

ABSTRACT
Splenic dysfunction is a major feature of sickle cell disease (SCD) and can manifest as acute splenic sequestration crisis (ASSC), which is the earliest life-threatening complication seen in patients with SCD. Aplastic crisis is another potentially deadly complication of sickle cell disease that develops when erythrocyte production temporarily drops. Infection with parvovirus B-19 frequently causes aplastic crises. These two complications are known to be mutually exclusive due to their classic presentation signs and symptoms but there have been few cases where a patient can have concomitant presentation of both phenomena, which can result in a fatal outcome. These few cases force us to rethink the etiology and subsequent management guidelines of these complications. We present to you a case of an unfortunate 23-year-old female who had both complications occurring at the same time, resulting in death.
KEYWORDS: Sickle cell disease, sickle cell trait, acute splenic sequestration, transient aplastic crisis, parvovirus B19 infection
1. Introduction
Acute splenic sequestration crisis (ASSC) usually occurs in children with sickle cell disease and is characterized by splenomegaly with supporting laboratory values of reticulocytosis and thrombocytopenia. TAC (transient aplastic crisis) usually occurs in older children or adults with sickle cell disease commonly due to acute human parvovirus B19 infection and is characterized by reticulocytopenia and mild neutropenia/thrombocytopenia. Both of these complications have striking differences in presentation which makes these diagnoses mutually exclusive. However, there have been few cases in the past where ASSC occurred in association with TAC due to acute human parvovirus B19 infection. This type of clinical picture is possible in older children or adults with sickle cell-hemoglobin C disease where the splenic function is commonly preserved. This clinical entity needs to recognized because patients inflicted with ASSC in the setting of TAC are at risk of quick decompensation and multi organ failure. We present a case of a 23-year-old female with this clinical picture that resulted in a fatal outcome.
2. Case report
The patient presented with fevers and neck pain which had started two days prior to admission, but her symptoms worsened, leading her to come to the hospital. On presentation, she had fevers and paraspinal tenderness on palpation. She had a history of the sickle cell disease trait and she had never been hospitalized with sickle cell crisis or other complications of sickle cell disease. The lab work showed anemia (Hb 8.9) with marked microcytosis (MCV 68.2) and low reticulocyte count (0.3%) and high lactate dehydrogenase levels. Also noticed was a sedimentation rate of 44, a C Reactive Protein of 6.54 and 11% bands. She was admitted for management of sickle cell crisis with concern for aplastic anemia and possible CNS infection. The next morning, a rapid response team was called for increasing lethargy and drowsiness. Stat ABG was done which showed Hb of 3.9 (pH/pCO2/pO2/HCO3: 7.1/84.03/32.7/16.7). Her blood smear was done which showed many nucleated RBCs, no reticulocytes, and no sickle cells, which was consistent with ASSC. Stat bedside ultrasound (Figure 1) and subsequent CT scan showed splenomegaly at 15 cm, suggesting ASSC (Figure 2), but her reticulocyte count was low (0.3%). Within a few hours the patient became hypotensive, requiring pressor support. She developed diffuse airspace opacities requiring intubation. The patient was being continuously transfused to maintain Hb levels around 8 and was also put on broad spectrum antibiotics to cover for any possible infection. A trial to put an arterial line showed clots in the bilateral femoral arteries. The patient went on to develop MODS (multi organ dysfunction syndrome), reflecting worsening liver function tests and renal failure. A few hours into the hospital course, the patient went into cardiac arrest and expired after an unsuccessful attempt to revive her. Later her parvovirus B19 serologies came back positive (IgM-13.6 and IgG 1.2), confirming TAC in the setting of acute human parvovirus B19 infection. Her serum electrophoresis showed HbF 1.1, HbA 54.6, HbS 21.2, HbC 19.6 and HbA2 3.5, confirming HbSC disease. Autopsy showed a marked congestion of spleen with expansion of the red pulp by sickled RBCs, confirming acute splenic sequestration syndrome.
Figure 2.
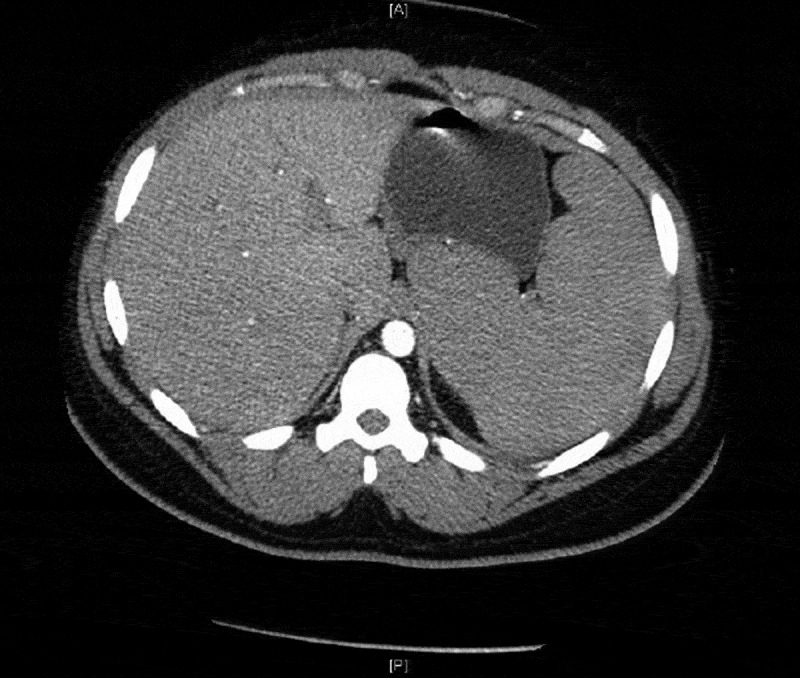
CT scan image confirming splenomegaly.
Figure 1.
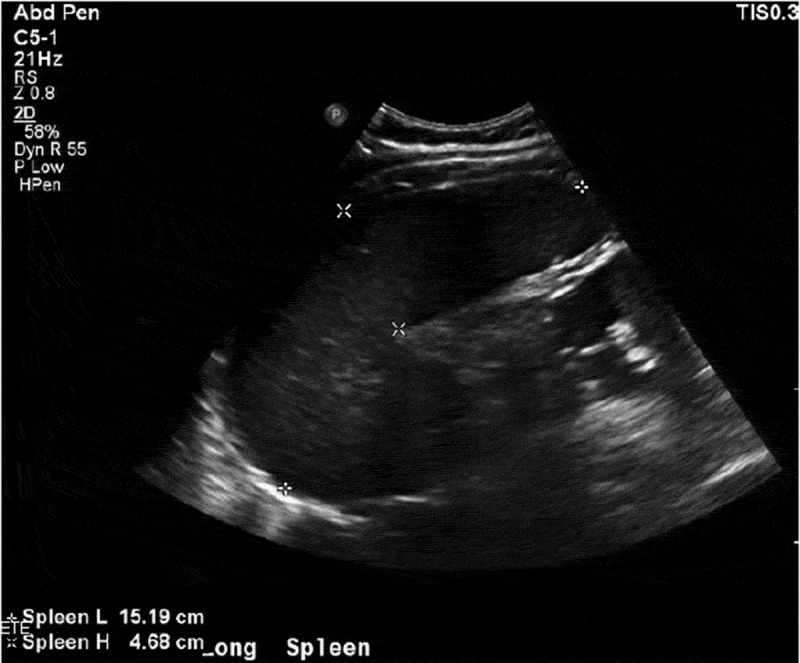
US image showing splenomegaly. Spleen length: 15.19 cm.
3. Discussion
A review of the literature has shown that HbSC patients usually have a clinically severe course of TAC.[1] There are not a lot of cases reported of this clinical phenomenon in adults but children with HbSC appear to be more likely to develop ASSC and acute chest syndrome during TAC than children with HbSS.[2] Concomitant ASSC and TAC is a rare occurrence and can easily be missed in differentials, resulting in a fatal outcome. This case emphasizes the need to monitor adults with HbSC disease with acute parvovirus B19 infection for development of splenomegaly and MODS. Since the differences in clinical and laboratory picture of ASSC and TAC makes them mutually exclusive, definition of ASSC should be modified to include reticulocytopenia in the case of concurrent TAC. Last but not least, we need more prospective studies investigating concurrent ASSC and TAC from B19V infection which can provide us more insight into the epidemiology of this event and appropriate treatment.
Disclosure statement
No potential conflict of interest was reported by the author.
References
- [1].Shao SH, Orringer EP.. Case report: splenic sequestration and multiorgan failure as the presenting manifestation of hemoglobin SC disease. Am J Med Sci. 1996;311(3):139–141. [PubMed]. [DOI] [PubMed] [Google Scholar]
- [2].Smith-Whitley K, Zhao H, Hodinka RL. Epidemiology of human parvovirus B19 in children with sickle cell disease. Blood. 2004;103(2):422–427. [PubMed]. [DOI] [PubMed] [Google Scholar]