

Abstract
A better understanding of the binding preference of amino acids for gold nanoparticles of different diameters could aid in the design of peptides that bind specifically to nanoparticles of a given diameter. Here we identify the binding preference of 19 natural amino acids for three gold nanoparticles with diameters of 1.0, 2.0, and 4.0 nm, and investigate the mechanisms that govern these preferences. We calculate potentials of mean force between 36 entities (19 amino acids and 17 side chains) and the three gold nanoparticles in explicit water using well-tempered metadynamics simulations. Comparing these potentials of mean force determines the amino acids’ nanoparticle binding preferences and if these preferences are controlled by the backbone, the side chain, or both. Twelve amino acids prefer to bind to the 4.0 nm gold nanoparticle, and seven prefer to bind to the 2.0 nm one. We also use atomistic molecular dynamics simulations to investigate how water molecules near the nanoparticle influence the binding of the amino acids. The solvation shells of the larger nanoparticles have higher water densities than those of the smaller nanoparticles while the orientation distributions of the water molecules in the shells of all three nanoparticles are similar. The nanoparticle preferences of the amino acids depend on whether their binding free energy is determined mainly by their ability to replace or to reorient water molecules in the nanoparticle solvation shell. The amino acids whose binding free energy depends mainly on the replacement of water molecules are likely to prefer to bind to the largest nanoparticle and tend to have relatively simple side chain structures. Those whose binding free energy depends mainly on their ability to reorient water molecules prefer a smaller nanoparticle and tend to have more complex side chain structures.
Graphical abstract
1. INTRODUCTION
Gold nanoparticles have been used in a variety of medical and biological applications such as photodynamic therapy, drug delivery, sensors, and diagnostics.1–7 More recently, researchers have begun to use peptides8–10 and protein cages11,12 to manufacture gold nanomaterials with desired properties. Since amino acids are the building blocks of peptides and proteins, a better understanding of how they interact with gold nano-particles could help us design peptide and protein materials that promote the production of gold nanoparticles with desired diameters and shapes.
One question that arises concerning amino acid–gold nanoparticle interactions is how the nanoparticle binding preference of amino acids depends on nanoparticle diameter. It is well-known that the function and performance of nanoparticles can be manipulated by tuning their diameter.13–19 Changing the diameter of a nanoparticle alters its curvature and the arrangement of surface atoms; it also changes the structural and dynamic properties of the solvent molecules surrounding it. These variations can, in turn, change how strongly a molecule binds to the nanoparticle and the binding mechanisms. For instance, Zuo et al.16 studied the binding affinity of villin headpiece on graphene, carbon nanotubes, and C60. They found that variation in the surface curvature changes the adsorption mechanism: π–π stacking interactions drive the binding of the villin headpiece on graphene while hydrophobic interactions drive binding on the carbon nanotube and C60. Monopoli et al.17 also found that the curvature of a nanoparticle influences the adsorption of proteins.
Current understanding of mechanisms that govern bindings of amino acids to substrates remains limited. Hrushikesh et al.20 investigated the binding strength of Lys and Asp on gold nanoparticles using isothermal titration calorimetry. They concluded that Lys binds to gold nanoparticles more strongly than Asp does, and they suggested that this binding difference is due to the polarity of the two amino acids. Feng et al.21 investigated the adsorption mechanisms of single amino acids to a gold (111) surface using molecular dynamics simulations. They suggested that the molecular size of amino acids rather than specific chemistry determines their binding ability on gold surfaces. Hoefling et al.22 also investigated the adsorption of single amino acids on the gold (111) surface, and they argued that the chemical natures of amino acids determine their binding ability on gold surfaces. Yu et al.23 investigated the influence of amino acid sequence on the binding process of peptide AYSSGAPPMPPF on gold surfaces using molecular simulations. They suggested that Tyr, Met, and Phe are the strongest binding residues on that peptide. These studies provide insight about the adsorption mechanisms of amino acids on gold materials.
The differences in physical and chemical properties of the 20 natural amino acids can lead to distinct nanoparticle preferences. A measure of the binding preference of amino acids on gold nanoparticles is their binding strength on gold surfaces. Atomistic simulation-based studies21,22,24 have shown that the binding abilities of the 20 amino acids on a gold (111) surface are different. This is of course expected, given the variety of structures on the different amino acids. Here we seek to identify the physical and chemical origin of the binding preferences of amino acids for different nanoparticles from two aspects: molecular structure of amino acids and effects of solvent molecules. Since a natural amino acid is composed of two parts: backbone and side chain, we first examine the respective roles of these two parts of amino acids in their nanoparticle binding preferences. We also examine if nanoparticle preferences of amino acids can be grouped based on the roles of backbone and side chain, and how this grouping is related to the structures of amino acids. Solvent molecules play an important role in governing adsorption of molecules on substrates.25,26 Some research27,28 even suggests that the solvent molecules determine the adsorption strength and mechanisms. We analyze the structural properties of solvent molecules around nanoparticles and how they vary with the nanoparticle diameter to seek how they may influence the nanoparticle preferences of amino acids.
The objectives of this work are to determine the binding preferences of amino acids for gold nanoparticles with different diameters and to explore the mechanisms that govern these preferences. We investigate the potentials of mean force of 36 entities (19 natural amino acids and 17 side chains) and three gold nanoparticles with diameters of 1.0, 2.0, and 4.0 nm in explicit water using well-tempered metadynamics simulations. The amino acid cysteine is not considered because it forms chemical bonds with gold materials. Comparison of these potentials of mean force allows us to identify which amino acids are most likely to bind to gold nanoparticles of different diameters, and to examine the respective roles of amino acid backbone and side chain in their binding preferences. We also investigate the structural properties of water molecules near gold nanoparticles to explore mechanisms that govern the nanoparticle preferences of amino acids. The rest of the paper is organized as follows: Section 2 presents the details of the molecular simulations and well-tempered metadynamics, section 3 presents the results and discussion, and section 4 gives the conclusions.
2. SIMULATION METHOD
2.1. Molecular Model
The atomistic configurations of gold nanoparticles, amino acids, and side chains are obtained in the following way. The Nanoparticle Builder Module of OpenMD29 is used to generate atomistic configurations for three gold nanoparticles of diameters 1.0, 2.0, and 4.0 nm, as shown in Figure 1. The positions of gold atoms are generated from a gold crystal (Face Centered Cubic, 0.408 nm) based on the nanoparticle diameter. A generated gold nanoparticle has (100), (110), and (111) facets and edges on it. The molecular structures of the amino acids are obtained from the Avogadro package.30 The amino acid’s N and C terminals are capped with an acetyl group and an N-methyl amide group, respectively. The protonated/deprotonated status of side chains of amino acids are determined by comparing their pKa values with pH = 7. The side chain of His is an imidazole group. The side chains are modeled by detaching them from the backbone and replacing the Cα atom with an H atom. The side chain of proline is not considered in this work because it is closely coupled with the backbone.
Figure 1.

Atomistic configurations of three gold nanoparticles with diameters (a) 1.0 nm, (b) 2.0 nm, and (c) 4.0 nm. The gold atoms (yellow) are shown in the VDW model.
The gold atoms are described using the force field parameters developed by the Heinz group31,32 since they do a good job of representing the interfacial properties of gold materials. The amino acids and their side chains are described by the GROMOS54a7 force field.33 The water molecules are described using the SPC model34 as recommended for the GROMOS force field.
The simulation box for well-tempered metadynamics simulations is created using the following steps. The initial length of the box is set to 3.0, 4.0, and 7.0 nm for the gold nanoparticles with diameters of 1.0, 2.0, and 4.0 nm, respectively, to ensure that there are enough water molecules in the bulk phase. The number of water molecules are ~800, ~2000, and ~6000 in the boxes for the 1.0, 2.0, and 4.0 nm nanoparticles. The gold nanoparticle is placed near the center of the box; the amino acid or side chain entity is placed randomly in the box, and the rest of the box is filled with water molecules at a density of 1.0 g/cm3. Figure 2 shows the initial configuration of a glycine and a 4.0 nm gold nanoparticle in a water box of length 7.0 nm.
Figure 2.
Initial configuration of a glycine and a gold nanoparticle with a diameter of 4.0 nm in a cubic box of a length 7.0 nm. Au atom: yellow, C atom: green, N atom: blue, O atom: red, and H atom: white. The gold nanoparticle and the glycine molecules are depicted in the VDW representation, and the water molecules are depicted in the CPK model. This configuration was generated using the VMD package.50
The simulation box for atomistic molecular dynamics (MD) simulations of gold nanoparticles in water is created in the same way as that in the well-tempered metadynamics simulations. The gold nanoparticle is placed in the center of the box and the rest of the box is filled with water molecules. The box length is the same as in the well-tempered metadynamics simulations.
2.2. Simulation Details
Well-tempered metadynamics35 is an alternative to metadynamics simulation method.36 The basic idea of well-tempered metadynamics is similar to that of metadynamics: the location of the system in the ensemble space is determined by calculating some predefined collective variables and a positive Gaussian potential is added to the system energy to discourage it from to coming back to the previous point. Eventually when enough Gaussian potentials have been added, the system explores every point of the energy landscape evenly. At this point, the energy landscape can be recovered as the opposite of the sum of all Gaussians. The magnitude of the Gaussian potentials added to the energy decreases as the simulation progresses to ensure smooth convergence of the final result
Well-tempered metadynamics simulations were conducted on nanoparticle-amino acid and nanoparticle-side chain systems to calculate 108 potentials of mean force (PMFs) between 36 entities (19 amino acids and 17 side chains) and three gold nanoparticles (diameters = 1.0, 2.0, and 4.0 nm) in explicit water. In each case, after energy minimization, a 10 ns isothermal–isobaric ensemble (NPT, T = 300 K, P = 1 bar) MD simulation is conducted with a 1 fs time step to ensure that the system potential energy fluctuates less than 1%. The Berendsen algorithm is used to couple temperature and pressure35 in the NPT MD simulation because this algorithm helps the system reach the desired temperature and pressure quickly. Then a well-tempered metadynamics simulation36 (bias factor = 4, initial Gaussian hill height = 1.0 kJ/mol, σ = 0.02 nm, and update frequency = 200 steps) is conducted in the canonical ensemble (NVT, T = 300 K) to calculate the amino acid/side chain-nanoparticle PMF. The collective variable (CV) is the distance between the amino acid Cα atom/side chain center of the mass and the center of the gold nanoparticle. The temperature is controlled using the stochastic global thermostat.37 The short-range van der Waals interactions are described using the Lennard–Jones 12–6 function with a 1.0 nm cutoff distance. The long-range electrostatic interactions are described using a Coulombic interaction and treated with the particle mesh Ewald sum method.38 Nonbonded interactions among gold atoms are not considered, and the positions of the gold atoms are fixed in the simulations. All of the covalent bonds in the water molecules, amino acids and side chains are free to move during the minimization and the 10 ns NPT MD simulations. The bonds attached to the hydrogen atoms are constrained to their equilibrated length using the LINCS algorithm.39 Periodic boundary conditions are applied to every case. The well-tempered metadynamics simulation times range from 60 to 200 ns, depending on how soon the PMF curve converges. Figure S1 of the Supporting Information (SI) shows PMF vs simulation time and Figure S2 shows CV values vs simulation time for the systems with the 19 amino acids and the 1.0 nm nanoparticle. Figures S1 and S2 illustrate that the PMF curves converge and that the well-tempered metadynamics simulations well sample the space of the selected CV. The MD simulations and energy minimization were conducted using GROMACS-4.6.5, and the well-tempered metadynamics simulations were conducted using PLUMED40-implemented GROMACS 4.6.5.41
Parrinello, Voth, and their colleagues36,42,43 investigated the accuracy and convergence of well-tempered metadynamics and other metadynamics. They found that well-tempered metadynamics converges to the final state it is designed to reach, regardless of the dynamic behavior of collective variables,43 and that longer simulation times improve the statistical accuracy of the results.36 Due to the large number of systems investigated, each systems is run once for a long simulation time to ensure that it converges and generates an accurate potential of mean force for an entity-nanoparticle pair. For this reason, there are no error bars in our simulation results.
Atomistic MD simulations of single-nanoparticle systems are conducted to analyze the structural properties of water molecules near three gold nanoparticles with diameters of 1.0, 2.0, and 4.0 nm. The simulation settings are the same as in the well-tempered metadynamics except for the simulation time allocated for the data collection. For each system, an energy minimization and a 10 ns NPT ensemble MD simulation are conducted to attain system equilibrium. After that, a 50 ns NVT ensemble MD simulation is conducted to collect data at a 1 ps frequency. The minimizations and MD simulations are conducted using GROMACS-4.6.5.
3. RESULTS AND DISCUSSION
3.1. Binding Preferences of Amino Acids for Gold Nanoparticles
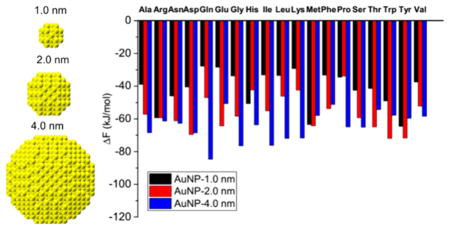
The binding preferences of amino acids on the three gold nanoparticles with diameters of 1.0, 2.0, and 4.0 nm can be determined by comparing the binding free energies. Figure 3 shows the binding free energies of 19 amino acids on the three gold nanoparticles. The binding free energies are defined to be the minimum values of the corresponding aminoacid–gold nanoparticle PMFs. The PMF curves are shown in Figures 4 and S3 and will be discussed in the next section. As shown in Figure 3, the 19 amino acids exhibit two types of nanoparticle preferences: 12 of them (Ala, Arg, Asn, Gln, Gly, His, Ile, Leu, Lys, Pro, Ser, and Val) prefer to bind to the 4.0 nm gold nanoparticle, and seven (Asp, Glu, Met, Phe, Thr, Trp, and Tyr) prefer to bind to the 2.0 nm nanoparticle. None prefers to bind to the 1.0 nm nanoparticle. The nanoparticle preferences of these two groups of amino acids are analyzed separately in detail below.
Figure 3.

Binding free energies of 19 natural amino acids on the three gold nanoparticles with diameters 1.0, 2.0, and 4.0 nm.
Figure 4.

Potentials of mean force of amino acids with three gold nanoparticles of diameters 1.0, 2.0, and 4.0 nm vs distance between Cα atom and the gold nanoparticle surface. (a) Ala, (b) Arg, and (c) Asn. The potentials of mean forces of the other 16 amino acids with the three gold nanoparticles are shown in Figure S3.
We first consider the 12 amino acids that prefer to bind to the 4.0 nm gold nanoparticle. They can be divided into three groups based on the order of their binding preferences for the three gold nanoparticles. The first group contains eight amino acids (Ala, Gln, Gly, Ile, Leu, Lys, Ser, and Val). Their nanoparticle preferences exhibit the order: 4.0 >2.0 >1.0 nm, as indicated by the order of their binding energies: ΔF4.0 nm < ΔF2.0 nm < ΔF1.0 nm. The differences between the binding free energies are at least 5.0 kJ/mol, two times kBT (2.5 kJ/mol) at room temperature. In the first group, six of the amino acids have relatively simple chemical structures (Ala, Gly, Ile, Leu, Ser, and Val); only Gln and Lys have relatively complex chemical structures. In the second group (Arg, Asn, and Pro) the chemical structures are in general more complex than those in the first group. The second group’s binding preferences for two of the three nanoparticles are less than kBT (2.5 kJ/mol), which could be easily erased by thermal fluctuations. As shown in Figure 3, Arg exhibits similar binding free energies to the 1.0 and 2.0 nm nanoparticles, Asn exhibits similar binding free energies to the 2.0 and 4.0 nm nanoparticles, and Pro exhibits similar binding free energies to the 1.0 and 2.0 nm nanoparticles. The third group contains only His. It exhibits a nanoparticle binding order of 4.0 >1.0 >2.0 nm. His is the only one among the 19 amino acids that prefers to bind to the 1.0 nm nanoparticle more than to the 2.0 nm nanoparticle. This nanoparticle preference may be related to the chemical groups of His which provide it with some unique features when designing peptides that can help to assemble small gold nanoparticles. For instance, His may play a key role in designing peptides that can be used to differentiate 1.0 nm nanoparticles from 2.0 nm ones.
We next consider the seven amino acids (Asp, Glu, Met, Phe, Thr, Trp, and Tyr) that prefer to bind to the 2.0 nm gold nanoparticle. They all have relatively complex side chains, indicating that the side chain may play an important role in their nanoparticle preference. We will investigate the respective roles of the backbone and the side chain in nanoparticle preference in the next section. These amino acids can be divided into two groups based on whether their binding preferences for the 2.0 nm gold nanoparticle is significant or not. The first group has Glu, Thr, Trp, and Tyr. The binding free energy for these four amino acids to the 2.0 nm gold nanoparticle is at least 5 kJ/mol lower than those on the other two nanoparticles. The second group contains Asp, Met, and Phe. As shown in Figure 3, Asp has similar binding free energies on the 2.0 and 4.0 nm nanoparticles; Met has similar binding free energies on the 1.0 and 2.0 nm nanoparticles; and Phe has similar binding free energies on the 2.0 and 4.0 nm nanoparticles. The preferences for the 2.0 nm nanoparticle of these three amino acids could easily be erased by the thermal fluctuations.
The analysis of nanoparticle binding preferences of the 19 amino acids suggests a way to design peptides that control nanoparticle size or recognize nanoparticles with certain diameters. Some amino acids (Ala, Gln, Gly, Glu, Ile, Leu, Lys, Ser, Thr, Trp Tyr, and Val) exhibit a distinct preference for the nanoparticle with a given diameter. These amino acids could be used to prepare peptides that bind to the nanoparticle with a given diameter more strongly than to other nano-particles. An ideal peptide would be composed of the amino acids that prefer to bind to a gold nanoparticle of a certain diameter more strongly than the others. Based on this criterion, the eight amino acids (Ala, Gln, Gly, Ile, Leu, Lys, Ser and Val) could be good components of peptides that bind to a gold nanoparticle with a diameter of 4.0 nm, while the three amino acids (Glu, Thr, Trp, and Tyr) could be good components of peptides that bind to a gold nanoparticle with a diameter of 2.0 nm.
It is also interesting that amino acids with similar chemical structures can present distinct nanoparticle preferences. For instance, Asn has only one methylene group less on the side chain than Gln yet Asn exhibits similar preferences to the 2.0 and 4.0 nm nanoparticles, while Gln exhibits a distinct nanoparticle preference order: 4.0 >2.0 >1.0 nm. A similar situation can be found for Asp and Glu. Asp only has one methylene group less on the side chain than Glu yet it has similar binding free energies on the 2.0 and 4.0 nm nanoparticles, while Glu exhibits a distinct preference for the 2.0 nm nanoparticle. These observations indicate that a small change in atomistic architecture may significantly influence the adsorption of amino acids on nanoparticles: the introduction of a methylene group on a side chain may change the flexibility of the side chain and change the nanoparticle preference of the amino acids.
Figure 3 also shows which amino acids bind most strongly to the 1.0, 2.0, and 4.0 nm nanoparticles. Among the 19 amino acids, Tyr binds to the 1.0 and 2.0 nm nanoparticle most strongly, consistent with experiments23 that show that Tyr binds to gold materials strongly. The aromatic ring on the Tyr side chain may contribute to this strong binding. The above analysis also shows that Tyr prefers to bind to the 2.0 nm nanoparticle, indicating the important role of the side chain in its nanoparticle binding. Gln binds to the 4.0 nm nanoparticle most strongly among the 19 natural amino acids indicating the important role of the backbone in its nanoparticle binding. However, the Gln side chain has a structure similar to amide bonds on peptides, which may contribute to the increase in binding energy as the nanoparticle diameter increases. As shown in Figure 3, Gln has the largest change in binding free energy as the nanoparticle diameters increases.
3.2. Binding Configurations of Amino Acids on Gold Nanoparticles
Figure 4 shows the PMFs for Ala, Arg, and Asn on the three gold nanoparticles vs the distance between the amino acid Cα atom and the gold nanoparticle surface. The PMFs for the other amino acids are shown in Figure S3. We plot PMFs vs the distance between the amino acid Cα atom and the gold nanoparticle surface instead of that between the amino acid Cα atom and the COM of the nanoparticle so that we can compare the binding positions of amino acids on three gold nanoparticles of different diameters in a straightforward way. The distance between the amino acid Cα atom and the gold nanoparticle surface is obtained in a straightforward fashion from knowledge of the radius of the gold nanoparticle and the distance between the amino acid Cα atom and the gold nanoparticle COM. We investigate the positions and numbers of minima in these PMFs to learn (1) how the binding configurations of amino acids on gold nanoparticles depend on nanoparticle diameter, and (2) if some amino acids have multiple adsorption configurations on gold nanoparticles.
To determine the binding configurations of the amino acids, we first analyze the positions of the global minima in the PMFs. As shown in Figures 4 and S3, the global minimum positions on the PMFs are 0.2–0.3 nm from the gold nanoparticle surface in the binding state. Such small values indicate that the amino acids bind to the gold nanoparticles directly instead of through a solvation shell. Atomistic simulations by Nawrocki et al.24 of the binding of amino acids to the gold (111) surface also indicate that amino acids bind directly to the gold surface.
We then investigate how the gold nanoparticle diameter influences the binding configurations of amino acids. As shown in Figure 4, the position of the global minimum for an amino acid is relatively insensitive to the nanoparticle diameter. For instance, the global minimum positions of the Ala–nanoparticle PMFs are 0.22, 0.20, and 0.18 nm on the nanoparticles with diameters of 1.0, 2.0, and 4.0 nm, respectively. This similarity suggests that the amino acids bind to all three gold nanoparticle surfaces directly. The slight change in the global minimum position could be because the amino acids change the orientation of their dipole moment or the side chain position. These small changes could influence the properties of the interfacial region on the nanoparticle in a subtle way.
We next determine if the amino acids have multiple binding configurations on nanoparticles. As shown in Figures 4 and S3, the PMFs of 13 amino acids (Arg, Asn, Asp, Gln, Glu, His, Ile, Leu, Lys, Met, Phe, Trp, and Tyr) have local minima in addition to the global minimum, indicating that these amino acids have multiple binding configurations on gold nano-particles. The PMFs of six amino acids (Ala, Gly, Pro, Ser, Thr, and Val) only have a global minimum, indicating that they have only one binding configuration on a gold nanoparticle. Comparing these two groups indicates that the amino acids having side chains with relatively complex structure are more likely to have multiple binding configurations, and the amino acids having side chains with relative simple structure are likely to have only a single binding configuration.
3.3. Roles of Backbone and Side Chains in Nanoparticle Binding Preferences of Amino Acids
It is interesting to consider which part of an amino acid, its backbone, side chain, or both, is more important in determining its binding preference for gold nanoparticles. The wide variation in the amino acid preference for nanoparticles indicates that the backbone and side chain may play different roles in binding the individual amino acids on gold nanoparticles. Here we compare the binding preferences of the amino acids, their side chains and their backbones for gold nanoparticles to see what is most important. The binding preference of the backbones are taken to be that of glycine because they have a similar chemical structure. Figure 5 shows the binding free energies of the 17 side chains on the three gold nanoparticles; these are extracted from the PMF curves using the same method as for the amino acid binding free energies. The binding preferences of the amino acid side chains are determined by comparing their binding free energies on the 1.0, 2.0, and 4.0 nm gold nanoparticles. Table 1 lists which gold nanoparticle the 17 amino acids and their side chains prefer.
Figure 5.

Binding free energy of 17 side chains on the three gold nanoparticles (diameters = 1.0, 2.0, and 4.0 nm).
Table 1.
Gold Nanoparticle Preferences of The 17 Amino Acids and Side Chainsa
| amino acid | side chain | |
|---|---|---|
| Ala | 4.0 | 4.0 |
| Arg | 4.0 | 4.0 |
| Asn | 4.0 | 2.0 |
| Asp | 2.0 | 2.0 |
| Gln | 4.0 | 2.0 |
| Glu | 2.0 | 2.0 |
| His | 4.0 | 4.0 |
| Ile | 4.0 | 2.0 |
| Leu | 4.0 | 1.0 |
| Lys | 4.0 | 2.0 |
| Met | 2.0 | 2.0 |
| Phe | 2.0 | 2.0 |
| Ser | 4.0 | 4.0 |
| Thr | 2.0 | 1.0 |
| Trp | 2.0 | 4.0 |
| Tyr | 2.0 | 4.0 |
| Val | 4.0 | 4.0 |
The gold nanoparticles are represented by their diameters (nm). The backbone prefers to bind to the 4.0 nm nanoparticle.
We first investigate what determines the binding preference of the amino acids that prefer to bind to the 4.0 nm gold nanoparticle. As stated above, the binding preference of glycine is taken to be that of the backbone. So the preference of these amino acids are the same as that of the backbone. These amino acids can be divided into two groups depending on whether or not the nanoparticle preference of their side chains is the same as their backbones. The first group contains five amino acids (Ala, Arg, His, Ser, and Val) whose side chains also prefer to bind to the 4.0 nm nanoparticle. Therefore, it does not matter whether the backbone or the side chain determines the nanoparticle preference of these amino acids. The second group contains five amino acids (Asn, Gln, Ile, Leu, and Lys). Their side chains prefer to bind to a gold nanoparticle other than the 4.0 nm one, which suggests that their preference is more likely to be determined by the backbone.
We then investigate what determines the binding preference of the amino acids that prefer to bind to the 2.0 nm gold nanoparticle. The binding preference of these amino acids is different from that of backbone. However, this does not necessarily mean that the side chains determine their preference. These amino acids can be divided into two groups concerning whether the nanoparticle preferences of the side chain and of the whole amino acid are the same or different. The first group contains four amino acids (Asp, Glu, Met, and Phe) that prefer to bind to the nanoparticle that their side chains prefer. The side chain likely determines the preferences of these amino acids. The second group contains three amino acids (Thr, Trp, and Tyr). They prefer to bind to a gold nanoparticle that neither their backbone nor side chain prefer: Thr prefers to bind to the 2.0 nm nanoparticle while its backbone and side chain prefer to bind to the 4.0 and 1.0 nm nanoparticles, respectively; Trp prefers to bind to the 2.0 nm nanoparticle while its backbone and side chain prefer to bind to the 4.0 nm nanoparticle; Tyr prefers to bind to the 2.0 nm nanoparticle while its backbone and side chain prefer to bind to the 4.0 nm nanoparticle.
The binding preference of Thr, Trp, and Tyr are apparently determined by the backbone and side chain together rather than separately. It is perhaps not surprising that the binding preference of Trp and Tyr are determined by the backbone and side chain together because they have relatively complex structures. The side chain of Trp is an indole and that of Tyr is a tyrosyl. As to Thr, as shown in Figure 5, the difference in the binding free energies of its side chain on the 1.0 and 2.0 nm nanoparticles is only 1 kJ/mol. Such a small difference is easily perturbed by the environment and the backbone.
In general, the amino acids can be divided into four groups based on the respective roles of the backbone and side chain in their nanoparticle preferences. The first group contains Ala, Arg, His, Ser, and Val; the nanoparticle binding preferences of the amino acid, backbone side chain are the same. The second group contains Asn, Gln, Ile, Leu, and Lys; their backbones determine the amino acids’ nanoparticle binding preferences. Both groups prefer to bind to the 4.0 nm gold nanoparticle. The third group contains Asp, Glu, Met, and Phe; the amino acid side chains determine their nanoparticle binding preferences. These amino acids prefer to bind to the 2.0 nm nanoparticles. The fourth group contains Thr, Trp, and Tyr; the backbones and the side chains play comparable roles in determining the nanoparticle binding preference of the amino acids. The nanoparticle binding preferences of these amino acids differ from those of their side chains and backbones, and so need to be examined case by case. The Gly and Pro are not in any of the four groups; the Gly side chain is only an H atom, and the Pro side chain is closely coupled with its backbone.
3.4. Influence of Solvation Shell on Nanoparticle Binding Preference of Amino Acids
Our above analysis has shown that amino acids exhibit various nanoparticle preferences, which do not show a close relationship with amino acids’ chemical structures. Here we investigate the structural properties of water molecules around the three gold nanoparticles in order to explore universal mechanisms that govern the adsorption of amino acids on nanoparticles and their nanoparticle preferences.
We first analyze the density distributions of the water molecules around a gold nanoparticle in the absence of an amino acid or a side chain. Figure 6a shows the density distributions of the water molecules around the gold nanoparticles versus the distance from the nanoparticle surface. The three curves all show a peak between 0.28 and 0.4 nm with heights of 4.5, 6.0, and 7.0 for the 1.0, 2.0, and 4.0 nm gold nanoparticles, respectively. There are valleys after the peaks, between 0.4 and 0.5 nm, and small peaks between 0.5 and 0.6 nm. The curves become flat at distances larger than 0.6 nm. Such density fluctuations are consistent with those observed for other solid substrates44 and indicate that the three gold nanoparticles all possess a stable solvation shell, which is 0.12 nm thick. An intermediate region exists between the solvation shell and the bulk water, which is 0.2 nm thick. The values of the peak height show that the water densities in the solvation shells of the three gold nanoparticles are several times that in the bulk phase, and become higher as the nanoparticle diameter increases.
Figure 6.

Structural properties of water molecules near the 1.0, 2.0, and 4.0 nm gold nanoparticles. (a) Reduced density of water molecules in the radial direction and distributions of (b) cos θ and (c) cos α in the [−1, 1] range. The two angles, θ and α, characterize the orientation of the water dipole moment and the water O—H bond relative to the radial direction from the nanoparticle.
We then analyze the distribution of the water molecule orientation in the solvation shells of the three gold nanoparticles in the absence of an amino acid or a side chain because the preferential orientation of water molecules around a substrate can determine if molecules bind to it.45 The orientation is measured in terms of two angles: θ and α. The former is the angle between the nanoparticle radial direction and the dipole moment of the water molecules, and the latter is the angle between the nanoparticle radial direction and the O—H bond of the water molecules. Figure 6b,c shows the distributions of cos θ and cos α for water molecules in the solvation shells of the three gold nanoparticles. The distributions of cos θ have a peak at around 0.1, and the distributions of cos α have two peaks at around −0.4 and 1.0. These peaks indicate that the water molecules in the solvation shells surrounding the gold nanoparticles prefer to have their dipole moment parallel to the nanoparticle surface and one O—H bond pointing to the gold nanoparticle surface. Observing water orientation around nanomaterials remains challenging in experiment. In simulations, the same type of orientation distribution for water molecules has been found for an SiO2 surface whose interactions with water molecules are parametrized via Lennard–Jones expression.44 The three curves in Figure 6b,c have similar shapes and peak heights, indicating that water molecules in the solvation shells of the three gold nanoparticles have the same orientation preferences, no matter how large the nanoparticle is.
The analysis of density and orientation distributions of water molecules around gold nanoparticles indicates the mechanisms that govern the nanoparticle preferences of amino acids. As discussed above, amino acids prefer to bind to gold nanoparticles with their Cα atoms 0.2–0.3 nm from the gold nanoparticle surface. This is where the solvation shells of the gold nanoparticles are. The amino acids need to replace the water molecules in the solvation shell to bind to the nanoparticle. Upon insertion into the solvation shell, the amino acids may perturb the orientation of water molecules around them. The density fluctuation and preferred orientation of water molecules near solutes contribute to the system stability44,46–48 and varying them changes the free energy of the system. The free energy for the amino acid binding on the nanoparticle can thus be broken into two terms related to the replacement of water molecules and the perturbation of the water molecules’ orientation. The value of the first term is expected to be larger as the gold nanoparticle diameter increases because an amino acid needs to replace more water molecules around a larger nanoparticle. The value of the second term depends on the chemical nature of the amino acid.
On the basis of this mechanism, an amino acid may prefer to bind to the 4.0 nm nanoparticle if its binding free energy is dominated by the first term and prefer to bind to a nanoparticle with a specific diameter if its binding free energy is dominated by the second term. The physicochemical properties of the amino acid determine whether the binding free energy depends on the first or second term. The amino acids are likely to have their binding free energies depend on the first term if they have a relatively simple molecular structure (such as Ala, Arg, His, Ser, and Val) or a side chain long enough that it has little influence on the orientation of water molecules in the solvation shell (e.g., Lys and Arg). The amino acids are likely to have their binding free energy depend on the second term if they have a side chain with a complex structure. For instance five of the amino acids that prefer to bind to the 2.0 nm nanoparticle have either an anionic group or an aromatic group; the side chain of Met has an S-methyl group, and the side chain of Thr is an alcohol-containing group.
We also observe an indirect amino acid–gold nanoparticle binding by comparing the solvation shell size and amino acid binding position. The solvation shell of the amino acid shares some water molecules with that of the nanoparticle in the indirect binding configuration. Here we take Glu as an example. As shown in Figure 4f, the Glu–gold nanoparticle PMFs have local minima at 0.4, 0.38, and 0.48 nm for the 1.0, 2.0, and 4.0 nm gold nanoparticles. These are near to the positions of the outer borders of the solvation shells of the three gold nanoparticles (0.4 nm). The proximity of the local minimum positions and the outer borders of the solvation shells indicates that Glu could bind to the gold nanoparticles through the solvation shell.
4. CONCLUSIONS
We investigate the binding preferences of 19 amino acids for three gold nanoparticles with diameters of 1.0, 2.0, and 4.0 nm using computer simulations. We determine the preference of amino acids for the three nanoparticles, the roles of the backbones and side chains in these preferences, and how the solvation shells of the gold nanoparticles affect the preferences. This is accomplished by using well-tempered metadynamics simulations to calculate the binding free energy of amino acids and their side chains on the gold nanoparticles, and standard molecular dynamics simulations to investigate the structural properties of water molecules around the gold nanoparticles. The simulation results show that 12 amino acids (Ala, Arg, Asn, Gln, Gly, His, Ile, Leu, Lys, Pro, Ser, and Val) prefer to bind to the gold nanoparticle with a diameter of 4.0 nm and seven (Asp, Glu, Met, Phe, Thr, Trp, and Tyr) prefer to bind to the nanoparticle with a diameter of 2.0 nm. Comparing the binding free energies of the amino acids and their backbones and side chains sheds light on the roles of the backbone and side chain in determining the binding of an amino acid on gold nanoparticles. The backbone determines the nanoparticle binding preferences of Asn, Gln, Ile, Leu, and Lys. The side chain determines the binding preferences of Asp, Glu, Met, and Phe. The backbone and side chain synergistically determine the binding preferences of Thr, Trp, and Tyr.
Our investigation suggests that the effect that an amino acid has on the solvation shell of a gold nanoparticle plays a major role in determining its nanoparticle preferences. The binding free energy of amino acids originates from two aspects: replacing the water molecules and changing their orientation in the nanoparticle’s solvation shell. The density of water molecules surrounding a gold nanoparticle increases as the nanoparticle diameter increases while the orientation distributions of the water molecules in the solvation shells of the three gold nanoparticles are similar. Therefore, the binding free energy related to the first aspect becomes larger as the nanoparticle diameter increases while that related to the second aspect is more likely to depend on the physical and chemical natures of an amino acid. The amino acids whose binding free energy depends on the replacement of water molecules are likely to prefer to bind to the 4.0 nm nanoparticle, including Ala, Arg, Asn, Gln, Gly, His, Ile, Leu, Lys, Pro, Ser, and Val. The amino acids whose binding free energy depends on the reorientation of water molecules are likely to prefer to bind a nanoparticle with a certain diameter, including Asp, Glu, Met, Phe, Thr, Trp, and Tyr. The amino acids that prefer to bind to the 2.0 nm gold nanoparticle generally possess more complex structure than those that prefer to bind to the 4.0 nanoparticle.
Another interesting aspect of the adsorption of amino acids on gold nanoparticles is their tendency to bind to certain regions on gold nanoparticles. As shown in Figure 1, the gold nanoparticle surfaces are not uniform and possess several types of facets and edges. The areas of the facets and edges depend on the nanoparticle shape and diameter. Amino acids may prefer to bind to some specific facets or edges, depending on their own chemical features and the characteristics of the facets and edges. Recently, Wright et al.49 investigated selectivity of peptide AuBP1 for gold facets (111), (100) (1 × 1), and (100) (5 × 1) using replica exchange with metadynamics simulations. Their work shows how computer simulations can help to understand mechanistic details of peptide–substrate interactions and rationalize the design of peptides that can be used to shape the formation of gold nanocrystals.
The nanoparticle preferences of amino acids determined in our simulations can be used to rationalize the design of peptides that bind specifically to nanoparticles with certain diameters. Peptides have been recognized as a powerful tool to manufacture inorganic nanomaterials. Our simulations suggest that one could rationally design specific peptides to promote the formation of nanoparticles with desired size or even shape. The potentials of mean force obtained in well-tempered metadynamics simulations can also be used to develop coarse-grained protein/nanoparticle models that enable us to study protein-nanoparticle interactions.
Supplementary Material
Acknowledgments
Funding
National Science Foundation (CBET-1236053), National Science Foundation (ACI-1053575), and National Institutes of Health (EB006006). This work was supported in part by the Research Triangle NSF-MRSEC on Programmable Soft Matter, DMR 1121107
This work was supported by National Science Foundation (CBET-1236053) and the National Institutes of Health (EB006006). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.langmuir.6b01693.
Figure S1 shows PMF vs simulation time. Figure S2 shows the value of CV (distance between Cα atoms and the center of the nanoparticle) vs simulation time for the systems with the 19 amino acids and the 1.0 nm nanoparticle. Figure S3 shows PMFs for 16 amino acids (Asp, Gln, Glu, Gly, His, Ile, Leu, Lys, Met, Phe, Pro, Ser, Thr, Trp, Tyr, and Val) on the three gold nanoparticles vs the distance between the amino acid Cα atom and the gold nanoparticle surface (PDF)
References
- 1.Daniel MC, Astruc D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology. Chem Rev. 2004;104:293–346. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 2.Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Gold Nanoparticles for Biology and Medicine. Angew Chem, Int Ed. 2010;49:3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alkilany A, Murphy C. Toxicity And Cellular Uptake Of Gold Nanoparticles: What We Have Learned so Far? J Nanopart Res. 2010;12:2313–2333. doi: 10.1007/s11051-010-9911-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Judy JD, Unrine JM, Bertsch PM. Evidence for Biomagnification of Gold Nanoparticles within a Terrestrial Food Chain. Environ Sci Technol. 2011;45:776–781. doi: 10.1021/es103031a. [DOI] [PubMed] [Google Scholar]
- 5.Murphy CJ, Gole AM, Stone JW, Sisco PN, Alkilany AM, Goldsmith EC, Baxter SC. Gold Nanoparticles in Biology: Beyond Toxicity to Cellular Imaging. Acc Chem Res. 2008;41:1721–1730. doi: 10.1021/ar800035u. [DOI] [PubMed] [Google Scholar]
- 6.Khlebtsov N, Dykman L. Biodistribution And Toxicity of Engineered Gold Nanoparticles: a Review of in vitro and in vivo Studies. Chem Soc Rev. 2011;40:1647–1671. doi: 10.1039/c0cs00018c. [DOI] [PubMed] [Google Scholar]
- 7.Love SA, Maurer-Jones MA, Thompson JW, Lin YS, Haynes CL. Assessing Nanoparticle Toxicity. Annu Rev Anal Chem. 2012;5:181–205. doi: 10.1146/annurev-anchem-062011-143134. [DOI] [PubMed] [Google Scholar]
- 8.Dickerson MB, Sandhage KH, Naik RR. Protein- and Peptide-Directed Syntheses of Inorganic Materials. Chem Rev. 2008;108:4935–4978. doi: 10.1021/cr8002328. [DOI] [PubMed] [Google Scholar]
- 9.Chen CL, Rosi NL. Peptide-Based Methods for the Preparation of Nanostructured Inorganic Materials. Angew Chem, Int Ed. 2010;49:1924–1942. doi: 10.1002/anie.200903572. [DOI] [PubMed] [Google Scholar]
- 10.Wright LB, Merrill NA, Knecht MR, Walsh TR. Structure of Arginine Overlayers at the Aqueous Gold Interface: Implications for Nanoparticle Assembly. ACS Appl Mater Interfaces. 2014;6:10524–10533. doi: 10.1021/am502119g. [DOI] [PubMed] [Google Scholar]
- 11.Loo L, Guenther RH, Basnayake VR, Lommel SA, Franzen S. Controlled Encapsidation of Gold Nanoparticles by a Viral Protein Shell. J Am Chem Soc. 2006;128:4502–4503. doi: 10.1021/ja057332u. [DOI] [PubMed] [Google Scholar]
- 12.Fan R, Chew SW, Cheong VV, Orner BP. Fabrication of Gold Nanoparticles Inside Unmodified Horse Spleen Apoferritin. Small. 2010;6:1483–1487. doi: 10.1002/smll.201000457. [DOI] [PubMed] [Google Scholar]
- 13.Ju SP. A Molecular Dynamics Simulation of the Adsorption of Water Molecules Surrounding an Au Nanoparticle. J Chem Phys. 2005;122:094718. doi: 10.1063/1.1854132. [DOI] [PubMed] [Google Scholar]
- 14.Elrad OM, Hagan MF. Mechanisms of Size Control and Polymorphism in Viral Capsid Assembly. Nano Lett. 2008;8:3850–3857. doi: 10.1021/nl802269a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCullagh M, Prytkova T, Tonzani S, Winter ND, Schatz GC. Modeling Self-Assembly Processes Driven by Nonbonded Interactions in Soft Materials. J Phys Chem B. 2008;112:10388–10398. doi: 10.1021/jp803192u. [DOI] [PubMed] [Google Scholar]
- 16.Zuo G, Zhou X, Huang Q, Fang HP, Zhou RH. Adsorption of Villin Headpiece onto Graphene, Carbon Nanotube, and C60: Effect of Contacting Surface Curvatures on Binding Affinity. J Phys Chem C. 2011;115:23323–23328. [Google Scholar]
- 17.Monopoli MP, Walczyk D, Campbell A, Elia G, Lynch I, Baldelli Bombelli F, Dawson KA. Physical–Chemical Aspects of Protein Corona: Relevance to in Vitro and in Vivo Biological Impacts of Nanoparticles. J Am Chem Soc. 2011;133:2525–2534. doi: 10.1021/ja107583h. [DOI] [PubMed] [Google Scholar]
- 18.Todorova N, Makarucha AJ, Hine NDM, Mostofi AA, Yarovsky I. Dimensionality of Carbon Nanomaterials Determines the Binding and Dynamics of Amyloidogenic Peptides: Multiscale Theoretical Simulations. PLoS Comput Biol. 2013;9:e1003360. doi: 10.1371/journal.pcbi.1003360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan S, Gupta A, Chaudhary A, Nandi CK. Orientational Switching of Protein Conformation as a Function of Nanoparticle Curvature and Their Geometrical Fitting. J Chem Phys. 2014;141:084704. doi: 10.1063/1.4893441. [DOI] [PubMed] [Google Scholar]
- 20.Joshi H, Shirude PS, Bansal V, Ganesh KN, Sastry M. Isothermal Titration Calorimetry Studies on the Binding of Amino Acids to Gold Nanoparticles. J Phys Chem B. 2004;108:11535–11540. [Google Scholar]
- 21.Feng J, Pandey RB, Berry RJ, Farmer BL, Naik RR, Heinz H. Adsorption Mechanism of Single Amino Acid and Surfactant Molecules to Au {111} Surfaces in Aqueous Solution: Design Rules for Metal-Binding Molecules. Soft Matter. 2011;7:2113–2120. [Google Scholar]
- 22.Hoefling M, Iori F, Corni S, Gottschalk KE. Interaction of Amino Acids with the Au(111) Surface: Adsorption Free Energies from Molecular Dynamics Simulations. Langmuir. 2010;26:8347–8351. doi: 10.1021/la904765u. [DOI] [PubMed] [Google Scholar]
- 23.Yu J, Becker ML, Carri GA. The Influence of Amino Acid Sequence and Functionality on the Binding Process of Peptides onto Gold Surfaces. Langmuir. 2012;28:1408–1417. doi: 10.1021/la204109r. [DOI] [PubMed] [Google Scholar]
- 24.Nawrocki G, Cieplak M. Aqueous Amino Acids and Proteins Near the Surface of Gold in Hydrophilic and Hydrophobic Force Fields. J Phys Chem C. 2014;118:12929–12943. [Google Scholar]
- 25.Pal SK, Zewail AH. Dynamics of Water in Biological Recognition. Chem Rev. 2004;104:2099–2124. doi: 10.1021/cr020689l. [DOI] [PubMed] [Google Scholar]
- 26.Tinberg CE, Khare SD, Dou J, Doyle L, Nelson JW, Schena A, Jankowski W, Kalodimos CG, Johnsson K, Stoddard BL, Baker D. Computational Design of Ligand-Binding Proteins with High Affinity and Selectivity. Nature. 2013;501:212–216. doi: 10.1038/nature12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng J, Li L, Tsao HK, Sheng YJ, Chen S, Jiang S. Strong Repulsive Forces between Protein and Oligo (Ethylene Glycol) Self-Assembled Monolayers: A Molecular Simulation Study. Biophys J. 2005;89:158–166. doi: 10.1529/biophysj.105.059428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He Y, Chang Y, Hower JC, Zheng J, Chen S, Jiang S. Origin of Repulsive Force and Structure/Dynamics of Interfacial Water in OEG-Protein Interactions: A Molecular Simulation Study. Phys Chem Chem Phys. 2008;10:5539–5544. doi: 10.1039/b807129b. [DOI] [PubMed] [Google Scholar]
- 29.Vardeman CF, Stocker KM, Gezelter JD. The Langevin Hull: Constant Pressure and Temperature Dynamics for Nonperiodic Systems. J Chem Theory Comput. 2011;7:834–842. doi: 10.1021/ct100670m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J Cheminf. 2012;4:1–17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinz H, Vaia RA, Farmer BL, Naik RR. Accurate Simulation of Surfaces and Interfaces of Face-Centered Cubic Metals Using 12–6 and 9–6 Lennard-Jones Potentials. J Phys Chem C. 2008;112:17281–17290. [Google Scholar]
- 32.Heinz H, Lin TJ, Kishore Mishra R, Emami FS. Thermodynamically Consistent Force Fields for the Assembly of Inorganic, Organic, and Biological Nanostructures: The INTERFACE Force Field. Langmuir. 2013;29:1754–1765. doi: 10.1021/la3038846. [DOI] [PubMed] [Google Scholar]
- 33.Schmid N, Eichenberger A, Choutko A, Riniker S, Winger M, Mark A, van Gunsteren W. Definition and Testing of The GROMOS Force-field Versions 54A7 and 54B7. Eur Biophys J. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- 34.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. SPC. Intermolecular Forces. Reidel; Dordrecht: 1981. [Google Scholar]
- 35.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular Dynamics with Coupling to An External Bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 36.Barducci A, Bussi G, Parrinello M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys Rev Lett. 2008;100:020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- 37.Bussi G, Donadio D, Parrinello M. Canonical Sampling Through Velocity Rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 38.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 39.Hess B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J Chem Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 40.Bonomi M, Branduardi D, Bussi G, Camilloni C, Provasi D, Raiteri P, Donadio D, Marinelli F, Pietrucci F, Broglia RA, Parrinello M. PLUMED: A Portable Plugin for Free-Energy Calculations with Molecular Dynamics. Comput Phys Commun. 2009;180:1961–1972. [Google Scholar]
- 41.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 42.Laio A, Rodriguez-Fortea A, Gervasio FL, Ceccarelli M, Parrinello M. Assessing The Accuracy of Metadynamics. J Phys Chem B. 2005;109:6714–6721. doi: 10.1021/jp045424k. [DOI] [PubMed] [Google Scholar]
- 43.Dama JF, Parrinello M, Voth GA. Well-Tempered Metadynamics Converges Asymptotically. Phys Rev Lett. 2014;112:240602. doi: 10.1103/PhysRevLett.112.240602. [DOI] [PubMed] [Google Scholar]
- 44.Giovambattista N, Debenedetti PG, Rossky PJ. Effect of Surface Polarity on Water Contact Angle and Interfacial Hydration Structure. J Phys Chem B. 2007;111:9581–9587. doi: 10.1021/jp071957s. [DOI] [PubMed] [Google Scholar]
- 45.Zangi R, Engberts JBFN. Physisorption of Hydroxide Ions from Aqueous Solution to a Hydrophobic Surface. J Am Chem Soc. 2005;127:2272–2276. doi: 10.1021/ja044426f. [DOI] [PubMed] [Google Scholar]
- 46.Vembanur S, Patel AJ, Sarupria S, Garde S. On the Thermodynamics and Kinetics of Hydrophobic Interactions at Interfaces. J Phys Chem B. 2013;117:10261–10270. doi: 10.1021/jp4050513. [DOI] [PubMed] [Google Scholar]
- 47.Sarupria S, Garde S. Quantifying Water Density Fluctuations and Compressibility of Hydration Shells of Hydrophobic Solutes and Proteins. Phys Rev Lett. 2009;103:037803. doi: 10.1103/PhysRevLett.103.037803. [DOI] [PubMed] [Google Scholar]
- 48.Chandler D. Interfaces and The Driving Force of Hydrophobic Assembly. Nature. 2005;437:640–647. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 49.Wright LB, Palafox-Hernandez JP, Rodger PM, Corni S, Walsh TR. Facet Selectivity in Gold Binding Peptides: Exploiting Interfacial Water Structure. Chem Sci. 2015;6:5204–5214. doi: 10.1039/c5sc00399g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.