

Abstract
DNA‐dependent activator of interferon regulatory factors/Z‐DNA binding protein 1 (DAI/ZBP1) is a crucial sensor of necroptotic cell death induced by murine cytomegalovirus (MCMV) in its natural host. Here, we show that viral capsid transport to the nucleus and subsequent viral IE3‐dependent early transcription are required for necroptosis. Necroptosis induction does not depend on input virion DNA or newly synthesized viral DNA. A putative RNA‐binding domain of DAI/ZBP1, Zα2, is required to sense virus and trigger necroptosis. Thus, MCMV IE3‐dependent transcription from the viral genome plays a crucial role in activating DAI/ZBP1‐dependent necroptosis. This implicates RNA transcripts generated by a large double‐stranded DNA virus as a biologically relevant ligand for DAI/ZBP1 during natural viral infection.
Keywords: DAI/ZBP1, IE3, murine cytomegalovirus, necroptosis, RIPK3
Subject Categories: Autophagy & Cell Death; Microbiology, Virology & Host Pathogen Interaction
Introduction
Necroptosis is a form of programmed cell death activated by death receptors, toll‐like receptors, interferon, certain viruses, and bacteria 1. The serine/threonine protein kinase, RIPK3, integrates signals from upstream receptors and adapters, resulting in oligomerization and kinase activation 2, 3, 4, 5. RIPK3 phosphorylates its downstream target, the necroptosis effector mixed lineage kinase‐like (MLKL), driving MLKL oligomerization, membrane insertion, and necroptotic pore formation leading to cell lysis 6, 7, 8, 9, 10. In addition to its kinase activity, RIPK3 employs RIP homotypic interaction motif (RHIM) interactions to initiate necroptosis signaling 11. Three RHIM‐containing adapters, RIPK1, TRIF, and DAI/ZBP1/DLM‐1, are known to act upstream of RIPK3. Once activated, RIPK3 oligomerization through RHIM‐RHIM interactions 1, 3 drives recruitment and phosphorylation of the pseudokinase MLKL to execute necroptosis 6, 8, 12.
Necroptosis plays an important role in the pathology of a variety of human diseases including ischemia reperfusion injury, cancer, neurodegeneration, and infection 13. We have previously demonstrated that murine cytomegalovirus (MCMV) encodes a RHIM‐containing inhibitor of necroptosis, the viral Inhibitor of RIP Activation (vIRA) encoded by the MCMV M45 gene 14. A recombinant mutant MCMV (MCMV‐M45mutRHIM) fails to inhibit RHIM‐mediated signal transduction and necroptosis, and this virus is severely attenuated both in vitro and in vivo in a DAI/ZBP1‐RIPK3‐dependent fashion 14, 15. In addition to MCMV, a number of diverse viruses including human cytomegalovirus (HCMV) 16, herpes simplex virus (HSV)1 and 2 17, 18, 19, vaccinia virus (VV) 4, 20, 21, reovirus 22, and influenza A virus (IAV) 23, 24, 25 have been shown to either induce or inhibit necroptosis during infection. While these studies clearly highlight necroptosis as an important intrinsic defense against viral pathogens, specific questions remain as to the natural ligands or signals that initiate antiviral necroptosis and how species restrictions impact this pathway 26. However, studies with MCMV established necroptosis as a bona fide host defense mechanism to infection in a natural host, making this virus an ideal system to study this pathway.
DAI/ZBP1 was first identified in cancer cells as an interferon‐induced protein that bound Z‐form nucleic acids and was later implicated in cytosolic sensing of double‐stranded DNA 27. More recently, DAI/ZBP1 has been shown to play a critical role in necroptosis induced by MCMV and IAV 23, 24, 28 as well as death initiated by the disruption of RIPK1 or RIPK1 RHIM signal transduction during development and in lethal inflammation 29, 30. DAI/ZBP1 contains two Z‐DNA‐binding domains in its N‐terminus, termed Zα1 and Zα2, two RHIMs, RHIM‐A and RHIM‐B, and a poorly characterized C‐terminal region 31, 32. Previously, RHIM‐A was identified as a bona fide RHIM that is absolutely required to mediate necroptotic signaling upon MCMV and IAV infection 15, 24. It has been hypothesized that DAI/ZBP1 recognizes incoming cytosolic viral genomic DNA through its Z‐DNA‐binding domains 33, 34; however, the exact mechanism by which DAI/ZBP1 senses infection in response to MCMV infection remains unknown. Evidence from IAV infection, during which DAI/ZBP1 binds viral genomic RNAs through its Zα2 domain, further raises questions as to the nature of the nucleic acid ligand during MCMV infection 24, 28. In addition, the MCMV genome, like all herpesviruses, is replicated in the nucleus of infected cells, and during its transport from the plasma membrane to the nucleus, the viral genome is protected by the capsid, precluding the presence of viral genomic DNA in the cytosol 35. This is supported by previous findings that UV‐inactivated MCMV lacking M45 fails to elicit cell death in sensitive cells 36. Therefore, significant questions remain regarding where and how DAI/ZBP1 senses MCMV in order to elicit necroptosis.
Here, we sought to systematically address the above question by using the M45mutRHIM virus to study DAI/ZBP1‐induced necroptosis. We find that MCMV genomic DNA either during the early stage of infection or following viral replication does not activate DAI/ZBP1. In contrast, capsid transport to the nucleus and viral immediate‐early protein 3 (IE3)‐dependent transcription are required for necroptosis. Structure function analysis of DAI/ZBP1 also reveals that the Zα2 domain, in addition to RHIM‐A, is required for MCMV‐induced necroptosis, indicating that this previously defined RNA‐binding domain 24 is essential for detecting MCMV infection. Our data suggest that DAI serves as an RNA sensor during MCMV infection and further challenge its assumed function as a cytosolic DNA sensor.
Results
MCMV‐induced necroptosis is induced prior to DNA replication, but requires capsid transport to the nucleus
In order to identify the step(s) of the viral lifecycle that activate DAI/ZBP1, we initially evaluated the kinetics of MLKL phosphorylation, a requisite step for necroptosis induction. Levels of phosphorylated MLKL (p‐MLKL) are detected as early as 5–7 h post‐infection (h.p.i.) (Fig 1A; long exposure) and peak by 9 h.p.i. with M45mutRHIM MCMV (Fig 1A; short exposure) when necroptosis is evident in infected cells 14, 15, suggesting that necroptosis initiation occurs early in infection. As expected, wild‐type (WT) MCMV blocks necroptosis, and infection with this virus results in lower levels of MLKL phosphorylation (Fig 1A). We next assessed the role of viral replication in the initiation of MCMV‐induced necroptosis. Phosphonoformic acid (PFA, Foscarnet) is a potent inhibitor of CMV DNA‐dependent DNA polymerases 37. As anticipated, treatment of MCMV‐infected cells with PFA decreased viral yields by more than four orders of magnitude (Fig 1B); however, PFA treatment failed to inhibit MCMV‐induced necroptosis in either SVEC4‐10 endothelial cells (Fig 1C) or 3T3‐SA cells (Fig EV1), indicating that necroptotic initiation occurs independently of viral DNA replication. This is consistent with findings that MCMV‐M45mutRHIM induces necroptosis in human fibroblasts, where MCMV DNA replication does not occur 16, as well as previous observations that this pathway is not initiated by UV‐inactivated virus 36. PFA failed to inhibit TNF‐induced necroptosis (Fig 1D), as expected.
Figure 1. MCMV‐induced necroptosis occurs prior to viral DNA replication.
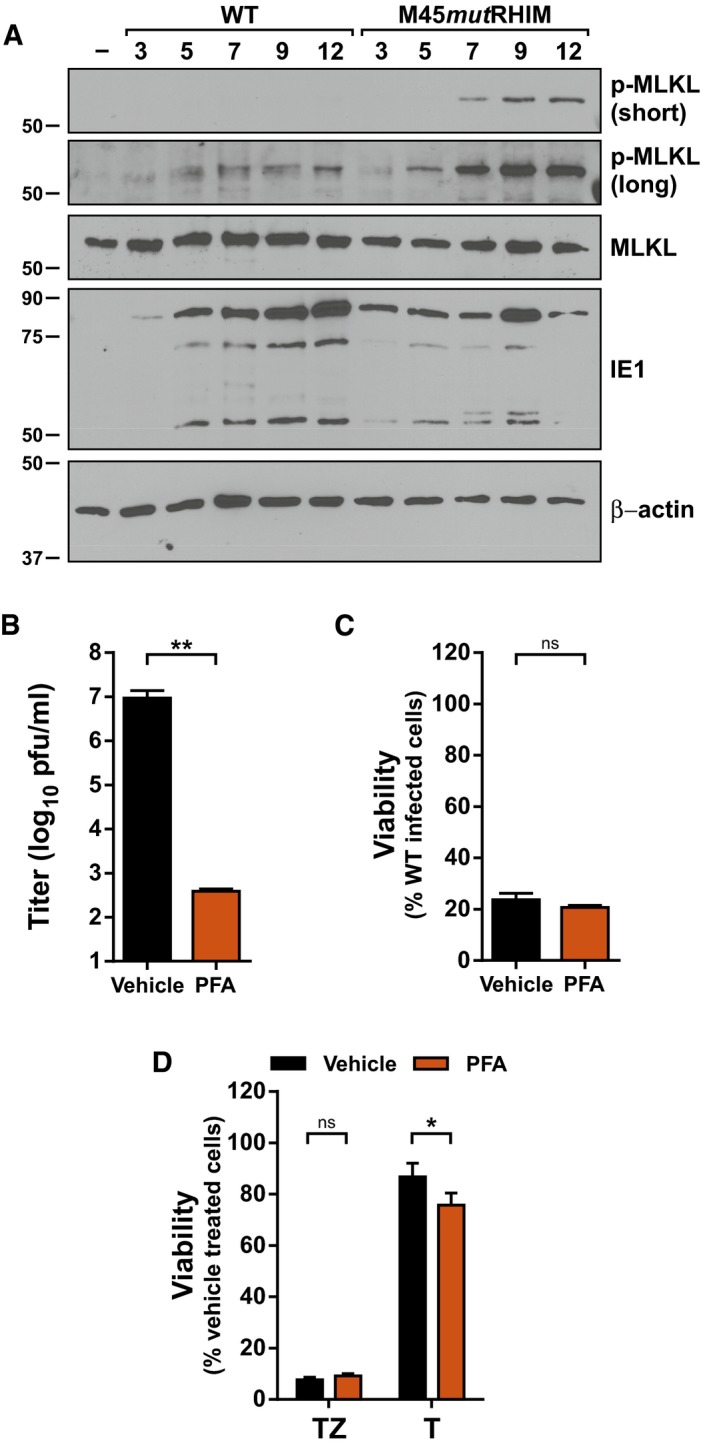
- Immunoblot (IB) analysis to detect p‐MLKL, total MLKL, IE1, and β‐actin from SVEC4‐10 cells infected with bacmid‐derived K181 (WT) or M45mutRHIM MCMV at a multiplicity of infection (MOI) of 5.
- Replication levels of WT MCMV in infected SVEC4‐10 cells 48 h post‐infection (h.p.i.) in the absence or presence of 200 μg/ml phosphonoformic acid (PFA; n = 3 biological replicates). Viral titers were determined by plaque assay.
- Relative viability of SVEC4‐10 cells infected with M45mutRHIM compared to WT MCMV (MOI = 5) in the presence or absence of 200 μg/ml PFA (n = 3 biological replicates).
- Relative viability of SVEC4‐10 cells treated with TNF (T) or TNF + ZVAD‐fmk (TZ) for 6 h in the presence or absence of 200 μg/ml PFA (n = 3 biological replicates).
Figure EV1. Murine cytomegalovirus DNA replication is not required for necroptosis in sensitive fibroblasts.
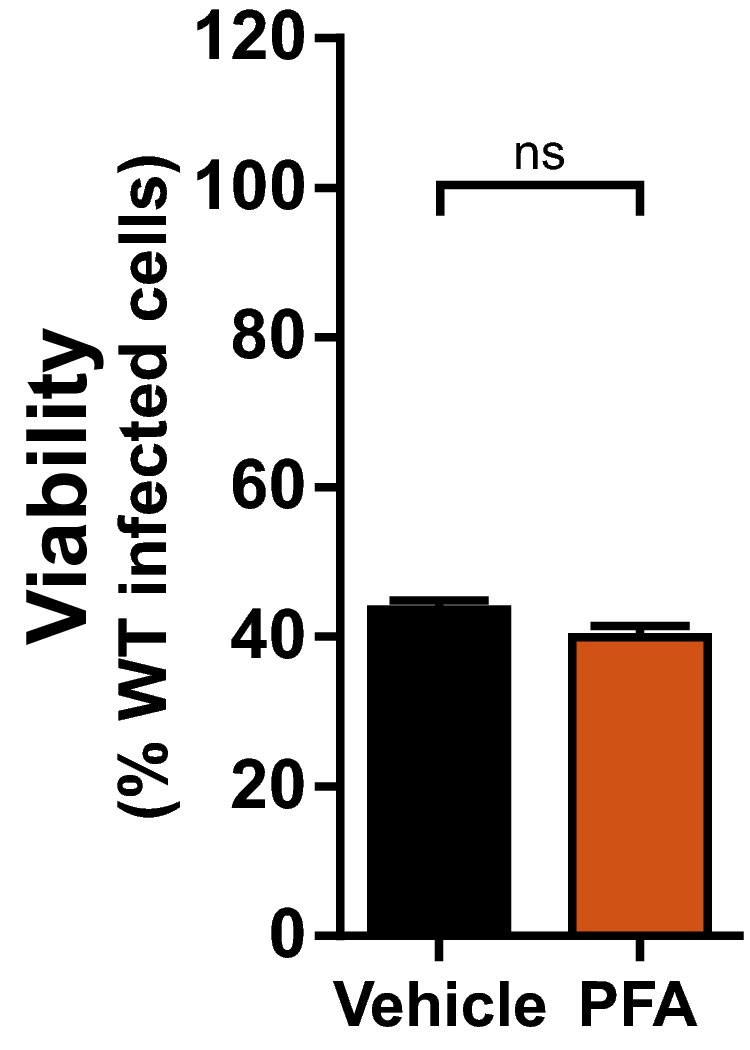
Relative viability of 3T3‐SA cells infected with M45mutRHIM compared to WT MCMV (MOI = 5) in the presence or absence of 200 μg/ml phosphonoformic acid (PFA). n.s., not significant (P > 0.05) by two‐tailed unpaired Student's t‐test. Error bars indicate SD (n = 3 biological replicates).
We next focused on the steps that precede DNA replication. Upon entry, herpesvirus capsids are transported to the cell nucleus via the microtubule network. Inhibition of microtubule polymerization with drugs like nocodazole prevents capsid transport, effectively trapping the virus in the cytoplasm 38. Treatment of infected cells with concentrations of nocodazole that blocked capsid transport [based on reduced expression of the immediate‐early protein 1 (IE1)] (Fig 2A) reversed cell death in M45mutRHIM‐infected cells (Fig 2B), commensurate with reduced MLKL phosphorylation (Fig 2C). Additionally, treatment of infected cells with Ciliobrevin‐D (CBD), a potent inhibitor of the AAA+ ATPase dynein motor, similarly rescued cells from MCMV‐induced necroptosis (Fig EV2A). Importantly, both drugs failed to inhibit TNF‐induced necroptosis (Figs 2D and EV2B), indicating the necroptotic signaling machinery is intact and that execution of TNF‐induced necroptosis does not require transport on the microtubule network. These data suggest that the initial stages of infection that occur before genome deposition in the nucleus, including receptor binding, entry, and uncoating, are insufficient to trigger DAI/ZBP1‐dependent necroptosis. Taken together, our results indicate that initiation of MCMV‐induced necroptosis occurs prior to viral DNA replication but requires trafficking of the capsid and genome to the nucleus of infected cells.
Figure 2. MCMV‐induced necroptosis requires microtubule‐based capsid transport to the nucleus, where DAI/ZBP1 accumulates in response to infection.
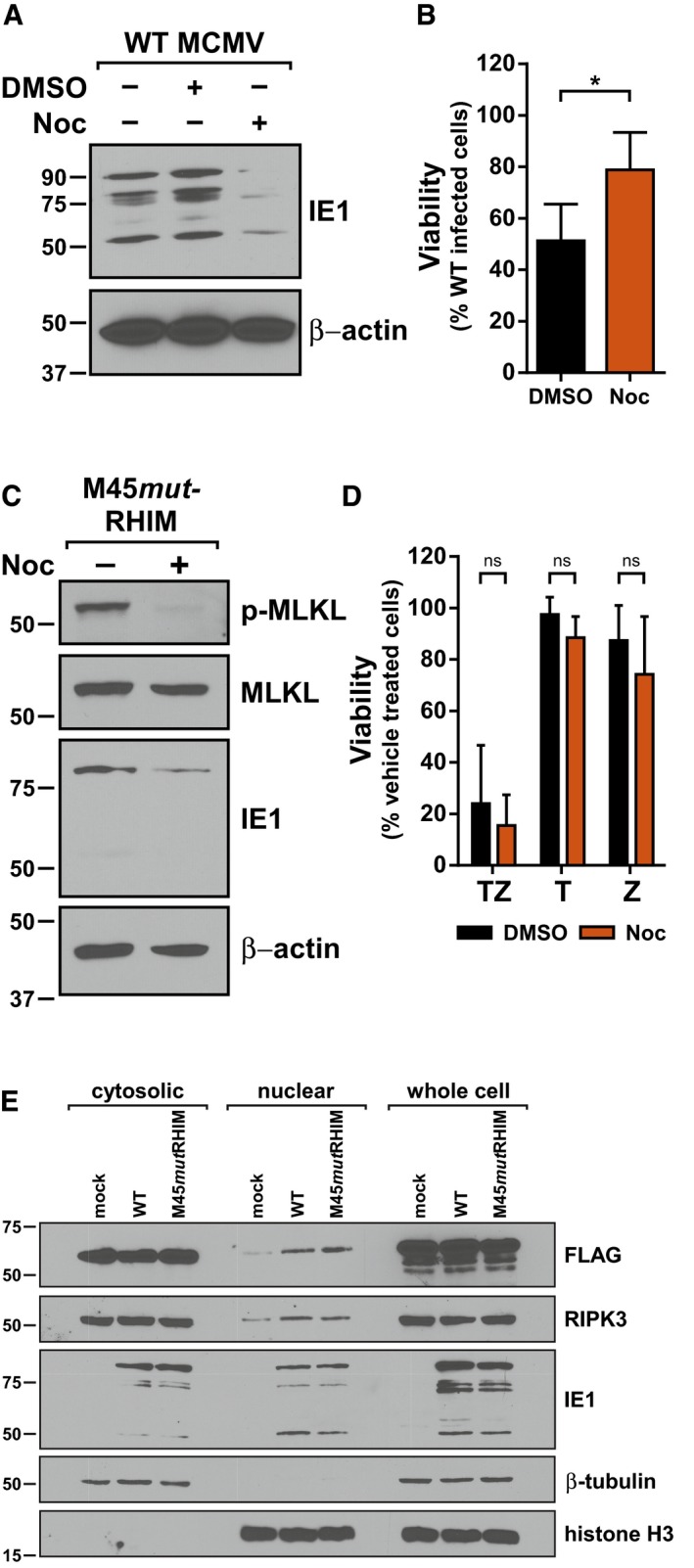
- IB analysis to detect IE1 and β‐actin from SVEC4‐10 cells infected with WT MCMV in the absence or presence of 1 μg/ml nocodazole (Noc).
- Relative viability of SVEC4‐10 cells infected with M45mutRHIM compared to WT MCMV (MOI = 5) in the presence or absence 1 μg/ml Noc (n = 4 biological replicates).
- IB analysis to detect p‐MLKL, total MLKL, IE1, and β‐actin from SVEC4‐10 cells infected with M45mutRHIM MCMV (MOI = 5) for 7 h in the presence or absence of 1 μg/ml Noc.
- Relative viability of SVEC4‐10 cells treated with TNF (T), zVAD (Z), or TNF + zVAD‐fmk (TZ) for 6 h in the presence or absence of 1 μg/ml Noc (n = 3 biological replicates).
- IB analysis to detect FLAG, RIPK3, and IE1 in subcellular fractions of 29‐11 cells stably reconstituted with FLAG‐epitope‐tagged WT DAI/ZBP1 and infected 7 h with WT or M45mutRHIM (MOI = 5). IB for β‐tubulin and histone H3 was used to assess quality of cytoplasmic and nuclear fractions, respectively.
Figure EV2. Ciliobrevin‐D treatment protects from MCMV‐induced necroptosis and characterization of DAI knockout 29‐11 cell lines.
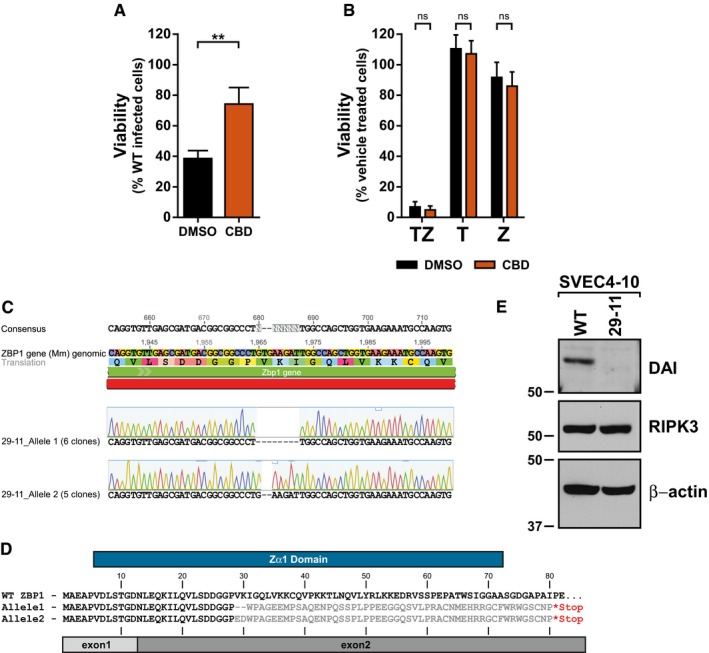
- Relative viability of SVEC4‐10 cells infected with M45mutRHIM compared to WT MCMV (MOI = 5) in the presence or absence of 100 μM Ciliobrevin‐D (CBD). **P < 0.01; two‐tailed unpaired Student's t‐test. Error bars indicate SD (n = 3 biological replicates).
- Relative viability of SVEC4‐10 cells treated with TNF (T), zVAD (Z), or TNF + zVAD‐fmk (TZ) for 6 h in the presence or absence of 100 μM CBD. n.s., not significant (P > 0.05) by two‐tailed unpaired Student's t‐test. Error bars indicate SD (n = 3 biological replicates).
- Representative results of sequencing and alignment of the DAI/ZBP1 exon 2 locus in 29‐11 cells. Amplicons generated from 29‐11 genomic DNA using ZBPSurveyor primers were cloned, and 11 independent clones sequenced.
- Amino acid alignment of protein products predicted from the sequencing results in (C). Gray bars denote the boundaries of exon 1 (light) and exon 2 (dark) of DAI/ZBP1, and blue bar represents the defined Zα1 domain.
- IB analysis to detect DAI/ZBP1, RIPK3, and β‐actin from SVEC‐10 parental and 29‐11 cell lines.
Although described as a cytosolic dsDNA sensor, DAI/ZBP1 has been shown to shuttle between the cytoplasm and nucleus 39, 40. Given the apparent requirement for delivery of the MCMV genome to the nucleus to induce necroptosis, we next sought to determine whether DAI/ZBP1 localization was influenced by MCMV infection. A knockout SVEC4‐10 cell line with a mutated genomic DAI/ZBP1 locus was generated using CRISPR‐CAS9 and confirmed for loss of endogenous DAI/ZBP1 expression (Fig EV2C–E). This cell line, designated 29‐11, was then reconstituted with epitope‐tagged WT DAI/ZBP1. Subcellular fractionation and immunoblotting of cells infected for 7 h with either WT or M45mutRHIM MCMV revealed that levels of DAI/ZBP1 in the nucleus increase following infection with either virus (Fig 2E). Thus, DAI accumulates in the nucleus at times post‐infection coincidently with the initiation of MCMV‐induced necroptosis. Nuclear accumulation of DAI/ZBP1 is observed in both WT and mutant virus infection, suggesting accumulation is a generalized host response to infection. Interestingly, RIPK3 also accumulates in the nuclear fraction of infected cells (Fig 2E). While the specific role of nuclear DAI and RIPK3 during MCMV infection remains to be determined, these results indicate that these two critical effectors, DAI/ZBP1 and RIPK3, accumulate in the nucleus of MCMV‐infected cells concurrently with the induction of necroptosis.
Early inhibition of gene transcription protects cells from MCMV‐induced necroptosis
We next sought to determine the events following genome deposition in the nucleus and prior to DNA replication that were involved in MCMV‐induced necroptosis. Herpesvirus lytic replication is characterized by a highly ordered and temporally controlled transcriptional cascade that can be generally divided into immediate‐early (IE), early (E), and late (L) phases 35. To investigate the role of transcription in MCMV‐induced necroptosis, infected cells were treated with α‐amanitin, a potent inhibitor of RNA polymerase II, in a time‐of‐addition experiment where α‐amanitin was added at specific times post‐infection (Fig 3A). Addition of the drug at the time of infection protected M45mutRHIM‐infected cells from necroptotic death (Fig 3B), indicating that nascent transcription is required for the initiation of MCMV‐induced necroptosis. Addition of α‐amanitin 2 h.p.i. was still sufficient to inhibit virus‐induced death. However, addition of α‐amanitin at 4 h.p.i. or later resulted in decreasing protection from necroptosis (Fig 3B). As expected, p‐MLKL was detected by 7 h.p.i. with M45mutRHIM, but not with WT virus (Fig 3C). Importantly, treatment with α‐amanitin eliminated this phosphorylation (Fig 3C), consistent with protection from necroptosis. Protein levels of DAI/ZBP1, RIPK3, and MLKL were not significantly affected by treatment or infection through 8 h.p.i. (Fig 3E), indicating that neither α‐amanitin treatment nor MCMV infection significantly alters levels of the major cellular effectors of MCMV‐induced necroptosis. Expression of a representative IE gene, IE1, was severely diminished when α‐amanitin was added at the time of infection. However, when added 2 h.p.i., a condition which still protected cells from death, IE1 levels were comparable to untreated infected cells. Levels of a representative early gene, E1, were severely diminished when α‐amanitin was added prior to 4 h.p.i., and increased in expression when α‐amanitin was added later during infection. Thus, as protection decreased with the time of α‐amanitin treatment, E1 expression increased, suggesting that a part of the viral life cycle associated with E gene expression is critical for virus‐induced necroptosis induction. As an important control, α‐amanitin did not have an inhibitory effect on TNF‐induced death (Fig 3D), indicating that nascent transcription is not essential for this pathway, further highlighting the difference in signal initiation between these arms of necroptosis. Thus, inhibition of nascent transcription at times immediately following infection protects cells from MCMV‐induced necroptosis. Although this protection closely correlates with inhibition of viral E gene expression, viral and/or cellular transcription may be involved in triggering necroptosis during MCMV infection.
Figure 3. MCMV‐induced necroptosis requires nascent transcription.
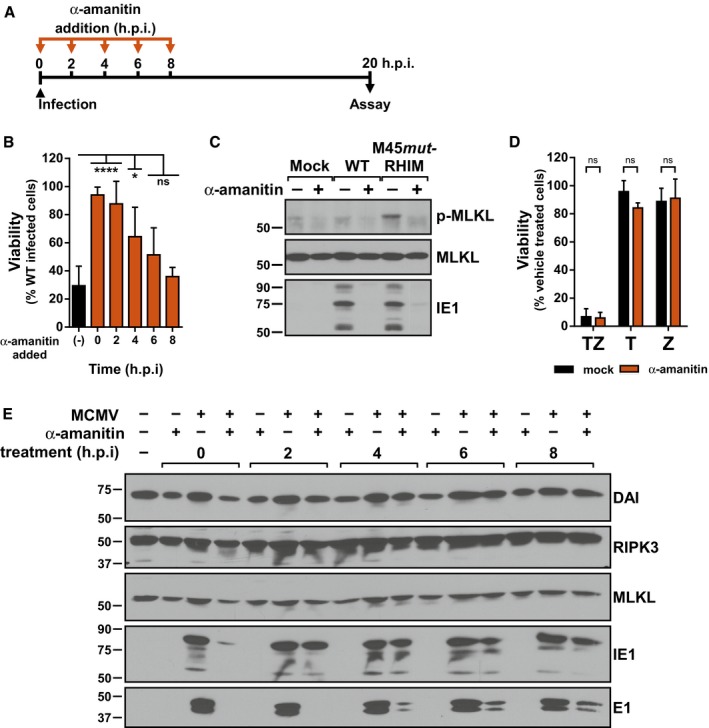
- Schematic representation of timed α‐amanitin experiment.
- Relative viability of SVEC4‐10 cells infected with M45mutRHIM compared to WT MCMV (MOI = 5) in the presence or absence of 50 μg/ml α‐amanitin according to the scheme in (A). ****P < 0.0001; *P < 0.05; n.s., not significant (P > 0.05) by one‐way ANOVA with Dunnett's multiple comparisons test, compared to untreated. Error bars indicate SD (n = 3 biological replicates).
- IB analysis to detect p‐MLKL, total MLKL, and IE1 from SVEC4‐10 cells infected 7 h with WT or M45mutRHIM MCMV (MOI = 5).
- Relative viability of SVEC4‐10 cells treated with TNF (T), zVAD (Z), or TNF + zVAD‐fmk (TZ) for 6 h in the presence or absence of 50 μg/ml α‐amanitin. n.s., not significant (P > 0.05) by two‐tailed unpaired Student's t‐test. Error bars indicate SD (n = 3 independent biological replicates).
- IB analysis to detect DAI/ZBP1, RIPK3, MLKL, IE1, and E1 from SVEC4‐10 cells infected with WT MCMV (MOI = 5) according to the scheme in (A).
Source data are available online for this figure.
The viral transcriptional activator, IE3, is required for necroptosis
To directly address the role of viral transcription in eliciting MCMV‐induced necroptosis, we targeted the major immediate‐early viral transactivator, IE3. IE3 is an essential gene required for early and late gene transcription that is alternately spliced from the major immediate‐early transcript, which also encodes IE1 41, 42, 43. Recombinant WT and M45mutRHIM MCMV viruses were generated from bacterial artificial chromosomes (BACs) in which the IE3 protein was regulated by fusion of the FKBP destabilization domain (DD) to its C‐terminus (Fig 4A). DD‐fusion proteins are inherently unstable and are rapidly degraded by the proteasome; however, in the presence of a chemical ligand, Shield‐1, the fusion proteins, are stabilized (Fig 4B) 44. The BACs were analyzed by RFLP and PCR/restriction analysis to ensure intact genomic features and the presence of the intended modifications (Fig EV3A–C). Immunoblot analysis of NIH3T3 cells infected with parental or IE3DD viruses revealed that the FKBP domain is expressed as a fusion with IE3 in cells and is not detected in parental WT or M45mutRHIM viruses (Fig EV3D). Interestingly, IE3DD fusion proteins can be detected in cells infected in the presence or absence of Shield‐1, suggesting that the robust expression of MCMV IE1/3 mediated by the major immediate‐early promoter (MIEP) may be sufficient for accumulation of IE3 in this system. Alternatively, MCMV IE3 downregulates the activity of MIEP similar to HCMV IE2 45, 46, and its functional deregulation may lead to aberrant IE protein expression. Importantly, in the absence of Shield‐1, E1 protein expression is not detected, consistent with the essential role of IE3 in promoting E1 expression 43 and suggesting that the DD fusion prevents IE3 from performing its transcriptional function (Fig EV3D). When analyzed for growth in NIH3T3 cells, both WT‐IE3DD and M45mutRHIM‐IE3DD were severely attenuated in the absence of Shield‐1 but were rescued in the presence of ligand (Fig 4C). Thus, tight control of IE3 function by the DD fusion makes these viruses important tools to study the role of IE3 in MCMV biology. We next assessed the ability of these recombinant viruses to induce necroptosis in the presence or absence of Shield‐1. Infection of SVEC4‐10 cells with WT‐IE3DD or M45mutRHIM‐IE3DD in the presence of Shield‐1 behaved as anticipated: M45mutRHIM‐IE3DD virus induced significant cell death as measured by Sytox Green incorporation, while WT‐IE3DD virus did not (Fig 4D). However, infection of cells with M45mutRHIM‐IE3DD in the absence of Shield‐1 resulted in a dramatic decrease in the levels of cell death, similar to WT‐IE3DD infection (Fig 4D), indicating that IE3 is critical to MCMV‐induced necroptosis. Importantly, death begins to become apparent by 9 h.p.i., concomitant with the detection of necroptotic biochemical markers (Fig 1A). Comparable results were obtained when measured by endpoint ATP‐based viability assays (Fig 4E), as well as infections using necroptosis‐sensitive 3T3‐SA fibroblasts (Fig EV3E). As expected, neither the growth of the parental WT and M45mutRHIM viruses (Fig EV3F) nor the viability of M45mutRHIM infected cells (Fig EV3G) are influenced by the addition of the stabilizing ligand, Shield‐1. Thus, we have resolved a long‐standing question regarding the biological requirements to elicit MCMV‐induced necroptosis. These results show an absolute requirement for either the IE3 protein itself or its function in controlling MCMV early transcription in M45mutRHIM‐induced DAI/ZBP1‐RIP3‐dependent necroptosis.
Figure 4. MCMV‐induced necroptosis requires IE3 function.
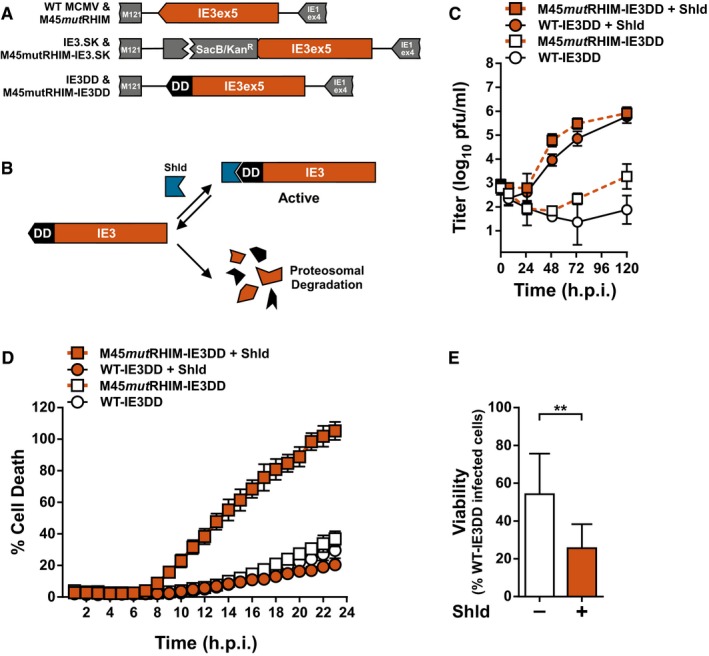
- Schematic representation of generation of recombinant WT‐IE3DD and M45mutRHIM IE3DD bacmids.
- Schematic representation of destabilizing domain (DD) function. An IE3‐DD fusion is stabilized and functional in the presence of Shield‐1 (Shld). DD fusions are rapidly degraded in the absence of ligand.
- Multistep replication levels of WT‐ and M45mutRHIM‐IE3DD viruses (MOI = 0.05) in NIH3T3 cells in the presence or absence of 1 μM Shld. Error bars indicate SD (n = 3 biological replicates).
- Kinetics of cell death of SVEC4‐10 cells infected with WT‐ or M45mutRHIM‐IE3DD viruses (MOI = 5) in the presence or absence of 1 μM Shld, measured in real time by Sytox green incorporation. Error bars indicate SD (n = 4 biological replicates).
- Relative viability of SVEC4‐10 cells infected with M45mutRHIM‐IE3DD compared to WT‐IE3DD (MOI = 5) in the presence or absence of 1 μM Shld. **P < 0.01; n.s., not significant (P > 0.05) by two‐tailed unpaired Student's t‐test. Error bars indicate SD (n = 3 biological replicates).
Figure EV3. Generation and characterization of IE3‐DD viruses.
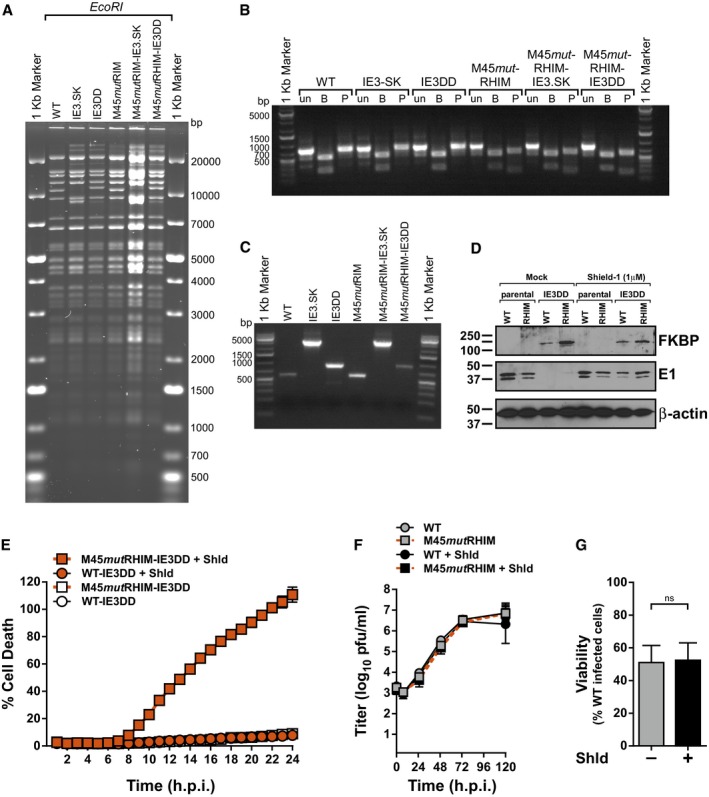
- RFLP analysis of parental WT, IE3.SK, IE3DD, parental M45mutRHIM, M45mutRHIM –IE3.SK, and M45mutRHIM‐IE3DD bacmids digested with EcoRI.
- Agarose gel analysis of PCR amplicons generated from bacmids described in (A) with primers HS36 and HS37. Amplicons were digested with the indicated restriction endonuclease to diagnose the M45mutRHIM locus. un, undigested; B, BamHI; P, PvuII.
- Agarose gel analysis of PCR amplicons generated from bacmids described in (A) with primers HS09 and HS10 to diagnose insertions at the end of IE3 exon 5.
- IB analysis to detect the DD‐domain (FKBP), E1, and β‐actin from NIH3T3 cells infected 13 h with parental or IE3DD WT or M45mutRHIM (RHIM) MCMV (MOI = 1) in the presence or absence of 1 μM Shield‐1.
- Kinetics of cell death of 3T3‐SA cells infected with WT or M45mutRHIM (MOI = 5) in the presence or absence of 1 μM Shield‐1 (Shld), measured in real time by Sytox green incorporation. Error bars indicate SD (n = 4 biological replicates).
- Multistep replication levels of parental WT and M45mutRHIM viruses (MOI = 0.05) in NIH3T3 cells in the presence or absence of 1 μM Shld. Error bars indicate SD (n = 3 biological replicates).
- Relative viability of SVEC4‐10 cells infected with M45mutRHIM compared to WT MCMV in the absence or presence of 1 μM Shld. n.s., not significant (P > 0.05) by two‐tailed unpaired Student's t‐test. Error bars indicate SD (n = 3 biological replicates).
Zα2 and RHIM‐A domains are crucial for DAI/ZBP1 activation by virus infection
In order to delineate how DAI/ZBP1 mediates MCMV‐induced necroptosis, its nucleic acid binding and RHIM functions were disrupted by either truncations or specific point mutations (Fig 5A). The 29‐11 DAI/ZBP1‐null cell line (Fig EV2C–E) was then stably reconstituted with the epitope‐tagged mutant DAI/ZBP1 alleles by retroviral transduction, and expression was confirmed by immunoblot (Fig 5B). Reconstituted cell lines were assessed for MCMV‐induced necroptosis by endpoint ATP viability assays (Fig 5C) as well as SYTOX green incorporation (Fig 5D–F). Wild‐type DAI/ZBP1‐reconstituted cells were susceptible to death, while empty vector reconstituted cells remained insensitive (Fig 5C and D), thus validating the system to interrogate the role of DAI/ZBP1 in this pathway. Cells reconstituted with DAI/ZBP1 containing a mutation in RHIM‐A (mRHIMA) remained insensitive to necroptosis as expected (Fig 5C and F). However, those reconstituted with DAI/ZBP1 containing a mutation in RHIM‐B (mRHIMB) or deletion of the C‐terminus (ΔC) continued to undergo necroptosis (Fig 5C and F), confirming that neither of these domains play an important role in necroptotic signaling upon MCMV infection. While deletion of Zα1 (ΔZα1) did not affect DAI/ZBP1's ability to induce necroptosis, additional deletion of Zα2 (ΔZα1α2) completely ameliorated sensitivity to necroptosis, indicating that the Zα2 domain of DAI/ZBP1 plays a critical role in MCMV‐induced, DAI/ZBP1‐dependent necroptotic signaling (Fig 5C and D). Importantly, the Zα1α2 double deletion does not interfere with DAI/ZBP1 binding to RIPK3, indicating the deletion does not significantly disrupt other functional domains or protein stability (Fig EV4A). Specific mutation of residues previously shown to mediate Zα1 and Zα2 (mutZα2, mutZα1α2) binding to nucleic acids 39 provided minimal protection from DAI/ZBP1's ability to cause necroptosis (Fig 5C and E). However, DAI/ZBP1 mutZα2‐expressing cells died with delayed kinetics compared to cells containing WT, ΔZα1, or ΔC DAI/ZBP1 (Fig EV4B). Death of mutZα2‐expressing cells begins by 10 h post‐infection, between 2 and 4 h after other mutants have initiated the process. Thus, while mutation of the nucleic acid‐binding residues of Zα2 fails to completely block MCMV‐induced necroptosis, it appears that nucleic acid binding by DAI/ZBP1 remains an important facet of DAI function. Together, these results suggest that while nucleic acid‐binding amino acids of Zα2 contribute to DAI/ZBP1's ability to sense MCMV infection, additional features of the Zα2 domain may play important roles.
Figure 5. MCMV‐induced necroptosis requires DAI/ZBP1's RNA‐binding domain.
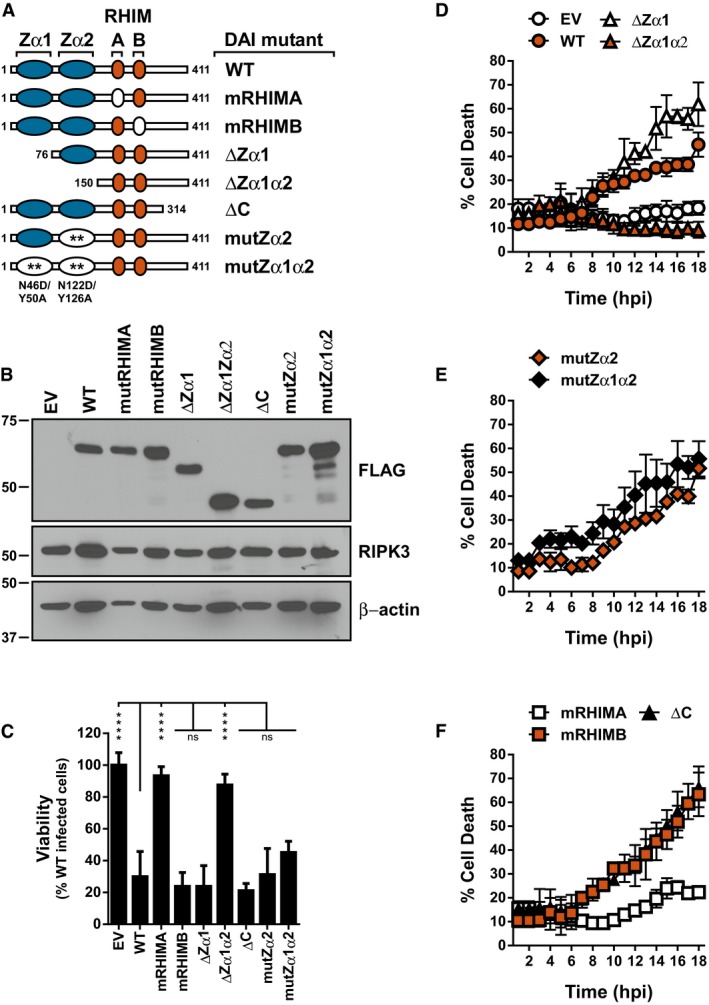
-
ASchematic representation of the DAI/ZBP1 truncations and mutants. Blue ovals depict Z‐DNA‐binding domains, orange ovals represent RHIMS, and white ovals represent mutations. Numbers indicate amino acid positions.
-
BIB analysis to detect FLAG‐DAI/ZBP1, RIPK3, and β‐actin from 29‐11 cell line reconstituted by retroviral transduction with indicated DAI/ZBP1 mutant.
-
CRelative viability of 29‐11 cell line reconstituted with indicated DAI/ZBP1 constructs infected with M45mutRHIM compared to WT MCMV (MOI = 5). ****P < 0.0001; n.s., not significant (P > 0.05) by one‐way ANOVA with Dunnett's multiple comparisons test, compared to WT. Error bars indicate standard deviation from the mean (SD; n = 3 biological replicates).
-
D–FKinetics of cell death of indicated DAI/ZBP1‐reconstituted 29‐11 cell lines infected with M45mutRHIM MCMV (MOI = 5), measured in real time by Sytox green incorporation. Error bars indicate SD (n = 4 biological replicates).
Source data are available online for this figure.
Figure EV4. Mutation of DAI's RNA binding domain delays necroptosis, and human DAI can mediate necroptosis.
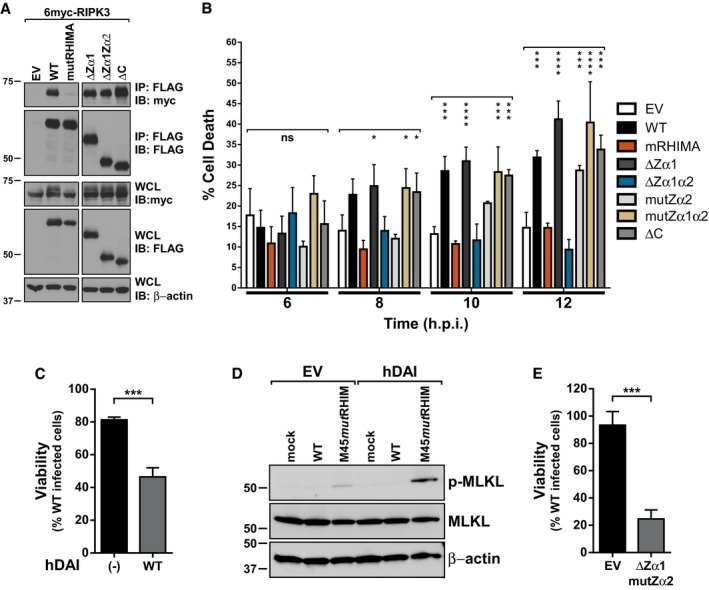
- Immunoprecipitation (IP) and IB analysis to detect FLAG‐DAI/ZBP1, myc‐RIPK3, and β‐actin from 293T cells transfected with vectors expressing 6myc‐RIPK3 and different 3XFLAG‐DAI/ZBP1 constructs, and subjected to IP with anti‐FLAG M2 agarose beads.
- Cell death of DAI/ZBP1‐reconstituted 29‐11 cell lines infected with M45mutRHIM MCMV (MOI = 5); 6, 8, 10, and 12 h post‐infection times were extracted from Fig 5D–F and replotted for clarity. ****P < 0.0001; ***P < 0.001; *P < 0.05; n.s., not significant (P > 0.05) by one‐way ANOVA with Dunnett's multiple comparisons test, compared to EV. Error bars indicate standard deviation from the mean (SD; n = 4 biological replicates).
- Relative viability of HT‐29 cells with or without hDAI expression infected with M45mutRHIM compared to WT MCMV. ***P < 0.001 by two‐tailed unpaired Student's t‐test. Error bars indicate SD (n = 3 biological replicates).
- IB analysis to detect p‐MLKL, total MLKL, and β‐actin from HT‐29 cells with or without hDAI expression infected with WT or M45mutRHIM for 10 h.
- Relative viability of 29‐11 cells reconstituted with empty vector (EV) or DAI‐ΔZα1mutZα2 infected with M45mutRHIM compared to WT MCMV. ***P < 0.001 by two‐tailed unpaired Student's t‐test. Error bars indicate SD (n = 3 biological replicates).
Differential requirements for DAI/ZBP1 in antiviral responses in human and murine cells have been reported 47, and little evidence thus far implicates DAI/ZBP1 as a mediator of necroptosis in human cells. MCMV is able to infect some human cell types, entering and progressing to gene expression but fails to productively replicate 48, 49. We have previously shown that M45mutRHIM induces necroptosis in RIPK3‐overexpressing human fibroblasts 16, but the role of DAI/ZBP1 was not assessed. To address whether DAI/ZBP1 can mediate this pathway in human cells, necroptosis‐sensitive HT‐29 cells were stably transduced with lentiviruses expressing human DAI/ZBP1 or a vector control. Infection of hDAI‐expressing HT‐29 cells with M45mutRHIM resulted in significantly enhanced levels of cell death compared to controls, and death correlated with elevated levels of p‐MLKL (Fig EV4C and D). Together, these data show that DAI/ZBP1 mediates antiviral RIPK3‐dependent necroptosis in response to infection in human cells, and indicate this pathway is conserved from mice to humans.
Discussion
Murine cytomegalovirus is a powerful tool to study virally induced, DAI/ZBP1‐dependent necroptosis in a natural setting. However, to date, the only proteins known to play a role in this pathway have been DAI/ZBP1, RIPK3, and MLKL. Here, we identify the viral IE3 protein as another important player in this pathway. We demonstrate a critical role for viral IE3‐dependent gene product(s) and their recognition by DAI/ZBP1's Zα2 domain in MCMV‐induced necroptosis. Simultaneously, these results add to a growing body of evidence that redefines the Zα2 domain of DAI/ZBP1 as an RNA‐binding domain during viral infection and challenge existing dogma that DAI/ZBP1 functions as a cytosolic DNA sensor by recognizing incoming viral genomes.
Using the M45mutRHIM recombinant virus, we demonstrate that microtubule‐based transport of MCMV early in infection is necessary for DAI/ZBP1 activation and virus‐induced necroptosis. Inhibition of these pathways does not influence TNF‐mediated necroptosis, confirming that MCMV‐ and TNF‐induced necroptosis are mediated by different pathways upstream of RIPK3. Although herpesvirus DNA has been detected in the cytoplasm of infected cells independent of capsid 50, our data suggest that trapping incoming virions in the cytoplasm prevents sensing and necroptotic signaling by DAI/ZBP1, simultaneously identifying the nucleus as a critical compartment needed for signal initiation. Consistent with this, we find both DAI/ZBP1 and RIPK3 localize to the nucleus upon MCMV infection. Herpesvirus DNA replication occurs in the nucleus and is followed by encapsidation and egress. However, our finding that chemical inhibition of viral DNA replication fails to inhibit DAI/ZBP1‐dependent death upon infection excludes a role for both nascent viral DNA synthesis and subsequent viral particle generation as the source for DAI/ZBP1 activation. Interestingly, we do detect a low level of p‐MLKL during early times of WT MCMV infection (Fig 1A: long exposure). Reports indicate that dynamic phosphorylation of MLKL both within and outside the activation loop “tunes” necroptosis signaling to execute death 51, suggesting that a threshold of MLKL phosphorylation must be achieved to drive death. Our results show that M45mutRHIM virus drives early and robust MLKL phosphorylation, while WT MCMV infection results in a lower, sustained level, consistent with prior reports of basal MLKL phosphorylation during HCMV infection 16. While this phenomenon currently remains unexplained, it is clear that the pro‐necroptotic phosphorylation of MLKL by M45mutRHIM occurs very early in infection and significantly precedes virus replication. Thus, events during MCMV infection prior to genome replication but after genome deposition in the nucleus must serve as the trigger for necroptotic signaling.
One critical event that occurs within this period is the transcription, translation, and function of MCMV IE and E genes. Surprisingly, we find that chemical inhibition of transcription at very early, but not later, times of infection renders M45mutRHIM‐infected cells resistant to necroptosis. Since the protein levels of the major cellular effectors of necroptosis remain relatively unchanged during the course of these experiments, and TNF‐induced necroptosis remains intact, it is likely that a viral transcriptional product serves as the DAI/ZBP1‐activating ligand. When we extended this analysis to specifically address the role of viral transcription, we find that MCMV IE3 is critical for the induction of DAI/ZBP1‐dependent necroptosis. IE3 is the major MCMV viral transactivating protein that controls the transcriptional cascade leading to productive infection and is therefore essential for MCMV replication 41, 52. Generation of recombinant WT and M45mutRHIM MCMVs in which IE3 protein stability is selectively controlled without affecting the other major IE transcript, IE1, affords a unique and powerful way to directly interrogate the role(s) of IE3 during infection. We find that in the absence of the stabilizing drug, Shield‐1, M45mutRHIM‐IE3‐DD no longer induces necroptosis, even though under conditions of IE3 depletion, vIRA is no longer expressed 42. This result implicates IE3 function in activating DAI/ZBP1‐dependent necroptosis and suggests a viral RNA as a ligand for DAI/ZBP1 during MCMV infection. However, the exact nature of this viral RNA remains unclear. MCMV has been shown to produce dsRNA during infection 53, which could serve as a potential DAI/ZBP1 ligand 54. While the Zα1 and Zα2 domains of DAI/ZBP1 have been shown to bind synthetic DNA, Zα2 also binds viral RNA during IAV infection 24, establishing it as a bona fide RNA‐binding domain. Moreover, other Z‐DNA‐binding domain containing proteins, such as ADAR1, also bind Z‐form dsRNA through their Zα domain 54, 55 presumably due to the similar structures adopted by Z‐form DNA and Z‐form RNA. It is also formally possible that DAI/ZBP1 recognizes sites of IE3‐dependent transcription, cellular, or viral, that are associated with the formation of Z‐form structures 56. Although the identity of the specific ligand remains undefined, we conclusively demonstrate a critical role for IE3‐dependent transcription in eliciting MCMV‐induced necroptosis during infection.
Recently, we and others 24, 28 have demonstrated a role for DAI/ZBP1 in necroptosis caused by infection with IAV, a negative‐sense RNA virus with no DNA step in its life cycle. In the case of IAV, DAI/ZBP1 binds viral RNA through its Zα2 domain, and mutation of this domain in DAI/ZBP1 renders IAV‐infected cells resistant to necroptotic death 24. Similarly to IAV, deletion of the Zα2 domain in DAI/ZBP1 abrogates its ability to promote MCMV‐induced necroptosis. However, unlike IAV, point mutations of residues within the Zα2 domain that have previously been shown to bind nucleic acid fail to significantly protect from necroptosis, suggesting additional features of the Zα2 domain may contribute to nucleic acid binding and are important for initiating necroptosis during MCMV infection. Our data also suggest that the Zα1 and Zα2 domains of DAI are likely not functionally redundant, since combined mutation of nucleic acid‐binding residues in both Zα1 and Zα2 minimally influences the level of MCMV‐induced necroptosis in reconstituted cells compared to WT infection (Fig 5C–E). It would be expected that, if redundant, the combined Zα1α2 point mutant DAI would show a phenotype similar to ΔZα1α2 and is supported by observations that a DAI mutant lacking Zα1 in combination with a mutant Zα2 continues to mediate viral necroptosis (Fig EV4E). These results argue for a similar, yet distinct mechanism of DAI/ZBP1 activation during MCMV infection as compared to that of IAV. It will be important to evaluate the role of DAI/ZBP1, viral RNAs and transcription in other scenarios of necroptosis induced by DNA viruses to gain a better understanding of the mechanisms of this pathway. Similar to MCMV, HSV‐1/2 17, 18, 19 induce necroptosis when lacking their virally encoded, RHIM‐containing necroptosis inhibitors, suggesting that HSV infection will likely provide important parallels with MCMV infection. Murine gammaherpesvirus‐68 and vaccinia virus are additional dsDNA viruses linked to necroptosis 4, 20, 21, 57, and future work will determine whether these viruses utilize similar or distinct mechanisms to elicit and inhibit necroptosis in infected cells.
DAI/ZBP1 and RIPK3 have been shown to shuttle between the nucleus and cytosol 39, 40, 58, 59, and in response to MCMV, both show enhanced accumulation in the nuclear fraction of infected cells by 7 h post‐infection (Fig 2E). Whether this defines the subcellular location of sensing and initiation of necroptosis during MCMV infection remains to be determined. Previous reports have demonstrated that DAI/ZBP1 mutants lacking Zα1α2 fail to shuttle between the nucleus and cytosol, while mutants lacking Zα1 or the C‐terminus retain this ability 40. This correlates with our findings that ΔZα1α2 fails to sensitize cells to M45mutRHIM infection while ΔZα1 and ΔC DAI appear to enhance necroptosis (Fig 5D–F). Engagement of the necroptotic signaling pathway by TNF results in the nuclear accumulation of RIPK3, RIPK1, and MLKL to potentially facilitate necroptosis 59. However, the accumulation of DAI/ZBP1 and RIPK3 in the nucleus occurs in response to both WT and mutant MCMV (Fig 2E) which may be indicative of a specific host response to infection. Furthermore, it is noteworthy that IAV, like herpesviruses, replicates in the nucleus of infected cells. Other RNA viruses that replicate in the cytoplasm do not activate DAI/ZBP1 23, suggesting a previously unappreciated nuclear role for DAI/ZBP1‐dependent necroptosis. Determining whether nuclear accumulation of DAI/ZBP1 and RIPK3 is seen in response to IAV infection as well as other RNA viruses should provide important mechanistic insight into the role of localization in virus‐induced necroptosis.
While our results point toward a RNA ligand for DAI/ZBP1 during MCMV infection, it remains possible that DAI/ZBP1 is antagonized by a viral gene product—DAI/ZBP1 could bind the IE3 protein itself or an early viral protein product of IE3‐dependent transcription. Precedence for this has been noted in the context of another herpesvirus HSV‐1, where ICP0, the major HSV‐1 IE transactivator and homolog of IE3, interacts with DAI/ZBP1 in infected cells via a region encompassing both the Zα2 and RHIM‐A 60. However, our data show that WT‐ or M45mutRHIM‐IE3DD viruses fail to express early genes in the absence of Shield‐1, despite detection of IE3 protein (Fig EV3D). This corresponds with the failure of M45mutRHIM‐IE3DD virus to induce necroptosis in the absence of Shield‐1 (Figs 4D and E, and EV3E), suggesting that IE3 itself is unlikely to directly elicit DAI/ZBP1‐dependent necroptosis.
Recently, two groups have reported a novel mechanism of DAI/ZBP1 activation 29, 30. Using RIPK1RHIM/RHIM mutant mice, these authors show that RIPK1 suppresses activation of DAI/ZBP1 during development and inflammation. Mutation of RIPK1's RHIM is able to relieve this suppression leading to DAI/ZBP1‐dependent activation of RIPK3. These studies indicate DAI/ZBP1 does not need a specific ligand, but rather is able to activate RIPK3 when RIPK1's RHIM is inhibited or mutated; however, RIPK1 knockdown or inhibition does not protect from MCMV‐induced necroptosis 14, though it remains to be seen whether RIPK1 plays a negative inhibitory role in suppressing DAI/ZBP1 during MCMV infection. Although vIRA inhibits RIPK1 signaling in a RHIM‐independent manner 61, it is tempting to speculate that MCMV may encode an additional IE3‐dependent product capable of inhibiting RIPK1.
In summary, this work redefines the role of DAI/ZBP1 as a sensor during MCMV infection. The essential role of IE3 in eliciting DAI/ZBP1‐dependent necroptosis suggests that a transcriptional product, not the DNA genome, serves as the activating ligand for DAI/ZBP1 during infection. This closely parallels the mechanism of DAI/ZBP1 in inducing necroptosis during IAV infection 24 and contributes to the accumulating evidence that DAI/ZBP1 is an RNA sensor for specific, diverse viruses.
Materials and Methods
Cells and reagents
SVEC4‐10 (ATCC CRL‐2181), NIH3T3 (ATCC CRL‐1658), 3T3‐SA (ATCC CCL92), and HT‐29 (ATCC HTB‐38) cell lines were obtained from ATCC, and maintained as previously described 14. All cell lines were confirmed and routinely monitored to be mycoplasma free (LookOut Mycoplasma PCR Detection Kit (MP0035); Sigma‐Aldrich). Cycloheximide (Sigma), zVAD‐fmk (Enzo life sciences), and murine TNF (PeproTech) were prepared and used as described previously 14. Nocodazole (Sigma), Ciliobrevin‐D (Calbiochem), Shield‐1 (Clontech), PFA (Sigma), and α‐amanitin (Sigma) were used at the concentrations indicated.
Plasmids, transfections, immunoblotting, and antibodies
6‐myc RIPK3 was previously described 31. Transfections and immunoprecipitations were performed as previously described 31. Immunoblotting was performed as previously described 14. The following antibodies were used: rabbit anti‐RIPK3 (IMG‐5523; Imgenex), rabbit anti‐DAI/ZBP1 (Clone M300; Santa Cruz), rabbit anti‐MLKL (ab172868; Abcam), rabbit anti‐phosphoS345‐MLKL (ab196436; Abcam), mouse anti‐m123/IE1 (CHROMA101; Center for Proteomics, University of Rijeka), mouse anti‐m112‐113/E1 (CHROMA103; Center for Proteomics, University of Rijeka), mouse anti‐FKBP12 (Clone 8; BD Biosciences), rabbit anti‐histone H3 (Clone D1H2; Cell Signaling Technology), rabbit anti‐β‐tubulin (Clone 9F3; Cell Signaling Technology), mouse anti‐β‐actin (Clone AC‐74; Sigma), rabbit anti‐ZBP1 (clone 887 62), mouse anti‐FLAG M2‐Peroxidase (Clone M2; Sigma‐Aldrich), mouse anti‐c‐Myc (Clone 9E10, Sigma), donkey anti‐mouse IgG‐HRP (Vector Laboratories), and donkey anti‐rabbit IgG‐HRP (Vector Laboratories). Blots were visualized using ECL Prime Western Blotting Detection Reagent (GE Healthcare) and exposure to film. Digital images were generated with a CanoSCAN LIDE 700F slide/film scanner (Cannon) and images processed with Canvas X16 software (ACD Systems International, Ft. Lauderdale, FL, USA). No digital enhancements were applied. All immunoblot data presented are representative of at least three independent experiments.
Generation of IE3DD mutants
Recombineering was performed as previously described 14, 63. Briefly, levansucrase (SacB) and kanamycin (Kan) genes were amplified from the pTBE100 plasmid with 50 nucleotide base pair overhangs corresponding to the MCMV genome flanking the end of exon 5 of IE3, with the primers HS01 and HS02. PCRs were digested with DpnI, gel purified, and electroporated into DH10B cell containing pSIM6 and either the parental WT K181 or M45mutRHIM bacmid 14. Recombinants were selected based on kanamycin resistance and sucrose sensitivity. For the second round of recombineering, a FKBP‐DD fragment was amplified from a plasmid expressing FKBP12 (provided by Edward Mocarski), with 50 nucleotide overhangs corresponding to the MCMV genome flanking the end of IE3‐Exon5 with the primers, HS09 and HS10. PCRs were DpnI treated, gel purified, and electroporated into DH10B cells containing pSIM6 and either IE3‐SacBKan mutants in WT or M45mutRHIM bacmid backgrounds. Recombinants were selected for kanamycin sensitivity and sucrose resistance. The genomic integrity of the BACs was confirmed after each step of recombination by RFLP analysis using EcoRI digest. PCR amplification of the IE3‐Exon5 junction was carried out with the primers, HS24 and HS29, and confirmed by sequence analysis. The mutated locus of M45mutRHIM contains a diagnostic PvuII site 14, and the presence of this site was confirmed with digestion of an amplicon of the RHIM locus, using the primers, HS36 and HS37. Oligonucleotide sequences are listed in Table EV1.
Viruses and infections
Parental WT and M45mutRHIM viruses have been previously described 14. WT‐IE3DD or M45mutRHIM‐IE3DD viruses were generated in NIH3T3 as previously described 64. Viruses were propagated, clarified and concentrated, and titered by plaque assay on NIH3T3 cells as previously described 14. For WT‐IE3DD and M45mutRHIM‐IE3DD viruses, where indicated, Shield‐1 was included at all stages of infection at a final concentration of 1 μM, and replenished every 2–3 days.
Cell death assays
Cell viability assay was performed as previously described 14 using Promega Cell Titer Glo kit at 18–22 h post‐infection. For assaying TNF‐induced necroptosis, cells were treated with combinations of 25 ng/ml TNF and 50 μM zVAD‐fmk for 6 h and cell viability determined as described above. Where indicated, infection or treatment with combinations of TNF and zVAD‐fmk was carried out in the presence of 1 μg/ml nocodazole (Noc) or 100 μM Ciliobrevin‐D (CBD) or 200 μg/ml PFA. For the α‐amanitin experiments, cells were infected or treated as above, and at the indicated time, α‐amanitin added to a final concentration of 50 μg/ml. Data are shown normalized to WT infection or vehicle control treatment. Cell death was also determined by monitoring cell integrity with the live cell impermeant nucleic acid stain‐Sytox Green (Invitrogen). Cells (104 cells/well) were seeded into Corning 96‐well tissue culture plates. After infection for 1 h, cells were cultured in the medium containing 50 nM Sytox Green and 1.25 μg/ml Hoechst 33342 (Thermo Fisher Scientific) and imaged on a Citation Cell Imaging Multi‐Mode Reader (Biotek). Two images of each well were collected. Sytox Green‐positive and Hoechst 33342‐positive cells per image were calculated. % Cell death = Sytox Green+ cells/Hoechst 33342+ cells.
Generation of DAI/ZBP1 knockout and reconstitution cells
Endogenous DAI/ZBP1 was targeted for knockout using CRISPR/Cas9, essentially as previously described 65. Briefly, sgRNAs were identified and designed with online bioinformatics tools (http://crispr.mit.edu and http://e-crisp.org). A target site was selected at nucleotides 173216940–173216959 of the NCBI Mus musculus Annotation Release 105 (NC_000068.7). This position corresponds to the 29th amino acid of DAI/ZBP1 located in exon 2. Two 60‐mers oligonucleotides, s‐aa‐29‐S and s‐aa‐29‐C, were annealed, extended, and cloned into AflII linearized gRNA_Cloning Vector 65 [a gift from George Church (Addgene plasmid # 41824)] by Gibson assembly. The cloned gRNA, hCas9 Vector 65 [a gift from George Church (Addgene plasmid # 41815)], and pQXCIP (Clontech) vector were co‐transfected into SVEC4‐10 cells, and cells selected with 2 μg/ml puromycin 24 h post‐transfection. Following outgrowth, single cells were subcloned by limiting dilution, and clonal populations surveyed for absence of DAI/ZBP1 expression by immunoblotting. A clonal line, designated 29‐11, was selected for continuing experiments. Sequence‐specific mutations in selected clones were confirmed by sequencing of PCR‐amplified genomic DNA. DNA was prepared and sequenced using the ZBP Surveyor primers. Generation of 3X‐FLAG‐DAI/ZBP1 and mutant retroviral expression vectors has been previously described 24. Retroviral production and reconstitution were performed as previously described 15, using 29‐11 cells. Oligonucleotide sequences are listed in Table EV1. Human DAI open reading frame (ORF) was inserted into pLV‐EF1α‐MCS‐IRES‐Puro lentiviral vector (Biosettia) and verified by DNA sequencing. Lentivirus stock was prepared from 293T cells as described previously 16. HT‐29 cells were transduced with lentiviral vector and selected with 2 μg/ml puromycin.
Cell fractionation
Subcellular fractionation was performed essentially as previously described 66. Briefly, 29‐11(WT DAI) cells were either mock‐infected or infected with WT or M45mutRHIM MCMV (MOI = 5). Cells were collected 7 h post‐infection, pelleted, and lysed on ice for 10 min in CSKT buffer [10 mM PIPES pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.5% Triton X‐100, 1× protease inhibitor (Sigma)]. Samples were centrifuged at 5,000 g for 3 min at 4°C. Supernatant (cytosolic fraction) and pellet (nuclear fraction) were separated, boiled in SDS sample buffer, and stored at 20°C until analysis.
Statistics
For all comparisons between mean values from control or treated samples, a two‐tailed t‐test was used to determine statistical significance. One‐way analysis of variance (ANOVA) followed by Dunnett's multiple comparisons test was used to determine significance across treatment groups. All statistical analysis was performed using Prism 6 (Graphpad, San Diego, CA, USA).
Author contributions
HS, KBR, HG, RPG, and VJL conducted experiments and analyzed data. HS, WJK, and JWU designed and oversaw experiments. HS, KBR, and JWU wrote the manuscript, and all authors participated in editing the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2A
Source Data for Figure 2C
Source Data for Figure 2E
Source Data for Figure 3C
Source Data for Figure 3E
Source Data for Figure 5
Acknowledgements
The authors thank Chris Sullivan, Bryan Davies, Ed Mocarski, Siddharth Balachandran, Jan Rehwinkel, and Jonathan Maelfait for insightful discussions, and Tillman Burckstummer and Giulio Superti‐Furga (CeMM) for reagents. Work was supported by National Institutes of Health Public Service Grant DP1 OD012198 to W.J.K., and Cancer Prevention & Research Institute of Texas (CPRIT) Scholar Award (R1202) and the William and Ella Owens Medical Research Foundation (201402505‐001) to J.W.U.
EMBO Reports (2017) 18: 1429–1441
References
- 1. Sridharan H, Upton JW (2014) Programmed necrosis in microbial pathogenesis. Trends Microbiol 22: 199–207 [DOI] [PubMed] [Google Scholar]
- 2. Cai Z, Liu ZG (2014) Execution of RIPK3‐regulated necrosis. Mol Cell Oncol 1: e960759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mocarski ES, Guo H, Kaiser WJ (2015) Necroptosis: the Trojan horse in cell autonomous antiviral host defense. Virology 479–480: 160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK (2009) Phosphorylation‐driven assembly of the RIP1‐RIP3 complex regulates programmed necrosis and virus‐induced inflammation. Cell 137: 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun X, Lee J, Navas T, Baldwin DT, Stewart TA, Dixit VM (1999) RIP3, a novel apoptosis‐inducing kinase. J Biol Chem 274: 16871–16875 [DOI] [PubMed] [Google Scholar]
- 6. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X et al (2012) Mixed lineage kinase domain‐like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227 [DOI] [PubMed] [Google Scholar]
- 7. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X et al (2013) Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res 23: 994–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF‐induced necroptosis. Nat Cell Biol 16: 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P et al (2014) Translocation of mixed lineage kinase domain‐like protein to plasma membrane leads to necrotic cell death. Cell Res 24: 105–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG (2012) Mixed lineage kinase domain‐like is a key receptor interacting protein 3 downstream component of TNF‐induced necrosis. Proc Natl Acad Sci USA 109: 5322–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM (2002) Identification of a novel homotypic interaction motif required for the phosphorylation of receptor‐interacting protein (RIP) by RIP3. J Biol Chem 277: 9505–9511 [DOI] [PubMed] [Google Scholar]
- 12. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez‐Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI et al (2013) The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453 [DOI] [PubMed] [Google Scholar]
- 13. Zhou W, Yuan J (2014) Necroptosis in health and diseases. Semin Cell Dev Biol 35: 14–23 [DOI] [PubMed] [Google Scholar]
- 14. Upton JW, Kaiser WJ, Mocarski ES (2010) Virus inhibition of RIP3‐dependent necrosis. Cell Host Microbe 7: 302–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Upton JW, Kaiser WJ, Mocarski ES (2012) DAI/ZBP1/DLM‐1 complexes with RIP3 to mediate virus‐induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11: 290–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES (2015) Suppression of RIP3‐dependent necroptosis by human cytomegalovirus. J Biol Chem 290: 11635–11648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES (2015) Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17: 243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Chen C, Li J et al (2015) RIP1/RIP3 binding to HSV‐1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17: 229–242 [DOI] [PubMed] [Google Scholar]
- 19. Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z et al (2014) Direct activation of RIP3/MLKL‐dependent necrosis by herpes simplex virus 1 (HSV‐1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA 111: 15438–15443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ (2003) A role for tumor necrosis factor receptor‐2 and receptor‐interacting protein in programmed necrosis and antiviral responses. J Biol Chem 278: 51613–51621 [DOI] [PubMed] [Google Scholar]
- 21. Li M, Beg AA (2000) Induction of necrotic‐like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus‐infected cells. J Virol 74: 7470–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Berger AK, Danthi P (2013) Reovirus activates a caspase‐independent cell death pathway. MBio 4: e00178‐00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH III, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O et al (2016) RIPK3 activates parallel pathways of MLKL‐driven necroptosis and FADD‐mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20: 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ et al (2016) DAI senses influenza A virus genomic RNA and activates RIPK3‐dependent cell death. Cell Host Microbe 20: 674–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodrigue‐Gervais IG, Labbe K, Dagenais M, Dupaul‐Chicoine J, Champagne C, Morizot A, Skeldon A, Brincks EL, Vidal SM, Griffith TS et al (2014) Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host Microbe 15: 23–35 [DOI] [PubMed] [Google Scholar]
- 26. Guo H, Kaiser WJ, Mocarski ES (2015) Manipulation of apoptosis and necroptosis signaling by herpesviruses. Med Microbiol Immunol 204: 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K et al (2007) DAI (DLM‐1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448: 501–505 [DOI] [PubMed] [Google Scholar]
- 28. Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, Kaneeganti TD (2016) ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 1: aag2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, Pasparakis M (2016) RIPK1 counteracts ZBP1‐mediated necroptosis to inhibit inflammation. Nature 540: 124–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, Lill JR, Roose‐Girma M, Warming S, Solon M et al (2016) RIPK1 inhibits ZBP1‐driven necroptosis during development. Nature 540: 129–133 [DOI] [PubMed] [Google Scholar]
- 31. Kaiser WJ, Upton JW, Mocarski ES (2008) Receptor‐interacting protein homotypic interaction motif‐dependent control of NF‐kappa B activation via the DNA‐dependent activator of IFN regulatory factors. J Immunol 181: 6427–6434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, Vazquez J, Benedict CA, Tschopp J (2009) DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF‐kappaB. EMBO Rep 10: 916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hornung V, Latz E (2010) Intracellular DNA recognition. Nat Rev Immunol 10: 123–130 [DOI] [PubMed] [Google Scholar]
- 34. Wang Z, Choi MK, Ban T, Yanai H, Negishi H, Lu Y, Tamura T, Takaoka A, Nishikura K, Taniguchi T (2008) Regulation of innate immune responses by DAI (DLM‐1/ZBP1) and other DNA‐sensing molecules. Proc Natl Acad Sci USA 105: 5477–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pellet PE, Roizman B (2007) The Family Herpesviridae: a brief introduction. pp 2479–2499. Philadelphia, PA: Lippincott‐Williams and Wilkins; [Google Scholar]
- 36. Brune W, Menard C, Heesemann J, Koszinowski UH (2001) A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 291: 303–305 [DOI] [PubMed] [Google Scholar]
- 37. Wagstaff AJ, Bryson HM (1994) Foscarnet. A reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs 48: 199–226 [DOI] [PubMed] [Google Scholar]
- 38. Bosse JB, Bauerfeind R, Popilka L, Marcinowski L, Taeglich M, Jung C, Striebinger H, von Einem J, Gaul U, Walther P et al (2012) A beta‐herpesvirus with fluorescent capsids to study transport in living cells. PLoS One 7: e40585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deigendesch N, Koch‐Nolte F, Rothenburg S (2006) ZBP1 subcellular localization and association with stress granules is controlled by its Z‐DNA binding domains. Nucleic Acids Res 34: 5007–5020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pham HT, Park MY, Kim KK, Kim YG, Ahn JH (2006) Intracellular localization of human ZBP1: differential regulation by the Z‐DNA binding domain, Zalpha, in splice variants. Biochem Biophys Res Commun 348: 145–152 [DOI] [PubMed] [Google Scholar]
- 41. Angulo A, Ghazal P, Messerle M (2000) The major immediate‐early gene ie3 of mouse cytomegalovirus is essential for viral growth. J Virol 74: 11129–11136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lacaze P, Forster T, Ross A, Kerr LE, Salvo‐Chirnside E, Lisnic VJ, Lopez‐Campos GH, Garcia‐Ramirez JJ, Messerle M, Trgovcich J et al (2011) Temporal profiling of the coding and noncoding murine cytomegalovirus transcriptomes. J Virol 85: 6065–6076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Messerle M, Buhler B, Keil GM, Koszinowski UH (1992) Structural organization, expression, and functional characterization of the murine cytomegalovirus immediate‐early gene 3. J Virol 66: 27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rakhit R, Navarro R, Wandless TJ (2014) Chemical biology strategies for posttranslational control of protein function. Chem Biol 21: 1238–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hermiston TW, Malone CL, Stinski MF (1990) Human cytomegalovirus immediate‐early two protein region involved in negative regulation of the major immediate‐early promoter. J Virol 64: 3532–3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tang Q, Li L, Maul GG (2005) Mouse cytomegalovirus early M112/113 proteins control the repressive effect of IE3 on the major immediate‐early promoter. J Virol 79: 257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lippmann J, Rothenburg S, Deigendesch N, Eitel J, Meixenberger K, van Laak V, Slevogt H, N'Guessan PD, Hippenstiel S, Chakraborty T et al (2008) IFNbeta responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM‐1/DAI). Cell Microbiol 10: 2579–2588 [DOI] [PubMed] [Google Scholar]
- 48. Jurak I, Brune W (2006) Induction of apoptosis limits cytomegalovirus cross‐species infection. EMBO J 25: 2634–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim KS, Carp RI (1972) Abortive infection of human diploid cells by murine cytomegalovirus. Infect Immun 6: 793–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB et al (2013) Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol 190: 2311–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tanzer MC, Tripaydonis A, Webb AI, Young SN, Varghese LN, Hall C, Alexander WS, Hildebrand JM, Silke J, Murphy JM (2015) Necroptosis signalling is tuned by phosphorylation of MLKL residues outside the pseudokinase domain activation loop. Biochem J 471: 255–265 [DOI] [PubMed] [Google Scholar]
- 52. Martinez FP, Cosme RS, Tang Q (2010) Murine cytomegalovirus major immediate‐early protein 3 interacts with cellular and viral proteins in viral DNA replication compartments and is important for early gene activation. J Gen Virol 91: 2664–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Budt M, Niederstadt L, Valchanova RS, Jonjic S, Brune W (2009) Specific inhibition of the PKR‐mediated antiviral response by the murine cytomegalovirus proteins m142 and m143. J Virol 83: 1260–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Placido D, Brown BA II, Lowenhaupt K, Rich A, Athanasiadis A (2007) A left‐handed RNA double helix bound by the Z alpha domain of the RNA‐editing enzyme ADAR1. Structure 15: 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brown BA II, Lowenhaupt K, Wilbert CM, Hanlon EB, Rich A (2000) The zalpha domain of the editing enzyme dsRNA adenosine deaminase binds left‐handed Z‐RNA as well as Z‐DNA. Proc Natl Acad Sci USA 97: 13532–13536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wittig B, Dorbic T, Rich A (1991) Transcription is associated with Z‐DNA formation in metabolically active permeabilized mammalian cell nuclei. Proc Natl Acad Sci USA 88: 2259–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schock SN, Chandra NV, Sun Y, Irie T, Kitagawa Y, Gotoh B, Coscoy L, Winoto A (2017) Induction of necroptotic cell death by viral activation of the RIG‐I or STING pathway. Cell Death Differ 24: 615–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang Y, Ma J, Chen Y, Wu M (2004) Nucleocytoplasmic shuttling of receptor‐interacting protein 3 (RIP3): identification of novel nuclear export and import signals in RIP3. J Biol Chem 279: 38820–38829 [DOI] [PubMed] [Google Scholar]
- 59. Yoon S, Bogdanov K, Kovalenko A, Wallach D (2016) Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ 23: 253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pham TH, Kwon KM, Kim YE, Kim KK, Ahn JH (2013) DNA sensing‐independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J Virol 87: 3076–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mack C, Sickmann A, Lembo D, Brune W (2008) Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1‐interacting protein. Proc Natl Acad Sci USA 105: 3094–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Karayel E, Burckstummer T, Bilban M, Durnberger G, Weitzer S, Martinez J, Superti‐Furga G (2009) The TLR‐independent DNA recognition pathway in murine macrophages: ligand features and molecular signature. Eur J Immunol 39: 1929–1936 [DOI] [PubMed] [Google Scholar]
- 63. Tandon R, Mocarski ES (2008) Control of cytoplasmic maturation events by cytomegalovirus tegument protein pp150. J Virol 82: 9433–9444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Redwood AJ, Messerle M, Harvey NL, Hardy CM, Koszinowski UH, Lawson MA, Shellam GR (2005) Use of a murine cytomegalovirus K181‐derived bacterial artificial chromosome as a vaccine vector for immunocontraception. J Virol 79: 2998–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA‐guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Reyes JC, Muchardt C, Yaniv M (1997) Components of the human SWI/SNF complex are enriched in active chromatin and are associated with the nuclear matrix. J Cell Biol 137: 263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2A
Source Data for Figure 2C
Source Data for Figure 2E
Source Data for Figure 3C
Source Data for Figure 3E
Source Data for Figure 5