

Abstract
Systemic drug delivery to a solid tumor involves a sequence of steps that determine efficacy and survival. Extravasation from circulation at the tumor site is a critical step in this sequence since it regulates how much of the drug accumulates in the tumor. Despite its importance in determining outcomes, extravasation from circulation remains a “black box.” The objective of this study is to develop predictive tools for optimization of drug delivery systems. By comparing pharmacokinetics of liposomal doxorubicin in tumor-free and tumor bearing mice we quantitatively assess the rate constants for distribution, elimination, and tumor accumulation. We then relate these rate constants to the tumor-type and drug delivery system. We compare tumor accumulation in three tumor types and show a 10-fold difference between a colorectal adenocarcinoma and a pancreatic adenocarcinoma. Finally, we show how quantitative predictions of changes in tumor accumulation can be used to optimize drug delivery systems.
Keywords: liposomes, tumor accumulation, extravasation, pharmacokinetics
Graphical abstract
Quantitative analysis of the tumor accumulation and pharmacokinetics in a solid tumor can be used to develop predictive tools for optimization of drug delivery systems. By comparing pharmacokinetics of liposomal doxorubicin in tumor-free and tumor bearing mice we quantitatively assess the rate constants for distribution, elimination, and tumor accumulation.
Background
The systemic delivery of a drug to a solid tumor involves several steps that occur in series and ultimately determine drug efficacy and survival. Extravasation from circulation at the tumor site is a critical step in the delivery process since it determines tumor accumulation (percentage of initial dose in the tumor, %ID) and yet it effectively remains a “black box.” There are two reasons for this. First, in pharmacokinetics, tumor accumulation is bundled into the rate constant for clearance (k10 in classic pharmacokinetic models), along with clearance by the kidneys, the mononuclear phagocyte system (MPS), and any other mechanisms.1 Combining tumor accumulation with other clearance mechanisms highlights the lack of understanding of the kinetics of this important process. Second, pre-clinical trials of drug delivery systems are usually designed to assess efficacy by measuring changes in tumor size and/or survival rates following administration in an animal model, rather than to identify the rate limiting steps.2, 3 Therefore, understanding the kinetics of tumor accumulation in different tumor types is key to enabling the development of new tools for optimization of drug delivery systems.
Extravasation from circulation and tumor accumulation are mediated by the Enhanced Permeability and Retention (EPR) effect, which describes the increased permeability of the tumor vasculature and increased retention associated with poor lymphatic drainage (Figure 1A).4, 5 Evidence from intravital microscopy studies suggests that the “leakiness” of tumor vasculature in mouse models is specific to the location, stage, and type of tumor.6–8 In other research, analysis of replicas of the tumor vasculature in mouse models has shown that the vascular structure is tumor specific.9–13 While a correlation has not yet been established, these studies suggest that the tumor accumulation of a drug delivery system is tumor specific. The development and translation of drug delivery systems can be accelerated by elucidating the factors that modulate tumor accumulation.
Figure 1.

The Enhanced Permeability and Retention (EPR) effect modulates accumulation in solid tumors. (A) The EPR effect mediates nanoparticle extravasation into a tumor. (B) Classic pharmacokinetics looks at two compartments: a central, vascular compartment accounting for the amount of drug in the blood, Nbl, and a peripheral, healthy tissue, compartment accounting for the amount of drug in healthy tissue, Np. The rate constants kp and kd are introduced to evaluate transport between the two compartments while the rate constant kel is used to evaluate elimination pathways. (C) The three-compartment model introduces a tumor compartment accounting for the amount of drug in the tumor, Nt. The rate constants kepr and kb evaluate transport between the vasculature and tumor compartments.
Chemotherapeutics are inherently toxic and hence treatment is invariably a balance between inducing cancer cell death and minimizing the adverse side effects associated with drug accumulation in normal tissue and other organs.14, 15 Combination products and drug delivery systems provide the possibility of allowing new approaches for reducing the unwanted side effects of systemic delivery, increasing tumor accumulation, and improving efficacy.16,17–19 However, the lack of quantitative insight into the parameters that control tumor accumulation is a major barrier to progress in the field. Tumor accumulation is an important parameter in developing drug delivery systems for two reasons: (1) it can be optimized by tuning physico-chemical properties, and (2) by optimizing tumor accumulation it is possible to decrease the dose and minimize unwanted side effects due to uptake in normal tissue. These considerations are not applicable for small molecule therapeutics since the intrinsic properties of the molecule itself determine pharmacokinetic properties and cannot usually be tuned independently.17, 20 Many small molecule chemotherapeutics are extremely toxic and are taken up in normal tissues as evidenced by large distribution volumes.20
The purpose of this work is to provide predictive tools for optimization of drug delivery systems, specifically: (1) to quantitatively assess the relationship between concentration in circulation (pharmacokinetics) and tumor accumulation in a murine model, (2) to quantitatively compare tumor accumulation in different tumor types, and (3) to demonstrate that the design of a drug delivery system can be optimized by making quantitative predictions of changes in tumor accumulation.
We begin by considering the rate constants for the three-compartment model in tumor free and tumor-bearing mice and note that the rate constants associated with intravasation into the tumor and extravasation from the tumor are significantly lower than the rate constants for the peripheral compartment and elimination. We assess the pharmacokinetics and tumor accumulation of liposomal doxorubicin in three subcutaneous xenograft models: a colorectal adenocarcinoma model, a pancreatic adenocarcinoma model, and a breast adenocarcinoma model, demonstrating the fastest tumor accumulation of liposomes in colorectal adenocarcinoma and the slowest tumor accumulation in pancreatic adenocarcinoma. We show how the concentration in circulation and tumor accumulation can be used to obtain the rate constants for extravasation from the vascular system, and intravasation back into circulation. We show that the rate constants for extravasation from the tumor back into circulation (kb) and the elimination rate constant (kel) have the highest impact on tumor retention of drug delivery systems, and we relate these rate constants to the physical properties as recommendations for future systems. We envision that in the future these tools could be extended to optimize drug delivery systems in the clinic.
Methods
Cell culture
All cells were cultured according to the cell culture guidelines put forth in American Type Cell Culture’s (ATCC’s) corresponding protocols. LS 174T colorectal adenocarcinoma cells (ATCC) were grown in Eagle’s Minimum Essential Medium (EMEM) (Quality Biological) with 10% fetal bovine serum (FBS) (Life Technologies) and 1% penicillin streptomycin (P/S) (Life Technologies). Capan-1 pancreatic adenocarcinoma cells (ATCC) were grown in Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies) with 20% FBS and 1% P/S. MDA-MD-231 breast adenocarcinoma cells (ATCC) were grown in DMEM with 10% FBS and 1% P/S. Cells were maintained in a humidified incubator at 5% CO2 and 37°C.
Animal models
All experiments were approved by the Animal Care and Use Committee at Johns Hopkins University. All experiments were performed in accordance the NIH Guide for the Care and Use of Laboratory Animals Athymic nu/nu mice 4–6 weeks old were purchased from Charles River.
Xenograft
Approximately 3 –5 million cells were suspended in 100 μL of 50% growth media and 50% growth-factor-reduced Matrigel (Corning). Cell suspensions were maintained on ice until subcutaneous inoculation. Xenografts were grown to 8 – 10 mm in diameter.
Drug administration
Liposomal doxorubicin (LipoCure, Avanti Polar Lipids) was injected at 6 mg/kg via tail vein injection.
Plasma and tumor collection
Mice were anesthetized via isoflurane for sample collection. Blood was collected via cardiac puncture and immediately centrifuged to separate and collect the plasma samples. Tumors were surgically removed promptly after mice were euthanized and then homogenized in 300 μL mouse plasma (Innovative Research) for 30 m using a cordless pestle motor (VWR). Cell matter was then removed via centrifugation. Mice were sacrificed at 5 min, 30 min, 2 h, 6 h, 12 h, 24 h, 48 h, and 72 h post injection.
Preparation for HPLC
1 μL of daunorubicin was added to plasma and tumor samples (100 μL mouse blood and the entire tumor liquid volume) as a standard control, and 10 μL of Triton-X was added to rupture the liposomes and release the doxorubicin for chemical evaluation. Acetonitrile (600 μL; HPLC grade, Sigma) was then added and vials were vigorously rocked for 30 min. Samples were then centrifuged for 10 min at 7000 rpm, and the supernatant extracted and subsequently dried under flow of argon in a water bath at 37 °C. When the sample appeared dry, it was placed in a vacuum chamber for 3 h or overnight to ensure complete removal of liquid. The sample was then resuspended in 30% methanol (HPLC grade, Fisher), 70% water for HPLC analysis. These samples were run at 1 mL min−1 using 70% methanol and 30% water with 0.1% formic acid through a C18 BDS Hypersil column. The output from these experiments yields doxorubicin concentration in blood and the tumor.
Modeling of pharmacokinetics and tumor accumulation
Experimental pharmacokinetic and tumor accumulation data were analyzed using a three compartment model (Figure 1C) described previously 21. We first used a two compartment model (Figure 1B) to obtain values for kp, kd, and kel from analysis of the drug concentration in blood for the tumor-free mice (see Supplementary Information). The experimental blood concentration and tumor accumulation data for the tumor-bearing mice were then fit to a three compartment model of the form N = Ae−αt + Be−βt + Ce−γt to determine values for A, B, C, α, β, and γ. We assume that the rate constants kp, kd, and kel, are the same for tumor-free and tumor-bearing mice and use a numerical error minimization algorithm written in MatLab to obtain values for values for kepr and kb (see Supplementary Information).
Statistical analysis
Statistical significance was determined using a Student’s t-test. *** p ≤ 0.001, ** p ≤ 0.01. * p ≤ 0.05.
Results
Assessing tumor accumulation
Accumulation of a drug delivery system in a solid tumor by the EPR effect Figure 1A) is dependent on the time-dependent concentration in blood, and hence requires knowledge of the pharmacokinetics. The standard two compartment model with central and peripheral compartments combines tumor accumulation with clearance by the kidneys and the MPS (Figure 1B).1 To distinguish tumor accumulation by the EPR effect from other elimination pathways, we have developed a model incorporating a tumor compartment and define rate constants specifying drug accumulation and removal from the tumor (Figure 1C).21 Drug extravasation into the tumor by the EPR effect is described by the rate constant kepr, and intravasation from the tumor back into circulation is described by kb. We define the rate constant kel to describe elimination by the kidneys, MPS, and any mechanisms other than tumor uptake. To avoid mass balance issues, we define the amount of drug in the different compartments, where Nbl, Np, and Nt represent the amount (in mg) of drug in blood, peripheral tissue, and tumor tissue, respectively.
Pharmacokinetics in tumor-free and tumor-bearing mice
To determine the kinetics for uptake by the EPR effect, we first determine the kinetics of elimination (kel) by all mechanisms excluding tumor accumulation. The concentration of liposomal doxorubicin tumor-free mice was determined at time points from 30 minutes to 48 h (Figure 2A). The concentration in circulation can be fit to a standard 2-compartment pharmacokinetic model with central and peripheral compartments with an initial distribution (Figure 2A). From fits to the data we can obtain values for the rate constants kel, kp, and kd (Table 1). Details of the fits to the data are provided in the Supplementary Information.
Figure 2.

(A) Pharmacokinetics of liposomal doxorubicin in tumor-free mice. Concentration of doxorubicin-loaded liposomes in blood following tail-vein administration in non-tumor-bearing mice. The solid line is a fit to the data using the 2-compartment model from which we can extract the rate constants kel, kp, and kd. (B) Concentration in blood and (C) tumor accumulation of of doxorubicin-loaded liposomes in LS 174T tumor-bearing mice. The rate constants kel, kp, and kd determined from fits to the tumor-free mice are held constant in fits to the LS 174T tumor-bearing mice. The values for kepr and kb found for the LS 174T tumor-bearing mice are insufficiently large to alter the pharmacokinetics of liposomal doxorubicin in a quantifiable manner. Solid lines are fits to the data. n = 10 mice per time point for tumor-free mice, and n = 4 – 8 mice per time point for LS 174T bearing mice. Data represents mean ± SE.
Table 1.
Rate constants for pharmacokinetic modeling of liposomal doxorubicin. The first order rate constants for modeling pharmacokinetic behavior of liposomal doxorubicin in mice. All values are in h−1. The elimination rate constant, kel, and the rate constants that describe movement between the vascular and peripheral compartments, kp and kd, are determined with fits to data obtained in tumor-free mice. These are then held constant to model data from LS 174T tumor-bearing mice to determine values for the rate constant to describe movement into the tumor compartment from the circulation and back, kepr and kb, respectively.
| Rate Constant | Tumor-free (s−1) | LS 174T tumor (s−1) |
|---|---|---|
| kp | 14.0 | |
| kd | 19.9 | |
| kel | 0.1 | 0.124 |
| kepr | 0.011 | |
| kb | 0.022 |
To evaluate the rate of tumor accumulation by the EPR effect, we next determined the pharmacokinetics and tumor accumulation in tumor-bearing mice with LS 174T colorectal adenocarcinoma subcutaneous xenografts following administration of liposomal doxorubicin (Figure 2B and 2C). The concentration in circulation is almost identical to the pharmacokinetics for non tumor-bearing mice (Figure 2A). The fact that the concentration in circulation is the same for non tumor-bearing and tumor-bearing mice indicates that: (1) the presence of the tumor does not significantly change the kinetics of uptake in peripheral tissue or elimination, and (2) that the amount of liposomes taken up in the tumor is small compared to other elimination pathways.
Tumor accumulation
The amount of liposomal doxorubicin in resected LS 174T tumors was determined from HPLC analysis of the doxorubicin concentration. The tumor accumulation increases over the first 12 hours to between 0.5 – 1.0% of the initial dose and then remains approximately constant (Figure 2C).
Kinetics of tumor accumulation in LS 174T tumors: determination of the rate constant for extravasation into the tumor (kepr) and the rate constant for intravasation back into circulation (kb)
To determine the rates of extravasation into the tumor by the EPR effect (kepr) and the rate of intravasation back into circulation (kb), we simultaneously fit the concentration in circulation (Figure 2B) and the tumor accumulation (Figure 2A) using our three-compartment model with a tumor compartment (Figure 1C). Since the pharmacokinetics are almost identical for tumor-free and tumor bearing mice, the fits are performed using values for kp, kd, and kel determined from analysis of the pharmacokinetics for tumor-free mice (Figure 2A and Table 1). The values of kepr and kb are determined from simultaneous optimization of the model to both the pharmacokinetic and tumor accumulation data (see Supplementary Information). From the fits (solid lines in Figures 2B and 2C) we obtain kepr = 0.011 s−1 and kb = 0.022 s−1 (Table 1).
Tumor accumulation in different tumor types
To assess the leakiness of different tumor types, we determined tumor accumulation in mice with subcutaneous colorectal adenocarcinoma (LS 174T), breast adenocarcinoma (MDA-MB-231), and pancreatic adenocarcinoma (Capan-1). Tumor accumulation was determined at 6 h and 24 h following administration of liposomal doxorubicin (Figure 3A). The tumor accumulation in LS174T xenografts at 6 hours post-injection (0.52% ± 0.18 %ID), is 8.5-fold higher than for a Capan-1 pancreatic adenocarcinoma (0.61 ± 0.01 %ID). The accumulation in MDA-MB-231 tumors is intermediate between the other two tumor types (0.12% ± 0.02 %ID). Similar trends are observed when comparing the normalized tumor accumulation (%ID/g) (Figure 3B). Tumor accumulation in LS174T xenografts (2.56 ± 1.1 %ID/g) at 6 h, is 11.6-fold higher than in Capan-1 tumors (0.22 ± 0.03 %ID/g). At 24 h, the normalized tumor accumulation in LS 174T tumors is 8.8-fold higher (%ID) and 10.4-fold higher (%ID/g) than in Capan-1 xenografts.
Figure 3.

Tumor accumulation is dependent on tumor type. Tumor accumulation in colorectal adenocarcinomas (LS 174T), breast adenocarcinomas (MDA-MB-231), and pancreatic adenocarcinomas (Capan-1) at 6 and 24 h post injection of liposomal doxorubicin. (A) Tumor accumulation, %ID. (B) Normalized tumor accumulation, %ID/g. N = 4 – 8. Data represent mean ± SE. Statistical significance was determined using a Student’s t-test. *** p ≤ 0.001, ** p ≤ 0.01, * p ≤ 0.05.
Predicting how modification of rate constants changes tumor accumulation: optimizing the design of drug delivery systems
A major challenge in the development of drug delivery systems is that design is largely empirical. Here we demonstrate how measurement of the rate constants for tumor accumulation (kepr and kb), distribution (kp and kd), and elimination (kel) can be used to predict how modifications of the drug delivery system will impact tumor accumulation, and hence the efficiency of delivery. Ultimately these tools could be used to optimize drug delivery systems for clinical efficacy.
In the context of the three-compartment model (Figure 1C), there are three general strategies to increasing tumor accumulation. First, designing a drug delivery system to increase the rate of extravasation to the tumor (kepr) and/or decrease the rate of intravasation back into circulation (kb) will increase tumor accumulation and decrease uptake in normal tissue. Second, modifying the delivery system to decrease the rate of uptake in normal tissue (kp) will increase tumor accumulation by increasing the concentration in circulation. Thirdly, designing the delivery system to reduce elimination by the mononuclear phagocyte system (MPS), clearance by the kidneys, or by any other mechanisms (kel), will increase tumor accumulation by increasing the concentration in circulation. Quantitative measurement of the rate constants along with our model can be used to assess the relative impact of different strategies to modify the design of drug delivery system to increase tumor accumulation. Here we independently investigate the influence of kb, kp, and kel on the concentration in circulation and tumor accumulation while keeping all other rate constants fixed. We keep kepr fixed since it is defined in large part by the vasculature of the LS 174T xenograft. Using this approach we can assess how different design strategies will influence pharmacokinetics and tumor accumulation.
Influence of the rate of intravasation from the tumor back into circulation (kb)
Decreasing the rate constant for intravasation of doxorubicin-loaded liposomes back into circulation by up to 10-fold has negligible effect on the concentration in circulation (Figure 4A). However, a 10-fold decrease in kb results in about a 3-fold increase in tumor accumulation (Figure 4A). At long times the rate of intravasation back into circulation can become significant when kbNt > keprNbl. Decreasing kb minimizes loss from the tumor and results in sustained tumor accumulation. In practice, this increase in tumor accumulation could be achieved by incorporating a ligand that binds to target tumor cells.22
Figure 4.

Optimizing the design of drug delivery systems: predicting how modification of rate constants changes tumor accumulation. The black lines represent fits to experimental data for the concentration in blood and tumor accumulation of liposomal doxorubicin in an LS174T xenograft model. The other curves show how changing the rate constants for a specific process would change the concentration in blood and tumor accumulation. (A, B) Role of the rate constant for intravasation back into circulation, kb. The amount of liposomal doxorubicin in blood and tumor accumulation from pre-clinical trial (kb = 0.05 h−1) along with simulations for kb/2, kb/5, and kb/10. (C, D) Role of the rate constant for uptake in normal tissue, kp. The amount of liposomal doxorubicin in blood and tumor accumulation from pre-clinical trial (kp = 14 h−1) along with simulations for kp/2, kp/5, and kp/10. (E, F) Role of the rate constant for elimination (excluding tumor accumulation), kel. The amount of liposomal doxorubicin in blood and tumor accumulation from pre-clinical trial (kel = 0.1 h−1) along with simulations for kel/2, kel/5, and kel/10. All other rate constants are held constant.
Influence of the rate of uptake in healthy tissue (kp)
Decreasing the rate constant for uptake in normal tissue (kp) has a complex effect on the concentration in circulation and tumor accumulation. At short times, decreasing kp results in an increase in the concentration in circulation (decreases the distribution phase) (Figure 4C). However, at longer times, the concentration in blood decreases due to the higher availability for elimination pathways. The corresponding tumor accumulation initially increases over the first 12 hours, but decrees significantly at longer times as the low concentration in circulation results in relatively fast intravasation back into circulation.
Influence of the rate of elimination (kel)
Decreasing the rate constant for elimination (kel) results in a progressive increase in the concentration in circulation (Figure 4E). The increase in circulation resulting from the 10-fold decrease kel is about 10-fold at 48 hours post injection (Figure 4F). Increasing the concentration in circulation results in about a 3-fold increase in tumor accumulation, very similar to the results for kb. Decreases in kel could be achieved by increasing the ability of the drug delivery system to evade the immune system.
Discussion
Kinetics of tumor uptake of liposomal doxorubicin in and the EPR effect in a colorectal adenocarcinoma model
The concentration of liposomal doxorubicin is the same in both tumor-free and a colorectal adenocarcinoma mouse model. These results highlight the fact that the rate of tumor accumulation is negligible in comparison to other elimination pathways (e.g. kidneys, MPS, and any mechanisms other than tumor uptake). In athymic nude mice, the impact of the immune response is suppressed due to blocking of thymus-derived T cells; however, natural killer (NK) cells still actively target circulating nanoparticles without markers-of-self. The similar concentrations of liposomal doxorubicin in tumor-bearing and tumor-free shows that any immune response is not sufficient to modulate the elimination half-time. The concentration in circulation in tumor free mice can be modeled by a conventional two-compartment model, commonly used for analysis of the pharmacokinetics of Doxil.23 We use a three-compartment model with an additional tumor compartment to assess the rate constants for tumor accumulation by the EPR effect keeping the rate constants associated with uptake in the peripheral compartment and the elimination rate constants constant. The rate constants associated with exchange with normal tissue are almost four orders of magnitude larger than the rate constants for exchange with the tumor compartment, in part reflecting the length of the vasculature. The total human vasculature is about 105 km whereas the total length of tumor vasculature is about 200 m for a 1 cm3 tumor.13, 24
The rate constants for exchange with the peripheral compartment and the rate constant for elimination can be directly compared to the rate constants obtained from clinical trials of Doxil and liposomal doxorubicin (kp = k12 and kd = k21).21 In a clinical trial of Doxil in patients with a variety of solid tumors, values of kp = 0.1 h−1, kd = 0.2 h−1, and kel = 0.02 h−1 were reported.23 In a trial of PEGylated liposomal doxorubicin where hydrogenated soy phosphatidylcholine (HSPC) was replaced by distearoyl phosphatidylcholine (DSPC) in patients with a variety of solid tumors, kp = 0.33 h−1, kd = 0.37 h−1, and kel = 0.06 h−1 were reported.25 The rate constants (kp and kd) in the colorectal adenocarcinoma mouse model are almost 100-fold higher than the clinical trials in various tumor types although the ratio kp/kd is about 1 in all cases. The rate constant for elimination kel, is only slightly higher in the mouse model, compared to the data from clinical trials.
Tumor accumulation of liposomal doxorubicin is strongly dependent on tumor type
Tumor accumulation in a colorectal adenocarcinoma mouse model is about 10-fold higher at both measured time points compared to a pancreatic adenocarcinoma model. Tumor accumulation in the breast adenocarcinoma model was intermediate between the two. These large differences highlight the significant differences in tumor leakiness in different tumor types.
These results highlight three important points. (1) The kinetics of tumor accumulation are modulated by tumor-specific vascular architecture. Therefore, elucidating the relationships between vascular structure, the EPR effect, and characteristics of the drug delivery system on tumor accumulation, evaluated in this research by the rate constant kepr, will be key to improved treatment of solid tumors. From a clinical perspective, if a goal of chemotherapy is to achieve a specific concentration in the tumor, then the leakiness of the tumor vasculature should be taken into account in assessing the dose. (2) The large differences in tumor accumulation between tumor models highlight the difficulties in comparing the performance and efficacy of drug delivery systems in pre-clinical trials 2. Comparison of drug delivery systems can only be made if pre-clinical trials are performed under identical conditions.3 (3) In addition, the numerical value of normalized tumor accumulation in units of %ID/g does not normalize variations in tumor type. This result is particularly important, since normalized values of tumor accumulation are generally thought to be a way to compare drug delivery systems.
Predicting how changes in the design of a drug delivery system will impact tumor accumulation
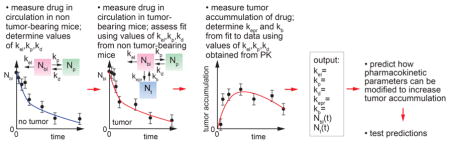
Quantitative assessment of the rate constants associated with tumor accumulation provides valuable insight into the kinetics of the EPR effect and competition with other sinks. Having determined the rate constants for a drug delivery system, the model can be used to predict how changes in the design will impact tumor accumulation. For the case of liposomal doxorubicin, we show how changes in design that influence kb, kp, and kel are predicted to change tumor accumulation. For the case of other drug delivery systems, such as nanoparticles, variations in the net rate of delivery to the tumor (kepr and kb) can be evaluated to determine efficacy in a given tumor system. This approach provides a powerful tool in optimizing drug delivery systems and developing design rules that can be applied to other systems (Figure 5).
Figure 5.

Workflow for quantitative analysis of the rate constants associated with the EPR effect.
We show that the largest increases in tumor accumulation for liposomal doxorubicin are predicted to be achieved by decreasing kel or kb. This insight can be used to optimize drug delivery systems to increase circulation time by evading the MPS (reducing kel) or to increase retention time with the inclusion of active targeting moieties (reducing kb). PEGylation is the current state-of-the-art in evading the MPS. PEGylated liposomes have relatively long clearance half-time, typically 1 - 4 days in patients,26, 27 but delays rather than suppresses opsonization and clearance by the MPS, and can eventually stimulate antibody generation.28, 29 Strategies for increasing circulation time and hence decreasing kel include the use of exosomes created from host red blood cells30, 31 and marker-of-self proteins such as CD47 or CD200.32–34 A hallmark of significant intravasation of a drug delivery system from the tumor back into circulation (large kb) is a time dependent decrease in tumor accumulation as the concentration in circulation decreases. Following extravasation from circulation, the concentration of a drug delivery system in the tumor increases. When other elimination pathways reduce the concentration in circulation below the concentration of free drug in the tumor, there is a net transport back into circulation. Therefore, strategies to efficiently bind a drug delivery system to target tumor cells reduces the concentration of free drug in the tumor and enhances uptake in tumor cells. While the success of active targeting has been very limited,2 it remains an important challenge in optimizing drug delivery and decreasing unwanted side effects.
The extravasation of a drug delivery system from circulation and accumulation in a solid tumor is a critical step in the delivery process. From measurements of tumor accumulation and concentration in circulation of liposomal doxorubicin we have determined the rate constants associated with extravasation form circulation and intravasation from the tumor using a three-compartment model with a tumor compartment. Tumor accumulation of liposomal doxorubicin is dependent on tumor type, with differences as large as 10-fold between a colorectal adenocarcinoma model and a pancreatic adenocarcinoma model. These results demonstrate that the kinetics of tumor accumulation are modulated by tumor-specific vascular architecture. The tumor-specific tumor accumulation highlights the difficulties in comparing and assessing the performance of drug delivery systems in pre-clinical trials. An important consequence of tumor-specific drug accumulation is that dosing of should be based, in part, on the leakiness of the tumor. Having determined the rate constants for the distribution and tumor accumulation of liposomal doxorubicin in a colorectal adenocarcinoma model, we demonstrate how a three-compartment tumor model can be used to optimize the design of a drug delivery system to increase tumor accumulation. By building upon this work to investigate how various drug delivery systems interact with the body and, in particular, with tumors, we can quantitatively establish design rules for how parameters such as composition and size will impact efficacy of delivery to the tumor.
Future directions
In the model described here, kb denotes extravasation back into circulation. In practice there are other sinks for a drug delivery system in the tumor, including drainage by the lymphatic system and diffusion into surrounding tissue. Studies of detail balance within the tumor microenvironment would enable refinement of these and other models.
While the difference in tumor accumulation between colorectal and pancreatic adenocarcinoma models is large, these results are obtained in a mouse xenograft model. The magnitude of the differences in orthotopic and heterotopic tumor models, and the relation to human tumors remains to be established. Nonetheless, this work provides a quantitative foundation to extend this analysis to other models.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health (U54CA151838).
Footnotes
The authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gibaldi M, Perrier D. Pharmacokinetics. 3. Dekker; New York: 2006. [Google Scholar]
- 2.Dawidczyk CM, Russell LM, Searson PC. Nanomedicines for cancer therapy: state-of-the-art and limitations to pre-clinical studies that hinder future developments. Front Chem. 2014;2:69. doi: 10.3389/fchem.2014.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawidczyk CM, Russell LM, Searson PC. Recommendations for Benchmarking Preclinical Studies of Nanomedicines. Cancer Res. 2015;75(19):4016–20. doi: 10.1158/0008-5472.CAN-15-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Advanced drug delivery reviews. 2011;63(3):136–51. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Advanced drug delivery reviews. 2013;65(1):71–9. doi: 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. P Natl Acad Sci USA. 1998;95(8):4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–6. [PubMed] [Google Scholar]
- 8.Jain RK. Determinants of Tumor Blood-Flow - A Review. Cancer Research. 1988;48(10):2641–2658. [PubMed] [Google Scholar]
- 9.Folarin AA, Konerding MA, Timonen J, Nagl S, Pedley RB. Three-dimensional analysis of tumour vascular corrosion casts using stereoimaging and micro-computed tomography. Microvascular research. 2010;80(1):89–98. doi: 10.1016/j.mvr.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Konerding MA, Fait E, Gaumann A. 3D microvascular architecture of pre-cancerous lesions and invasive carcinomas of the colon. British journal of cancer. 2001;84(10):1354–62. doi: 10.1054/bjoc.2001.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konerding MA, Malkusch W, Klapthor B, van Ackern C, Fait E, Hill SA, Parkins C, Chaplin DJ, Presta M, Denekamp J. Evidence for characteristic vascular patterns in solid tumours: quantitative studies using corrosion casts. British journal of cancer. 1999;80(5–6):724–32. doi: 10.1038/sj.bjc.6690416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Less JR, Skalak TC, Sevick EM, Jain RK. Microvascular architecture in a mammary carcinoma: branching patterns and vessel dimensions. Cancer Res. 1991;51(1):265–73. [PubMed] [Google Scholar]
- 13.Hilmas DE, Gillette EL. Morphometric analyses of the microvasculature of tumors during growth and after x-irradiation. Cancer. 1974;33(1):103–10. doi: 10.1002/1097-0142(197401)33:1<103::aid-cncr2820330116>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 14.Chabner BA, Roberts TG., Jr Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5(1):65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 15.DeVita VT, Jr, Chu E. A history of cancer chemotherapy. Cancer research. 2008;68(21):8643–53. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 16.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338(6109):903–10. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergs JW, Wacker MG, Hehlgans S, Piiper A, Multhoff G, Rodel C, Rodel F. The role of recent nanotechnology in enhancing the efficacy of radiation therapy. Biochim Biophys Acta. 2015;1856(1):130–43. doi: 10.1016/j.bbcan.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Youkhanna J, Syoufjy J, Rhorer M, Oladeinde O, Zeineldin R. Toward nanotechnology-based solutions for a particular disease: ovarian cancer as an example. Nanotechnol Rev. 2013;2(4):473–484. [Google Scholar]
- 20.Dawidczyk CM, Kim C, Park JH, Russell LM, Lee KH, Pomper MG, Searson PC. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J Control Release. 2014;187:133–44. doi: 10.1016/j.jconrel.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong AD, Ye M, Ulmschneider MB, Searson PC. Quantitative Analysis of the Enhanced Permeation and Retention (EPR) Effect. PLoS One. 2015;10(5):e0123461. doi: 10.1371/journal.pone.0123461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. International journal of nanomedicine. 2006;1(3):297–315. [PMC free article] [PubMed] [Google Scholar]
- 23.Gabizon A, Catane R, Uziely B, Kaufman B, Safra T, Cohen R, Martin F, Huang A, Barenholz Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994;54(4):987–92. [PubMed] [Google Scholar]
- 24.Aird WC. Phenotypic heterogeneity of the endothelium I. Structure, function, and mechanisms. Circ Res. 2007;100(2):158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 25.Hong RL, Tseng YL. Phase I and pharmacokinetic study of a stable, polyethylene-glycolated liposomal doxorubicin in patients with solid tumors: the relation between pharmacokinetic property and toxicity. Cancer. 2001;91(9):1826–33. [PubMed] [Google Scholar]
- 26.Barenholz Y. Doxil(R)--the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–34. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 27.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal doxorubicin - Review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419–436. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 28.Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew Chem Int Edit. 2010;49(36):6288–6308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- 29.Vllasaliu D, Fowler R, Stolnik S. PEGylated nanomedicines: recent progress and remaining concerns. Expert Opin Drug Del. 2014;11(1):139–154. doi: 10.1517/17425247.2014.866651. [DOI] [PubMed] [Google Scholar]
- 30.Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proceedings of the National Academy of Sciences. 2011 doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kevin N. Marker-of-self’functionalization of nanoscale particles through a top-down cellular membrane coating approach. Nanoscale. 2013;5(7):2664–2668. doi: 10.1039/c3nr00015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oldenborg P-A, Zheleznyak A, Fang Y-F, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288(5473):2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal “Self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339(6122):971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nature Reviews Drug Discovery. 2010;9(8):615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.