EM, Cosgrove KP, Staley JK, Bois F, Frohlich EB, Tamagnan GD, Dyck CH (2009). Age‐related decline in nicotinic receptor availability with [123I]5‐IA‐85380 SPECT. Neurobiol Aging 30, 1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney DM, Zhang L, Basile C, Senatorov VV, Ngsee J, Omar A & Hu B ( (2003). Distinct forms of cholinergic modulation in parallel thalamic sensory pathways. Proc Natl Acad Sci U S A 101, 320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newhouse PA, Potter A & Levin ED (1997). Nicotinic system involvement in Alzheimer's and Parkinson's diseases. Drugs Aging 11, 206–228. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Alafuzoff I & Winblad B (1992). Nicotinic and muscarinic subtypes in the human brain: changes with aging and dementia. J Neurosci Res 31, 103–111. [DOI] [PubMed] [Google Scholar]
- Ouda L, Profant O & Syka J (2015). Age‐related changes in the central auditory system. Cell Tissue Res 361, 337–358. [DOI] [PubMed] [Google Scholar]
- Parker MJ, Beck A & Luetje CW (1998). Neuronal nicotinic receptor β2 and β4 subunits confer large differences in agonist binding affinity. Mol Pharmacol 54, 1132–1139. [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998). The Rat Brain in Stereotaxic Coordinated. Academic Press, London. [Google Scholar]
- Perry DC & Kellar KJ (1995). [3H] epibatidine labels nicotinic receptors in rat brain: an autoradiographic study. J Pharmacol Exp Ther 275, 1030–1034. [PubMed] [Google Scholar]
- Picciotto M, Higley M & Mineur Y (2012). Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76, 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW & Lester RA (2002). Desensitization of neuronal nicotinic receptors. J Neurobiol 53, 457–478. [DOI] [PubMed] [Google Scholar]
- Radcliffe KA, Dani JA (1998). Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci 18, 7075–7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson BD, Ling LL, Uteshev VV & Caspary DM (2011). Extrasynaptic GABAA receptors and tonic inhibition in rat auditory thalamus. PLoS ONE 6, e16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne JO, Myllykyla T & Lo P (1991). A postmortem study of brain nicotinic receptors in Parkinson's and Alzheimer's disease. Brain Res 547, 155–158. [DOI] [PubMed] [Google Scholar]
- Sarter M, Givens B & Bruno JP (2001). The cognitive neuroscience of sustained attention: where top‐down meets bottom‐up. Brain Res Rev 35, 146–160. [DOI] [PubMed] [Google Scholar]
- Schliebs R & Arendt T (2011). The cholinergic system in aging and neuronal degeneration. Behav Brain Res 221, 555–563. [DOI] [PubMed] [Google Scholar]
- Schofield B, Motts S & Mellott J (2011). Cholinergic cells of the pontomesencephalic tegmentum: connections with auditory structures from cochlear nucleus to cortex. Hear Res 279, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurden DP, Court JA, Lloyd S, Oakley A, Perry R, Pearson C, Pullen RG & Perry EK (1997). Nicotinic receptor distribution in the human thalamus: autoradiographical localization of [3H] nicotine and [125I] α‐bungarotoxin binding. J Chem Neuroanat 13, 105–113. [DOI] [PubMed] [Google Scholar]
- Terry AV & Buccafusco JJ (2003). The cholinergic hypothesis of age and Alzheimer's disease‐related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther 306, 821–827. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Bertrand D, Marguerat A & Raggenbass M (2004). Comparative distribution of nicotinic receptor subtypes during development, adulthood and aging: an autoradiographic study in the rat brain. Neuroscience 124, 405–420. [DOI] [PubMed] [Google Scholar]
- Turner JG & Caspary DM (2005). Comparison of two rat models of aging In Plasticity and Signal Representation in the Auditory System, Ed: Syka Josef. and Merzenick Michael M. pp. 217–225. Springer Science, New York, NY. [Google Scholar]
- Wang H, Turner JG, Ling L, Parrish JL, Hughes LF & Caspary DM (2009a). Age‐related changes in glycine receptor subunit composition and binding in dorsal cochlear nucleus. Neuroscience 160, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Brozoski TJ, Turner JG, Ling L, Parrish JL, Hughes LF & Caspary DM (2009b). Plasticity at glycinergic synapses in dorsal cochlear nucleus of rats with behavioral evidence of tinnitus. Neuroscience 164, 747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker P, Sharples CG & Wonnacott S (1998). Agonist‐induced up‐regulation of α4β2 nicotinic acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol Pharmacol 53, 950–962. [PubMed] [Google Scholar]
- Winer JA (1992). The functional architecture of the medial geniculate body and the primary auditory cortex In The Mammalian Auditory Pathway: Neuroanatomy, Ed: Webster Douglas B., Popper Arthur N., Fay Richard R., pp. 222–409. Springer, New York, NY. [Google Scholar]
- Wonnacott S (1997). Presynaptic nicotinic ACh receptors. Trends Neurosci 20, 92–98. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Meyer EL, Thompson JM, Surin A, Wroblewski J & Kellar KJ (1998). Rat α3/β4 subtype of neuronal nicotinic acetylcholine receptor stably expressed in a transfected cell line: pharmacology of ligand binding and function. Mol Pharmacol 54, 322–333. [DOI] [PubMed] [Google Scholar]
- Xiao Y & Kellar KJ (2004). The comparative pharmacology and up‐regulation of rat neuronal nicotinic receptor subtype binding sites stably expressed in transfected mammalian cells. J Pharmacol Exp Ther 310, 98–107. [DOI] [PubMed] [Google Scholar]
- Zoli M, Picciotto MR, Ferrari R, Cocchi D & Changeux JP (1999). Increased neurodegeneration during ageing in mice lacking high‐affinity nicotine receptors. EMBO J 18, 1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]