

Abstract
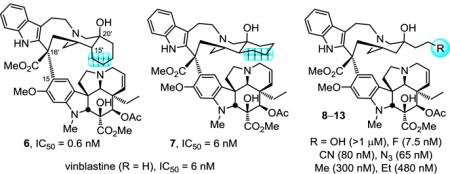
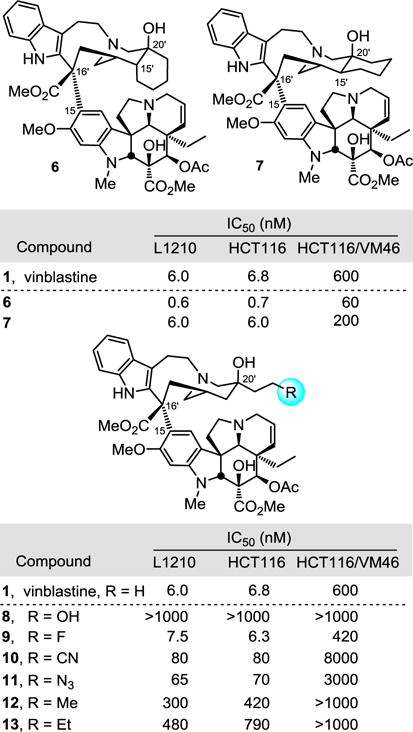
A key series of vinblastine analogs 7–13, which contain modifications to the C20′ ethyl group, was prepared with use of two distinct synthetic approaches that provide modifications of the C20′ side chain containing linear and cyclized alkyl groups or added functionalized substituents. Their examination revealed the unique nature of the improved properties of the synthetic vinblastine 6, offers insights into the origins of its increased tubulin binding affinity and 10-fold improved cell growth inhibition potency, and served to probe a small hydrophobic pocket anchoring the binding of vinblastine with tubulin. Especially noteworthy were the trends observed with substitution of the terminal carbon of the ethyl group that, with the exception of 9 (R = F vs H, equipotent), led to remarkably substantial reductions in activity (>10-fold): R = F (equipotent with H) > N3, CN (10-fold) > Me (50 fold) > Et (100-fold) > OH (inactive). This is in sharp contrast to the maintained (7) or enhanced activity (6) observed with its incorporation into a cyclic C20′/C15′-fused six-membered ring.
Graphical abstract
The Vinca alkaloids represent a group of natural products that continue to have a remarkable impact on the field of anticancer drug discovery and treatment.1 They were originally isolated as trace constituents of the Madagascar periwinkle plant (Catharanthus roseus (L.) G.Don).2 The most prominent member, vinblastine (1), was among the first natural products used in the clinic for the treatment of cancer.3 It along with the related natural product vincristine (2) and three recent semi-synthetic analogs are integral oncology drugs employed today in highly successful combination drug therapies. Even today, their mode of action, which involves disruption of tubulin assembly during mitosis, remains one of the most successful approaches for inhibiting cancer cell growth.4
Pioneering total syntheses of vinblastine have been developed5,6 and an especially effective approach relies on the late-stage biomimetic Fe(III)-promoted oxidative coupling of catharanthine with vindoline.5d,6 When combined with a unique subsequent in situ Fe(III)-mediated hydrogen atom transfer (HAT) free radical oxidative installation of the C20′ tertiary alcohol,6,7 this has provided a powerful approach to access a variety of vinblastine analogs that contain systematic modifications within either the lower vindoline-derived or upper catharanthine-derived subunits in routes as short as 8 steps. As a result of these developments, several series of key analogs have been prepared recently, systematically exploring and defining the impact individual structural features and substituents have on tubulin binding affinity and tumor cell growth inhibition.8 Complementary to the studies that probed the vindoline C4 acetate,9 C5 ethyl substituent,10 C6–C7 double bond,11 and the vindoline core structure itself,12 herein we detail a systematic study of the C20′ ethyl substituent found in the upper catharanthine-derived subunit. In preceding studies, we have shown that whereas replacement of the C20′-OH with extended C20′ ureas led to remarkably potent analogs as much as 100-fold more active than vinblastine (IC50 as low as 50–75 pM),13 C16′ methyl ester modification,14 C10′ or C12′ indole substitution,15 or alteration of the C20′ ethyl group appear much less forgiving with respect to the substituent presence, nature, and size. The upper catharanthine-derived (velbanamine) subunit is deeply imbedded in the tubulin binding site located at the α/β-tubulin dimer-dimer interface.16 In contrast to the C20′ ureas that reside at a site that permits their extension along and further disruption of the continuing protein–protein interaction, the indole group and ethyl substituent each are anchored tightly in small hydrophobic pockets found on the α- or β-tubulin subunits, respectively. Each occupies the opposite top corners of a T-shaped tubulin-bound conformation of vinblastine with the core of the velbanamine subunit filling the intervening space and serving as a rigid scaffold that fixes the placement of these two anchoring groups. To date, only two very specific modifications, one at each site, have led to improved biological potency, 10′-fluorovinblastine (5)13 and the 15′/20′ cis-fused cyclohexyl analog 6 that incorporates the C20′ ethyl group (Figure 1).17 Like the small hydrophobic pocket that surrounds the uniquely active C16′ methyl ester, the binding sites appear to display very little spatial tolerance for changes in vinblastine. Herein, we report a key set of vinblastine analogs 7–13 systematically modified at the C20′ ethyl site that were prepared to define its impact and establish the unique improvement initially designed and observed with 6.
Figure 1.
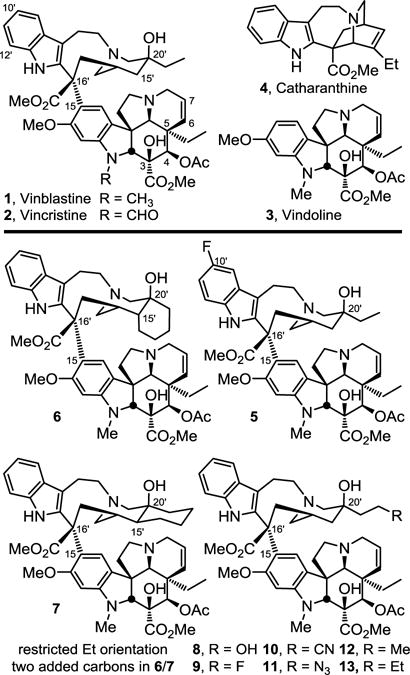
Structures of the natural products, two prior key analogs 5 and 6, and analogs 7–13 disclosed herein.
The syntheses detailed herein enlisted the Fe(III)-promoted coupling/oxidation cascade, which couples an appropriately modified catharanthine derivative with vindoline that proceeds with exclusive generation of the natural C16′ stereochemistry at the critical coupling site followed by subsequent in situ Fe(III)–NaBH4/O2 oxidation of the intermediate anhydrovinblastine analog to install the C20′ alcohol in a single operation. The corresponding modified catharanthine substrates were all prepared from 14, which in turn was derived from commercially available catharanthine sulfate in 4 steps (44% overall) as detailed previously.17,18
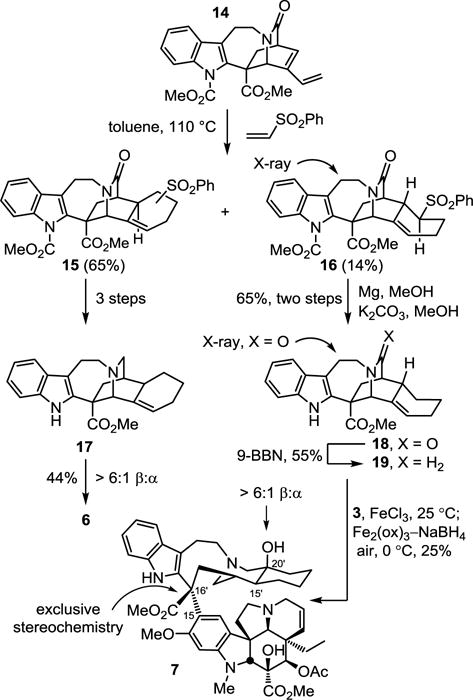
Two complementary strategies produced the reported compounds from this common intermediate 14. The first provided the fused six-membered rings through a Diels–Alder reaction of 14, whereas the second strategy relied on functionalization of its terminal vinyl substituent. The trans C15′/C20′ cyclohexyl-fused derivative 7 was prepared according to the route detailed for the isomeric C15′/C20′ cis analog 6,17 enlisting the minor Diels–Alder product 16 obtained from the thermal cycloaddition of 14 and phenyl vinyl sulfone (toluene, 110 °C, 45 h) (Scheme 1). This minor product 16, in which the newly formed six-membered ring adopts a twist boat conformation, was isolated as a single regioisomer and single diastereomer derived from exo addition to the sterically more encumbered lower face of the diene. Its structure and stereochemistry were confirmed in a single crystal X-ray structure determination conducted on the free indole following removal of the methyl carbamate (CCDC 1544466). Subjection of 16 to mild reductive removal of the sulfone by magnesium (10 equiv, MeOH, 23°C, 4h) and completion of the indole carbamate deprotection by treatment with K2CO3 (6.5 equiv, MeOH, 23 °C, 1 h) provided 18, the structure of which was established by X-ray crystallography (CCDC 1540549). Reduction of the amide (5 equiv 9-BBN, THF, 23°C, 8h, 55%) produced the catharanthine analog 19. Compound 19 was coupled with vindoline with application of the Fe(III)-promoted single electron oxidative coupling (0.05 N aq. HCl/TFE, 10:1, 5 equiv FeCl3, 23°C, 3h) followed by in situ Fe(III)-promoted free radical HAT oxidation of the intermediate trisubstituted alkene (Fe2(ox)3, NaBH4, air, 0°C, 30 min) and afforded 7 isomeric with 6 at C15′. Notably, 19 bears an alkene exocyclic (vs endocyclic) to the catharanthine skeleton and incorporates the added fused six-membered ring and additional C15′ stereochemistry. Its coupling with vindoline exclusively provided the natural C16′ stereochemistry and its participation in the HAT oxidation reaction, which generates the same C20′ tertiary radical as an endocyclic olefin, was trapped in situ by O2 with a >6:1 diastereoselection for introduction of the C20′ alcohol.
Scheme 1.
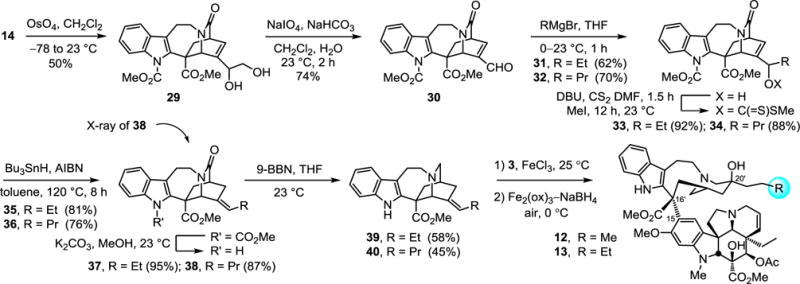
A hydroboration–oxidation of 14 (2.5 equiv 9-BBN dimer, 23°C, 2h; 20 equiv NaBO3, H2O, 0–23°C, 1h, 63%) simultaneously reduced the amide and selectively generated the primary alcohol 20 from the common intermediate 14. Deprotection of the indole carbamate (K2CO3, MeOH, 23°C, 6h) and coupling of 25 with vindoline provided 8, bearing a primary alcohol at the terminus of the C20′ ethyl group. In order to replace the primary alcohol with additional probative or reactive functional groups, 20 was treated with MsCl (1.3 equiv, 2.6 equiv i-Pr2NEt, CH2Cl2, 0°C, 30 min, 60%) to generate the mesylate 21. Without optimization, treatment of 21 with Bu4NF, NaCN, or NaN3 provided the corresponding fluoride (22), nitrile (23) or azide (24), which upon indole deprotection with K2CO3 (MeOH, 23°C, 4h) provided the three catharanthine derivatives 26, 27 and 28. Notably, the nitrile 23 underwent indole deprotection under the conditions of mesylate displacement, providing 27 directly. Their subjection to the coupling with vindoline and in situ HAT oxidation sequence produced the vinblastine analogs 9, 10 and 11.
Because initial efforts to utilize alkyl-based nucleophiles to displace the mesylate in 21 could not be applied successfully, an alternative approach was adopted to prepare the catharanthine analogs that contain extended C20′ alkyl side chains. We targeted the compounds that contain a propyl and butyl side chain, representing addition of one and two more carbons to the ethyl group, respectively. Key of these, the latter represents an analog of the fused cyclohexyl derivatives, containing the same two carbon chain extension but lacking the cyclic structure. Dihydroxylation of the terminal olefin in 14 with use of stoichiometric OsO4 (CH2Cl2, −78 to 23°C, 12h; aq. HCl, MeOH, 0–23°C, 2h, 50%) afforded the diol 29. Subsequent glycol cleavage was achieved upon treatment with sodium periodate deliberately conducted at high concentration (2 equiv of NaIO4, aq. NaHCO3, CH2Cl2, 0.7 M, 23°C, 2h, 74%) to provide aldehyde 30. Treatment of 30 with ethyl or n-propyl magnesium bromide (THF, 0°C) afforded the corresponding addition products 31 and 32, a strategy that can be extended to a variety of Grignard reagent additions. The resulting alcohols reacted cleanly with carbon disulfide and methyl iodide (3 equiv of DBU, 10 equiv CS2, DMF, 23°C, 75 min; 10 equiv MeI, 23°C, 12h) to provide the xanthates 33 and 34. A key Barton–McCombie reductive deoxygenation (0.5 equiv AIBN, 2.5 equiv Bu3SnH, toluene, reflux, 8h) proceeded smoothly to afford 35 and 36, bearing the unfunctionalized side chains with migration of the double bond to the exocyclic olefin in a reaction that proved surprisingly dependent on the quality of Bu3SnH. The compounds 35 and 36 were produced largely as a single positional and geometrical isomer and only small quantities (<10% each) of alternative isomers were detected. Removal of the indole carbamate with K2CO3–MeOH (23°C, 2h) and reduction of the lactams 37 and 38 (4 equiv 9-BBN, THF, 23°C, 3h) gave the catharanthine derivatives 39 and 40. The structure of 38 and both the position and stereochemistry of the double bond were established in a single crystal X-ray structure determination (CCDC 1544467). Without optimization, final coupling with vindoline and in situ oxidation provided the vinblastine analogs 12 and 13 that contain a C20′ propyl and butyl substituent, respectively.
Both of the latter two synthetic pathways (Schemes 2 and 3) may be generalized for the preparation of additional vinblastine analogs, where the first route is more concise and the second allows extensive diversification at the terminal carbon of C20′ ethyl substituent.
Scheme 2.

Scheme 3.
The new analogs, alongside vinblastine and the earlier cis fused cyclohexyl analog 6, were tested for cell growth inhibition against two tumor cell lines routinely used to initially examine vinblastine analogs, HCT116 (human colon) and its isogenic vinblastine-resistant HCT116/VM46 (via Pgp overexpression) cell line and L1210 (mouse leukemia). The results are presented below and displayed remarkable trends. Little free space is present in the hydrophobic pocket surrounding the ethyl group in the X-ray crystal structures16 of vinblastine bound to tubulin (Figure 2). The earlier and remarkable improvements in potency and tubulin binding affinity for 6, which incorporates the ethyl group constrained in a six-membered ring adding only two sp3 methylenes and the C15′ stereocenter, may be attributed to its ability to more favorably occupy and fill the ethyl binding site without changing the intrinsic conformational or structural features of the natural product. Consistent with its design but stunning nonetheless, compound 6 proved to be 10-fold more potent than vinblastine, displaying an IC50 of 600–700 pM in the cell growth inhibition assays. Compound 6 was also found to displace BODIPY-vinblastine bound to tubulin more effectively than vinblastine itself, confirming that it binds the same site and with a higher affinity.17
Figure 2.
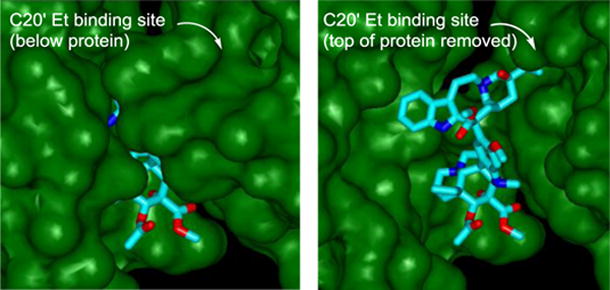
X-ray crystal structure of tubulin-bound vinblastine (pdb 1Z2B)16a highlighting the C20′ ethyl binding site at the dimer–dimer interface where vinblastine binds (left) and site of binding with top of proteins removed to visualize bound vinblastine (right).
By contrast and highlighting the remarkable behavior of 6, the trans fused cyclohexyl analog 7, which is identical in all respects to 6 with the exception of the C15′ stereochemistry, matched but did not exceed the activity of vinblastine and proved to be 10-fold less potent than 6 (Figure 3). Compound 7 constrains the C20′ ethyl side chain to an extended conformation likely preferred in free, but not tubulin-bound vinblastine. This likely results in a combination of offsetting effects derived from non-optimal conformational restriction of the ethyl side chain versus additional stabilizing interactions in the constrained binding pocket derived by more favorably filling the hydrophobic pocket (vs Et). Just as remarkable, the replacement of the C20′ ethyl group with a propyl (12) or butyl (13) group led to large progressive losses in activity (ca. 50-fold and 100-fold, respectively) and this has been independently observed in the recent efforts of others.19 Notably, 13 contains the same two added carbons as 6 and 7, but not the cyclization with C15′ to form the six-membered rings. Although sufficient space is available to accommodate the butyl side chain of 13, its forced adoption of a compact, folded conformation that mimics that found in either 6 or 7 destabilizes binding and substantially exceeds any stabilization that might be derived from the added hydrophobic interactions. Clearly, both the conformational restriction as well as the added hydrophobic interactions combine in 6 to enhance its target binding affinity and functional cell growth inhibition activity.
Figure 3.
Tumor cell growth inhibition by 6–13.
Consistent with both the behavior of the propyl analog 12 and the hydrophobic nature of the binding pocket, addition of an alcohol at the terminus of the ethyl group with 8 resulted in a complete loss in activity. More significantly, addition of a fluorine with 9 provided an analog that matched the activity of vinblastine. This superb activity is likely attributable to the smaller size of the added group (F vs Me), its intrinsic hydrophobic character (F vs OH), and its propensity for adopting a gauche versus extended conformation. Finally, the nitrile (10) and azide (11) analogs, incorporating sterically small, polarized and longer linear substituents, experience a 10-fold loss in potency. Although substantial, it is a notably smaller loss in activity than observed with the added methyl group in 12, likely attributable to more favorable adoption of gauche versus extended conformations (vs Me).
It is a remarkable set of observations in which a series of seemingly benign substitutions at the terminus of the C20′ ethyl group with 8–13, even addition of a methyl group, led to >10-fold or larger losses in activity (50-fold for Me). Thus, it is especially notable that only 9, containing a terminal fluorine, matched the activity of vinblastine, making it a key analog deserving of further study. Clearly, the C20′ ethyl group in vinblastine represents an integral substituent contributing to the properties of vinblastine in an especially substantial manner. Moreover, these comparisons highlight how stunning the behavior of 6 is, improving the activity of vinblastine 10-fold, and just how significant even the behavior of its isomer 7 is, matching the activity of vinblastine. The analog 6 in which the ethyl group is constrained in a C15′/C20′ cis fused six-membered ring is not only 10-fold more potent than vinblastine, but is 1000-fold more potent than 13, differing in structure only in cyclization of the butyl substituent. As noted earlier,17 rarely does one consider adding benign molecular complexity to the underlying core structure of a complex natural product and yet that is what the added six-membered ring in 6 represents. This may be attributed to its ability to more favorably occupy and fill the ethyl binding site without changing the intrinsic conformational or structural features of not only the core of the natural product, but that of the added six-membered ring as well.
A key series of vinblastine analogs containing modifications on the C20′ ethyl group was prepared and assessed. The compounds examined were accessible only by chemical synthesis and presently are inaccessible by natural product derivatization, late-stage functionalization, or biosynthetic methods. Two distinct synthetic routes were developed that provide potential for further analog exploration, providing modifications of the C20′ side chain that contain linear and cyclized alkyl groups and added substituents. Even with a substituent as fundamental to the expression of the properties of vinblastine as the anchoring C20′ ethyl group, the studies highlight both how remarkable the improved properties of 6 are, and that it is possible to use added molecular complexity, even added benign complexity (ABC),17 to enhance target binding affinity and functional biological activity. Our combined use of the powerful synthetic approach detailed herein along with rational design principles that may improve on the properties of a complex natural product continue, targeting additional key structural features of vinblastine, and will be reported in due course.
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA042056 and CA115526, D.L.B.), the Skaggs Institute for Chemical Biology, the Swiss National Science Foundation for fellowship support (O.A.; PZZHP2_158958) and the American Cancer Society for fellowship support (R.M.C.). We especially wish to thank Dr. K. K. Duncan for early cell growth inhibition data in the series and Professor A. L. Rheingold, Dr. Curtis E. Moore and Dr. Milan Gembicky (UCSD) for the single crystal X-ray structure determinations of the free indole derived from 16, compound 18 and compound 38.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information. Experimental details associated with this article can be found in the online version at http://dx.doi.org/10.10167j.bmcl.xxxxxxx.
References
- 1.Neuss N, Neuss MN. Therapeutic use of bisindole alkaloids from Catharantheus. In: Brossi A, Suffness M, editors. The Alkaloids. Vol. 37. Academic Press; 1990. [Google Scholar]
- 2.(a) Noble RL, Beer CT, Cutts JH. Ann N Y Acad Sci. 1958;76:882. doi: 10.1111/j.1749-6632.1958.tb54906.x. [DOI] [PubMed] [Google Scholar]; (b) Noble RL. Lloydia. 1964;27:280. [Google Scholar]; (c) Noble RL. Biochem Cell Biol. 1990;68:1344. [PubMed] [Google Scholar]; (d) Svoboda GH, Neuss N, Gorman M. J Am Pharm Assoc Sci Ed. 1959;48:659. doi: 10.1002/jps.3030481115. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kuehne ME, Marko I. Synthesis of vinblastine-type alkaloids. In: Brossi A, Suffness M, editors. The Alkaloids. Vol. 37. Academic Press; 1990. [Google Scholar]; (b) Borman LS, Kuehne ME. Functional hot spot at the C-20′ position at vinblastine. In: Brossi A, Suffness M, editors. The Alkaloids. Vol. 37. Academic Press; 1990. [Google Scholar]; (c) Pearce HL. Medicinal chemistry of bisindole alkaloids from Catharanthus. In: Brossi A, Suffness M, editors. The Alkaloids. Vol. 37. Academic Press; 1990. [Google Scholar]
- 4.(a) Timasheff S, Andreu J, Gorbunoff M, Medranot F, Prakash V. Cell Pharmacol. 1993;1:S27. [Google Scholar]; (b) Jordan MA, Wilson L. Nat Rev Cancer. 2004;4:253. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 5.(a) Langlois N, Gueritte F, Langlois Y, Potier P. J Am Chem Soc. 1976;98:7017. doi: 10.1021/ja00438a046. [DOI] [PubMed] [Google Scholar]; (b) Mangeney P, Andriamialisoa RZ, Langlois N, Langlois Y, Potier P. J Am Chem Soc. 1979;101:2243. doi: 10.1021/jo01336a006. [DOI] [PubMed] [Google Scholar]; (c) Kutney JP, Hibino T, Jahngen E, Okutani T, Ratcliffe AH, Treasurywala AM, Wunderly S. Helv Chim Acta. 1976;59:2858. doi: 10.1002/hlca.19760590824. [DOI] [PubMed] [Google Scholar]; (d) Kutney JP, Choi LSL, Nakano J, Tsukamoto H, McHugh M, Boulet CA. Heterocycles. 1988;27:1845. [Google Scholar]; (e) Kuehne ME, Matson PA, Bornmann WG. J Org Chem. 1991;56:513. [Google Scholar]; (f) Bornmann WG, Kuehne ME. J Org Chem. 1992;57:1752. [Google Scholar]; (g) Kuehne ME, Zebovitz TC, Bornmann WG, Marko I. J Org Chem. 1987;52:4340. [Google Scholar]; (h) Magnus P, Stamford A, Ladlow M. J Am Chem Soc. 1990;112:8210. [Google Scholar]; Magnus P, Mendoza JS, Stamford A, Ladlow M, Willis P. J Am Chem Soc. 1992;114:10232. [Google Scholar]; (i) Yokoshima S, Ueda T, Kobayashi S, Sato A, Kuboyama T, Tokuyama H, Fukuyama T. J Am Chem Soc. 2002;124:2137. doi: 10.1021/ja0177049. [DOI] [PubMed] [Google Scholar]
- 6.(a) Ishikawa H, Colby DA, Boger DL. J Am Chem Soc. 2008;130:420. doi: 10.1021/ja078192m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2009;131:4904. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gotoh H, Sears JE, Eschenmoser A, Boger DL. J Am Chem Soc. 2012;134:13240. doi: 10.1021/ja306229x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ishikawa H, Elliott GI, Velcicky J, Choi H, Boger DL. J Am Chem Soc. 2006;128:10596. doi: 10.1021/ja061256t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Leggans EK, Barker TJ, Duncan KK, Boger DL. Org Lett. 2012;14:1428. doi: 10.1021/ol300173v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Barker TJ, Boger DL. J Am Chem Soc. 2012;134:13588. doi: 10.1021/ja3063716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sears JE, Boger DL. Acc Chem Res. 2015;48:653. doi: 10.1021/ar500400w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Yang S, Sankar K, Skepper CK, Barker TJ, Lukesh JC, Brody DM, Brütsch MM, Boger DL. Chem Sci. 2017;8:1560. doi: 10.1039/c6sc04146a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sears JE, Barker TJ, Boger DL. Org Lett. 2015;17:5460. doi: 10.1021/acs.orglett.5b02818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Va P, Campbell EL, Robertson WM, Boger DL. J Am Chem Soc. 2010;132:8489. doi: 10.1021/ja1027748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Sasaki Y, Kato D, Boger DL. J Am Chem Soc. 2010;132:13533. doi: 10.1021/ja106284s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kato D, Sasaki Y, Boger DL. J Am Chem Soc. 2010;132:3685. doi: 10.1021/ja910695e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Schleicher KD, Sasaki Y, Tam A, Kato D, Duncan KK, Boger DL. J Med Chem. 2013;56:483. doi: 10.1021/jm3014376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Campbell EL, Skepper CK, Sankar K, Duncan KK, Boger DL. Org Lett. 2013;15:5306. doi: 10.1021/ol402549n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Leggans EK, Duncan KK, Barker TJ, Schleicher KD, Boger DL. J Med Chem. 2013;56:628. doi: 10.1021/jm3015684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Barker TJ, Duncan KK, Otrubova K, Boger DL. ACS Med Chem Lett. 2013;4:985. doi: 10.1021/ml400281w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Carney DW, Lukesh JC, Brody DM, Brütsch MM, Boger DL. Proc Natl Acad Sci USA. 2016;113:9691. doi: 10.1073/pnas.1611405113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tam A, Gotoh H, Robertson WM, Boger DL. Bioorg Med Chem Lett. 2010;20:6408. doi: 10.1016/j.bmcl.2010.09.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gotoh H, Duncan KK, Robertson WM, Boger DL. ACS Med Chem Lett. 2011;2:948. doi: 10.1021/ml200236a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Gigant B, Wang C, Ravelli RBG, Roussi Fanny, Steinmetz MO, Curmi PA, Sobel A, Knossow M. Nature. 2005;435:519. doi: 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]; (b) Wang Y, Benz FW, Wu Y, Wang Q, Chen Y, Chen X, Li H, Zhang Y, Zhang R, Yang J. Mol Pharmacol. 2016;89:233. doi: 10.1124/mol.115.100149. [DOI] [PubMed] [Google Scholar]
- 17.Allemann O, Brütsch MM, Lukesh JC, Brody DM, Boger DL. J Am Chem Soc. 2016;138:8376. doi: 10.1021/jacs.6b04330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giovanelli E, Leroux S, Moisan L, Carreyre H, Thuéry P, Buisson D-A, Meddour A, Coustard J-M, Thibaudeau S, Rousseau B, Nicolas M, Hellier P, Doris E. Org Lett. 2011;13:4116. doi: 10.1021/ol201637m. [DOI] [PubMed] [Google Scholar]
- 19.(a) Zhang Y, Xue Y, Li G, Yuan H, Luo T. Chem Sci. 2016;7:5530. doi: 10.1039/c6sc00932h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kuehne ME, Bornmann WG, Marko I, Qin Y, LeBoulluec KL, Fraiser DA, Xu F, Mulamba T, Ensinger CL, Borman LS, Huot AE, Exon C, Bizzarro FT, Cheung JB, Bane SL. Org Biomol Chem. 2003;1:2120. doi: 10.1039/b209990j. [DOI] [PubMed] [Google Scholar]