

Abstract
Intra-tumoral genetic heterogeneity has been characterized across cancers by genome sequencing of bulk tumors, including chronic lymphocytic leukemia (CLL). In order to more accurately identify subclones, define phylogenetic relationships, and probe genotype–phenotype relationships, we developed methods for targeted mutation detection in DNA and RNA isolated from thousands of single cells from five CLL samples. By clearly resolving phylogenic relationships, we uncovered mutated LCP1 and WNK1 as novel CLL drivers, supported by functional evidence demonstrating their impact on CLL pathways. Integrative analysis of somatic mutations with transcriptional states prompts the idea that convergent evolution generates phenotypically similar cells in distinct genetic branches, thus creating a cohesive expression profile in each CLL sample despite the presence of genetic heterogeneity. Our study highlights the potential for single-cell RNA-based targeted analysis to sensitively determine transcriptional and mutational profiles of individual cancer cells, leading to increased understanding of driving events in malignancy.
The unbiased characterization of mutational landscapes by massively parallel sequencing of bulk tumor samples has been transformative across cancers (Garraway and Lander 2013). For chronic lymphocytic leukemia (CLL), large-scale DNA-level characterizations have provided unexpected and clinically important insights (Wang et al. 2011; Landau et al. 2015; Puente et al. 2015). These studies not only have revealed the spectrum of key somatic mutations in CLL but also have uncovered clonal heterogeneity within individual samples that appear to impact clinical outcomes (Landau et al. 2013; Jeromin et al. 2014; Nadeu et al. 2016). While bulk DNA-level data provide a framework to begin characterizing clonal heterogeneity, the cancer cell phenotype is undoubtedly controlled by both genetic composition and gene expression and, hence, understanding this relationship mandates integration of genetic with transcript information at the single-cell level.
The recurrence of particular somatic single-nucleotide variants (sSNVs) in CLL implies positive selection and suggests that these mutations affect key cellular pathways (Landau et al. 2015; Puente et al. 2015). In many cases, though, the functional etiology of these mutations is unknown. The emergence of single-cell transcriptome sequencing for analyzing cancer highlights the potential to discover novel cellular subpopulations and states (Patel et al. 2014; Tirosh et al. 2016a). These studies identified single cells with large chromosomal arm–level alterations and detected aberrant expression of cellular pathways impacted by genes within these deleted regions (Patel et al. 2014; Tirosh et al. 2016a). It has not been clear, however, whether smaller focal alterations, including sSNVs, can be reliably inferred and analyzed in an analogous fashion. While these questions could be addressed in simultaneously extracted DNA and RNA from single cells, these efforts are still nascent (Dey et al. 2015; Macaulay et al. 2015; Hou et al. 2016).
This study examines the relationship between subclonal architecture and phenotype at the single-cell level in a series of CLL samples previously characterized by bulk genomic sequencing using three experimental approaches: targeted DNA, whole transcriptome, and targeted RNA (Fig. 1A). Our targeted RNA-based approach reliably detects subclonal mutations and enables recapitulation of single-cell DNA information, including phylogenetic structure. Integrative analysis to correlate genotype and phenotype revealed phenotypic convergence between distinct subclones and unexpectedly found drivers of CLL not evident through analysis of bulk samples. Overall, we demonstrate the ability to robustly integrate DNA- and RNA-level information in order to dissect the impact of somatic mutations on cellular phenotype.
Figure 1.

Detection of somatic alterations and gene expression patterns in single CLL cells. (A) Workflow of DNA and RNA analysis at the single-cell level. Viable leukemia cells from CLL patients were flow sorted either into 96-well plates or processed initially as bulk populations. DNA (left) and RNA (right) plate-based approaches were used for phylogeny reconstruction and integration of genotype and phenotype, respectively. Bulk cells (middle) were applied to C1 integrated fluidic circuits (IFCs) for single-cell capture and cDNA library generation (see Methods). Sequencing libraries were generated and sequenced on an Illumina HiSeq system. (B) Number of single-cell DNA-based detection assays designed (top) and the cancer cell fraction (CCF) of all the alterations (bottom panel) for five CLL samples. Each point is an alteration with specific alterations indicated by colors as noted. (C) Correlation between mutation and chromosomal abnormalities detected by single-cell DNA analysis and CCF inferred from bulk tumor whole-exome sequencing (WES).
Results
Establishing the precise subclonal architecture of CLL
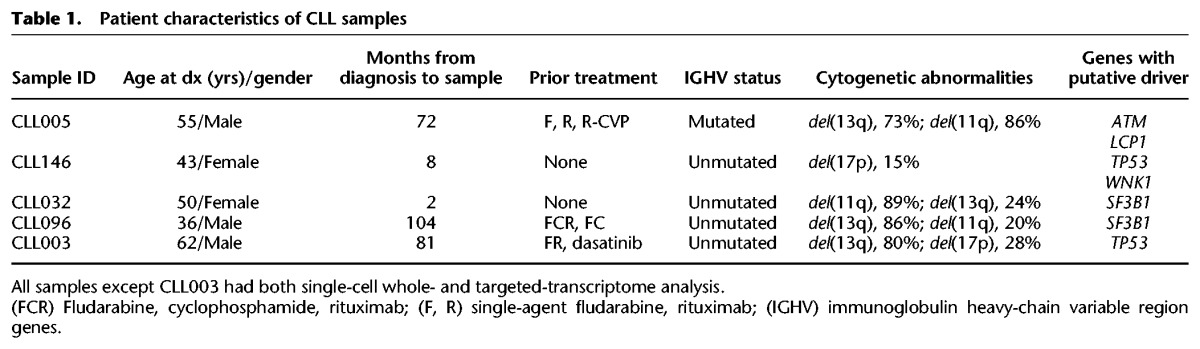
Single-cell targeted DNA sequencing was used to define the clonal structure for five CLL samples previously characterized by bulk WES. The five specimens had clonal or subclonal deletions corresponding to recurrent CLL-associated copy number alterations: del(13q) in four patients, del(17p) in two, and del(11q) in three (Table 1; Fig. 1B). The samples also had known CLL driver mutations, such as TP53 (CLL003, CLL146), ATM (CLL005), and SF3B1 (CLL096, CLL032). Our single-cell targeted DNA sequencing approach comprised whole-genome amplification (WGA) from flow-sorted, viable CD19+CD5+ single cells; multiplex PCR to amplify segments containing single-nucleotide alterations identified by the bulk WES; and deep sequencing.
Table 1.
Patient characteristics of CLL samples
Primers were designed to generate 90 amplicons for sSNVs and 111 amplicons for single-nucleotide polymorphisms (SNPs) in chromosomal regions corresponding to somatic copy number alterations (sCNAs). A median of 10 SNP sites (range, six to 17) was selected for each focal sCNA. Low-depth whole-genome sequencing of the WGA products from 96 single CLL005 cells confirmed even coverage across the genome (Supplemental Fig. S1). Of 1152 cells analyzed from the five samples, 86% (991 cells) passed the quality metric of sufficient DNA quality (100 ng) after WGA. For the amplicons, 89% were successfully amplified from the single cells (Supplemental Tables S1, S2). Following sequencing of the amplicon libraries, >85% of the reads aligned to target regions, and there was a median depth of 5160 reads per target region (Supplemental Fig. S2).
In order to address the complication of allelic dropout, a novel probabilistic algorithm was developed that is robust against bias from WGA and allelic amplification (see Supplemental Methods, Supplemental Fig. S3). This method uses information from all sSNVs and SNPs data to infer missing data in order to determine allelic imbalance and sCNAs. For all five samples, the proportion of single cells harboring genetic alterations was highly concordant with the cancer cell fraction (CCF) calls inferred from bulk WES (r2 = 0.937) (Fig. 1C; Supplemental Table S3) using ABSOLUTE (Carter et al. 2012; Landau et al. 2013), suggesting that our single-cell DNA sequencing and sCNA inference accurately characterizes overall genetic heterogeneity. To discover the relationship between different subpopulations, we performed clustering analysis and phylogenetic reconstruction. Four samples exhibited branched evolution, with only CLL032 showing a linear evolution structure. For CLL003, the majority of somatic mutations were clonal, including mutations in the putative CLL drivers DDX3X and del(13q). One subclone of 130 cells harbored a MYH1 mutation. Within this subclone, a subset of 55 cells (35% of total cells) had del(17p) (minimal deleted region contains TP53). Separate from the MYH1 subclone, a set of 24 cells (16% of total) had a TP53 mutation. The relative proportions of these subpopulations were confirmed using fluorescence in situ hybridization (FISH) with probes specific for Chromosomes 13q and 17p (Fig. 2A). Thus, consistent with convergent evolution, two separate subclones with heterozygous mutations leading to reduced TP53 expression were evident, rather than homozygous inactivation of TP53 within the same clone of cells.
Figure 2.

Reconstruction of tumor phylogeny in CLL from single-cell DNA analysis. (A–C) Detection and clustering of somatic mutations and chromosomal deletions (rows) for single viable CD19+CD5+ cells (columns) from CLL003 (A), CLL146 (B), CLL005 (C; for analysis of CLL096 and CLL032, see Supplemental Fig. S4A,B). (Left) Blue indicates the presence of mutations (sSNVs) or chromosomal deletion; beige, the absence of sSNVs; red, the absence of chromosomal deletion. (Middle) Clonal architecture for CLL003, CLL146, and CLL005 derived from single-cell DNA analysis. (Right) FISH hybridization (A,B) and karyotyping images (C) as validation of the single-cell chromosomal deletion analysis with percentage of positive FISH cells enumerated from 100 cells. Sensitivity of probes used in the study was confirmed by hybridization with PBMC from a normal donor (Supplemental Fig. S4C).
By WES, CLL146 was known to have a series of subclonal chromosome abnormalities. Single-cell DNA results identified one branch with three sCNAs (del(6p), del(17p), and del(18p)) all within one subclone of 55 cells (40%), supported by FISH analysis (Fig. 2B). A separate branch (60%) had a mutation in the critical kinase domain of WNK1 (V430F). For CLL005, although del(13q) is a common early event in CLL, this was clearly a subclonal event in this patient, and co-occurred with del(11q) and a MTOR mutation (Fig. 2C). These alterations were not present in a separate subclone defined by a mutation in LCP1. Copy number variation was not analyzed in CLL096, but the sSNV data defined four subclones, with one having a known CLL driver SF3B1 mutation (Supplemental Fig. S4A). In contrast, CLL032 demonstrated linear evolution with clonal events, including del(11q), del(13q), and del(1q), and with a subclonal SF3B1 mutation (Supplemental Fig. S4B).
CLL single-cell transcriptome analysis reveals transcriptional heterogeneity
Single-cell whole-transcriptome sequencing was performed for four of the five CLL samples (up to 96 cells per sample). Of the 384 cells analyzed, 289 cells (75%) passed quality-control filtering. A median of 3,781,092 reads (range, 664,548–6,889,700) was generated per cell, with a median of 2473 unique transcripts (range, 1741–4255) detected per cell (Supplemental Table S4).
In order to characterize transcriptional heterogeneity within these samples, we applied pathway and gene set overdispersion analysis (PAGODA) (Fan et al. 2016) to identify significant aspects of coordinated variability within annotated pathways and correlated gene sets (Fig. 3A,B). As detailed in the Methods, PAGODA uses error models and variance normalization (Supplemental Fig. S5A,B) in order to accurately quantify biological variability in a manner that is robust to drop-out, expression-magnitude dependence, and other technical factors (Munsky et al. 2012; Brennecke et al. 2013; Kharchenko et al. 2014). Over 8000 annotated pathways from the Molecular Signatures Database (MSigDb) (Subramanian et al. 2005) were analyzed to identify significantly overdispersed pathways (P < 0.05) that were further clustered to reduce redundancy. For each sample, two to four significant aspects of transcriptional heterogeneity corresponded to cellular processes previously implicated in CLL, such as cell cycle and immune signaling (Fig. 3A,B; Supplemental Fig. S6A,B). Other processes not previously highlighted by mutational studies, such as antigen presentation, phospholipid binding, and protein folding (Supplemental Table S5), were also identified. Joint analysis of single cells from all four CLL samples revealed significant aspects of transcriptional heterogeneity shared across all samples, such as mitochondrial and ribosomal processes (Supplemental Fig. S6C; Supplemental Table S5).
Figure 3.

CLL transcriptional heterogeneity revealed by single-cell transcriptome sequencing. Pathway and gene set overdispersion analysis (PAGODA) was used to identify transcriptionally defined subpopulations for CLL005 (A) and CLL146 (B) (for analysis of CLL096 and CLL032, see Supplemental Fig. S6A,B). Based on gene sets defined by MSigDB annotations, significantly overdispersed pathways group cells into coherent and distinct aspects of transcriptional heterogeneity. Aspect scores (Cell PC score) are oriented so that high values generally correspond to increased expression of associated gene sets. Also shown are expression patterns of select genes driving each aspect of transcriptional heterogeneity along with their loading contributions to the aspect scores (left). Mutation information inferred from single-cell transcriptome sequencing is also shown at the bottom. CCF for each mutation derived from bulk tumor WES and the number of reads (log10 transformed) from single-cell transcriptome sequencing are also indicated.
In order to connect genotype and phenotype, we attempted to identify mutations in the single-cell RNA-seq data that would define distinct subclonal branches based on the prior DNA analysis (i.e., WNK1, MTOR, LCP1, PURG, and SF3B1). However, scarcity of coverage at the mutation sites of interest limited our ability to confidently call mutations in the majority of cells (Fig 3A,B; Supplemental Fig. S6A,B, bottom; Supplemental Table S4). Thus, although single-cell RNA-seq identified clear transcriptional heterogeneity based on pathway-driven gene expression analysis, these data could not be used to confidently resolve the genetic subclone structure or to assess the correspondence of genetic structure with the observed transcriptional heterogeneity.
Integrated single-cell targeted gene expression and mutation detection is robust and sensitive
The sparseness of coverage in single-cell RNA-seq data prompted us to develop a targeted RNA approach to assess transcript expression profiles and mutational status in the same single cells. By leveraging the Biomark microfluidic technology that has been successfully used for analysis of single-cell RNA expression (Guo et al. 2010, 2013; Buganim et al. 2012; Wills et al. 2013), we implemented a two-stage RT-PCR amplification strategy (Fig. 1A) to include mutation detection. In the first stage, multiplex preamplification generated cDNA libraries from individual cells that contain both targets for RNA quantification and segments encompassing sSNVs or SNPs diagnostic for sCNAs. In the second stage, high-throughput qPCR was performed on matched integrated fluidic circuits (IFCs) using assays nested relative to the preamplification primers. One IFC was used to quantify RNA expression; the second, to detect single-nucleotide alterations.
This approach was tested in seven CLL samples, including the five analyzed above. For RNA expression, qPCR assays for 96 genes were used per cell. As an initial characterization, we assessed detection as a function of the number of cells analyzed, titrating from 40 cells to single cells (Fig. 4A; Supplemental Fig. S7). As the cell number per sample decreases, the number of genes detected decreases and the variation across replicate samples increases. Up to 384 single cells from each of the seven CLL samples and from normal CD19+ B cells were analyzed. Based on the expression of the housekeeping genes ACTB and B2M, we detected robust expression in 1951 of 2112 cells (92.4%). Across these samples, the highly expressed genes in bulk RNA were consistently highly expressed in single cells (Fig. 4B; Supplemental Fig. S7B). However, genes with a lower expression in bulk RNA exhibited bimodal expression. Many single cells showed high expression, while others exhibited lower or undetected expression. Notably, for those genes with undetected expression in the bulk specimen, 3% had high expression in a subset of single cells. Principal component analysis (PCA) (Fig. 4C) and hierarchical clustering (Supplemental Fig. S7A) discerned expression heterogeneity across 354 single CLL cells and clearly discriminated these cells from 174 normal B cells. The CLL cells from two patients were also distinguished from each other.
Figure 4.

Establishment of a targeted RNA-based approach to perform integrated, targeted, and multiplexed detection of somatic mutations and gene expression in single cells. (A) Number of genes detected from 96 genes using cell numbers ranging from one to 40 cells per well by the targeted approach illustrated in Figure 1A, right panel. (B) Correlation between gene expression derived from a bulk RNA-seq and single-cell targeted approach in CLL005 (analyses of other individual CLL samples and in aggregates in Supplemental Fig. S7). (C) Gene expression of a set of 96 genes in single cells distinguishes normal (CD19+) and CLL-B cells by principal component analysis (PCA). Single cells were derived from one normal healthy donor and two CLL patients. (D, top) SuperSelective primer design (Vargas et al. 2016) was used for mutation detection. The primer contains a long 5′-anchor sequence that binds strongly to template strands, a short 3′-foot sequence that includes an interrogating nucleotide complementary to the corresponding nucleotide in a mutant template (but mismatches the corresponding nucleotide in a wild-type template), and a linking bridge sequence. (Bottom) Schema of a successful targeted mutation detection assay, in which distinct paired assays (wild-type and mutant allele) were designed for detection of a single mutation. (E) Heat map of seven patient-specific mutation detection assays (with seven matched wild-type assays) performed on cDNA derived from 20 single cells from normal CD19+ B cells or two CLL-B cells, with detection measured as the number of cycles to achieve the detection threshold (Cq). (F) Examples for mutation calls in single cells with or without mutations LCP1 and WNK1. Cells were called wild-type (WT; blue), mutant (red), or unclear (gray).
In parallel, paired assays interrogating alternative alleles for sSNVs and SNPs were used to analyze the same single cells. These assays utilized qPCR detection with SuperSelective allele-specific primers (Fig. 4D; Vargas et al. 2016). Testing on artificial templates or bulk cDNA, the designs of the allele-specific primers were refined to achieve a ΔCq delay of at least seven cycles for a mutant assay on wild-type template or a wild-type assay on mutant template (Fig. 4D; Supplemental Fig. S7C). When this assay design was applied to CLL samples, wild-type alleles were readily distinguished from mutant alleles. Mutant alleles were consistently observed in the originating patient but not in unrelated CLLs or in nonleukemic B cells (Fig. 4E,F).
Phylogeny reconstruction and inference of convergent evolution from single-cell RNA data
Inclusion of SNP assays enabled detection of chromosomal deletions in single cells because complete allelic imbalance at germline heterozygous SNPs signifies the presence of a deletion. SNP assays were chosen from highly expressed genes (mean counts, more than 30 from single-cell RNA-seq) and within candidate deletion regions identified from bulk WES data (Fig. 5A,B; Supplemental Table S6). By use of allele-specific assays to detect sSNVs and SNPs related to sCNAs in four of the five samples, the RNA-based estimates of allele frequency from single cells were highly concordant with single-cell, targeted DNA-based detection of somatic alterations, as well as with CCF inferred from bulk WES (Fig. 5C). Through the combined RNA-based targeted analysis of sSNVs and sCNAs, we could reconstruct phylogeny in a manner consistent with our prior DNA-based analysis. The assessment of 316 single cells from CLL005 confirmed the co-occurrence of deletions in Chromosomes 11q and 13q and IFNAR2 mutation in a subpopulation of cells (cluster 1) that did not co-occur with expression of mutated LCP1 (cluster 2) (Fig. 5D). As expected, single cells inferred to harbor deletions consistently had lower expression of genes within the putative deleted regions (Fig. 5E,F). PCA on the expression of genes within the deletion regions separated the two genetic subpopulations, confirming that our approach was able to distinguish cells with and without deletions of interest (Fig. 5G; Supplemental Fig. S4C).
Figure 5.

Integrated analysis of DNA- and RNA-level information by simultaneous detection of mutation, deletion, and gene expression in single cells. (A) Schema of deletion call based on expression of heterozygous SNPs. Dashed line indicates deleted region. A deletion was called when a cell expresses only one allele in the deleted region while expressing similar levels of both alleles for SNPs outside the deleted region. (B) Examples for deletion calls in CLL005 single cells for subclonal deletions in Chromosomes 11 and 13. Each point represents a cell. Orange indicates cells with no detectable expression from the deleted allele are inferred to harbor the deletion; blue, cells with expression from both alleles or the deleted allele are inferred to lack the deletion. (C) Mutation and chromosomal deletion frequency detected from single-cell RNA from five CLL samples show high correlation with CCF (r2 = 0.921) and with mutation frequency per single-cell DNA analysis (r2 = 0.923). (D) Mutation and chromosomal deletion detection in single-cell RNA from sample CLL005 enables reconstruction of phylogeny. (E,F) Genes within the chromosomal deletion regions exhibit significantly lower expression based on a one-sided Wilcoxon rank-sum test in cells inferred to harbor the deletions (cluster 1; orange) compared with cells not harboring the deletions (cluster 2; blue). Housekeeping genes are not significantly differentially expressed among single cells from the two clusters. (G) PCA of single cells from CLL005 or CLL146 based on gene expression for genes within chromosomal deletion regions leads to separation of cells by genetic subpopulation. Linear discriminant analysis achieves elevated ROC AUCs of 0.774 for CLL005 and 0.771 for CLL146. (H) PCA of single cells from CLL005 or CLL146 based on gene expression for genes driving aspects of transcriptional heterogeneity (23 genes for CLL005, 33 for CLL146) fails to separate cells by genetic subpopulation. Linear discriminant analysis does not perform substantially better than random, achieving ROC AUC of 0.516 for CLL005 and 0.595 for CLL146.
Based on the targeted single-cell RNA annotations of the mutation and deletion status of each cell, we tested if the transcriptionally defined subpopulations mapped to the genetically defined subclonal branches for CLL005 and CLL146. In both cases, genes previously identified from the single-cell RNA-seq analysis to be variable and driving aspects of transcriptional heterogeneity do not exhibit significant expression differences between the two genetic subpopulations (Supplemental Fig. S5C). PCA on the expression of driving aspect genes also did not distinguish cells in the different subclonal branches (Fig. 5H). In addition, genes associated with overexpression of wild-type or mutant LCP1 in HEK293 and K562 cells were identified using RNA-seq (Supplemental Fig. S6D,E). We did not observe a correlation between expression of these genes and LCP1 mutation status in CLL005 single cells (data not shown). This lack of correspondence between prominent aspects of transcriptional heterogeneity and subclonal branches prompted us to consider that perhaps each sample has phenotypically similar leukemic subpopulations despite different genetic identities.
Mutated LCP1 and WNK1 have potential CLL driving function
Although the subclonal structures of CLL005 and CLL146 had clear genetically defined subclonal branches, one prominent branch lacked a recognizable driver alteration. Experiments were performed to test the premise that knowledge of the phenotype of one branch can inform us about functions of previously uncharacterized somatic mutations in the other branch.
In CLL005, one subclonal branch had the known drivers del(13q), which encompasses the MIR15-16 locus, whose knockdown accelerates the cell cycle in a mouse model (Klein et al. 2010), and del(11q), whose minimally deleted region includes the DNA damage response gene ATM. The other branch had a truncating mutation in LCP1, encoding the actin-binding L-plastin. Expressed in normal cells of hematopoietic lineage and across malignant human cells of nonhematopoietic origin, LCP1 has been previously associated with del(11q) in CLL (Aalto et al. 2001) and reported to play a role in niche homing (Dubovsky et al. 2013). Because of the phenotypes associated with focal deletions in Chromosomes 11 and 13, we tested if the LCP1 mutation could accelerate cell proliferation or impact DNA damage sensing. Stable HEK293 cell lines that express either wild-type or truncated LCP1 were generated (Fig. 6A,B). The expression of mutated LCP1 did not impact rate of cell growth (Supplemental Fig. S8A) but did lead to an altered DNA damage response. This was measured by comparing phosphorylated H2AX expression following gamma irradiation in cells expressing mutant or wild-type LCP1 (Fig. 6B). As expected, irradiation led to phosphorylation of the DNA damage sensor and transducer ATM in cells expressing wild-type LCP1. In contrast, cells with truncated LCP1 exhibited weak phosphorylation of ATM but strong activation of DNA-PK phosphorylation. Thus, L-plastin encoded by LCP1 likely participates in the sensing and transducing of DNA damage signals. Consistent with this idea, immunoprecipitation detected direct physical interaction between ATM and wild-type LCP1, as well as the truncated LCP1 (Fig. 6C), irrespective of radiation exposure (Supplemental Fig. S8B,C). Finally, LCP1-mutant cells had higher survival following exposure to gamma-irradiation compared with wild-type LCP1-expressing cells, even in the presence of ATM inhibitors (Fig. 6D). Altogether, these results suggest that LCP1 mutation confers resistance to DNA insult.
Figure 6.

Mutations in LCP1 and WNK1 increase cell fitness and favor cell growth and survival. (A) Schema of the nonsense mutation in LCP1 found in CLL005. (B) LCP1-mutant cells have altered DNA response upon gamma-irradiation. HEK293 cells stably expressing wild-type, mutant, or control constructs were treated with 10 Gy. Levels of gamma-H2AX and phosphorylated forms of ATM, ATR, DNA-PK, and GAPDH were assessed using immunoblot. (C) Both wild-type and mutant LCP1 physically interact with ATM protein. HEK293 cells stably expressing wild-type, mutant, or control constructs were transiently transfected with an ATM-expressing construct (with N-terminal His tag). Forty-eight hours after transfection, cells were either treated or not treated with irradiation. Immunoprecipitations were performed on protein lysates using anti-His beads or anti-GFP as well as isotype control antibodies followed by immunoblot against GFP or ATM protein. Shown are immunoprecipitates from untreated cells. Irradiation does not change the physical association between these proteins (Supplemental Fig. S8). (D) Cells with mutant LCP1 have greater overall survival after gamma-irradiation compared with cells with wild-type LCP1 with or without ATM inhibitor treatment. The survival rate was calculated by normalization to each group of nonirradiated cells using colony assays performed on six-well plates. Mean ± SD; n = 3. (E) Crystal structure of WNK1 kinase domain (PDB entry 4Q2A). Val403 is located underneath the activation loop of WNK1. Mutation of Val403 to Phe potentially disrupts activation loop folding and affects kinase activity. (F) Cells with mutant WNK1 demonstrate a faster progression from G1 to S phase. HEK293 cells were induced to express either wild-type or mutant WNK1 for 48 h. After synchronization by serum deprivation for 24 h, cell cycle was assessed by EdU assay 24 h after return to complete media. The mean percentage of cells (±SD; n = 3) in different phases of the cell cycle is shown.
For CLL146, the WNK1-V403F mutation was present in a subclone distinct from a subclone carrying multiple chromosomal aberrations (in 6p, 17p, and 18p). A member of the WNK subfamily of serine/threonine protein kinases, WNK1 has known roles in the regulation of osmotic stress, cell cycle progression, metabolic tumor cell adaptation, and evasion of metastasis (Moniz and Jordan 2010). Based on crystal structure, WNK1-V403F localizes to the catalytic domain of WNK1 kinase and physically interacts with its critical T-loop region (Fig. 6E). PolyPhen scoring (Adzhubei et al. 2013) predicted a strong function-altering effect for this amino acid substitution based on its position in the protein crystal structure. This was confirmed by showing that the WNK1-V403F mutation completely ablates catalytic activity of a recombinant fragment of bacterially expressed WNK1 that encompasses the kinase domain (Supplementary Fig. S9A). To explore the effect of this mutation in mammalian cells, we knocked out endogenous WNK1 in HEK293 cells and then introduced inducible expression of wild-type and mutant WNK1 (Supplemental Fig. S9B). When these cells were exposed to osmolar stress followed by WNK1 immunoprecipitation, drastically reduced kinase activity was observed on the target OSR1 D164A, although the effects on WNK1 signaling cascades were not obvious (likely due to compensation of other WNK isoforms that are highly expressed in HEK293 cells) (Supplemental Fig. S9C). Thus, it is not clear if the effect of the mutation on the osmolar stress pathway contributes to a cancer phenotype. On the other hand, we observed a faster transition from G1 to S phase in WNK1-V403F–mutant cells, suggesting that the mutation favors cell proliferation and growth (Fig. 6F).
Discussion
For a population of malignant cells to expand as a subclone, a driver event is presumably present. Although large-scale sequencing studies have uncovered a spectrum of recurrently mutated driver genes across diverse malignancies (Lawrence et al. 2013, 2014), it has been estimated that ∼10% of CLLs have no identifiable driver (Landau et al. 2015; Puente et al. 2015). This highlights a lack of completeness in our catalog of driving events in cancer and in our full understanding of biologic features underlying the cancer process. Thus, a continued effort in identifying driver events in CLL (and cancer in general) remains warranted. We posited that a detailed analysis of the genetic structure of CLL could enable better identification of novel somatic mutations in CLL and, in turn, understanding of their function.
The molecular study of tumors generally requires a discovery phase to establish the breadth of heterogeneity. For the five CLL patients studied here, the discovery of genetic heterogeneity was provided by previously published bulk WES results. Although some information about clonality can be inferred from bulk WES, it cannot be used to unambiguously determine subclonal structure (Paguirigan et al. 2015). For this, we used targeted single-cell DNA sequencing to detect sSNVs and sCNAs to reconstruct phylogeny. Discovery of transcriptional heterogeneity was provided by single-cell RNA-seq and established key transcripts and pathways that contribute to the phenotype of each particular sample. Because the analytical power of genetics depends on the association of phenotype with genotype, we intended to link the transcriptional heterogeneity with underlying genetic alterations. However, due to the sparseness of coverage in the single-cell RNA-seq data, genotype/phenotype correlation was accomplished by targeted RNA qPCR. The selection of targets for both the gene expression assays and the sSNV and SNP assays depended on abundance measurements from bulk and single-cell RNA-seq. Surprisingly, subclones were detected that had no apparent driver but did have a phenotype similar to subclones with known drivers. By using the phenotypes of the subclones with known drivers as a guide, functional studies indicated that mutations in LCP1 and WNK1 may be drivers of CLL that have not previously been identified.
The single-cell DNA analysis revealed high levels of genetic complexity in each CLL, with branching subclones observed more commonly than not. There was a strong concordance between inferred CCF estimates from bulk analysis and the number of single cells comprising a genetically defined subclone. The single-cell information made it possible to clearly distinguish branched from linear tumor evolution. Even within this small cohort, however, we observed deviations from the trends observed at the population level. For example, although bulk CLL sequencing studies have delineated chromosomal aberrations such as del(13q) as early clonal events (as in CLL003 and CLL032), we clearly identified CLL cells with sSNVs as earlier events than chromosomal deletions [i.e., CLL005 and CLL146 with clonal mutations in ATM and TP53 and subclonal del(13q)]. These observations reinforce the idea that there is a not a dogmatic order per se in acquiring chromosomal changes before point mutations, or vice versa, in CLL, consistent with the notion that heterogeneous evolutionary trajectories can produce malignancy.
Single-cell RNA profiling, via qPCR (Guo et al. 2010, 2013; Dalerba et al. 2011; Buganim et al. 2012; Wills et al. 2013) or sequencing (Shalek et al. 2013, 2014), has emerged as a powerful technology for establishing cell state (Treutlein et al. 2014; Klein et al. 2015; Zeisel et al. 2015). Our single-cell RNA-seq data clearly delineated transcriptional states but could not be used to reliably detect mutations. To provide one example, the clonal ATM mutation in CLL005 was detected in only two of 96 single cells (2.1%) using RNA-seq data due to coverage limitations. In contrast, we could detect 203 of 288 single cells (70.0%) with ATM mutation using targeted qPCR. Similarly, Tirosh et al. (2016b) found a particular CIC mutation in seven of 1056 single cells (0.66%) using RNA-seq reads but improved sensitivity to 28 of 467 (3.9%) using a targeted qPCR assay. Such large discrepancies in detection frequency cannot be made up by merely sequencing deeper, without even considering the prohibitive cost of sequencing hundreds of single cells to great depth. Clearly, the targeted RNA-based approach we described is a reliable, cost-effective method for sSNV or SNP detection in single cells.
Thus, our strategy transitioned to the use of targeted RNA analysis to determine genotype and phenotype in the same single cell, with qPCR as the readout. Advantages of using qPCR rather than targeted sequencing include simpler workflow, faster data acquisition, and more facile data processing. RNA-based genotyping can improve sensitivity as transcript copy number is higher than genomic copy number. This advantage is tempered by the occurrence of random monoallelic expression (Deng et al. 2014; Reinius and Sandberg 2015). Despite this complication, our targeted qPCR analysis of single-cell RNA recapitulated the clonal structures detected by targeted, single-cell DNA sequencing and provided RNA profiles consistent with the data from single-cell whole-transcriptome sequencing. The versatility of this microfluidics-based qPCR approach has been further demonstrated by its use to associate alternative splicing with somatic mutations (Wang et al. 2016).
Our single-cell analysis consistently revealed convergent evolution as a driving force in CLL. At the genetic level, this was evident in the example of CLL003 wherein we observed del(17p) and TP53 mutation present in distinct subclones, rather than as co-occurring, such that the majority of cells were impacted by TP53 inactivation. Likewise, at the transcriptional level, although we observed expression heterogeneity within individual samples, the overall phenotypic picture was one of expression coherence. Within this framework, we delved deeper into examining the functional effects of two candidate CLL drivers: LCP1 mutation (CLL005) and WNK1 mutation (CLL146). Indeed, we found evidence of mutation in LCP1 impacting the DNA damage response and in WNK1 on cellular proliferation.
Intra-tumoral heterogeneity at multiple levels (DNA, RNA, epigenetics, protein) provides the fuel for tumor evolution, which forms the basis of disease progression, treatment response, relapse, and metastasis (Burrell et al. 2013; Mazor et al. 2016; Turajlic and Swanton 2016). Distinct genetic or epigenetic subclones may compete or collaborate for better fitness in response to microenvironmental or developmental changes (Tirosh et al. 2016b). Our observation that distinct subclones within the same CLL sample had similar transcriptome profiles suggests multiple levels of heterogeneity contribute to phenotype, consistent with other published findings (Tirosh et al. 2016b). Although our study is limited to only five CLL samples, our findings are in line with accumulating evidence across cancers. For example, a recent pan-cancer analysis of more than 1100 whole exomes from 12 different types of solid tumors has shown intra-tumoral genetic heterogeneity to exist in all tumor types examined and is linked to poor prognosis (Andor et al. 2016). Our study results are also consistent with two recent analyses of acute lymphocytic leukemia, in which branched phylogenies were demonstrated as common (Potter et al. 2013; Gawad et al. 2014). We anticipate integration of genetic, epigenetic, transcriptome, and proteomic information from the same single cell will ultimately provide the path for unraveling the complexity of tumor heterogeneity.
Methods
Patient samples
CLL-B and normal B cells were collected from patients and healthy adult volunteers enrolled on clinical research protocols at the Dana-Farber/Harvard Cancer Center (DF/HCC), approved by the DF/HCC Human Subjects Protection Committee. Heparinized blood was collected, and peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll/Hypaque density-gradient centrifugation, cryopreserved with 10% DMSO, and stored in vapor-phase liquid nitrogen until the time of analysis. Informed consent on DFCI IRB-approved protocols for genomic sequencing of patients’ samples was obtained prior to the initiation of sequencing studies.
Processing of normal B and CLL-B samples to single cells
Cryopreserved peripheral blood CLL samples were thawed and stained with anti-CD19 FITC and anti-CD5 PE antibodies (Beckman Coulter). 7-AAD (Invitrogen) was added before FACS as a viability control. Live single or multiple CD19+CD5+ tumor cells were flow sorted directly into 96-well plates with either 5 µL of phosphate-buffered saline (for DNA analysis) or 5 µL of a lysis buffer (for RNA analysis) consisting of 10 mM Tris-HCl (pH 8.0); 0.1 mM EDTA; 0.5% NP-40 (Thermo Scientific), and 0.1 U/µL SUPERase-In (Life Technologies). Following sorting, each plate of cells was gently vortexed, quickly spun down at 1500 rpm, and flash frozen on dry ice. To minimize the in vitro exposure time for the single cells, the time from cell thawing to flow-sorting was standardized to be within 90 min.
Targeted mutation detection from DNA of single cells
Genomic DNA amplification from plates of single cells was carried out using a REPLI-g amplification (WGA) kit (Qiagen) (Zhang et al. 2015). WGA DNA yield was determined by picogreen quantification (Life Technologies). DNA samples (40 ng) were processed using targeted multiplex PCR assays with sample-specific primers that were designed based on WES data (GeneRead, Qiagen). WES, SNP array information, and RNA-seq of the bulk tumor samples from patients have been previously reported and data deposited in dbGaP (phs000435.v2.p1). Multiplex PCR was performed for 22 cycles following the manufacturer's suggested conditions. Sequencing libraries were generated using the Qiagen GeneRead library preparation protocol following the manufacturer's recommended conditions. The resulting libraries were quantified (Library Quantification, Kapa Biosystems), pooled, and sequenced on an MiSeq instrument using a 300 cycle v2 kit (MiSeq reagent kit, Illumina). For mutation analysis related to single-cell DNA targeted PCR data, please refer to the Supplemental Methods.
Single-cell transcriptome sequencing and analysis
Single-cell CLL RNA libraries were generated from viable CD19+CD5+ tumor cells obtained by flow cytometry. Bulk cells were adjusted to 250 cells/µL and applied to the C1 system for single-cell capture with a 5–10 micron IFC (capture rate >80%) (Fluidigm). In the C1, whole-transcriptome amplification (WTA) was performed with the SMARTer kit (Clontech), and the product was converted to Illumina sequencing libraries using Nextera XT (Illumina). RNA-seq was performed on a HiSeq instrument (Illumina). Reads were aligned to the human reference genome (hg19) using TopHat v1.4 (Trapnell et al. 2009) with GENCODE v12 gene annotations.
Quality control was performed using Picard (http://broadinstitute.github.io/picard/index.html). Rarely expressed genes (detected in fewer than four cells) were removed prior to SCDE model fitting. Poor cells or empty wells (estimated library size <1 × 106, mode mapping quality = 0, passed filter aligned bases <1.5 × 108) were removed prior to model fitting. PAGODA was performed using the SCDE package (v1.99.1) (see the Supplemental Methods; Fan et al. 2016). We filtered 14,024 annotated pathways from MSigDB (major collections GO [C5v4], manually curated [C2v4], and oncogenic [C6v4]) to pathway gene sets with greater than 10 and less than 100 genes, resulting in 8437 final gene sets used.
Single-cell targeted mutation, gene expression, and chromosomal deletion analysis
Integrated detection of somatic mutations, SNPs within regions of chromosomal deletion, and gene expression in the same single cell was performed on a Biomark HD instrument (Fluidigm) (Livak et al. 2013; Vargas et al. 2016). cDNA was generated using reverse transcription master mix (100-6299, Fluidigm), and preamplification was performed with PreAmp master mix (100-5744, Fluidigm) and 10× preamplification primer mix (500 nM each primer). For gene expression, allele-specific mutation and SNP detection, qPCR was performed using 96.96 dynamic array IFCs (Fluidigm) as previously described (Livak et al. 2013; Burger et al. 2016). Data were analyzed with the Fluidigm real-time PCR analysis software using the linear (derivative) baseline correction method and the auto (global) Ct threshold method. The Cq values determined were exported, and data were processed as the Cq threshold (set to 28) minus the experimental Cq values. The transformation yields values equivalent to a log2 transformation of sequencing read count. For more details on primer design, cDNA generation, and preamplification conditions, please see the Supplemental Methods.
Mutation and deletion calling from RNA targeted analysis
Mutation calling relied on detection of significant levels of mutant allele expression above the expected background level. We did not differentiate between homozygous and heterozygous mutants. Because the primers for mutant and wild-type allele assays differed by only one nucleotide, increased expression of the wild-type allele leads to a nonzero background of mutant allele detection (“cross-talk”). To determine the expected background level of mutant allele detection as a function of wild-type allele detection, we used negative controls (i.e., cells known not to have the mutation of interest) and applied linear regression to model the expected degree of cross-talk. The details of this process are in the Supplemental Methods. A similar logic was applied to heterozygous SNPs within chromosomal regions affected by deletion.
Linking gene expression with genetic branches
To establish genetic branches, we applied hierarchical clustering with Ward.D linkage and Jaccard distance metric to the confident calls for relevant mutations and deletions. The resulting tree was cut into two primary branches to establish the two expected genetic branches. Association of genetic branch with gene expression was performed by binning cells into classes based on the inferred genetic branch, either cluster 1 or cluster 2. Then the distribution of gene expression levels was directly compared between the two classes. A two-sided Wilcoxon rank-sum test was used to assess statistical significance. Linear discriminant analysis, with performance quantification by ROC AUC, was also used to assess the extent to which the two classes could be distinguished by the first two principal components with PCA.
Functional evaluation of LCP1 and WNK1 mutation
Full-length LCP1 construct in pCMV6-AC-GFP vector was purchased (OriGene Technologies). Mutant LCP1 plasmid was generated by PCR amplification and subcloned at the AscI/MluI site in pCMV6-AC-GFP vector. Stable HEK293 cell lines expressing wild-type and mutant LCP1 were generated by transfection of these constructs and cultured in the presence of G418 (0.6 mg/mL) to maintain GFP positivity of >90%. DNA damage response and cell survival in relation to expression of wild-type and mutant LCP1 was assessed by immunoblot and colony assay, respectively. Interaction of LCP1 and ATM protein was assessed by immunoprecipitation using cell lysate generated from HEK293 cell lines that were transiently transfected with ATM-expressing construct (N-terminal His tag, gift from Dr. Brendan Price). The antibodies used in the study are detailed in the Supplemental Methods.
Functional effects of WNK1 mutation were evaluated in HEK293 cells that had inducible expression of either wild-type or mutant WNK1 without endogenous WNK1 expression. Generation of the WNK1 knockout cell line and inducible lines is detailed in the Supplemental Methods. WNK1 kinase activity and its activation of downstream signaling in relation to osmotic stress were assessed by immunoblot. Cell cycle in the cells with inducible WNK1 protein expression was profiled with the Click-iT Edu assay (Thermo Fisher Scientific). Detailed experimental information is provided in the Supplemental Methods.
Statistical analysis
The data in Figure 6F were analyzed using an unpaired two-tailed Student's t-test. A P-value <0.05 was considered significant.
Data access
The single-cell RNA sequencing, targeted mutation detection, as well as gene expression data have been submitted to NCBI's Database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap) under study number phs001372.v1.p1.
Supplementary Material
Acknowledgments
We thank Drs. Jonathan Duke-Cohan, Brendan D. Price, and Adrian M. Dubuc from Dana-Farber Cancer Institute and Brigham and Women's Hospital for helpful discussions and reagents. We thank Greg Harris of Fluidigm for help with the design of the qPCR assays for measuring gene expression. We also thank the excellent technical support of the MRC Protein Phosphorylation and Ubiquitylation Unit (PPU) Reagents team for DNA sequencing, cloning, and antibody production from University of Dundee. DRA's research is supported by the Medical Research Council (MC_UU_12016/2). We also thank the pharmaceutical companies supporting the Division of Signal Transduction Therapy Unit (Boehringer-Ingelheim, GlaxoSmithKline, Merck KGaA, DRA). L.W. was supported by the Lymphoma Research Foundation (LRF) postdoctoral fellowship. J.F. was supported by the National Science Foundation Graduate Research Fellowship (DGE1144152) and the NIH (F31CA206236-01). C.J.W. acknowledges support from the Blavatnik Family Foundation, ISF-Broad Foundation (P15439), the LRF, NHLBI (1R01HL103532-01; 1R01HL116452-01) and NCI (1R01CA155010-01A1; 1U10CA180861-01; R01CA182461) and is a recipient of a LLS Translational Research Program and Scholar Award and of an AACR SU2C Innovative Research Grant.
Author contributions: L.W., J.F., and C.J.W. designed the study. L.W., R.G., S.L., and K.J.L. performed single-cell RNA experiments. L.W. and S.H. examined the functional impact of LCP1 and WNK1 mutations. G.G. generated WNK1 construct and knockout cell lines and performed WNK1-related protein analysis. J.M.F. and C.-Z.Z. designed the DNA sequencing experiments. J.M.F. and L.W. performed single-cell DNA amplification and target enrichment. C.-Z.Z. performed the single-cell targeted DNA sequencing analysis. J.F. performed RNA computational analysis. C.W.Z. and M.B. generated the HEK293 and K562 LCP1 overexpression RNA library. C.X.Y., S.X., and P.D.C. performed FISH experiments. D.N. performed statistical analysis. D.K. and S.A.S. provided computational help. J.R.B., D.R.A., P.V.K., and K.J.L. provided reagents and constructive suggestions. C.J.W. supervised the study. Both co-first authors prepared the manuscript with help from all coauthors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.217331.116.
References
- Aalto Y, El-Rifa W, Vilpo L, Ollila J, Nagy B, Vihinen M, Vilpo J, Knuutila S. 2001. Distinct gene expression profiling in chronic lymphocytic leukemia with 11q23 deletion. Leukemia 15: 1721–1728. [DOI] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, Sunyaev SR. 2013. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7: Unit 7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C, Ji HP, Maley CC. 2016. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med 22: 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke P, Anders S, Kim JK, Kolodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC, et al. 2013. Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods 10: 1093–1095. [DOI] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. 2012. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150: 1209–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger JA, Landau DA, Taylor-Weiner A, Bozic I, Zhang H, Sarosiek K, Wang L, Stewart C, Fan J, Hoellenriegel J, et al. 2016. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun 7: 11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell RA, McGranahan N, Bartek J, Swanton C. 2013. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 501: 338–345. [DOI] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. 2012. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30: 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA, Sim S, Okamoto J, Johnston DM, Qian D, et al. 2011. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol 29: 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Ramskold D, Reinius B, Sandberg R. 2014. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343: 193–196. [DOI] [PubMed] [Google Scholar]
- Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. 2015. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol 33: 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubovsky JA, Chappell DL, Harrington BK, Agrawal K, Andritsos LA, Flynn JM, Jones JA, Paulaitis ME, Bolon B, Johnson AJ, et al. 2013. Lymphocyte cytosolic protein 1 is a chronic lymphocytic leukemia membrane-associated antigen critical to niche homing. Blood 122: 3308–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, Kaper F, Fan JB, Zhang K, Chun J, et al. 2016. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat Methods 13: 241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Lander ES. 2013. Lessons from the cancer genome. Cell 153: 17–37. [DOI] [PubMed] [Google Scholar]
- Gawad C, Koh W, Quake SR. 2014. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. Proc Natl Acad Sci 111: 17947–17952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Huss M, Tong GQ, Wang C, Li Sun L, Clarke ND, Robson P. 2010. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell 18: 675–685. [DOI] [PubMed] [Google Scholar]
- Guo G, Luc S, Marco E, Lin T-W, Peng C, Kerenyi MA, Beyaz S, Kim W, Xu J, Das PP. 2013. Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell Stem Cell 13: 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, Wu X, Wen L, Tang F, Huang Y, et al. 2016. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 26: 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeromin S, Weissmann S, Haferlach C, Dicker F, Bayer K, Grossmann V, Alpermann T, Roller A, Kohlmann A, Haferlach T, et al. 2014. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 28: 108–117. [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Silberstein L, Scadden DT. 2014. Bayesian approach to single-cell differential expression analysis. Nat Methods 11: 740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi-Impiombato A, Califano A, Migliazza A, Bhagat G, et al. 2010. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17: 28–40. [DOI] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW. 2015. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161: 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, et al. 2013. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152: 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M, Bottcher S, et al. 2015. Mutations driving CLL and their evolution in progression and relapse. Nature 526: 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. 2013. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499: 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. 2014. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Wills QF, Tipping AJ, Datta K, Mittal R, Goldson AJ, Sexton DW, Holmes CC. 2013. Methods for qPCR gene expression profiling applied to 1440 lymphoblastoid single cells. Methods 59: 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, Goolam M, Saurat N, Coupland P, Shirley LM, et al. 2015. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods 12: 519–522. [DOI] [PubMed] [Google Scholar]
- Mazor T, Pankov A, Song JS, Costello JF. 2016. Intratumoral heterogeneity of the epigenome. Cancer Cell 29: 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniz S, Jordan P. 2010. Emerging roles for WNK kinases in cancer. Cell Mol Life Sci 67: 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munsky B, Neuert G, van Oudenaarden A. 2012. Using gene expression noise to understand gene regulation. Science 336: 183–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeu F, Delgado J, Royo C, Baumann T, Stankovic T, Pinyol M, Jares P, Navarro A, Martin-Garcia D, Bea S, et al. 2016. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1 and ATM mutations in chronic lymphocytic leukemia. Blood 127: 2122–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paguirigan AL, Smith J, Meshinchi S, Carroll M, Maley C, Radich JP. 2015. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Sci Transl Med 7: 281re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. 2014. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344: 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter NE, Ermini L, Papaemmanuil E, Cazzaniga G, Vijayaraghavan G, Titley I, Ford A, Campbell P, Kearney L, Greaves M. 2013. Single cell mutational profiling and clonal phylogeny in cancer. Genome Res 23: 2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, Munar M, Rubio-Perez C, Jares P, Aymerich M, et al. 2015. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 526: 519–524. [DOI] [PubMed] [Google Scholar]
- Reinius B, Sandberg R. 2015. Random monoallelic expression of autosomal genes: stochastic transcription and allele-level regulation. Nat Rev Genet 16: 653–664. [DOI] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, et al. 2013. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498: 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, Chen P, Gertner RS, Gaublomme JT, Yosef N, et al. 2014. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510: 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH II, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. 2016a. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352: 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. 2016b. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539: 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. 2014. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509: 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S, Swanton C. 2016. Metastasis as an evolutionary process. Science 352: 169–175. [DOI] [PubMed] [Google Scholar]
- Vargas DY, Kramer FR, Tyagi S, Marras SA. 2016. Multiplex real-time PCR assays that measure the abundance of extremely rare mutations associated with cancer. PLoS One 11: e0156546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, et al. 2011. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365: 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S, Hergert S, Yin S, Freeman SS, Levin JZ, et al. 2016. Transcriptomic characterization of SF3B1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Cancer Cell 30: 750–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills QF, Livak KJ, Tipping AJ, Enver T, Goldson AJ, Sexton DW, Holmes C. 2013. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat Biotechnol 31: 748–752. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, et al. 2015. Brain structure: cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347: 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. 2015. Chromothripsis from DNA damage in micronuclei. Nature 522: 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.