

Abstract:
Multiple sclerosis is a heterogenous disease. Although several EMA-approved disease-modifying treatments including biopharmaceuticals are available, their efficacy is limited, and a certain percentage of patients are always nonresponsive. Drug efficacy monitoring is an important tool to identify these nonresponsive patients early on. Currently, detection of antidrug antibodies and quantification of biological activity are used as methods of efficacy monitoring for interferon beta and natalizumab therapies. For natalizumab and alemtuzumab treatments, drug level quantification could be an essential component of the overall disease management. Thus, utilization and development of strategies to determine treatment response are vital aspects of multiple sclerosis management given the tremendous clinical and economic promise of this tool.
Key Words: biopharmaceuticals, therapeutic drug monitoring, drug efficacy monitoring, health economics
INTRODUCTION
Multiple sclerosis (MS) is an autoimmune, inflammatory, and degenerative disease of the central nervous system (CNS) that affects more than 2 million people worldwide. MS is characterized by chronic inflammation leading to CNS damage that results in neurological deterioration along with a multitude of other symptoms.
Depending upon the pattern of the progression of disease, 3 subtypes have been characterized: (1) relapsing remitting MS (RRMS): this is the most common disease course, characterized by the appearance of new or increasing neurological symptoms. These attacks, known as relapses, are followed by periods of partial or complete remissions, during which the symptoms may disappear, or may continue and become permanent. However, there is no continuous progression of the disability. Approximately 85% of all patients with MS are initially diagnosed with RRMS; (2) primary progressive MS (PPMS): this subtype is characterized by the worsening of neurological functions (accumulation of disability) right from the onset of the symptoms, without early relapses or remissions. Approximately 15% of patients are diagnosed with PPMS; (3) secondary progressive MS (SPMS) subtype follows an initial relapsing-remitting course. Most patients diagnosed with RRMS eventually evolve in to a SPMS which is characterized by progressive worsening of neurological functions with accumulation of disability. Here, evidence of disease activity as indicated by relapses or changes on magnetic resonance imaging (MRI) may or may not be present.1
Over the past decade, the landscape of care for MS has changed tremendously due to the advent of multiple disease modifying treatments (DMTs). Till date, 15 pharmaceutical formulations have been approved (Table 1) for RRMS. Amongst these, only mitoxantrone and IFNβ1-b are approved for SPMS as well. These DMTs differ with respect to the efficacy, formulation, method and schedule of administration, and possible adverse drug reactions (ADRs) in addition to cost. These latest formulations also include biopharmaceuticals such as different formulations of IFNβ, monoclonal antibodies (MAbs) against α4/β1 and β7 integrin (NAT) and anti-CD52 (alemtuzumab). Many of these drugs are associated with serious ADRs such as cardiac events, opportunistic infections, and secondary autoimmunity.2 Therefore, the selection of the right drug for the right patient, or personalized treatment, is highly desirable. Consistent progress has been made towards the identification of pharmacogenomic markers of DMT response3 in MS. However, limited pharmacogenetic or pharmacogenomic tests are available to predict the efficacy of a treatment till date, and as a result, predicting patient response to DMT in advance is very difficult. The general approach is to weigh benefits and risks taking into consideration factors such as the aggressiveness of the disease, the efficacy of the drug, and the possibility of ADRs. In addition, several other factors including tolerability, planning of pregnancy, preference and life style of the patient, previous treatments, adherence to treatment, clinical, and MRI examinations along with the cost may play an equally important role in the selection of the right drug. In most cases, the neurologists and patients must rely on a “trial and error” approach. This is inadequate and risky because a treatment failure can cause an irreversible damage of CNS functions. Thus, an approach like drug efficacy monitoring is important to enable the physician to detect nonresponsive patients as early as possible. Monitoring of drug efficiency can essentially include any biochemical, clinical or genetic evaluations that could aid in modulation of drug type, dosage or schedule of administration to optimally benefit the patient and minimize the possibility of ADRs. On the other hand, the concept of therapeutic drug monitoring (TDM) essentially involves measurement of the concentration of the drug in the serum. In the context of MS, TDM alone may not be sufficient to provide enough information regarding drug response to enable the physician to effectively individualize the treatment. Therefore, drug efficacy monitoring in MS must include other components such as the quantification of antidrug antibodies (ADA) (induced by IFNβ or NAT), and evaluation of biological activity in addition to TDM in order to predict the efficacy of biopharmaceuticals. However, the measurement of biological activity can be useful in clinical practice only if a biomarker is specifically up- or down-regulated after the drug administration.4,5
TABLE 1.
EMA and FDA Approved DMTs for Multiple Sclerosis

In the present review, attempts have been made to explore the available literature with respect to 2 of the most commonly used biopharmaceuticals in MS, namely, IFNβ and NAT, in addition to a newer drug such as alemtuzumab, and delineate the available methods for drug efficacy monitoring in detail.
IFNβ
Mechanism of Action of IFNβ
Natural IFNβ, the type I IFN, is secreted by fibroblasts. It binds to the IFN receptor and activates the JAK/STAT pathway to phosphorylate STAT1 and STAT2.6 These factors dimerize and associate with IFN regulatory factor-3, and bind to IFN-stimulated response elements in the cell nucleus. This, in turn, activates hundreds of IFN-stimulated genes and leads to the production of antiviral, antiproliferative, and antitumor products.7 The mechanism of action of IFNβ is complex. It balances the expression of antiinflammatory and proinflammatory cytokines, reduces the trafficking of inflammatory cells across the blood-brain-barrier, and increases the production of nerve growth factor. Moreover, in the peripheral blood, it increases the number of natural killer cells, which are producers of antiinflammatory mediators. In MS, IFNβ acts via decreasing annualized relapse rate (ARR), the risk of sustained disability progression, reducing MRI lesion activity and brain atrophy. It might also delay the onset of clinically definite MS after the first appearance of neurological symptoms.8
Drug Level
To evaluate IFNβ serum level an “antibody sandwich” ELISA has been developed, which involves coating the plates with a mouse monoclonal antihuman IFNβ antibody.9,10 However, drug level has never been used as a parameter to monitor the efficacy of any form of IFNβ, because of relatively short half-lives (range: 5–78 hours).
A pharmacokinetic study carried out in a group of 6 patients with MS receiving 6 MU of non-PEGylated IFNβ-1a intramuscular (IM) once a week, demonstrated that the IFNβ-1a levels become detectable at 4 hours, and peak at 8 hours postinjection. IFNβ-1a levels became undetectable in serum 24 hours postinjection. Peak serum levels range from 92 to 102 IU/mL, with a mean of 94.8 IU/mL.9 Additionally, other recent studies conducted on a new formulation of PEGylated IFNβ-1a (PEG-IFNβ) have shown that the concentration peak, measured using an ELISA, occurs later in this form of IFN as compared with the non-PEGylated IFNβ-1a (∼36 hours).11 After subcutaneous doses of PEG-IFNβ-1a in patients with MS, the mean Cmax is 280 pg/mL, and the peak of serum concentration occurs between 1 and 1.5 days. The pharmacokinetics (PK) profile of PEGylated form in a study involving 1512 patients with RRMS was consistent with that in healthy subjects. In healthy volunteers, the median area under the curve (AUC) from time 0 to 168 hours postdose [AUC (0, 168 hours)] was reported to be 27.2 ng·mL−1·h, while in patients with MS the same AUC (0, 168 hours) ranged from 23.5 to 32.0 ng·mL−1·h.12
A dosing regimen of PEG-IFNβ-1a once every 2 weeks provides 4.5-fold higher cumulative AUC, as compared with non-PEGylated IFNβ-1a administered weekly. Although definitive exposure–efficacy relationships are yet to be established, the increased cumulative exposure potentially explains the maintained efficacy of PEG-IFNβ-1a despite its reduced dosing frequency. However, such pharmacokinetic studies have only helped to define the best route and frequency of administration, and have not been utilized so far in the individualization of the treatment.
Pharmacogenomics: Identification of Biomarkers
Quantification of biological activity of IFNβ allows the early identification of patients who are not responsive to the treatment. The biological activity of IFNβ is investigated by evaluating a number of IFN-stimulated genes, induced by IFNβ injection, including myxovirus-resistance protein A (MxA) at the level of protein or mRNA, b2-microglobulin, oligo-adenylate-synthetase, TRAIL, viperin, IFI27, CCL2, and CXCL10.13 A strong risk of relapses in the absence of biological activity has been found.14 The European recommendations suggest the combined evaluation of MxA mRNA and ADA to assess the continuing efficacy of IFNβ therapy.15
Anti-IFNβ Antibodies
Several studies have reported the occurrence of binding antibodies (BAbs) and neutralizing antibodies (NAbs) against IFNβ during the treatment.15 A majority of the patients develop BAbs, however, only NAbs interfere with the biological activity of IFNβ, and they are present in a smaller proportion of patients with ADA. NAbs inhibit the binding between IFNβ and IFN receptors, abolishing its biological activity and, consequently, the therapeutic effect. The development of BAbs occurs during the first months of IFNβ treatment, whereas the occurrence of NAbs requires several months. Most patients become positive for NAbs during the first 18 months of therapy and rarely during the second or third year of treatment as well.
The importance of quantification of the NAbs and of the biological activity of IFNβ in the management of patients with MS is underlined by the European and Italian National Guidelines,16,17 and by international expert consensus17 that provide recommendations for timing of measurement and therapeutic consequences of NAbs against IFNβ, and of absence of biological activity (Fig. 1).
FIGURE 1.
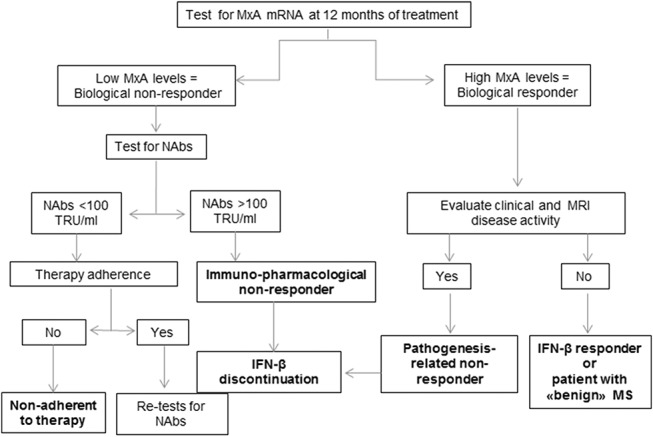
Clinical and biological flow chart for identification of subsets of IFN-β responder and nonresponder patients using pharmacogenomics and anti-IFNβ ADAs quantification.48
ELISA, both with or without a capture antibody, is the most commonly used method for BAbs measurement.18 For NAbs measurement, 3 methods are used based on the antiviral MxA protein: (1) cytopathic effect assay, considered as “gold standard” and recommended by both the World Health Organization and European Guidelines18; (2) MxA protein assay,19 and (3) MxA gene expression assay.20 Another type of assay based on the evaluation of luciferase expressed after sera incubation on cells transfected with an IFN-regulated luciferase reporter-gene construct has been proposed.21
ADA abolishes the biological activity of IFNβ, but also other factors such as noncompliance and soluble circulating IFNβ receptors could contribute to the lack of biological activity.13 Many evidences indicate that NAbs reduce or abolish the therapeutic efficacy of IFNβ in preventing relapses, independently of the type of IFN used.15–18 In fact, MRI, clinical disease activity,15 and the risk of disability progression are higher in NAbs-positive patients.22 The risk of development of NAbs varies between <1% and 31% for different IFNβ formulations.23,24 This immunogenicity difference is intensely influenced by excipients, route, and timing of administration and drug composition that differ among the various formulations.
Neurologists face 2 options during management of MS in patients. Multiple weekly injections offer more clinical efficacy than once a week injection. However, in this approach, many more patients are at risk of becoming NAbs positive than patients treated once a week. As a result, they will lose the clinical benefits of IFNβ. Moreover, they must be switched to another category of DMT, as NAbs are cross-reactive against all types of IFNβ.25
NATALIZUMAB
Mechanism of Action of Natalizumab
NAT is a humanized MAb that binds to the α4-subunit of β1 integrin, also called CD49d antigen, which is highly expressed on all leukocytes, except neutrophils. Specifically, after binding to the α4β1 integrin, NAT blocks the interaction of this integrin with its receptor, vascular cell adhesion molecule-1 (VCAM-1), and other ligands. Disruption of these interactions avoids transmigration of leukocytes across the blood-brain-barrier, and recruitment of activated T lymphocytes into inflamed tissue, and may suppress inflammation in the CNS. Normally, VCAM-1 is not expressed in the brain. However, in the presence of proinflammatory cytokines, it is upregulated in endothelial cells and possibly in glial cells close to the sites of inflammation.
A phase III placebo-controlled study26 showed the efficacy of NAT in reducing ARR and preventing disability progression which might be higher or comparable with IFNβ. These findings were confirmed by another independent trial that compared NAT plus IFNβ-1a against IFNβ-1a alone.27
Drug Level
Population-based modeling of the relationship between dose, concentration, and effects, that is, PK and pharmacokinetics-pharmacodynamics (PK-PD) of NAT, could help to precisely quantify individual sources of variability based on dynamic biomarkers and considering the onset of adverse events. Readers are encouraged to see Chapter 6 of this TDM special issue and the article by Ternant et al28 for the rationale of developing PK-PD modeling of monoclonal antibodies in TDM of inflammatory diseases. From an initial phase I study,29 it was concluded that doses from 0.03 to 3 mg/kg were safe, despite minor side effects. The approved 300 mg dose every 4 weeks leads to a mean half-life of 16 ± 4 days and a mean clearance of 13.1 ± 5 mL/h (file EMA/H/C/000603), depending on weight and anti-NAT antibodies. This dose was chosen to achieve 70% of α4-integrin saturation throughout the 28-day dosing interval. In the MS221 study from Biogen (reported in FDA clinical pharmacology and biopharmaceutics review, application number 125104), cytometry analysis of receptor occupancy was nearly saturated at all tested doses ranging from 1 to 6 mg/kg, however, the duration of saturation increased with increasing dose levels. As the noncompartmental analysis performed in this study does not adequately describe the nonlinear elimination of PK and, therefore, receptor saturation, it could be relevant to describe PK profile by compartmental approach, using, for example, a Michaelis–Menten-type elimination to address this problem. However, PML was found to be a major safety concern. Khatri et al30 developed a plasma-exchange strategy to swiftly reduce concentrations of circulating NAT to restore immune surveillance in the brain; VLA-4 desaturation appears to take place below 1 mcg/mL of circulating NAT. However, the rate of wash-out may vary considerably between patients, which suggests that measurement of NAT concentrations may be helpful to guide plasma exchange strategy.31 Evaluation of serum NAT concentrations is complicated since NAT can exchange Fab arms with endogenous human IgG4.32 Several immunoassays were developed to quantify serum NAT concentrations accurately33,34 without interference by Fab arm exchange nor IgG4 Fc interactions. Interestingly it has been shown that both low NAT concentration, below 1 mcg/mL, and high antibody titers, are associated with a lack of therapeutic efficacy.35
Utilizing paired CSF and serum samples, a recent study shows that it would be helpful to measure free and cell-bound NAT to determine the optimal individual NAT dosing regimen for patients.36 DELIVER study37 suggests that NAT will probably lead to similar efficacy whatever the administration route (intravenous (IV), subcutaneous (SC) or IM). PK profiles were quite similar with variations in Cmax: SC and IM were about 40% lower than IV, and mean bioavailability relative to IV was about 50% with SC or IM administration. Mean trough serum concentrations were lower with IM administration.
Pharmacodynamics of Natalizumab
Apart from ADA, current data available in the literature do not allow clinicians to design a personalized dosing regimen. However, Defer et al38 found a 55% decrease of CD49d expression on circulating T and B lymphocytes after NAT infusion. This low level remained stable for the entire period of treatment, except for patients who are ADA positive, in whom CD49d levels reverted to pretreatment levels. Thus, this antigen expression could be used to monitor the effectiveness of NAT. Millonig et al39 confirmed this finding, suggesting that CD49d is decreased on T cells, but also on B cells and NK cells. Moreover, they showed a significant decrease of serum sVCAM-1 concentration in ADA-negative patients. sVCAM-1 concentration reverts to pretreatment levels in case of ADA development. CD49d and sVCAM-1 could be useful in establishing a personalized timing of NAT administration.
Anti-Natalizumab Antibodies
Clinical trials with NAT have demonstrated the possibility of ADA generation with this treatment.40 ADAs induce a loss of efficacy with a higher risk of adverse events.26,35,40 The proposed mechanism of loss of clinical outcomes is the formation of NAT-ADAs immune complexes that lead to enhanced clearance and decreased functional serum concentration of NAT.35 As per current data, 9%–12% of NAT-treated patients develop ADA, out of which 6% remain persistently positive and 3%–6% are transiently positive for ADA.40 The treatment is discontinued if the measures reveal persistent ADAs. Patients with infusion reactions, or with disease activity should be tested for ADAs. The assay currently used to evaluate the presence of anti-NAT antibodies is a standardized bridging ELISA method developed by Biogen Idec (Cambridge, MA); protocol “Assay procedure to determine Natalizumab (Tysabri) immunogenicity (CST02-180AP-R.2).”40 The combined measurements of ADA, NAT serum level, and CD49d could be utilized to tailor a personalized infusion regimen. These measurements could also be useful to determine the withdrawal of NAT in patients with persistently high levels of ADA.
ALEMTUZUMAB
Mechanism of Action of Alemtuzumab
Alemtuzumab is a MAb of the IgG1 subclass that selectively binds to the CD52 protein, present in large amounts on the surface of T and B cells, and to a lesser extent on other cells. The treatment with this drug induces the depletion of circulating T and B cells, followed by repopulation. The repopulation phenomenon is faster for B cells and slower for T lymphocytes. Alemtuzumab action in MS is therefore attributable not only to the destruction of T and B cells, but also to the way in which the repopulation occurs. This treatment has minimal impact on other immune cells, ensuring the protection of the innate immune system. Clinical studies41,42 comparing alemtuzumab and IFNβ SC 3 times a week, demonstrated that the former reduces both ARR and disability progression more efficiently than IFNβ.
Drug Level
From the EMA approval of alemtuzumab for leukemia in 2001 till its approval for MS in 2014, all pharmacokinetic studies have been carried out only in patients with leukemia. ELISA and FACS have been the assays used in these studies for the assessment of the alemtuzumab serum concentration.43 In MS, the approved treatment strategy is 12 mg IV daily for 5 consecutive days, and 12 mg IV daily for 3 consecutive days administered 12 months after the first treatment course. This treatment regimen results in a mean Cmax of 3014 ng/mL on day 5 of the initial treatment course, and 2276 ng/mL on day 3 of the second treatment course. The half-life of this drug is approximately 4–5 days, and is comparable between courses. The serum concentration of alemtuzumab reaches low or undetectable levels within approximately 30 days following each treatment course (http://www.ema.europa.eu/docs/en_GB/document_library/EPAR-Product_Information/human/003718/WC500150521.pdf). In addition, attempts have been made in patients with chronic lymphocytic leukemia to delineate the pharmacokinetics of alemtuzumab. A two-compartment model with nonlinear elimination has been proposed by Mould et al. In this study performed in 2007, they demonstrate that the maximal trough concentrations range from 3.6 to 21.0 mg/mL with a mean of 10.2 mg/mL in responders, and below the limit of quantification to 26.8 mg/mL with a mean of 5.9 mg/mL in nonresponders. Additionally, a direct relationship between maximal trough concentrations and clinical outcomes was also described, with increasing alemtuzumab exposure resulting in a greater probability of positive tumor response.44 Data from any such studies in patients with MS are so far unavailable. Therefore, it would be interesting to design future prospective studies in MS to model dose-concentration-effects relationships of alemtuzumab, and investigate if indeed it is similar to that observed in chronic lymphocytic leukemia. Such studies of alemtuzumab pharmacokinetics in patients with MS would also aid in the implementation of TDM strategies and further individualization of treatment with this drug.
Anti-Alemtuzumab Antibodies
Alemtuzumab-binding antibodies have been shown to be present in 29% of patients immediately before the second course of treatment, and in 86% of patients 1 month after the second course of treatment.41 The percentage of patients whose test results were considered positive for antibodies to alemtuzumab using an ELISA and confirmed by a competitive binding assay. The presence and concentration of antialemtuzumab antibodies do not seem to influence either the efficacy or the safety of the MAb41 nor the pharmacodynamics at the beginning of treatment course. However, their impact after many doses remains to be established.
It has been shown that, during the first 5 years of treatment, almost one-third of the patients develop a secondary autoimmunity, in particular thyroid autoimmunity (30%), and idiopathic thrombocytopenic purpura (2%). Some studies have suggested that the pretreatment evaluation of IL-21 serum level could predict the development of posttreatment autoimmunity. However, currently available ELISA kits to evaluate IL-21 level seem to fail as predictive tests to evaluate this potential biomarker of secondary autoimmunity.45
Economic Impact of Drug Efficacy Monitoring
Very few studies have investigated the economic impact of drug efficacy monitoring in MS, and all of them have so far focused only on IFNβ. An Austrian study showed that testing for ADA against IFNβ, according to the European guidelines, is cost effective because it reduces total direct costs by approximately 34 million € in 5 years. Translated to the whole of Europe, the reduction of total direct costs would amount to approximately 594 million €.46
An Italian study has estimated the annual cost of managing patients with RRMS, with and without NAbs. The results have shown an increase of 3100 € per patient-year as the consequence of the onset of NAbs. Considering the patients with MS treated with IFNβ in Italy and the percentage of NAbs development, the evaluation of ADA could allow a better allocation of approximately 10 million €/year.47
For the other DMTs, no study related to the drug efficacy monitoring exists to date, although considering their cost, relapses and disability progression in young patients, it would be surprising if drug efficacy monitoring strategies would not be more cost effective.
Footnotes
Le Studium Loire Valley Institute for Advanced Studies; Ministero Salute Project Code: RF-2013-02357497.
M. Caldano received speaker honoraria from Biogen Idec, Merck Serono and Teva. T. Rispens received payments for lectures from Pfizer, AbbVie, Regeneron, and a research grant from Genmab. A. Bertolotto served on the scientific advisory boards of Almirall, Bayer, Biogen, and Genzyme; received speaker honoraria from Biogen, Genzyme, Novartis, Sanofi-Aventis, and Teva. His institution has received grant support from Bayer, Biogen, Merck, Novartis, Teva, the Italian Multiple Sclerosis Society, Fondazione Ricerca Biomedica ONLUS, and San Luigi ONLUS. The remaining author declares no conflict of interest.
REFERENCES
- 1.Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.English C, Aloi JJ. New FDA-approved disease-modifying therapies for multiple sclerosis. Clin Ther. 2015;37:691–715. [DOI] [PubMed] [Google Scholar]
- 3.Grossman I, Knappertz V, Laifenfeld D, et al. Pharmacogenomics strategies to optimize treatments for multiple sclerosis: insights from clinical research. Prog Neurobiol. 2016. 10.1016/j.pneurobio.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Bertolotto A, Gilli F, Sala A, et al. Evaluation of bioavailability of three types if IFN beta in multiple sclerosis patients by a new quantitative-competitive-PCR method for MxA quantification. J Immunol Methods. 2001;256:141–152. [DOI] [PubMed] [Google Scholar]
- 5.Pachner AR, Bertolotto A, Deisenhammer F. Measurement of MxA mRNA or protein as a biomarker of IFNβ bioactivity: detection of antibody-mediated decreased bioactivity (ADB). Neurology. 2003;61:S24–S26. [DOI] [PubMed] [Google Scholar]
- 6.Dhib-Jalbut S. Mechanisms of action of interferons and glatiramer acetate in multiple sclerosis. Neurology. 2002;58:S3–S9. [DOI] [PubMed] [Google Scholar]
- 7.Bekisz J, Sato Y, Johnson C, et al. Immunomodulatory effects of interferons in malignancies. J Interferon Cytokine Res. 2013;33:154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Severa M, Rizzo F, Giacomini E, et al. IFN-β and multiple sclerosis: cross-talking of immune cells and integration of immunoregulatory networks. Cytokine Growth Factor Rev. 2015;26:229–239. [DOI] [PubMed] [Google Scholar]
- 9.Khan OA, Dhib-Jalbut SS. Serum interferon beta-1a (Avonex) levels following intramuscular injection in relapsing-remitting MS patients. Neurology. 1998;51:738–742. [DOI] [PubMed] [Google Scholar]
- 10.Khan OA, Xia Q, Bever CT, Jr, et al. Interferon beta-1b serum levels in multiple sclerosis patients following subcutaneous administration. Neurology. 1996;46:1639–1643. [DOI] [PubMed] [Google Scholar]
- 11.Hu X, Miller L, Richman S, et al. A novel PEGylated interferon beta-1a for multiple sclerosis: safety, pharmacology, and biology. J Clin Pharmacol. 2012;52:798–808. [DOI] [PubMed] [Google Scholar]
- 12.Hu X, Cui Y, White J, et al. Pharmacokinetics and pharmacodynamics of peginterferon beta-1a in patients with relapsing-remitting multiple sclerosis in the randomized ADVANCE study. Br J Clin Pharmacol. 2015;79:514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertolotto A, Granieri L, Marnetto F, et al. Biological monitoring of IFNβ therapy in multiple sclerosis. Cytokine Growth Factor Rev. 2015;26:241–248. [DOI] [PubMed] [Google Scholar]
- 14.Malucchi S, Gilli F, Caldano M, et al. Predictive markers for response to interferon therapy in patients with multiple sclerosis. Neurology. 2008;70:1119–1127. [DOI] [PubMed] [Google Scholar]
- 15.Polman CH, Bertolotto A, Deisenhammer F, et al. Recommendations for clinical use of data on neutralising antibodies to interferon-beta therapy in multiple sclerosis. Lancet Neurol. 2010;9:740–750. [DOI] [PubMed] [Google Scholar]
- 16.Sorensen PS, Ross C, Clemmesen KM, et al. Clinical importance of neutralising antibodies against interferon beta in patients with relapsing-remitting multiple sclerosis. Lancet. 2003;362:1184–1191. [DOI] [PubMed] [Google Scholar]
- 17.Bertolotto A, Capobianco M, Amato MP, et al. Guidelines on the clinical use for the detection of neutralizing antibodies (NAbs) to IFN beta in multiple sclerosis therapy: report from the Italian Multiple Sclerosis Study group. Neurol Sci. 2014;35:307–316. [DOI] [PubMed] [Google Scholar]
- 18.Sorensen PS, Deisenhammer F, Duda P, et al. Guidelines on use of anti-IFN-beta antibody measurements in multiple sclerosis: report of an EFNS Task Force on IFN-beta antibodies in multiple sclerosis. Eur J Neurol. 2005;12:817–827. [DOI] [PubMed] [Google Scholar]
- 19.Wadhwa M, Subramanyam M, Goelz S, et al. Use of a standardized MxA protein measurement-based assay for validation of assays for the assessment of neutralizing antibodies against interferon-b. J Interferon Cytokine Res. 2013;33:660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertolotto A, Sala A, Caldano M, et al. Development and validation of a real time PCR-based bioassay for quantification of neutralizing antibodies against human interferon-beta. J Immunol Methods. 2007;321:19–31. [DOI] [PubMed] [Google Scholar]
- 21.Lam R, Farrell R, Aziz T, et al. Validating parameters of a luciferase reporter gene assay to measure neutralizing antibodies to IFNbeta in multiple sclerosis patients. J Immunol Methods. 2008;336:113–118. [DOI] [PubMed] [Google Scholar]
- 22.Paolicelli D, D'Onghia M, Pellegrini F, et al. The impact of neutralizing antibodies on the risk of disease worsening in interferon β-treated relapsing multiple sclerosis: a 5 year post-marketing study. J Neurol. 2013;260:1562–1568. [DOI] [PubMed] [Google Scholar]
- 23.Bertolotto A, Malucchi S, Sala A, et al. Differential effects of three interferon betas on neutralising antibodies in patients with multiple sclerosis: a follow up study in an independent laboratory. J Neurol Neurosurg Psychiatry. 2002;73:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giovannoni G, Barbarash O, Casset-Semanaz F, et al. Safety and immunogenicity of a new formulation of interferon beta-1a (Rebif New Formulation) in a Phase IIIb study in patients with relapsing multiple sclerosis: 96-week results. Mult Scler. 2009;15:219–228. [DOI] [PubMed] [Google Scholar]
- 25.Bertolotto A, Malucchi S, Milano E, et al. Interferon beta neutralizing antibodies in multiple sclerosis: neutralizing activity and cross-reactivity with three different preparations. Immunopharmacology. 2000;48:95–100. [DOI] [PubMed] [Google Scholar]
- 26.Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. [DOI] [PubMed] [Google Scholar]
- 27.Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911–923. [DOI] [PubMed] [Google Scholar]
- 28.Ternant D, Bejan-Angoulvant T, Passot C, et al. Clinical pharmacokinetics and pharmacodynamics of monoclonal antibodies approved to treat rheumatoid arthritis. Clin Pharmacokinet. 2015;54:1107–1123. [DOI] [PubMed] [Google Scholar]
- 29.Sheremata WA, Vollmer TL, Stone LA, et al. A safety and pharmacokinetic study of intravenous natalizumab in patients with MS. Neurology. 1999;52:1072–1074. [DOI] [PubMed] [Google Scholar]
- 30.Khatri BO, Man S, Giovannoni G, et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology. 2009;72:402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vennegoor A, Rispens T, Van Oosten BW, et al. Application of serum natalizumab levels during plasma exchange in MS patients with progressive multifocal leukoencephalopathy. Mult Scler. 2015;21:481–484. [DOI] [PubMed] [Google Scholar]
- 32.Labrijn AF, Buijsse AO, van den Bremer ET, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. 2009;27:767–771. [DOI] [PubMed] [Google Scholar]
- 33.Rispens T, Leeuwen Av, Vennegoor A, et al. Measurement of serum levels of natalizumab, an immunoglobulin G4 therapeutic monoclonal antibody. Anal Biochem. 2011;411:271–276. [DOI] [PubMed] [Google Scholar]
- 34.Shapiro RI, Plavina T, Schlain BR, et al. Development and validation of immunoassays to quantify the half-antibody exchange of an IgG4 antibody, natalizumab (Tysabri®) with endogenous IgG4. J Pharm Biomed Anal. 2011;55:168–175. [DOI] [PubMed] [Google Scholar]
- 35.Vennegoor A, Rispens T, Strijbis EM, et al. Clinical relevance of serum natalizumab concentration and anti-natalizumab antibodies in multiple sclerosis. Mult Scler. 2013;19:593–600. [DOI] [PubMed] [Google Scholar]
- 36.Sehr T, Proschmann U, Thomas K, et al. New insights into the pharmacokinetics and pharmacodynamics of natalizumab treatment for patients with multiple sclerosis, obtained from clinical and in vitro studies. J Neuroinflammation. 2016;13:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plavina T, Fox EJ, Lucas N, et al. A randomized trial evaluating various administration routes of natalizumab in multiple sclerosis. J Clin Pharmacol. 2016;56:1254–1262. [DOI] [PubMed] [Google Scholar]
- 38.Defer G, Mariotte D, Derache N, et al. CD49d expression as a promising biomarker to monitor natalizumab efficacy. J Neurol Sci. 2012;314:138–142. [DOI] [PubMed] [Google Scholar]
- 39.Millonig A, Hegen H, Di Pauli F, et al. Natalizumab treatment reduces endothelial activity in MS patients. J Neuroimmunol. 2010;227:190–194. [DOI] [PubMed] [Google Scholar]
- 40.Calabresi PA, Giovannoni G, Confavreux C, et al. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69:1391–1403. [DOI] [PubMed] [Google Scholar]
- 41.Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing–remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380:1819–1828. [DOI] [PubMed] [Google Scholar]
- 42.Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380:1829–1839. [DOI] [PubMed] [Google Scholar]
- 43.Elter T, Molnar I, Kuhlmann J, et al. Pharmacokinetics of Alemtuzumab and the relevance in clinical practice. Leuk Lymphoma. 2008;49:2256–2262. [DOI] [PubMed] [Google Scholar]
- 44.Mould DR, Baumann A, Kuhlmann J, et al. Population pharmacokinetics-pharmacodynamics of Alemtuzumab (Campath) in patients with chronic lymphocytic leukaemia and its link to treatment response. Br J Clin Pharmacol. 2007;64:278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Azzopardi L, Thompson SA, Harding KE, et al. Predicting autoimmunity after alemtuzumab treatment of multiple sclerosis. J Neurol Neurosurg Psychiatry. 2014;85:795–798. [DOI] [PubMed] [Google Scholar]
- 46.Walter E, Deisenhammer F. Socio-economic aspects of the testing for antibodies in MS-patients under interferon therapy in Austria: a cost of illness study. Mult Scler Relat Disord. 2014;3:670–677. [DOI] [PubMed] [Google Scholar]
- 47.Paolicelli D, Iannazzo S, Santoni L, et al. The cost of relapsing-remitting multiple sclerosis patients who develop neutralizing antibodies during interferon beta therapy. PLoS One. 2016;11:e0159214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertolotto A. Evaluation of the impact of neutralizing antibodies on IFNβ response. Clin Chim Acta. 2015;449:31–36. [DOI] [PubMed] [Google Scholar]