

Abstract
Key points
The present study demonstrates that electromyograms (EMGs) obtained during locomotor activity in mice were effective for identification of early physiological markers of amyotrophic lateral sclerosis (ALS). These measures could be used to evaluate therapeutic intervention strategies in animal models of ALS.
Several parameters of locomotor activity were shifted early in the disease time course in SOD1G93A mice, especially when the treadmill was inclined, including intermuscular phase, burst skew and amplitude of the locomotor bursts.
The results of the present study indicate that early compensatory changes may be taking place within the neural network controlling locomotor activity, including spinal interneurons.
Locomotor EMGs could have potential use as a clinical diagnostic tool.
Abstract
To improve our understanding of early disease mechanisms and to identify reliable biomarkers of amyotrophic lateral sclerosis (ALS), a progressive neurodegenerative disease, we measured electromyogram (EMG) activity in hind limb muscles of SOD1G93A mice. By contrast to clinical diagnostic measures using EMGs, which are performed on quiescent patients, we monitored activity during treadmill running aiming to detect presymptomatic changes in motor patterning. Chronic EMG electrodes were implanted into vastus lateralis, biceps femoris posterior, lateral gastrocnemius and tibialis anterior in mice from postnatal day 55 to 100 and the results obtained were assessed using linear mixed models. We evaluated differences in parameters related to EMG amplitude (peak and area) and timing (phase and skew, a measure of burst shape) when animals ran on level and inclined treadmills. There were significant changes in both the timing of activity and the amplitude of EMG bursts in SOD1G93A mice. Significant differences between wild‐type and SOD1G93A mice were mainly observed when animals locomoted on inclined treadmills. All muscles had significant effects of mutation that were independent of age. These novel results indicate (i) locomotor EMG activity might be an early measure of disease onset; (ii) alterations in locomotor patterning may reflect changes in neuronal drive and compensation at the network level including altered activity of spinal interneurons; and (iii) the increased power output necessary on an inclined treadmill was important in revealing altered activity in SOD1G93A mice.
Keywords: amyotrophic lateral sclerosis, electromyogram activity, locomotion, SOD1 mouse
Key points
The present study demonstrates that electromyograms (EMGs) obtained during locomotor activity in mice were effective for identification of early physiological markers of amyotrophic lateral sclerosis (ALS). These measures could be used to evaluate therapeutic intervention strategies in animal models of ALS.
Several parameters of locomotor activity were shifted early in the disease time course in SOD1G93A mice, especially when the treadmill was inclined, including intermuscular phase, burst skew and amplitude of the locomotor bursts.
The results of the present study indicate that early compensatory changes may be taking place within the neural network controlling locomotor activity, including spinal interneurons.
Locomotor EMGs could have potential use as a clinical diagnostic tool.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- BFP
biceps femoris posterior
- EMG
electromyogram
- FF
fast fatigable
- FR
fast fatigue resistant
- LG
lateral gastrocnemius
- P
postnatal day
- S
slow
- TA
tibialis anterior
- VL
vastus lateralis
- WT
wild‐type
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease marked by muscle wasting, cortical and spinal motoneuron dysfunction, eventually leading to a complete loss of motor control (Simon et al. 2014). Although finding treatments for ALS has proven difficult, new therapies could be on the horizon (Vucic et al. 2014; Wainger et al. 2014; Bartus et al. 2016; Riancho et al. 2016). Unfortunately, the effectiveness of these new treatments may be constrained by limitations of diagnostic protocols. By the time that patients are diagnosed, motor unit loss is well underway (Sobue et al. 1983). Both earlier diagnosis and more effective treatments are crucial for preventing the loss of motor function in ALS.
The present study aimed to identify early markers of ALS in animal models by looking for functional changes in EMGs. Although fasciculations, fibrillations and positive sharp waves in electromyogram (EMG) traces from quiescent ALS patients are diagnostically significant (de Carvalho et al. 2008), much less is known about how EMG activity is altered during co‐ordinated locomotor activity, such as walking or running. One previous study found an abnormality in activity in tibialis anterior (TA) starting at postnatal day (P)84 in SOD1G93A mice (Akay, 2014), and another study described functional abnormalities in stance, paw placement and grip strength (Vinsant et al. 2013a, b), although neither study performed a full analysis of locomotor‐evoked EMG activity. Although comprising a more invasive technique, the EMG approach used in the present study increases the sensitivity of our results and provides additional insight into changes in neuromuscular drive. There is a wealth of evidence indicating that both upper and lower motor neuron properties are significantly altered before loss of motor units (Kuo et al. 2004, 2005; Bories et al. 2007; Amendola & Durand, 2008; Pambo‐Pambo et al. 2009; Pieri et al. 2009, 2013; Quinlan, 2011; Filipchuk & Durand, 2012; Jara et al. 2012; Martin et al. 2013; Leroy et al. 2014; Fogarty et al. 2015; Quinlan et al. 2015; Saba et al. 2015). Even after the first motor units fail, overt symptoms such tremors do not appear in the SOD1G93A mouse for over 1 month (Hegedus et al. 2007). Thus, changes within the neurons and networks controlling motor output begin long before gross functional losses. It appears that the nervous system is able to compensate for the loss of a great number of motor units before functional decline. Our hypotheses are (i) that the delay between motor unit loss and functional decline is provided by compensatory shifts in motor patterning and (ii) that these shifts in patterning will be detectable using EMGs. In particular, muscles that are predominately composed of the most vulnerable fast fatigable (FF) and fast fatigue resistant (FR) motor units (Pun et al. 2006; Hegedus et al. 2007) may progressively carry less of the burden of force production, whereas an increased burden may be taken on by muscles that are predominantly composed of slow (S) fibres, and thus are more resistant. Such a compensatory shift in activity would not necessarily be observable from gross locomotor kinematics but requires EMG recordings. Our results show EMG patterns during locomotor activity are altered in ALS, potentially reflecting early changes in the central patterning of motor activity and, importantly, providing new tools for experimental evaluations of ALS.
Methods
Ethical approval
Northwestern University Animal Care and Usage Committee approved all animal procedures, and all studies were conducted in accordance with the United States Public Health Service's Policy on Humane Care and Use of Laboratory Animals. All steps were taken to minimize any pain and suffering of animals used in the present study. The authors understand the ethical principles under which The Journal of Physiology operates and ensure that this work complies with all points in the animal ethics checklist (Grundy, 2015).
SOD1 mouse model
We examined the hind limb EMG activity in a commonly used animal model of ALS overexpressing a point‐mutated form of the human SOD1 gene (Gurney et al. 1994). Transgenic mice (SOD1G93A; www.jax.org/strain/002726) were compared with a control group that overexpresses wild‐type (WT) human SOD1 gene (SOD1WT; www.jax.org/strain/002297). We used 12 adult B6SJL SOD1G93A and SOD1WT transgenic mice of both sexes: four G93A male, four G93A female, two WT male and two WT female. Food and water was provided ad libitum and mice were housed singly. Recordings were made in a period including pre‐symptomatic and symptomatic time points [post‐embryonic day (P)55–100], overlapping the onset of overt symptoms in these animals at P90. Each mouse was implanted with eight EMG electrodes (four per hind limb).
Implantation of EMG electrodes
Electrodes were surgically implanted into the muscles as described previously (Tysseling et al. 2013). Briefly, mice were anaesthetized with the inhalation anaesthetic, isoflurane (1–3%) and prepared for aseptic surgery. Each set of electrodes was placed in the thick portion of the muscle with these specifications: distal muscle belly of vastus lateralis (VL), mid‐muscle belly of biceps femoris posterior (BFP) and TA and distal muscle belly of lateral gastrocnemius (LG) to best avoid interference from BFP. Mice were given Meloxicam (1 mg kg−1) prior to the end of surgery and daily thereafter for 48 h after surgery and after treadmill running. After surgery, mice recovered in cages warmed with heating pads placed below and were monitored for any signs of distress. Implantation of EMG electrodes was well tolerated in all 12 mice. The study was terminated after P100 when the SOD1G93A mice had difficulty keeping up with the treadmill. Animals were then killed by i.p. injection of 150 mg kg−1 Euthasol (Virbac, Carros, France).
Data collection during treadmill locomotion
After recovery from the implantation (1 week), animals were placed on a motorized treadmill (Columbus Instruments, Columbus, OH, USA), locomoted at 30 cm s−1 on a flat (0°) and inclined (20°) treadmill for 3 min (each condition), during which the entire session was recorded. The back connector was attached to a cable and the signals were amplified (X5000, Model 1700 Differential Amplifier; A‐M Systems, Carlsborg, WA, USA) and bandpass filtered (30–1000 Hz) prior to being sampled (2500 Hz; 16 bit, PCI‐6229; National Instruments, Austin, TX, USA) using custom routines created in LabVIEW (National Instruments). Analyses were performed in Matlab (Mathworks, Natick, MA) using customized routines and Merlin software (Rossignol Laboratory, University of Montreal, Montreal, QC, Canada). All recordings underwent high pass digital filtering (second order Butterworth, 50 Hz cut‐off) to minimize movement artefact prior to analysis. Quality control of the EMGs was performed at several stages. During experimentation, mice were monitored for damaged wires, and channels were removed from analysis as necessary. Mice were examined post mortem to verify proper electrode placement and channels were removed from analysis as necessary. Finally, a blind assessment was performed on the quality of raw EMG data: each channel was rated for the signal quality from 1 to 6 (1–2 is best quality: signal to noise ratio is high, there is no movement artefact and bursts are clearly defined; 3–4: signal to noise ratio is not high but bursting is still clearly distinguishable; 5–6: locomotor bursts cannot be distinguished from noise or wires were damaged; often channels were turned off during data collection in the last case). Any channel rated below 4 was dropped from the analysis. Typical raw data are shown in Fig. 1. In Fig. 1 A, all channels were rated 2 except for RLG, which was rated 5 at both time points and was not included in the analysis. In Fig. 1 B, at P69, all channels were rated as 2, except for RTA (rated 3) and RLG (rated 4). At P100, the SOD1G93A channels were rated 2 (LVL and LLG), 3 (LBFP, LTA, RBFP), 5 (RTA and RLG, these channels were not included in the final analysis) and 6 (RVL, as a result of a disconnected wire, this channel was turned off). All experimenters were blinded during data collection and analysis. All analysis was performed by a laboratory member with EMG expertise.
Figure 1. In vivo EMG recording.
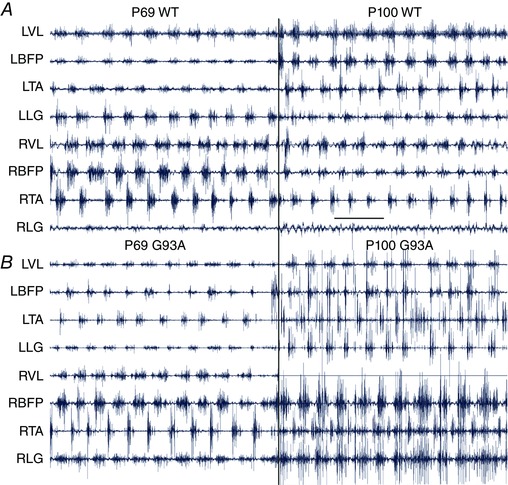
Raw EMG traces (prior to digital filtering, rectification and averaging) from two mice: one SOD1WT (A) and one SOD1G93A (B). At P69, G93A mice are pre‐symptomatic (left traces) and, at P100, mice are symptomatic (right traces). Muscle labels apply to traces on both left and right. Scale bar in (A) = 500 ms applies to all. [Color figure can be viewed at wileyonlinelibrary.com]
Data analysis
Primary analyses aimed to explore the relationship between disease status (i.e. mutation) and several outcome measures related either to burst amplitude (burst peak, burst area) or to burst timing (phase, skew). Each parameter was determined from the averaged activity over about 30 locomotor cycles (using onset of activity in VL as the marker of cycle onset). Both left and right legs were used for analysis if both VLs were of acceptable quality (defined above). Onsets of muscles were correlated with their ipsilateral VL to determine phase and skew. If the ipsilateral VL was not of sufficient quality, muscles from that particular leg were not used for analysis of phase and skew. Cycle duration was measured in a preliminary data set without averaging together locomotor cycles and no differences were found between G93A and WT. Burst peak was calculated as the maximum amplitude of the averaged activity for each muscle. Burst area was calculated as the integrated amplitude of the averaged activity. The mean phase was calculated as the centre of the averaged activity for each muscle, as described previously (Klein et al. 2010). Circular skew of bursts was calculated from the averaged activity with the Circular Statistics Toolbox in Matlab (Berens, 2009), using the formula:
where b is the circular skew value, N is the number of phase samples used, m i is the EMG activity for the ith sample, ᾱ is the mean phase of the EMG activity and αi is the phase of the ith sample. Circular skew is directly analogous to standard measures of skew in non‐circular distributions and reflects the symmetry of the distribution around its mean as shown in Fig. 2. Negative values of skew indicate that the EMG activity is mainly concentrated early in its burst; positive values of skew indicate that the EMG activity is mainly concentrated late in its burst. For simplicity, we refer to this measure as burst skew or skew.
Figure 2. Schematic of methods for measurement of burst parameters.
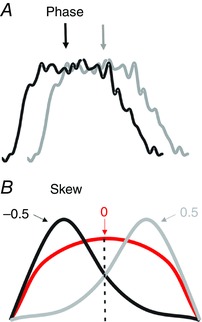
All measurements were taken from rectified and averaged traces. A, mean phase was determined from the timing of activity within the locomotor cycle (using onset of activity in VL as the marker of cycle onset). B, idealized traces illustrating three bursts with different measures of skew. All three bursts have the same mean phase. The red trace has activity that is symmetrically located around its mean phase and so corresponds to a skew value of 0. The black trace has most of its activity located to the left of its mean and so corresponds to a negative skew value. The grey trace has most of its activity located to the right of its mean, corresponding to a positive skew value.
Statistical analysis
We analysed each outcome measure (peak, area, skew, mean phase) for each muscle group (VL, BFP, TA, LG) separately using a series of linear mixed models implemented in Matlab. Because amplitude measures (peak, area) were non‐negative, distributions of these measures usually violated normality assumptions; we therefore natural log‐transformed these measures prior to analysis. Inspection of the distribution of mean phases for each muscle confirmed that they were well clustered within the locomotor cycle, justifying our use of linear models to analyse this outcome measure. Although linear mixed models for circular statistics would be more appropriate for this measure, such statistical methods are not well developed at present. All distributions were inspected for normality and occasional outliers that violated this assumption were excluded (3/205 data points for LG peak amplitude and 2/121 data points for TA mean phase). To account for correlated multiple observations for each animal, we included random intercept (animal) and slope (time or age) effects as appropriate. To account for variability in outcomes due to differences in experimental conditions (i.e. mutant vs. WT, incline vs. flat) and age, we included the following fixed terms in an initial model: age, incline, mutation‐by‐age interaction, mutation‐by‐incline interaction, age‐by‐incline interaction and a quadratic age term. Age was treated as a continuous variable; all other variables were treated as nominal. To avoid convergence issues because of multicolinearity of the potential predictors involving age, we centred the age variable for model building purposes (i.e. subtracted the mean age from each value). We started with an initial model containing all fixed terms then eliminated variables one at a time, refitting the model each time, to obtain the most parsimonious model. The higher order (interaction and quadratic) terms were first examined and removed according to descending magnitude of model type III P value (a criterion of P < 0.10 was used to decide whether to include a variable in the final model). Any first‐order fixed terms remaining in the model were then removed one‐at‐a‐time according to descending magnitude of model type III P value until the final, most parsimonious model contained only variables significant at P = 0.10. This procedure was repeated for each outcome measure and for each muscle. All analyses were implemented in Matlab using fitlme (with parameters selecting maximum likelihood model fits and using ‘effects’ to code dummy variables) and anova (using the Satterthwaite estimate for degrees of freedom).
Results
Stable recordings from hind limb muscles were obtained for BFP, VL, LG and TA (443, 332, 234 and 206 locomotor sessions, respectively). Typical EMG traces are shown in Fig. 1. Smaller numbers of usable observations in LG and TA were probably a result of their relatively small size and accessibility of the distal electrodes to the mice. To capture overall activity, 20–30 locomotor bursts in each session were rectified and averaged for analysis as shown in Fig. 2 A. A mixed model was used for statistical analysis to detect changes in burst characteristics. Analysis incorporated three independent variables (age, G93A mutation and incline of treadmill) and four dependent variables (burst peak, area, mean intermuscular phase and skew). Schematics of the calculation of phase and skew are shown in Fig. 2.
Age and treadmill incline increase EMG amplitude and area
Burst area and peak amplitude significantly increased in all muscles with both age and treadmill incline (Table 1). The parameter of time was considered as both Age and Age2 because it was unknown whether the effects would be linear during disease onset. There was also an increase in area and amplitude when the treadmill was inclined. Inclining the treadmill served to increase the amount of work required from each muscle: more motor units had to be activated.
Table 1.
Statistical significance for measured parameters
| Variable | Incline | Age × Incline | Age | Age2 | Mutation | Mutation × Age | Mutation × Incline | |
|---|---|---|---|---|---|---|---|---|
| Amplitude | VL | < 0.0001 | 0.0010 | 0.0036 | ||||
| BFp | < 0.0001 | 0.0014 | 0.0089 | |||||
| LG | 0.0008 | 0.0008 | 0.0213 | < 0.0001 | ||||
| TA | 0.0001 | 0.0097 | 0.0006 | 0.0153 | ||||
| Area | VL | 0.0025 | 0.0011 | 0.0357 | ||||
| BFp | < 0.0001 | 0.0197 | ||||||
| LG | < 0.0001 | 0.0034 | 0.0046 | 0.0046 | 0.0147 | |||
| TA | 0.0137 | 0.0068 | < 0.0001 | |||||
| Phase | VL | < 0.0001 | 0.0136 | < 0.0001 | ||||
| BFp | 0.0388 | 0.0076 | 0.0002 | |||||
| LG | 0.0010 | 0.0035 | 0.0271 | |||||
| TA | 0.0002 | |||||||
| Skew | VL | 0.0130 | ||||||
| BFp | 0.0012 | 0.0354 | ||||||
| LG | 0.0006 | 0.0418 | 0.0244 | |||||
| TA | 0.0027 | < 0.0001 | 0.0022 | |||||
Burst amplitude increased in SOD1G93A mice
As shown in Fig. 3, TA EMG bursts reached a larger peak amplitude in G93A mice. This was also the case in LG, where, along with TA, the effect of mutation was significant in interaction with incline. The interaction of mutation and incline indicates that significantly larger burst amplitudes appeared when G93A mice ran on an inclined treadmill. For example, in Fig. 3 A, there is no increase in peak amplitude in age‐matched WT and G93A mice running on a level treadmill. However, there is a marked increase in TA amplitude of G93A compared to WT on the inclined treadmill (Fig. 3 B). Thus, there is significant interaction of mutation and incline (Table 1). Measurements of peak amplitude are shown for the other three muscles in Fig. 4, and the ratio of peak amplitude on an inclined treadmill to peak amplitude on a flat treadmill is shown in Fig. 5. In LG, there was a significant interaction of mutation and age, indicating that G93A mice showed an increased peak burst amplitude at older ages. TA and BFP in G93A mice had larger amplitude bursts independent of age. Thus, the effects of SOD1G93A mutation were most pronounced when the treadmill was inclined, and were often independent of age. By contrast to burst amplitude, burst area was not greatly affected by G93A mutation. The only significant mutation effect found for burst area was in LG in interaction with incline. Perhaps burst area is not a sensitive measurement for detecting changes occurring in EMG activity of G93A mice.
Figure 3. Peak amplitude of bursts increases in G93A mice and on an inclined treadmill.
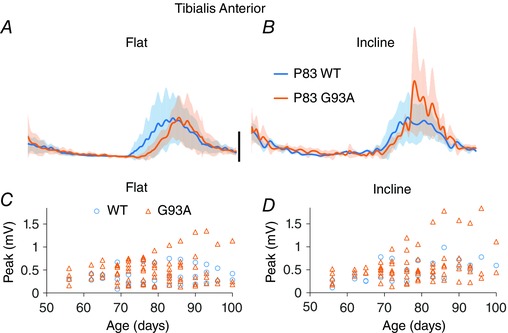
A and B, EMG traces from TA obtained at P83. All WT TA data (blue traces) and G93A TA data (orange traces) are averaged across mice. SD of averaged traces is indicated by the semitransparent shading above and below the averaged traces. The significant interaction of treadmill incline and G93A is clear when comparing peak of bursts in (A) and (B). Scale bar = 0.1 mV applied to all averages. C and D, measurements of peak burst amplitude for TA from all mice, plotted by age.
Figure 4. Peak amplitude of bursts plotted by age.
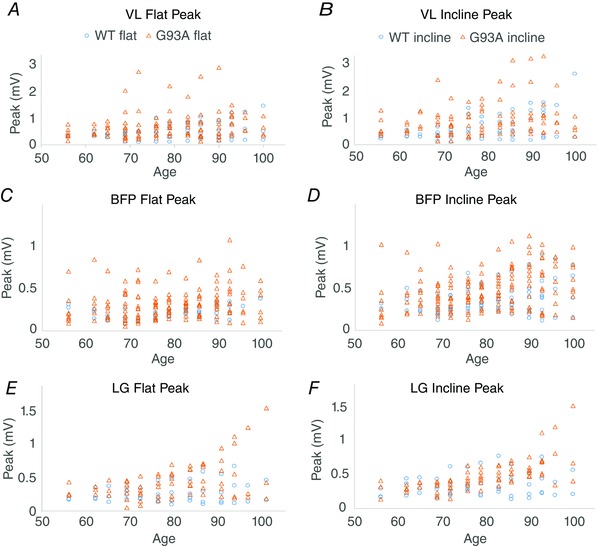
A and B, VL data from a flat treadmill (left) and from an inclined treadmill (right) by age. Peak burst amplitude is not significantly changed in G93A VL. C and D, plots of peak BFP amplitude by age. G93A BFP peak amplitude was significantly increased. E and F, plots of peak LG amplitude by age. G93A LG peak amplitude was significantly increased in interaction with age and incline.
Figure 5. The ratio of peak amplitude of bursts from inclined treadmill : flat treadmill.
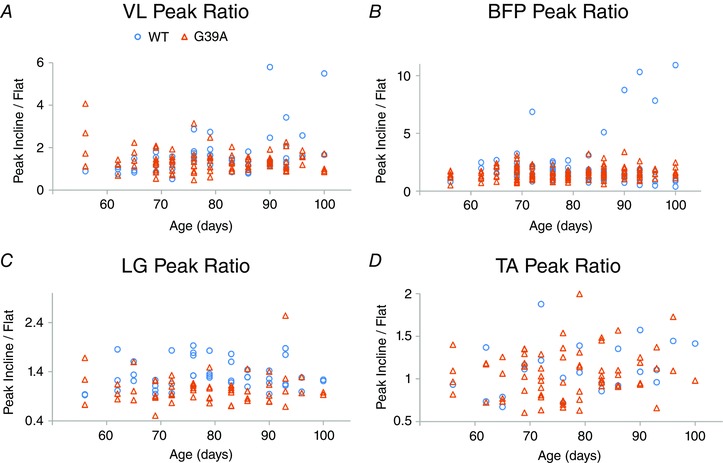
Peak amplitude data plotted by age VL, BFP, LG and TA in (A) to (D). LG and TA both show a significant interaction of mutation and incline with respect to peak burst amplitude.
Intermuscular phasing and burst skew are affected by G93A mutation
The primary aim of the present study was to determine whether changes in locomotor patterning were taking place presymptomatically. Early differences in both skew and phase in G93A mice show this was indeed the case. Burst skew indicates the shape of the distribution of EMG activity within its burst; activity that is symmetric around its mean has a skew of 0, whereas activity with a large asymmetry will have a large skew. As shown for LG in Fig. 6, the skew was significantly shifted to negative values by the G93A mutation because there was increased EMG activity earlier in the burst. This particular example is from the LG of mice aged P76, well before overt symptom onset at P90. G93A BFP also showed a significant shift in skew, from values near 0 to more negative values in G93A, in interaction with age (Fig. 7 and Table 1). G93A VL showed a significant change in skew in interaction with incline. In G93A VL, the skew values shifted closer to 0 from more positive values in WT VL, indicating that WT VL bursts were less symmetric. In all cases, the change in skew indicated that there was more EMG activity earlier in the burst in G93A muscles.
Figure 6. Skew in locomotor bursts is significantly shifted.
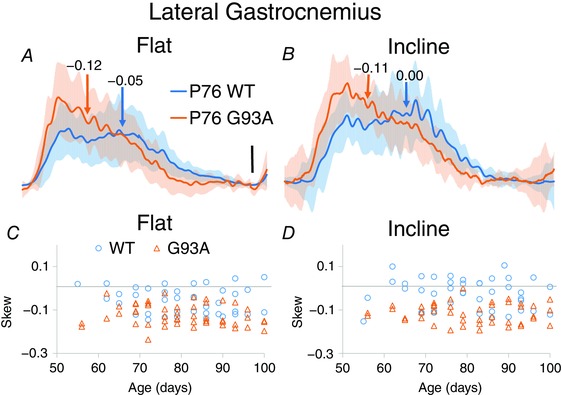
A and B, averaged data from LG from a flat treadmill (left) and an inclined treadmill (right). SD of averaged traces is shown in semitransparent shading above and below the averaged traces. C and D, plots of skew for LG by age. Horizontal lines at skew = 0 shows that LG tends to be skewed slightly earlier in the burst. Skew is shifted in G93A LG in interaction with treadmill incline.
Figure 7. Skew in locomotor bursts is significantly shifted in all G93A muscles.
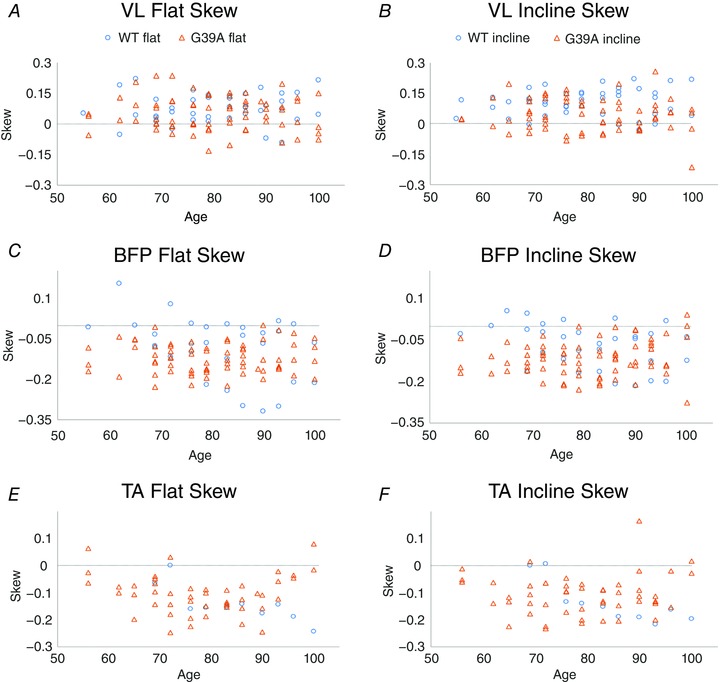
A and B, plots of skew for VL by age. Skew is shifted in G93A VL in interaction with treadmill incline. C and D, plots of skew for BFP by age. Skew in G93A BFP was shifted in interaction with age. E and F, plots of skew for TA by age. Skew in G93A TA was shifted in interaction with age. Horizontal lines at skew = 0 shows that VL tends to be skewed towards more activity later in the burst, whereas BFP and TA are skewed early in the burst.
Intermuscular phase was advanced at an early age in G93A mice. The examples in Fig. 8 were taken from P65, showing possibly the earliest behavioural signs of dysfunction in G93A mice. Phase shifts were observed in BFP (Fig. 8) and LG (Fig. 9), as well as in G93A VL in interaction with incline. In each case, there was a tendency for muscles to be activated earlier in the locomotor cycle in G93A mice.
Figure 8. Intermuscular phasing measurements.
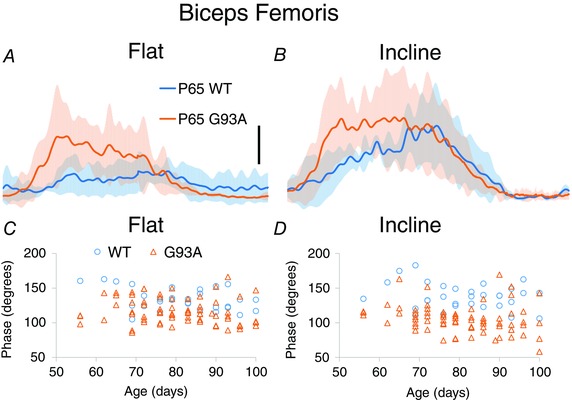
A and B, averages of BFP showing phase advance at P65. SD of averaged traces is indicated by the semitransparent shading above and below the averaged traces. C and D, individual measurements of phase in BFP, plotted by age. Phase significantly advances in G93A in interaction with incline.
Figure 9. Intermuscular phasing measurements.
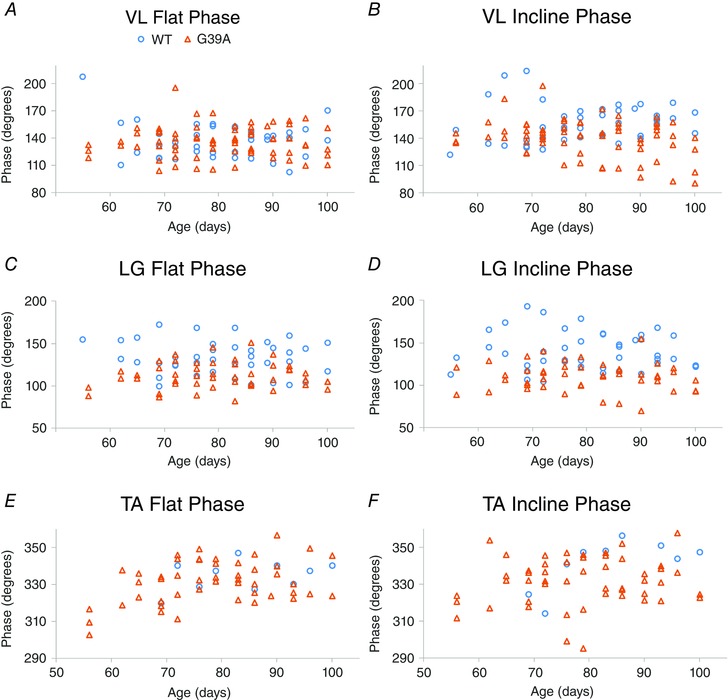
A and B, individual measurements of phase in VL, plotted by age. Phase advanced in G93A VL in interaction with incline. C and D, plots of phase for LG by age. Note the separation in phase in LG, even at early ages. G93A LG was advanced by mutation and in interaction with incline. E and F, plots of the phase values for TA by age. Phase is not significantly changed in TA.
In summary, these changes indicated that both motoneuron firing (burst amplitude characteristics) and locomotor network activity (patterning) were affected before overt symptom onset in G93A mice. These markers could be used as experimental targets for the assessment of early treatments.
Discussion
We have identified novel early biomarkers produced by an ALS‐causing mutation. These markers can help identify disease mechanisms and experimental efficacy of early treatments for ALS. These biomarkers could be particularly helpful for testing new therapies in animal models. Early changes in the SOD1G93A mouse model of ALS were manifest in the amplitude of EMG bursts (BFP, LG and TA), intermuscular phasing (VL, BFP and LG) and skew (VL, BFP, LG and TA). BFP and LG appeared to be the best early disease indicators because they were significantly altered in G93A mutants for each of the above measurements. These two muscles stand out for the greatest vulnerability based on muscle fibre type (LG; see below) and the greatest number of observations obtained (BFP), suggesting that the significance we found might be the result of a combination of statistical power and vulnerability to the disease. The majority of these changes were independent of age. Inclined locomotion was especially effective in revealing effects of G93A mutation.
Motor unit type and disease progression
Motor units are selectively vulnerable to ALS. In SOD1 mice, FF motor units are lost first, followed by FR and finally S motor units (Frey et al. 2000; Pun et al. 2006; Hegedus et al. 2007). All of the muscles investigated in the present study have a high proportion of FF motor units. The highest percentage of FFs in mice is the lateral gastrocnemius, with values reported in the literature at 83–94% FF, 5–17% FR and 0–6% slow (Wang & Kernell, 2001; Guderley et al. 2008). TA in mice reportedly has 60% FF, 30% FR and 5% slow (Hegedus et al. 2007). In rats, which presumably are comparable to mice, BFP has 49% FF, 47% FR and slow 4% (Ariano et al. 1973) and VL has 42% FF, 56% FR and 2% slow (Ariano et al. 1973). Thus, each of the muscles investigated in the present study had a high likelihood of showing early deficits in response to denervation of FF motor units.
Plasticity of locomotor patterning in G93A mice
Changes in intermuscular phase and burst skew could be brought about by two factors: (i) changing the intrinsic properties of the motoneurons and (ii) altering the synaptic drive to motoneurons.
Although sprouting of motoneuron axons could contribute to the observed changes in burst skew and intermuscular phase, intrinsic properties of motoneurons probably do not play a large role in these alterations. Evidence for hyperexcitability, glutamate excitotoxicity and increased Ca2+ permeability of glutamate receptors in motoneurons has been obtained from ALS patients and animal models (Williams et al. 1997; Lin et al. 1998; Trotti et al. 1999; Guo et al. 2003; Van Damme et al. 2005; Pardo et al. 2006; Vucic & Kiernan, 2006; Vucic et al. 2008; Pieri et al. 2009; Meehan et al. 2010; Fuchs et al. 2013); however, recent studies suggest that, in the lead up to neurodegeneration, there is a more nuanced progression than simple overactivity of motoneurons (Saxena et al. 2013; Delestree et al. 2014). Indeed, motoneuron firing has been shown to be relatively unchanged until failure (de Carvalho et al. 2014; Delestree et al. 2014) and the primary determinants of motoneuron recruitment order do not change substantially in ALS (Heckman & Binder, 1993). EMGs may, however, be shaped by the progressive loss of fast motor units and re‐sprouting of S motoneuron axons to the denervated muscle fibres during the course of the present study (Pun et al. 2006; Krysciak et al. 2014; Jensen et al. 2016). These new larger motor units would be composed of both S and FF muscle fibres and activated by S motoneurons. Because S motoneurons require less drive to activate than FFs, they would be active throughout the entire burst (i.e. S motor units are the first to be activated and the last to turn off). Skew values driven by these units would be close to 0 because they are symmetrical. Thus re‐sprouting could be a factor in skew measurements decreasing from a positive number towards values near 0 because, normally, FF motor units turn on toward the end of the burst but, as they are lost and re‐innervated by S motoneurons, their activity would become more symmetrical. However in LG, BFP and TA, there is significant progression from neutral values to more negative values, which is not easily explained by re‐sprouting of motor units.
Altering synaptic drive to motoneurons is another potential explanation of changes in skew and phase. Organization of intermuscular activity is achieved in vivo by (and can be modulated through alterations in) neuromodulatory descending drive (Ghosh & Pearse, 2014), local spinal circuits (Zhang et al. 2008; Crone et al. 2009) and sensory feedback loops (Akay et al. 2014). Neural circuits modelled to recapitulate locomotor activity indicates a two‐layer circuit in which patterning changes (such as those reported in the present study) can be brought about by altering pre‐motor drive (McCrea & Rybak, 2008). Perhaps compensation at the network level could help to temporarily mask functional motor deficits during the time between initial motoneuron denervation at P40 and overt symptom onset at P90 in the mouse model.
The idea that spinal premotor activity is altered in ALS is not new, although it is not well‐explored. In ALS patients, a longer and less complete mixed nerve silent period, evoked by electrical stimulation of a mixed nerve, suggests dysfunction of inhibitory interneurons (Shefner & Logigian, 1998). Studies in SOD1G93A mice show changes in spinal inhibitory circuits beginning around 1 month and progressing until symptom onset (Martin et al. 2007; Chang & Martin, 2009; Casas et al. 2013; Wootz et al. 2013). Furthermore, in the isolated sacral cord, activity patterns suggest increased activity of spinal interneurons (Jiang et al. 2009). Additional evidence suggesting that the activity of spinal neurons resistant to degeneration can affect the survival of vulnerable neuron populations comes from mouse lines with impaired afferent input and a loss of γ motoneurons (Kieran et al. 2005; Chen et al. 2007; Ilieva et al. 2008; Lalancette‐Hebert et al. 2016). Synaptic drive from afferents has been shown to contribute to loss of vulnerable α motoneurons and negatively affect lifespan (Lalancette‐Hebert et al. 2016). These studies suggest that spinal circuits play a role in disease mechanisms in ALS.
In summary, although increased burst amplitude indicates changes in the number of active motoneurons or the size of motor unit potentials, changed phase and skew indicate an altered activity of the locomotor circuitry. In other words, the neural network co‐ordinating locomotor activity in the spinal cord may be changing intramuscular co‐ordination. Perhaps a better understanding of changes in the synaptic drive to motoneurons will aid in the identification of improved treatments for ALS.
Overall lack of effect of age on EMG activity in G93A mutants
There was a surprising lack of interaction of age and G93A mutation (Table 1), despite known progressive denervation during this time. It might be expected that, as G93A mice became more obviously affected by the disease around P80–90, behavioural effects would worsen and all EMG changes would have an age‐mutation interaction. Instead, the scarcity of age‐mutation interactions may indicate that the detected changes were already present at P55 in G93A mice and remained present throughout the course of this study from P55 to 100. This suggests that changes in EMG amplitude, phase and skew may represent the first behavioural signs of ALS.
From Table 1, it is clear that both WT and G93A animals showed progressive increases in peak amplitude and area of EMG bursts during the course of the present study. This increase was also noted in the original methods study (Tysseling et al. 2013), although the underlying reason for this interaction is still unclear. Muscle fibre volume in mice has been shown to reach a plateau at P56 and muscle fibre types are settled by P21; thus, these factors are probably not driving changes in amplitude or the area of EMGs observed in the present study (Wirtz et al. 1983a, b; White et al. 2010). These increases could be artefacts that arise from the EMG implantation procedure, which may involve a greater interaction of the EMG wires with the nearby muscle fibres over time. The increase could also arise from different mechanisms in the G93A mice vs. the SOD1WT mice. For example, the transgenic SOD1WT mice were free from disease and normally continue to gain weight up to P120 (Koschnitzky et al. 2014). Age‐dependent increases in amplitude and area of EMG bursts thus could be a result of increasing weight, which would require more work from muscles during locomotion. By contrast, SOD1G93A mice do not increase in weight in a similar way as their control counterparts; their weight plateaus around P80 (Koschnitzky et al. 2014). Accordingly, although increasing the amplitude and area of bursts could be a result of weight gain in WT mice, this would not be the case in G93A mice and, instead, the same phenomenon could be a result of a re‐sprouting of S motoneurons to re‐innervate denervated FF and FR muscle fibres, creating larger motor units in G93A mice (Thompson & Jansen, 1977; Bodine‐Fowler et al. 1993; Gordon et al. 1993; Pun et al. 2006; Tysseling et al. 2013).
New measure for the effectiveness of intervention strategies
Our strategy was to identify early physiological markers of ALS, although another merit of the present study is the results suggesting that EMGs can be used as a sensitive measure for evaluating therapeutic intervention strategies in animal models of ALS. Most motor units are already lost by the time of ALS diagnosis (Sobue et al. 1983) and so it is clear that, to be more effective, clinical interventions and diagnoses have to start even earlier. The results of the present study suggest that our locomotor protocol, particularly the task of running on the inclined treadmill, and the parameters of burst amplitude, phase and skew comprise a sensitive method for early diagnosis in mice. Future studies are needed to directly evaluate the predictive ability of these measures. Although our EMG electrodes were implanted in mice, future studies in patients could employ multielectrode surface EMGs to non‐invasively collect similar data (Farina & Negro, 2012). Better and earlier treatments for ALS are desperately needed along with better diagnostic markers, such that patients who are diagnosed have the hope of retaining motor functions and preserving their current quality of life. Based on the results of the present study, future investigations investigating new diagnostic methods for ALS in patients might consider focusing on activities driven by vulnerable neural circuits rather than on a single neuronal element in isolation.
Additional information
Competing interests
The authors declare that they have no competing financial interests.
Author contributions
This work was performed in the laboratories of MCT and CJH. KAQ, MCT, CJH and VMT are responsible for the conception and design of the study. KAQ and RDM are responsible for data acquisition. KAQ, EK are JDC are responsible for data analysis. KAQ is responsible for interpretation of the data. EK, JDC and RDM helped draft the manuscript. KAQ is responsible for writing the manuscript. KAQ, MCT, CJH and VMT are responsible for the intellectual content of the study. All authors approved the final version of the manuscript and agreed to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify are listed.
Funding
Funding was provided by NINDS NS077863 (CJH); NINDS F32 NS063535 (KAQ), Target ALS Springboard Fellowship (KAQ); NINDS R01NS086973 (MCT), NIH NIAMS R01AR053608 (MCT); and Craig H. Neilsen Postdoctoral Fellowship (VMT), NICHD K01HD084672 (VMT).
Acknowledgements
The authors gratefully acknowledge Philippe Drapeau, Alain Frigon and the laboratory of Serge Rossignol for allowing us to use their customized software for the EMG analysis.
References
- Akay T (2014). Long‐term measurement of muscle denervation and locomotor behavior in individual wild‐type and ALS model mice. J Neurophysiol 111, 694–703. [DOI] [PubMed] [Google Scholar]
- Akay T, Tourtellotte WG, Arber S & Jessell TM (2014). Degradation of mouse locomotor pattern in the absence of proprioceptive sensory feedback. Proc Natl Acad Sci USA 111, 16877–16882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola J & Durand J (2008). Morphological differences between wild‐type and transgenic superoxide dismutase 1 lumbar motoneurons in postnatal mice. J Comp Neurol 511, 329–341. [DOI] [PubMed] [Google Scholar]
- Ariano MA, Armstrong RB & Edgerton VR (1973). Hindlimb muscle fiber populations of five mammals. J Histochem Cytochem 21, 51–55. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Betourne A, Basile A, Peterson BL, Glass J & Boulis NM (2016). beta2‐Adrenoceptor agonists as novel, safe and potentially effective therapies for amyotrophic lateral sclerosis (ALS). Neurobiol Dis 85, 11–24. [DOI] [PubMed] [Google Scholar]
- Berens P (2009). CircStat: a MATLAB toolbox for circular statistics. J Stat Softw 31, 1–21. [Google Scholar]
- Bodine‐Fowler SC, Unguez GA, Roy RR, Armstrong AN & Edgerton VR (1993). Innervation patterns in the cat tibialis anterior six months after self‐reinnervation. Muscle Nerve 16, 379–391. [DOI] [PubMed] [Google Scholar]
- Bories C, Amendola J, Lamotte d'Incamps B & Durand J (2007). Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci 25, 451–459. [DOI] [PubMed] [Google Scholar]
- Casas C, Herrando‐Grabulosa M, Manzano R, Mancuso R, Osta R & Navarro X (2013). Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav 3, 145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q & Martin LJ (2009). Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a quantitative confocal analysis. Am J Pathol 174, 574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Levedakou EN, Millen KJ, Wollmann RL, Soliven B & Popko B (2007). Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. J Neurosci 27, 14515–14524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone SA, Zhong G, Harris‐Warrick R & Sharma K (2009). In mice lacking V2a interneurons, gait depends on speed of locomotion. J Neurosci 29, 7098–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, Mills K, Mitsumoto H, Nodera H, Shefner J & Swash M (2008). Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol 119, 497–503. [DOI] [PubMed] [Google Scholar]
- de Carvalho M, Eisen A, Krieger C & Swash M (2014). Motoneuron firing in amyotrophic lateral sclerosis (ALS). Front Hum Neurosci 8, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delestree N, Manuel M, Iglesias C, Elbasiouny SM, Heckman CJ & Zytnicki D (2014). Adult spinal motoneurones are not hyperexcitable in a mouse model of inherited amyotrophic lateral sclerosis. J Physiol 592, 1687–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina D & Negro F (2012). Accessing the neural drive to muscle and translation to neurorehabilitation technologies. IEEE Rev Biomed Eng 5, 3–14. [DOI] [PubMed] [Google Scholar]
- Filipchuk AA & Durand J (2012). Postnatal dendritic development in lumbar motoneurons in mutant superoxide dismutase 1 mouse model of amyotrophic lateral sclerosis. Neuroscience 209, 144–154. [DOI] [PubMed] [Google Scholar]
- Fogarty MJ, Noakes PG & Bellingham MC (2015). Motor cortex layer V pyramidal neurons exhibit dendritic regression, spine loss, and increased synaptic excitation in the presymptomatic hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Neurosci 35, 643–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W & Caroni P (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 20, 2534–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Kutterer S, Muhling T, Duda J, Schutz B, Liss B, Keller BU & Roeper J (2013). Selective mitochondrial Ca2+ uptake deficit in disease endstage vulnerable motoneurons of the SOD1G93A mouse model of amyotrophic lateral sclerosis. J Physiol 591, 2723–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M & Pearse DD (2014). The role of the serotonergic system in locomotor recovery after spinal cord injury. Front Neural Circuits 8, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon T, Yang JF, Ayer K, Stein RB & Tyreman N (1993). Recovery potential of muscle after partial denervation: a comparison between rats and humans. Brain Res Bull 30, 477–482. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . Exp Physiol 100, 755–758. [DOI] [PubMed] [Google Scholar]
- Guderley H, Joanisse DR, Mokas S, Bilodeau GM & Garland T, Jr (2008). Altered fibre types in gastrocnemius muscle of high wheel‐running selected mice with mini‐muscle phenotypes. Comp Biochem Physiol B Biochem Mol Biol 149, 490–500. [DOI] [PubMed] [Google Scholar]
- Guo H, Lai L, Butchbach ME, Stockinger MP, Shan X, Bishop GA & Lin CL (2003). Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet 12, 2519–2532. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX et al (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. [DOI] [PubMed] [Google Scholar]
- Heckman CJ & Binder MD (1993). Computer simulations of the effects of different synaptic inputs systems on motor unit recruitment. J Neurophysiol 70, 1827–1840. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT & Gordon T (2007). Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 28, 154–164. [DOI] [PubMed] [Google Scholar]
- Ilieva HS, Yamanaka K, Malkmus S, Kakinohana O, Yaksh T, Marsala M & Cleveland DW (2008). Mutant dynein (Loa) triggers proprioceptive axon loss that extends survival only in the SOD1 ALS model with highest motor neuron death. Proc Natl Acad Sci USA 105, 12599–12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jara JH, Villa SR, Khan NA, Bohn MC & Ozdinler PH (2012). AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol Dis 47, 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen L, Jorgensen LH, Bech RD, Frandsen U & Schroder HD (2016). Skeletal muscle remodelling as a function of disease progression in amyotrophic lateral sclerosis. Biomed Res Int 2016, 5930621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Schuster JE, Fu R, Siddique T & Heckman CJ (2009). Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J Neurosci 29, 15031–15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Hafezparast M, Bohnert S, Dick JR, Martin J, Schiavo G, Fisher EM & Greensmith L (2005). A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol 169, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DA, Patino A & Tresch MC (2010). Flexibility of motor pattern generation across stimulation conditions by the neonatal rat spinal cord. J Neurophysiol 103, 1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschnitzky JE, Quinlan KA, Lukas TJ, Kajtaz E, Kocevar EJ, Mayers WF, Siddique T & Heckman CJ (2014). Effect of fluoxetine on disease progression in a mouse model of ALS. J Neurophysiol 111, 2164–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysciak K, Grieb P & Celichowski J (2014). Changes in motor unit properties in SOD1 (G93A) rats. Muscle Nerve 50, 577–586. [DOI] [PubMed] [Google Scholar]
- Kuo JJ, Schonewille M, Siddique T, Schults ANA, Fu RG, Bar PR, Anelli R, Heckman CJ & Kroese ABA (2004). Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol 91, 571–575. [DOI] [PubMed] [Google Scholar]
- Kuo JJ, Siddique T, Fu R & Heckman CJ (2005). Increased persistent Na+ current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol 563, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette‐Hebert M, Sharma A, Lyashchenko AK & Shneider NA (2016). Gamma motor neurons survive and exacerbate alpha motor neuron degeneration in ALS. Proc Natl Acad Sci USA 113, E8316‐E8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy F, Lamotte d'Incamps B, Imhoff‐Manuel RD & Zytnicki D (2014). Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CL, Bristol LA, Jin L, Dykes‐Hoberg M, Crawford T, Clawson L & Rothstein JD (1998). Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20, 589–602. [DOI] [PubMed] [Google Scholar]
- Martin E, Cazenave W, Cattaert D & Branchereau P (2013). Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 54, 116–126. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA & Golden WC (2007). Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase‐1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol 500, 20–46. [DOI] [PubMed] [Google Scholar]
- McCrea DA & Rybak IA (2008). Organization of mammalian locomotor rhythm and pattern generation. Brain Res Rev 57, 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan CF, Moldovan M, Marklund SL, Graffmo KS, Nielsen JB & Hultborn H (2010). Intrinsic properties of lumbar motor neurones in the adult G127insTGGG superoxide dismutase‐1 mutant mouse in vivo: evidence for increased persistent inward currents. Acta Physiol (Oxf) 200, 361–376. [DOI] [PubMed] [Google Scholar]
- Pambo‐Pambo A, Durand J & Gueritaud JP (2009). Early excitability changes in lumbar motoneurons of transgenic SOD1G85R and SOD1G(93A‐Low) mice. J Neurophysiol 102, 3627–3642. [DOI] [PubMed] [Google Scholar]
- Pardo AC, Wong V, Benson LM, Dykes M, Tanaka K, Rothstein JD & Maragakis NJ (2006). Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp Neurol 201, 120–130. [DOI] [PubMed] [Google Scholar]
- Pieri M, Caioli S, Canu N, Mercuri NB, Guatteo E & Zona C (2013). Over‐expression of N‐type calcium channels in cortical neurons from a mouse model of Amyotrophic Lateral Sclerosis. Exp Neurol 247, 349–358. [DOI] [PubMed] [Google Scholar]
- Pieri M, Carunchio I, Curcio L, Mercuri NB & Zona C (2009). Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol 215, 368–379. [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L & Caroni P (2006). Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9, 408–419. [DOI] [PubMed] [Google Scholar]
- Quinlan KA (2011). Links between electrophysiological and molecular pathology of amyotrophic lateral sclerosis. Integr Comp Biol 51, 913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan KA, Lamano JB, Samuels J & Heckman CJ (2015). Comparison of dendritic calcium transients in juvenile wild type and SOD1(G93A) mouse lumbar motoneurons. Front Cell Neurosci 9, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riancho J, Berciano MT, Ruiz‐Soto M, Berciano J, Landreth G & Lafarga M (2016). Retinoids and motor neuron disease: potential role in amyotrophic lateral sclerosis. J Neurol Sci 360, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba L, Viscomi MT, Caioli S, Pignataro A, Bisicchia E, Pieri M, Molinari M, Ammassari‐Teule M & Zona C (2015). Altered functionality, morphology, and vesicular glutamate transporter expression of cortical motor neurons from a presymptomatic mouse model of amyotrophic lateral sclerosis. Cereb Cortex. [DOI] [PubMed] [Google Scholar]
- Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros‐Louis F & Caroni P (2013). Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80, 80–96. [DOI] [PubMed] [Google Scholar]
- Shefner JM & Logigian EL (1998). The mixed nerve silent period in normal subjects and patients with amyotrophic lateral sclerosis. Electromyogr Clin Neurophysiol 38, 505–510. [PubMed] [Google Scholar]
- Simon NG, Turner MR, Vucic S, Al‐Chalabi A, Shefner J, Lomen‐Hoerth C & Kiernan MC (2014). Quantifying disease progression in amyotrophic lateral sclerosis. Ann Neurol 76, 643–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobue G, Hashizume Y, Sahashi K, Takahashi A, Mukai E, Matsuoka Y & Mukoyama M (1983). Amyotrophic lateral sclerosis. Lack of central chromatolytic response of motor neurocytons corresponding to active axonal degeneration. Arch Neurol 40, 306–309. [DOI] [PubMed] [Google Scholar]
- Thompson W & Jansen JK (1977). The extent of sprouting of remaining motor units in partly denervated immature and adult rat soleus muscle. Neuroscience 2, 523–535. [DOI] [PubMed] [Google Scholar]
- Trotti D, Rolfs A, Danbolt NC, Brown RH Jr. & Hediger MA (1999). SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci 2, 427–433. [DOI] [PubMed] [Google Scholar]
- Tysseling VM, Janes L, Imhoff R, Quinlan KA, Lookabaugh B, Ramalingam S, Heckman CJ & Tresch MC (2013). Design and evaluation of a chronic EMG multichannel detection system for long‐term recordings of hindlimb muscles in behaving mice. J Electromyogr Kinesiol 23, 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Braeken D, Callewaert G, Robberecht W & Van Den Bosch L (2005). GluR2 deficiency accelerates motor neuron degeneration in a mouse model of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 64, 605–612. [DOI] [PubMed] [Google Scholar]
- Vinsant S, Mansfield C, Jimenez‐Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, Prevette D, Caress J, Oppenheim RW & Milligan C (2013a). Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part I, background and methods. Brain Behav 3, 335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinsant S, Mansfield C, Jimenez‐Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, Prevette D, Caress J, Oppenheim RW & Milligan C (2013b). Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav 3, 431–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S & Kiernan MC (2006). Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 129, 2436–2446. [DOI] [PubMed] [Google Scholar]
- Vucic S, Nicholson GA & Kiernan MC (2008). Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131, 1540–1550. [DOI] [PubMed] [Google Scholar]
- Vucic S, Rothstein JD & Kiernan MC (2014). Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci 37, 433–442. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH Jr, Cudkowicz ME, Bean BP, Eggan K & Woolf CJ (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient‐derived motor neurons. Cell Rep 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LC & Kernell D (2001). Fibre type regionalisation in lower hindlimb muscles of rabbit, rat and mouse: a comparative study. J Anat 199, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RB, Bierinx AS, Gnocchi VF & Zammit PS (2010). Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev Biol 10, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TL, Day NC, Ince PG, Kamboj RK & Shaw PJ (1997). Calcium‐permeable alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole propionic acid receptors: a molecular determinant of selective vulnerability in amyotrophic lateral sclerosis. Ann Neurol 42, 200–207. [DOI] [PubMed] [Google Scholar]
- Wirtz P, Loermans HM, Peer PG & Reintjes AG (1983a). Postnatal growth and differentiation of muscle fibres in the mouse. I. A histochemical and morphometrical investigation of normal muscle. J Anat 137, 109–126. [PMC free article] [PubMed] [Google Scholar]
- Wirtz P, Loermans HM, Peer PG & Reintjes AG (1983b). Postnatal growth and differentiation of muscle fibres in the mouse. II. A histochemical and morphometrical investigation of dystrophic muscle. J Anat 137, 127–142. [PMC free article] [PubMed] [Google Scholar]
- Wootz H, Fitzsimons‐Kantamneni E, Larhammar M, Rotterman TM, Enjin A, Patra K, Andre E, Van Zundert B, Kullander K & Alvarez FJ (2013). Alterations in the motor neuron‐renshaw cell circuit in the Sod1(G93A) mouse model. J Comp Neurol 521, 1449–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Narayan S, Geiman E, Lanuza GM, Velasquez T, Shanks B, Akay T, Dyck J, Pearson K, Gosgnach S, Fan CM & Goulding M (2008). V3 spinal neurons establish a robust and balanced locomotor rhythm during walking. Neuron 60, 84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]