

Abstract
Serum amyloid A1 (SAA1) is an apolipoprotein that binds to the high‐density lipoprotein (HDL) fraction of the serum and constitutes the fibril precursor protein in systemic AA amyloidosis. We here show that HDL binding blocks fibril formation from soluble SAA1 protein, whereas internalization into mononuclear phagocytes leads to the formation of amyloid. SAA1 aggregation in the cell model disturbs the integrity of vesicular membranes and leads to lysosomal leakage and apoptotic death. The formed amyloid becomes deposited outside the cell where it can seed the fibrillation of extracellular SAA1. Our data imply that cells are transiently required in the amyloidogenic cascade and promote the initial nucleation of the deposits. This mechanism reconciles previous evidence for the extracellular location of deposits and amyloid precursor protein with observations the cells are crucial for the formation of amyloid.
Keywords: FRET, live‐cell imaging, macrophages, protein aggregation, serum amyloid A
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport, Protein Biosynthesis & Quality Control
Introduction
Amyloid fibrils are filamentous polypeptide aggregates with a cross‐β structure 1. They occur inside the human body associated with neurodegenerative diseases, systemic amyloidosis, type II diabetes and “normal” ageing 2, 3. Biophysical analysis of the mechanisms by which chemically pure polypeptide chains self‐assemble inside a test tube provided insights into the kinetics and chemical steps of protein self‐assembly 4, 5. But how fibril formation occurs within a native cellular environment, which is obviously more complex than the conditions of dilute polypeptide chains in simple chemically buffered solutions, is less well understood. To determine the cellular mechanism of protein misfolding, we here focus on AA amyloidosis, the prototype of a systemic amyloidosis.
AA amyloidosis shows a worldwide distribution, and it occurs in humans, mammals and birds 6. The disease is associated with massive amyloid deposits that primarily affect spleen, liver and kidneys. Renal amyloid is the major cause of debilitations in AA amyloidosis, which frequently develop proteinuria if not an end‐stage kidney disease with sometimes fatal outcome 7. The pathology of systemic amyloidosis also depends to a much greater extent on fibrils than on the toxic activity of fibrillation intermediates 8, the major pathogenic culprits in many neurodegenerative diseases 9. Human and murine AA amyloid fibrils consist of C‐terminally truncated SAA1 protein, an acute‐phase apolipoprotein that is secreted by the liver in response to a strong inflammatory stimulus 10.
Acute‐phase SAA1 concentrations in the serum can be more than 1 mg/ml compared to about 0.001 mg/ml under normal conditions 10, indicating that high blood SAA1 levels are crucial for the onset of disease. Blood‐borne SAA1 is mainly associated with the plasma HDL fraction, where it has been associated with multiple biological functions 11. The protein interacts functionally and physically with macrophages and modulates the physiological activity and lipid homeostasis of these cells under inflammatory conditions 12, 13. Lipid‐free SAA1 possesses a random coil‐like conformation at 37°C 14, 15, whereas lowering the temperature to 4°C or addition of sodium dodecyl sulphate (SDS), HDL or lipids may stabilize the protein in an α‐helical conformation 14, 15, 16.
AA amyloid deposits develop first within the reticuloendothelial system 17, suggesting that macrophages are involved in the biogenesis of amyloid deposits in vivo. Amyloid deposits can be infiltrated by macrophages 18, and analysis of histological sections of diseased tissue with transmission electron microscopy revealed amyloid fibrils within the lysosomes 19. Extraction of peritoneal macrophages from AA amyloidotic mice and their injection into inflamed recipient mice can transfer AA amyloidosis from animal to animal 20. Clodronate depletion of splenic macrophages, specifically within the marginal zone, antagonizes the development of murine AA amyloidosis 21, 22. Addition of HDL‐SAA1 to cultures of primary macrophages or macrophage‐like cell lines leads to the internalization of SAA1 into the cells and to the deposition of amyloid within the culture dish 20, 23, 24. The formed amyloid consists of extracellular fibrils, lipids, glycosaminoglycans and serum amyloid P component and thus of key components of AA amyloid deposits in vivo 24, 25, 26. In the present study, we have used this cell model for analysing the cellular mechanism of amyloid fibril formation from SAA1 protein.
Results
Formation of amyloid deposits in the cell culture model
To induce the formation of amyloid, we incubated murine J774A.1 cells, a widely used model of monocytic activity and macrophage‐like function, with soluble SAA1 protein and HDL (Appendix Fig S1). SAA1 was added from a stock of the protein in pure water in which the protein was non‐native and exhibited a low α‐helical content of ~10%, as demonstrated by far‐ultraviolet (UV) circular dichroism (CD) spectroscopy (Appendix Fig S2A and C). Addition of the protein to medium lacking HDL and foetal bovine serum (FBS) increases the α‐helical content to about 25% (Appendix Fig S2A and C), which is less than the α‐helical content of available crystal structure homologues (70–74%), as defined by the protein data bank files 27, 28. It is also less than the α‐helical content that we and others obtained by CD with murine SAA1 or its homologues at 4°C (Appendix Fig S2B and C) or in the presence of SDS or HDL 14, 15, 16. Soluble SAA1 lacks amyloid structure and interacts only weakly with amyloid binding dyes, such as thioflavin T (ThT) and Congo red (CR; Appendix Fig S2D–F). It is polydisperse, specifically in medium containing HDL and FBS, and forms a range of differently sized HDL‐bound states as well as monomeric protein as indicated by size exclusion chromatography (SEC) (Appendix Fig S2G).
The formed amyloid deposits, in contrast to soluble SAA1, bind Congo red (CR) and give rise to green birefringence, the gold standard of amyloid diagnosis in pathology 29. Varying the SAA1 concentration from 0.1 to 1.0 mg/ml shows that the amyloid forms faster and more extensively at higher SAA1 concentrations (Fig 1) which reproduces the natural requirement of high SAA1 levels for disease in vivo 6. It further follows the general laws of mass action, according to which assembly reactions become favoured if the protomer concentration is increased.
Figure 1. Effect of the SAA1 concentration on the kinetics of amyloid formation.
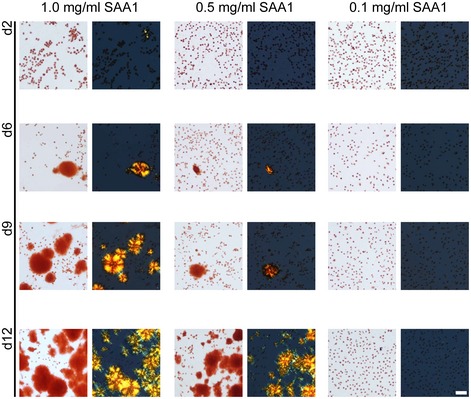
Bright field and dark field polarizing microscopy images of CR‐stained J774A.1 cells exposed for 2, 6, 9 and 12 days to 1.0, 0.5 or 0.1 mg/ml SAA1 and HDL. Scale bar: 50 μm.
Generation of a fluorescence sensor to monitor SAA1 self‐assembly
To follow the fate of SAA1 protein within the cell model, we generated a Förster resonance energy transfer (FRET) sensor, which monitors the self‐assembly of the protein into fibrils. We produced a recombinant variant of SAA1 protein in which residue 101 of the otherwise cysteine (Cys)‐free protein was replaced with Cys (Appendix Fig S3A). The resulting protein, termed SAA1101Cys, was modified at its single thiol group with the maleimide‐conjugated fluorescent dyes Alexa Fluor 488 (AF488) or Alexa Fluor 594 (AF594). The single site labelled proteins will hereafter be referred to as SAA1101Cys‐AF488 and SAA1101Cys‐AF594. Control experiments confirmed that the attachment of a fluorescent group at position 101 did not substantially modify the assembly of SAA1 fibrils in vitro. The fluorescent protein fibrillated similarly in a 1:49 mixture with wild‐type SAA1 as the unmodified bulk protein (Appendix Fig S3B–D). There was no evidence that the incorporation of fluorescently labelled SAA1 notably alters the morphology of the formed fibrils (Appendix Fig S3E–H).
Incubation of a 48:1:1 mixture of SAA1, SAA1101Cys‐AF488 and SAA1101Cys‐AF594 for 70 h in vitro and measurement of the fluorescence spectra before and after incubation show in the spectra recorded with the aged samples additional bands arising from FRET (Appendix Fig S4A and B). FRET depends on dipolar interactions between nearby fluorophores (closer than 10 nm) and has been used, for example, to provide information about the assembly of α‐synuclein protein from Parkinson's disease 30, 31. Following the temporal development of FRET shows a similar kinetic behaviour as bulk fibril formation, which we monitor here by centrifugation and measurement of the protein content in pellet and supernatant (Appendix Fig S4D). We conclude that a 48:1:1 mixture of SAA1, SAA1101Cys‐AF488 and SAA1101Cys‐AF594 represents a FRET sensor for the fibrillation and aggregation of SAA1 protein.
FRET demonstrates SAA1 self‐assembly to start inside of the cell
Addition of the three protein variants together with HDL to J774A.1 cells leads to FRET within a 24‐h incubation period as shown by LSM (Fig 2). All FRET signals seem to be associated with focal spots within the cytoplasm of the cells, suggesting their presence in a vesicular compartment. A 4‐h incubation period is not sufficient to produce FRET (Fig 2B) although the signals recorded in the donor or acceptor channels show that SAA1 is already internalized at this early time point (Fig 2A). In other words, cells take up the protein for a long time before the first assemblies can be detected by FRET, which occurs much earlier than the deposition of CR green birefringent amyloid, which is not seen within 4 days (Fig 2C), but can only be observed under comparable conditions from 5 days onwards (Figs 1 and 4B).
Figure 2. FRET demonstrates fibril formation to start inside the cell.
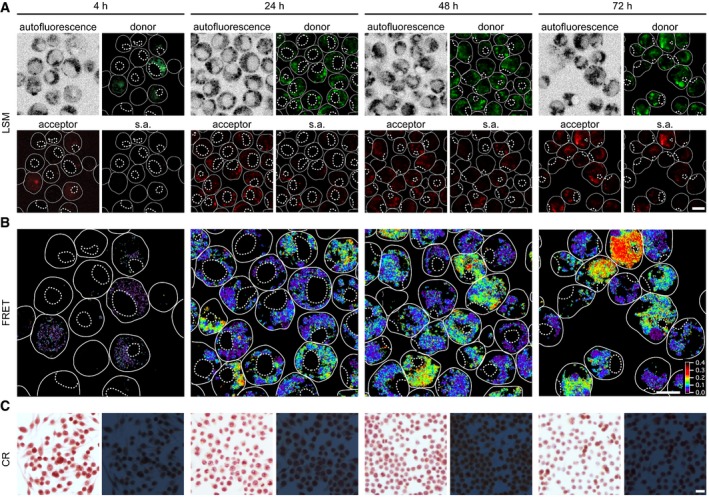
- LSM images of living cells exposed to a 48:1:1 mixture of SAA1, SAA1101Cys‐AF488, SAA1101Cys‐AF594 (total SAA1 concentration 50 μM) and HDL for 4, 24, 48 or 72 h. Images show autofluorescence, donor, acceptor and sensitized acceptor (s.a.) fluorescence. Scale bar: 10 μm.
- The FRET images were calculated from the s.a. fluorescence images as described in the Materials and Methods. Scale bar: 10 μm.
- Bright field (left) and dark field polarizing microscopy images (right) of CR‐stained cell cultures exposed to 0.5 mg/ml SAA1 and HDL for 4 h, 1 day, 2 days or 3 days. Scale bar: 20 μm.
Preventing the internalization of SAA1 protein reduces FRET
Uptake of fluorescently labelled SAA1 is potently blocked by addition of monodansyl cadaverine (MDC) or chlorpromazine (Fig 3A), two well‐established inhibitors of clathrin‐dependent endocytosis as shown by flow cytometry. By contrast, none of the compounds targeting phagocytosis (cytochalasin B, latrunculin A), pinocytosis (colchicine, nocodazole), macropinocytosis (rottlerin, dimethylamiloride) and caveolae‐dependent uptake pathways (nystatin, genistein) discernibly alter cellular uptake (Fig 3A) although we see a slight, but non‐significant reduction with inhibitors of dynamin‐dependent endocytosis (dynasore, dynole) which is consistent with previous observations that dynamin‐dependent uptake is linked to clathrin‐dependent endocytosis 32. The used inhibitor concentrations and incubation times were not significantly cell toxic as confirmed with the MTT assay (Fig 3B). LSM shows the co‐localization of fluorescently labelled SAA1 with fluorescently labelled transferrin, a marker of clathrin‐dependent endocytosis (Fig 3C). Addition of MDC reduces the formation of intracellular FRET (Appendix Fig S5B). None of the inhibitors affecting the other internalization pathways reduced the obtained FRET signals in a statistically significant fashion (Appendix Fig S5B), consistent with their lack of activity on SAA1 uptake (Fig 3A).
Figure 3. Fluorescently labelled SAA1 is mainly internalized through clathrin‐dependent endocytosis.
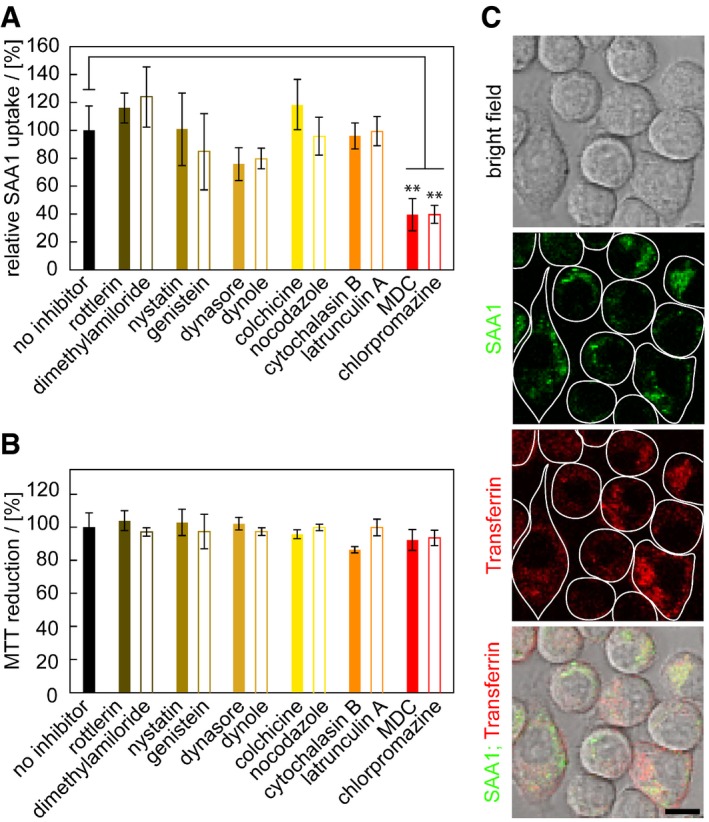
- Flow cytometric analysis of cells exposed to 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF647, HDL and different uptake inhibitors as indicated (n = 4).
- MTT‐based cell viability measurement of cells incubated with or without inhibitors (n = 4).
- LSM images of J774A.1 cells incubated with 0.5 mg/ml SAA1, 0.01 mg/ml fluorescent SAA1, HDL and AF647‐labelled transferrin, a fluorescent marker for clathrin‐dependent endocytosis. Scale bar: 10 μm.
Intracellular SAA1 aggregation is toxic to the cells
Following the kinetics of aggregation of fluorescently labelled SAA1 over a period of 6 days reveals the progressive clustering of the cells (Appendix Fig S6A–D). The clusters often enclose large‐sized spots of fluorescent SAA1 (Appendix Fig S6A), and the cell death marker propidium iodide (PI) demonstrates inclusion of dead cells in such clusters (Fig 4A). TEM analysis of ultrathin sections confirms this conclusion and demonstrates a core of extracellular material that is surrounded by a cluster of dead cell bodies (Appendix Fig S6D). Based on the MTT, we obtain the first signs of an altered cellular metabolism or toxicity as early as on day 1 (Fig 4B), corresponding to the intracellular accumulation of SAA1 aggregates as revealed by the FRET assay (Fig 2B). FRET‐positive aggregates form much earlier than CR‐positive amyloid deposits that emerge, based on the amyloid score (Appendix Fig S7), only from day 5 onwards (Fig 4B). The mechanism of cell death involves apoptosis, as demonstrated by an enhanced caspase‐3/7 activity (Fig 4C) and a positive TUNEL staining that arises from the apoptotic fragmentation of the cellular DNA (Fig 4D). Co‐localization studies with the lysosomal marker LysoTracker® Red DND‐99 reveal the internalization of SAA1 into the lysosomes (Fig 5A). The pH‐dependent reporter dye acridine orange (AO) additionally provides evidence for lysosomal defects (Fig 5B). AO can be internalized into the lysosomes and shows a reduced fluorescence when transferred to the cytoplasm. Quantified by flow cytometry and measurement of the AO fluorescence we find lysosomal leakage after an incubation period of 24 h but not after 5 h (Fig 5B), indicating that toxicity is triggered by the intracellular SAA1 aggregates (Fig 4B–D).
Figure 4. Fibril formation leads to the formation of cell clusters and toxicity.
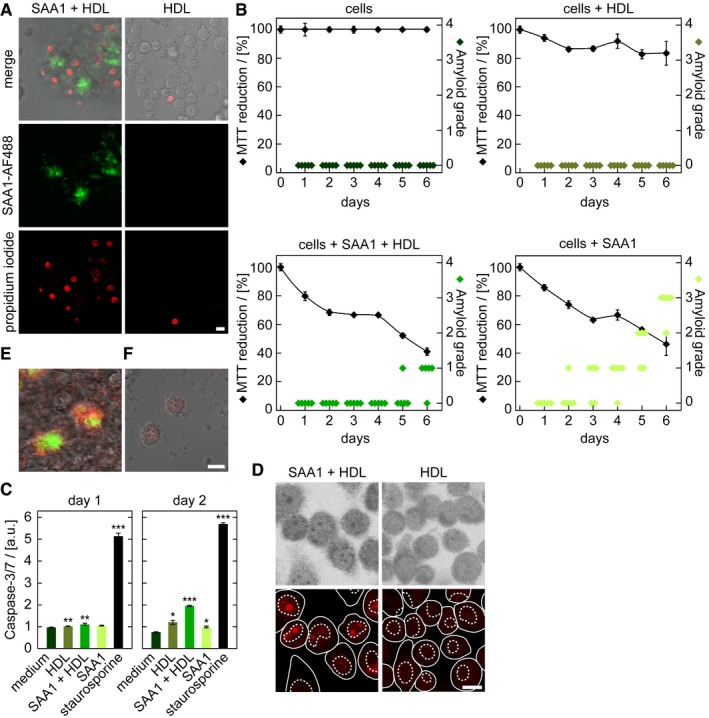
-
ALSM images of PI‐stained J774A.1 cells exposed to HDL or HDL together with 0.5 mg/ml SAA1 and 0.01 mg/ml SAA1‐AF488 and HDL for 6 days. Scale bar: 10 μm.
-
BMTT‐based cell viability measurements (black, n = 4) and quantitative estimation of amyloid load by CR staining (green, n = 5) of cells incubated with or without 0.5 mg/ml SAA1 and HDL as indicated. The sample incubated only with medium was set to 100%.
-
CCaspase‐3/7 activity of cells incubated with 0.5 mg/ml SAA1, HDL or the cell toxin staurosporine for up to 2 days (n = 3). a.u.: arbitrary units.
-
DLSM images after applying the TUNEL assay to monitor apoptosis in J774A.1 cells incubated for 3 days with 0.5 mg/ml SAA1 and HDL as indicated. Scale bar: 10 μm.
-
E, FLSM images of cells exposed for 6 days to HDL (F) or 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF488 (green) and HDL (E). Afterwards, cells were killed by methanol fixation and the culture dish was further incubated with 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF647 (red) and HDL for 1 day. Scale bar: 10 μm.
Figure 5. Intracellular fibril formation induces lysosomal leakage.
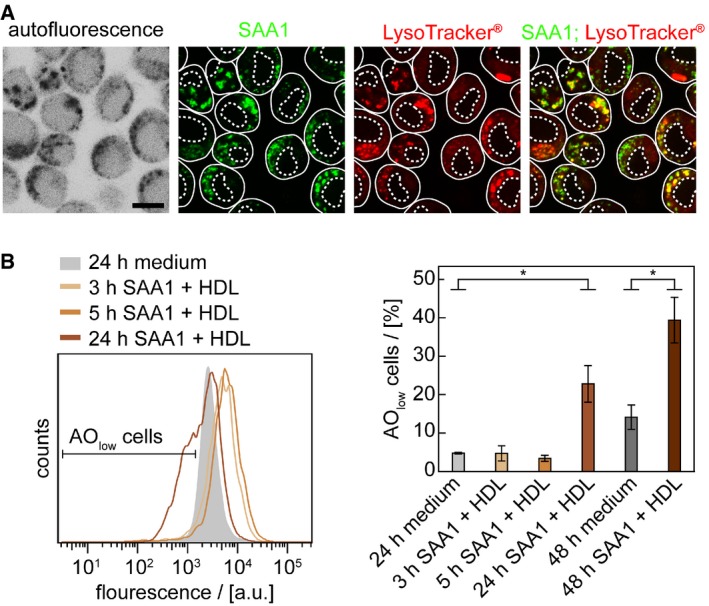
- LSM images of cells incubated with 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF488 and HDL for 24 h and stained with LysoTracker® Red DND‐99 (red), a marker of acidic organelles. Scale bar: 10 μm.
- Cells were incubated with or without 0.5 mg/ml SAA1 and HDL for up to 48 h, stained with pH‐sensitive dye AO, analysed by flow cytometry using an EX wavelength of 488 nm and an EM filter from 673 to 727 nm (n = 3). Data are presented as mean ± SD. *P < 0.05 (Student's t‐test).
The intracellular SAA1 becomes released into the extracellular space as demonstrated by an experiment in which cells were incubated with biotinylated SAA1 for 1 day to induce the formation of intracellular aggregates. We then removed all extracellular biotin‐SAA1 with trypsin and incubated the cells further with unlabelled SAA1. Analysis of the resulting deposits with SEM shows extracellular fibrils that bind gold‐conjugated streptavidin (Appendix Fig S8)1, indicating that previously intracellular biotin‐SAA1 protein is present in the extracellular fibril. The formed amyloid is able to grow further even in the absence of living cells, as killing the cells with methanol fixation did not stop their further growth. However, this effect was only seen if the cells were preincubated for 6 days with SAA1 and HDL to generate initial deposits, but not with cells kept for 6 days in the absence of SAA1 that did not produce any nascent deposit (Fig 4E and F). These data imply that cells are transiently involved in amyloid biogenesis and help to form a deposit nucleus, which then seeds further amyloid growth.
HDL prevents the fibrillation of SAA1 protein in the absence of cells
Spontaneous fibrillation of SAA1 protein is blocked in solution by HDL as we show here by real time fibrillation measurements, in which we monitored the formation of SAA1 fibrils by the fluorescence intensity of the fibril binding dye ThT (Fig 6A). TEM confirms the absence of fibrils under these conditions, sharply contrasting to the filamentous morphologies obtained in the absence of HDL (Fig 6A). Similar observations are made in the cell model, where we find a retarded amyloid deposition (Fig 4B) and a reduced uptake of N‐fluorescently labelled SAA1 into J774A.1 cells in the presence of HDL (Fig 6B). Taken together with the previous observation that HDL helps to fold SAA1 into an α‐helical conformation 15, we conclude that the lipid particles sequester the protein in a conformation that is not competent for fibril formation, while internalization of the protein overcomes this inhibitory activity.
Figure 6. HDL‐binding sequesters SAA1 protein and prevents fibril formation.
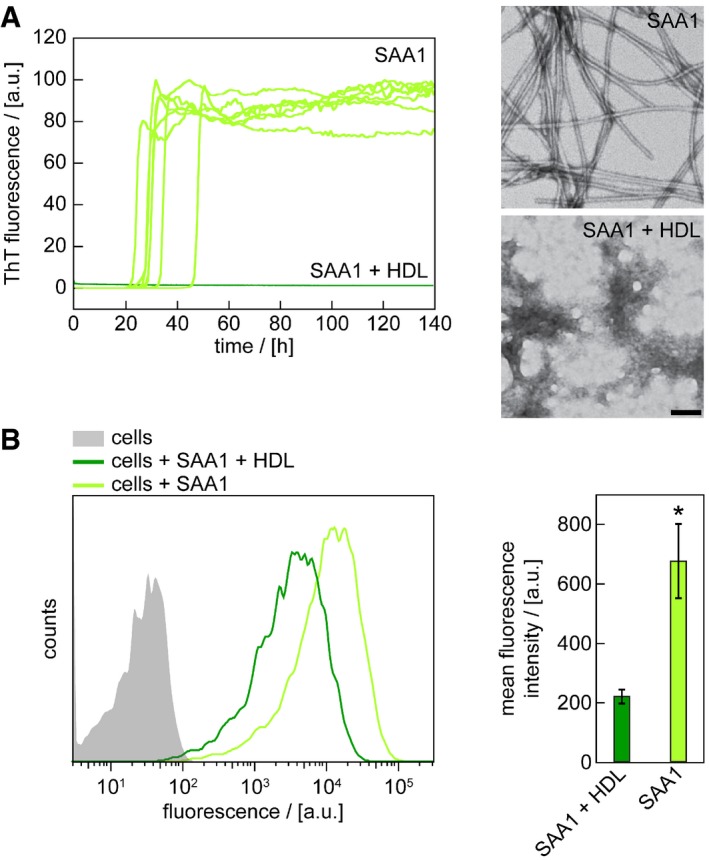
- Time‐dependent ThT fluorescence measurements of 50 μM SAA1 in the presence or absence of HDL. Seven replicates per condition are shown. The lag time is 31.5 ± 6.8 h in the absence of HDL. TEM images were taken from samples after 140 h. Scale bar: 100 nm.
- Flow cytometric analysis of cells incubated with 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF488 and HDL as indicated for 5 h (n = 3). Data are presented as mean ± SD. *P < 0.05 (Student's t‐test).
Discussion
Analysis of the cellular formation of amyloid from SAA1 protein yielded a mechanism that consists of six steps (Fig 7). The first step is the internalization of the extracellular protein by clathrin‐dependent endocytosis. This result is obtained here by the use of twelve different pharmacological uptake inhibitors, sampling a total of six different uptake pathways (Fig 3A). Fluorescence microscopy additionally demonstrates that SAA1 co‐localizes with transferrin, a marker of clathrin‐dependent endocytosis (Fig 3C). Internalized SAA1 protein may be passed on to neighbouring cells via direct cell–cell interactions 33 or may be trafficked, unlike transferrin that enters endocytic recycling compartments, to the lysosomes 34 where it co‐localizes with the lysosomal marker LysoTracker® Red DND‐99 (Fig 5A). Taken together with observations that the lysosomal proteases as cathepsins B and L cleave SAA1 protein 35, these data suggest that SAA1 is targeted for degradation.
Figure 7. Schematic view of the mechanism of fibril formation.
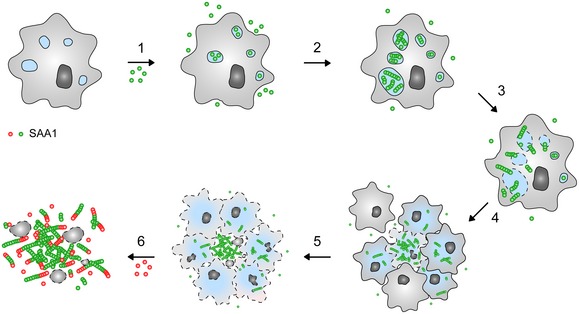
According to our data cellular amyloid formation consists of six steps: (1) Uptake of SAA1 protein into the cells, (2) intracellular fibril nucleation, (3) leakage of intracellular compartments, (4) formation of cellular clusters, (5) release of intracellular SAA1 into the extracellular space and (6) further extracellular growth of the nucleated deposit.
The second step in the cellular formation of amyloid is the intracellular accumulation of SAA1 aggregates (Fig 2B). While amyloid fibrils were previously observed in lysosomes or other endocytic compartments of amyloidotic tissues 18, 19 and of cell models 34, 36, 37, it remained unclear whether protein assembly took place inside the cell or whether the cells had internalized the preformed fibrils. Indeed, several studies demonstrated that mononuclear phagocytes can internalize and degrade amyloid fibrils 26, 38, 39. Our FRET assay, which is considerably more sensitive than previous CR‐based methods, detects the first aggregates inside the cell (Fig 2B). These data lend strong support to the relevance of an intracellular route of fibril formation in the course of disease. However, amyloid deposition is only seen if the cells had internalized the protein for more than 20 h (Fig 2); that is, fibril formation occurs under conditions when the influx of SAA1 into the cells exceeds their proteolytic capacity. Further support for an intracellular origin of fibril formation is provided by data showing that the genetic knockout of lysosomal proteases modulates the development of AA amyloidosis in mice 35.
The next two steps in the cellular formation of amyloid are the disruption of lysosomal membranes and the development of cellular toxicity. Both events are seen after the cells were incubated with SAA1 for 24 h; that is, they appear concomitantly with intracellular SAA1 aggregates. Non‐degradable lysosomal cargo in the form of organic nanoparticles was previously shown to induce lysosomal leakage, mitochondrial disturbances and apoptotic death in a monocytic cell line 40. This damage could be attributed to the surface charge and was only seen with positively charged, amino‐functionalized but not with negatively charged carboxyl‐functionalized nanoparticles 40. A similar mechanism might also account for SAA1 protein, which has an isoelectric point of 5.89 and thus carries a positive net charge in the lysosomes that possess typical pH values of 4.5–5.0. Other mechanisms leading to membrane damage could be the formation of an amyloid pore, which has been demonstrated for numerous amyloid‐forming systems, including human SAA1, with artificial lipids in vitro 41, 42, or the physical disruption of the vesicular lipid bilayer by the fibrils. The latter mechanism was found in a cell model of Aβ amyloid formation, in which intracellular fibrils distorted the compartment structure or pricked through the membrane to extend into the cytoplasm 43.
The last two steps in the generation of amyloid deposits are the formation of cellular clusters and the release and further extracellular growth of the initial deposits. The clusters frequently contain cells that are affected by cell death, as shown by PI staining (Fig 4A) and TEM (Appendix Fig S6D). They often enclose a core of extracellular material or SAA1 protein, as indicated by TEM and fluorescence microscopy (Fig 4A and Appendix Fig S6D). Cell clusters may thus represent sites where amyloid becomes released to the extracellular space due to cell death. Once the deposits are able to grow further even in the absence of living cells, as SAA1 protein directly enters an already existing deposit even if the cells are killed by methanol fixation (Fig 4E). This extracellular growth is only seen, however, if living cells were there initially and had sparked off the process of amyloid deposition, as cultures preincubated without SAA1 could not form initial deposits and did not promote the aggregation of SAA1 (Fig 4F). We conclude that cells need to be present at least transiently to support the nucleation of nascent deposits, which are then able to grow further even without the participation of cells.
This mechanism explains previous observations that amyloid deposits and fibril precursor protein are extracellular in systemic AA amyloidosis and that macrophages are required for the formation of amyloid deposits in vivo 21, 22 and in cell culture 23, 24, 26. Fibril formation is known to be a nucleation‐dependent process 44 that is kinetically controlled by the slow and thermodynamically unfavourable step of nucleus formation. This step is in the case of SAA1 protein additionally retarded by binding to HDL, as we show here both in the absence (Fig 6A) and in the presence of cells (Fig 4B). HDL sequesters SAA1 protein in an α‐helical conformation 15 that is not competent for amyloid formation, as the hydrogen bond donor and acceptor groups of the backbone are no longer available to form an intermolecular β‐sheet. Internalization of SAA1 protein into living cells helps to overcome this barrier, and possible explanations for this effect include relatively high local concentrations of intracellular SAA1, the exposure of SAA1 to glycans or confined space conditions 45, 46 or a disruption of the interactions between SAA1 and HDL as the protein becomes degraded 23. It will thus be interesting to learn how fibrils or amyloid‐enhancing factor, which was shown to promote fibril formation in vivo and in the cell model 23, 47, modulate the encountered cellular mechanism. One possible effect could be the uptake of AEF such that it encounters SAA1 protein inside the cell.
Several other types of systemic and localized amyloidosis have previously been reported to involve either mononuclear phagocytes, endocytotic uptake or a proteolytic processing of the amyloid precursor protein during fibril formation. Examples hereof are the misfolding of light chains in systemic AL amyloidosis 48, 49, the formation of pancreatic amyloid deposits from islet amyloid polypeptide in type II diabetes mellitus 50, the misfolding of β2‐microglobulin in dialysis‐related amyloidosis 51, the formation of apolipoprotein A‐I amyloid deposits in ApoA‐I amyloidosis 52 and the deposition of amyloid plaques from Aβ peptide or prion protein in Alzheimer's disease or transmissible spongiform encephalopathy 41, 42, 46, 53, 54. Furthermore, several amyloid precursor proteins are known to aggregate in vitro under acidic conditions, resembling the pH conditions of lysosomal compartments. Examples hereof are light chains, transthyretin and β2‐microglobin 4, 55, 56. Hence, elements of the presently described mechanism may be relevant for other protein misfolding diseases as well.
Materials and Methods
Generation of SAA1 and SAA1101Cys proteins
Murine full‐length SAA1.1 protein (Appendix Fig S3A) was recombinantly expressed in Escherichia coli RV308 cells as described elsewhere 25. In brief, the coding region of SAA1 was cloned to the 5′‐end of the gene of maltose‐binding protein in the pMAL‐c2X vector (New England Biolabs). A His‐tag and a tobacco etch virus cleavage site were inserted in between the genes for maltose binding protein and SAA1 to aid in purification. Protein purification from the cell lysate was based on column chromatography and consisted of the following steps: (i) chromatography via amylose high flow (New England Biolabs) resin, (ii) chromatography via fast flow nickel–Sepharose (GE Healthcare) medium, (iii) cleavage of the fusion protein with tobacco etch virus protease (overnight incubation at 34°C), (iv) a second nickel chelate chromatography step to separate SAA1 from the His‐tagged maltose binding protein, (v) reversed‐phase chromatography (RPC) with Source 15 RPC (GE Healthcare) medium. The purified protein was finally lyophilized with an Alpha 2–4 LD plus freeze dryer (Christ) and stored at −80°C. The gene for SAA1101Cys was generated by site‐directed mutagenesis from that of SAA1 using a QuikChange XL Site‐Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer's protocol. Recombinant expression and purification of SAA1101Cys were performed as described above for SAA1.
Generation of SAA1‐AF488 and SAA1‐AF647
2 mg of recombinant SAA1 was dissolved in 500 μl 100 mM sodium carbonate buffer pH 8. 50 μl of Alexa Fluor® 488 (AF488) or Alexa Fluor® 647 (AF647) succinimidyl ester (4 mg/ml, in dimethyl sulfoxide (DMSO), Thermo Fisher Scientific) was added to the solution and incubated in a thermostated mixer (Eppendorf) under continuous shaking (300 rpm) at room temperature (RT) for 1 h. The labelling reaction was stopped by addition of 100 μl 1.5 M hydroxyl amine pH 8.5. The solution was centrifuged (16,900 × g, 30 min), and the pellet was dissolved in 7.5 M guanidine hydrochloride and 25 mM sodium phosphate buffer pH 7.4. The supernatant and the pellet were purified separately with a 3 ml Resource RPC column (GE Healthcare) using a linear gradient from 0 to 86% (v/v) acetonitrile in 0.1% (v/v) trifluoroacetate. The concentration of fluorescently labelled SAA1 protein in the eluted fraction was determined by absorbance at 500 nm (SAA1‐AF488, ε = 71,000 M−1 cm−1) or 653 nm (SAA1‐AF647, ε = 239,000 M−1 cm−1). The protein purified from supernatant and pellet was lyophilized and stored separately at −80°C. The fluorescently labelled SAA1 variants were dissolved in DMSO and added to the medium without any filtration.
Generation of SAA1101Cys‐AF488 and SAA1101Cys‐AF594
Lyophilized SAA1101Cys protein was dissolved at a concentration of 0.4 mg/ml in 1 mM tris(2‐carboxyethyl)phosphine to prevent the formation of disulphide bonds. The pH was adjusted to a value of 5–6 with 1 M NaOH, and the sample was incubated for 45 min. The labelling reaction was started by the addition of 150 μl 60 mM sodium phosphate buffer pH 7.6 and 20 μl of AF488 or AF594 maleimide (Thermo Fisher Scientific) dyes that were dissolved in DMSO at a concentration of 10 mg/ml. After an incubation period of 2.5 h at RT, in which the sample was agitated continuously at 300 rpm, the reaction was stopped by the addition of 13 μl 0.1 M glutathione. The sample was centrifuged (16,900 × g, 30 min), and the pellet was dissolved in 7.5 M guanidinium chloride, 25 mM sodium phosphate buffer pH 7.4. The supernatant and the pellet were purified separately with a 3 ml Resource RPC column (GE Healthcare) using a linear gradient from 0 to 86% (v/v) acetonitrile in 0.1% (v/v) trifluoroacetate. The labelled protein concentration in the eluted fraction was estimated by absorbance at 500 nm (SAA1101Cys‐AF488, ε = 71,000 M−1 cm−1) or 594 nm (SAA1101Cys‐AF594, 92,000 M−1 cm−1). The protein purified from supernatant and pellet was lyophilized and stored separately at −80°C. The protein identity was verified by mass spectrometry using a Bruker REFLEX mass spectrometer (Bruker Daltonik). For SAA1101Cys‐AF488, the calculated mass/charge (m/z) ratio was 12,337 Da, while the observed m/z was 12,339 Da. For SAA1101Cys‐AF594, the calculated m/z ratio was 12,526 Da, while the observed m/z was 12,527 Da. All solutions were thoroughly degassed by purging them with argon for 30 min. The fluorescently labelled SAA1 variants were dissolved in DMSO and added to the medium without any filtration.
Generation of biotinylated SAA1
2 mg of recombinant SAA1 was dissolved in 500 μl of 100 mM sodium carbonate buffer (pH 8) and supplemented with 50 μl of 32 mg/ml EZ‐Link™ NHS‐Biotin (Thermo Scientific) in DMSO. This solution was incubated for 30 min at RT in a thermostated mixer under continuous shaking (250 rpm). The solution was centrifuged (16,900 × g, 30 min), and the pellet was dissolved in 500 μl 7.5 M guanidinium chloride, 25 mM sodium phosphate buffer pH 7.4. The supernatant and the pellet were purified separately with a 3 ml Resource RPC column (GE Healthcare) using a linear gradient from 0 to 86% (v/v) acetonitrile in 0.1% (v/v) trifluoroacetate. The protein purified from supernatant and pellet were lyophilized and stored separately at −80°C. The biotinylated SAA1 was dissolved in water and added to the medium without any filtration.
General cell culture methods
J774A.1 cells (Sigma‐Aldrich) were plated out at a density of 350,000 cells/ml in 96‐well plates (Greiner Bio‐One) unless indicated otherwise. Cells were grown on glass cover slips (Thermo Fisher Scientific) or sapphire discs (diameter: 3 mm; Engineering Office Wohlwend) depending on whether they were further analysed by light microscopy or electron microscopy. Sapphire discs were coated with a 20‐nm carbon layer using a Balzers BAF 300 instrument (Bal‐Tec), incubated overnight in an oven (120°C) and sterilized by UV irradiation at 320 nm for 10 min immediately prior to use. Cells dedicated for flow cytometry were plated out in 24‐well plates (Greiner Bio‐One) and at a density of 400,000 cell/ml, while cells dedicated for LSM experiments were incubated in 8‐well Nunc™ Lab‐Tek™ II slides (Thermo Fisher Scientific). All cells were cultured at 37°C in an atmosphere of 5% (v/v) CO2 using Dulbecco's modified Eagle's medium (Life Technologies) which we supplemented with 10% (v/v) heat‐inactivated FBS (Life Technologies) and 1% (v/v) antibiotic–antimycotic solution (Life Technologies). After plating out the cells, they were incubated without SAA1 and HDL for 24 h unless stated otherwise, before the medium was replaced with fresh medium containing soluble SAA1 and HDL that were added from the stock solutions as indicated in the respective experiments. Medium and supplements were generally replenished every 2–3 days until the end of an experiment.
Preparation of the SAA1 stock
The SAA1 stock was prepared by dissolving the recombinant protein at a concentration of 10 mg/ml in pure water. Residual trifluoroacetate from the purification was removed by two filtration steps through a 3‐kDa membrane filter (Amicon Ultra‐0.5 ml 3K, Merck Millipore) and centrifugation at 14,000 × g for 10 min at 4°C. The retentate was filled up with water to the original volume, and the protein was eluted from the membrane by inversion of the filter and centrifugation step at 1,000 × g for 1 min at 4°C. The stock solution was stored in frozen aliquots that were thawed immediately prior to addition to the cell culture medium. The added amount was adjusted to reach the desired final SAA1 concentration.
Preparation of the HDL stock
The HDL stock was obtained commercially (Applichem) and stored in frozen aliquots. Its concentration was estimated based on the triglyceride content provided by the vendor. The aliquots were thawed immediately prior to their addition to the medium. The added amount was adjusted such that the final triglyceride concentration (in mg/ml) in the medium corresponded to 9% (w/w) of the SAA1 protein concentration also expressed in mg/ml.
General flow cytometry methods
After plating out the cells, we kept them without SAA1 and HDL for 42 h, before the medium was replaced with 350 μl medium containing 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF647 and HDL as indicated in the experiment. At the end of each experiment, the medium was removed and the cells were trypsinized. Afterwards, the cells were scraped off from each well with a cell scraper (TPP) and transferred into a separate 1.5‐ml tube. The cell suspension was centrifuged at 200 × g for 5 min at 4°C, and the supernatant was discarded. The cell pellet was resuspended in 1 ml flow cytometry buffer containing 0.5% (w/v) bovine serum albumin, 0.1% (w/v) sodium azide and PBS (137 mM NaCl, 2.7 mM KCl, 8 mM di‐sodium hydrogen phosphate, 2 mM potassium dihydrogen phosphate‐buffered saline pH 7.4; Thermo Fisher Scientific) and centrifuged once more for 5 min at 200 × g and 4°C. The cell pellet was resuspended in 200 μl flow cytometry buffer containing 2% (w/v) paraformaldehyde (Carl Roth) and incubated for 15 min at RT to fix the cells. Afterwards, the cells were centrifuged again and resuspended in 200 μl flow cytometry buffer. They were stored at 4°C overnight and transferred on the next day into flow cytometry tubes (Sarstedt) for analysis in a BD FACSVerse™ flow cytometer (BD Biosciences) using an excitation (EX) wavelength of 633 nm and an emission (EM) filter with a range of 655–665 nm. For each sample, 10,000 events were measured if not indicated otherwise. The data were analysed using FlowJo (FlowJo, LLC) software.
General laser scanning microscopy (LSM) methods
After plating out the cells, we kept them without SAA1 and HDL for 42 h, before the medium was replaced with 200 μl fresh medium containing 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF488 and HDL as indicated in each experiment. Immediately before LSM analysis, the medium was removed and the extracellular proteins were degraded by trypsinization. After addition of 200 μl PBS, we imaged the cells at 37°C in an atmosphere of 5% (v/v) CO2 using a LSM710 confocal microscope (Carl Zeiss) using an EX wavelength of 405 nm and an EM filter range from 410 to 460 nm to monitor autofluorescence and an EX wavelength of 488 nm and an EM filter range from 494 to 553 nm to monitor SAA1‐AF488, or using an Eclipse Ti‐E LSM (Nikon) and an EX wavelength of 488 and an EM filter range from 500 to 550 nm to monitor SAA1‐AF488. Bright field images were recorded using a transmission detector. Uptake of fluorescently labelled SAA1 into cells was quantified by using ImageJ software (National Institutes of Health).
Measurement of cellular FRET levels with LSM
After plating out the cells, we kept them without SAA1 and HDL for 24 h, before the medium was replaced with 200 μl medium containing 48 μM SAA1, 1 μM SAA1101Cys‐AF488, 1 μM SAA1101Cys‐AF594 and HDL. The medium was removed after 4, 24, 48 or 72 h. Extracellular proteins were removed by trypsinization. Cells were analysed at 37°C in 200 μl fresh medium and in an atmosphere of 5% (v/v) CO2. A LSM710 confocal microscope (Carl Zeiss) was used to subsequently scan a given area of the culture using an EX wavelength of 488 nm and an EM filter range from 500 to 545 nm (donor; I D 488), an EX wavelength of 488 nm and an EM filter range from 610 to 645 nm (s.a. sensitized acceptor; I A 488), an EX wavelength of 561 nm and an EM filter range from 610 to 645 nm (acceptor; I A 561) or an EX wavelength of 405 nm and an EM filter range from 410 to 460 nm (autofluorescence). The original magnification was 63‐fold. The FRET efficiency E was calculated pixel by pixel from the I A 488 image according to following formula (1) using IGOR Pro (Wavemetrics):
| (1) |
In this equation, γ is a correction factor, while I A was calculated from the images , and according to formula (2).
| (2) |
In this equation, c CT accounts for the donor fluorescence cross talk, while c AE corrects for the direct acceptor excitation at 488 nm. To obtain c CT, we prepared fibrils in vitro by incubation of 49 μM SAA1 and 1 μM SAA1101Cys‐AF488 in 10 mM Tris pH 8 for 6 days at 300 rpm and at 37°C. We then recorded LSM images of this sample using the four EX/EM settings described above. The factor cCT represents the ratio of and as determined from the in vitro fibrils:
| (3) |
To calculate this ratio, we excluded all pixels where either or was below 2% of the dynamic range of the detector. The ratios of the intensities of the remaining pixels were averaged. To obtain c AE , we prepared fibrils in vitro by incubation of 49 μM SAA1 and 1 μM SAA1101Cys‐AF594 in 10 mM Tris pH 8 for 6 days at 300 rpm and at 37°C and recorded LSM images using the above EX and EM channel settings. The factor cAE represents the ratio of and as determined from the second sample of in vitro fibrils:
| (4) |
To calculate c AE, we excluded all pixels where either or was below 2% of the dynamic range of the detector. The ratios of the intensities of the remaining pixels were averaged. The correction factor γ was calculated according to the following equation:
| (5) |
In this formula, ϕ D and ϕ A represent the quantum yields of donor and acceptor. T D and T A represent the spectral transmission of the donor and acceptor channel, while c Det accounts for the different detection efficiencies in both channels. The quantum yields of donor (ϕ D = 0.38) and acceptor (ϕ A = 0.48) were determined by comparison with the quantum yield of the standard Rhodamine 6G in ethanol (ϕR6G = 0.95) 57. T D and T A were calculated from the fluorescence emission spectra of 1 μM freshly dissolved SAA1101Cys‐AF488 and SAA1101Cys‐AF594, respectively. The spectra were measured within a LS55 fluorescence spectrometer (Perkin Elmer) at RT and normalized such that the area integral intensity below the spectrum becomes 1. The normalized spectrum of SAA1101Cys‐AF488 was integrated between 500 and 545 nm to obtain T D and of SAA1101Cys‐AF594 between 610 and 645 nm to obtain T A. The factor c Det was calculated using the following formula:
| (6) |
represents the area integral between 610 and 645 nm of the normalized spectrum of SAA1101Cys‐AF488.
To analyse the effects of uptake inhibitors on the intracellular formation of fibrils as determined by FRET, the medium was replaced 24 h after plating the cells with 200 μl medium containing 48 μM SAA1, 1 μM SAA1101Cys‐AF488, 1 μM SAA1101Cys‐AF594 and HDL. This medium was replaced after 24 h with 200 μl fresh medium containing the respective uptake inhibitors without SAA1 and HDL according to the concentration and incubation time as described in the section “Use of pharmacological inhibitors to block SAA1 uptake”. The medium was replaced with 200 μl fresh medium containing 48 μM SAA1, 1 μM SAA1101Cys‐AF488, 1 μM SAA1101Cys‐AF594, HDL and the respective inhibitor. After 5 h, the medium was exchanged with 200 μl fresh medium containing SAA1 and HDL and incubated for another 17 h. The medium was discarded. The extracellular proteins were removed by trypsinization, and cells were analysed in 200 μl fresh medium at 37°C in an atmosphere containing 5% (v/v) CO2 using a LSM710 confocal microscope (Carl Zeiss).
Measurement of in vitro fibril assembly by FRET and centrifugation
A 48:1:1 μM mixture of SAA1, SAA1101Cys‐AF488 and SAA1101Cys‐AF594 in 10 mM Tris pH 8.0 was incubated at 37°C in a thermostatic mixer (Eppendorf) with continuous agitation (300 rpm). At different time points, aliquots (160 μl) were withdrawn from this mixture and analysed using a LS55 fluorescence spectrometer (Perkin Elmer). All EX spectra from 350 to 625 nm were recorded at an EM wavelength of 630 nm, and all EM spectra were recorded from 490 to 730 nm using an EX wavelength of 480 nm in a microcell cuvette at 37°C. Subsequently, 120 μl of each aliquot was centrifuged (16,000 × g, 30 min), and the supernatant was removed and diluted with 180 μl 10 mM Tris pH 8.0. The pellet was dissolved in 300 μl 9 M urea, 10 mM Tris pH 8.0. Afterwards, absorbance spectra at 280 nm of the supernatant and pellet sample were recorded between 200 and 800 nm using a Lambda 35 spectrometer (Perkin Elmer).
Lysosomal co‐localization
To test for lysosomal co‐localization, cells were incubated for 1 day with 200 μl fresh medium containing 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF488 and HDL. Extracellular proteins were removed by trypsinization. Afterwards, the cells were fixed and stained for 1 h at RT with 50 nM LysoTracker® Red DND‐99 (Life Technologies) in PBS. The staining solution was removed, and the cells were imaged in 200 μl PBS using a LSM710 confocal microscope (Carl Zeiss) and an EX wavelength of 405 nm and an EM filter range from 410 to 460 nm to monitor autofluorescence, an EX wavelength of 488 nm and an EM filter range from 494 to 553 nm to monitor SAA1‐AF488 as well as an EX wavelength of 561 nm and EM filter range from 574 to 620 nm to monitor LysoTracker® Red DND‐99 fluorescence.
Removal of extracellular protein by trypsinization
The medium of the cells was replaced with 100 μl of a commercial available trypsin–ethylenediaminetetraacetic acid (trypsin–EDTA) solution (Invitrogen). After 10 s, the trypsin–EDTA solution was removed, and the cells were incubated for 5 min at 37°C. The trypsinization reaction was stopped by the addition of 200 μl medium.
Use of pharmacological inhibitors to block SAA1 uptake
After plating out the cells, we kept them in the absence of SAA1, HDL and inhibitors. After 42 h, the cells were further incubated with fresh medium containing inhibitors as detailed here: 9 μg/ml chlorpromazine for 30 min 58, 100 μg/ml colchicine for 2 h 59, 3 μg/ml cytochalasin B for 2 h 60, 10 μM dimethylamiloride for 30 min 61, 80 μM dynasore for 30 min 59, 10 μM dynole® 34‐2 for 30 min 62, 100 μM genistein for 30 min 63, 25 ng/ml latrunculin A for 30 min 64, 100 μM MDC for 10 min 65, 10 μg/ml nocodazole for 1 h 66, 50 μg/ml nystatin for 15 min 59 and 2 μM rottlerin for 30 min 59. All inhibitors were purchased from Sigma, Applichem or Abcam and initially dissolved in DMSO as 200‐fold concentrated stocks. After this initial incubation step with the inhibitors that varied in length, we replaced the medium with fresh medium containing the respective inhibitors as well as 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF488 and HDL and incubated the cells for another 5 h.
Co‐localization of SAA1 with endocytic uptake pathway
After plating out the cells, we kept them without SAA1 and HDL for 24 h, before the medium was replaced with 200 μl medium containing 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF488, HDL and 25 μg/ml AF647 labelled transferrin (Life Technologies) for 30 min. Afterwards the cells were washed with 200 μl PBS and imaged in 200 μl fresh PBS using an Eclipse Ti‐E LSM (Nikon) and EX wavelength of 488 nm and an EM filter range from 500 to 550 nm to monitor SAA1‐AF488 and an EX wavelength of 640 nm and an EM filter range from 650 to 1,000 nm to monitor transferrin. Bright field images were recorded using a transmission detector.
Terminal deoxynucleotidyl transferase‐mediated deoxyuridine 5′‐triphosphate nick end labelling (TUNEL) assay
After plating out the cells, we kept them without SAA1 and HDL for 24 h, before the medium was replaced with 200 μl medium containing 0.5 mg/ml SAA1 and HDL for 3 days. The TUNEL assay was performed using the Cell Meter™ TUNEL Apoptosis Assay Kit (ATT Bioquest®) according to the manufacturer's protocol. Samples were imaged using a LSM710 confocal microscope (Carl Zeiss) and using an EX wavelength of 405 nm and an EM filter range from 410 to 460 nm to monitor autofluorescence and an EX wavelength of 461 nm and EM filter range of 574–608 nm to monitor the TUNEL fluorescence.
Lysosomal leakage assay
After plating out the cells, we kept them without SAA1 and HDL for 24 h, before the medium was replaced with 100 μl fresh medium supplemented with 0.5 mg/ml SAA1 and HDL. This medium was removed after 3, 5, 24 and 48 h and replaced with 40 μl fresh medium containing no FBS but 5 μg/ml AO (Sigma‐Aldrich). After a 15‐min incubation step at RT, the medium was discarded and the cells were washed twice with 100 μl PBS for 5 s, trypsinized, scraped off and transferred into flow cytometry tubes. AO fluorescence was analysed by using the BD VACSVerse™ flow cytometer (10,000 events) and an EX wavelength of 488 nm and an EM filter2 with a range of 673–727 nm. Cells exhibiting lysosomal leakage show low AO fluorescence (AOlow).
Propidium iodide (PI) staining
After plating out the cells, we kept them without SAA1 and HDL for 24 h, before the medium was replaced with 200 μl fresh medium containing 0.5 mg/ml SAA1, 0.01 mg/ml SAA1‐AF488 and HDL. After 6 days of incubation, 4 μl of a 1 mg/ml PI stock solution was added to the medium to reach a final concentration of 20 μg/ml. Fluorescence images were captured immediately after addition of PI for a maximum of 5 min. Images were gathered at 37°C in an atmosphere of 5% (v/v) CO2 using an Eclipse Ti‐E LSM (Nikon) and an EX wavelength of 488 nm and EM filters with a range of 500–550 nm to monitor SAA1‐AF488 and 570–620 nm to monitor PI fluorescence. Bright field images were recorded using a transmission detector.
Caspase‐3/7 activity assay
After plating out the cells, we kept them without SAA1 and HDL for 24 h, before the medium was replaced with 100 μl fresh medium supplemented with 0.5 mg/ml SAA1 and HDL as indicated. The caspase‐3/7 activity was determined after incubation for 1 or 2 days using the SensoLyte Homogeneous Rh110 Caspase‐3/7 Assay Kit (AnaSpec) according to the manufacturer's protocol. Fluorescence was measured with a FLUOstar OMEGA plate reader (BMG Labtech) using an EX/EM combination of 485/520 nm.
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay
After plating out the cells, we kept them without SAA1 and HDL for 24 h, before the medium was replaced with 100 μl medium containing 0.5 mg/ml SAA1 and HDL as indicated. The medium was replenished, together with all additives, every 48 or 72 h. To analyse the effect of uptake inhibitors on the cellular viability, cells were seeded at a density of 400,000 cells/ml in a 96‐well plate. After an incubation period of 24 h, the medium was replaced with 100 μl medium containing the uptake inhibitors using the respective concentrations and incubations times. The cell viability was determined after different time points as indicated in the experiment using an MTT assay (Cell Proliferation Kit I, Roche) according to the manufacturer's protocol.
Transmission electron microscopy (TEM)
5 μl of the sample solution was placed onto a formvar–carbon‐coated copper grid (Plano) for 1 min. The grid was washed trice with 50 μl water and stained trice with 50 μl 2% (w/v) uranyl acetate. Samples were examined at 120 kV using a JEM‐1400 electron microscope (Jeol) with a VELETA 2k × 2k side‐mounted TEM camera (Olympus).
TEM analysis of ultrathin sections
Sapphire discs with attached cells/amyloid deposits were removed from the 96‐well plates and plunged into 95% (v/v) 1‐hexadecene (Sigma‐Aldrich). Two sapphire discs were stacked up face‐to‐face, separated by a gold ring (3.05 mm diameter, central bore 2 mm, Plano) and mounted in a holder (Engineering Office M. Wohlwend). The holder was inserted into a HPF Compact 01 high‐pressure freezer (Engineering Office M. Wohlwend). After disassembling the stack, each sapphire disc was placed in a precooled (−90°C) 1.5‐ml tube. To each tube, we added 1 ml of a −90°C cold freeze substitution solution (0.2% (w/v) osmium tetroxide, 0.1% (w/v) uranyl acetate and 5% (v/v) water in acetone). The temperature of the tubes was raised from −87°C to 0°C over a period of 24 h and then further to RT within 1 h using a special designed computer‐controlled substitution apparatus 67. Each sample was washed with 1 ml acetone, and the cells were embedded into resin by placing them consecutively for 1 h each in 1 ml 33% (v/v), 50% (v/v) and 66% (v/v) epoxy resin (Fluka) in acetone and 100% (v/v) epoxy resin. After a 24‐h incubation period at RT, each sapphire disc was transferred into a new 0.5‐ml tube containing 250 μl 100% (v/v) fresh epoxy resin and incubated at 60°C for 24 h to polymerize the resin. 70‐nm‐thick sections from each resin block were prepared using a Ultracut UCT ultra microtome (Leica) with a diamond knife (Diatome). The slices were collected on formvar–carbon‐coated copper grids (200 mesh, Plano) and analysed at 120 kV using a JEM‐1400 electron microscope (Jeol).
Scanning electron microscopy (SEM)
In the experiment with the biotinylated fibrils (see Appendix Fig S8), after plating out the cells, we kept them without SAA1 and HDL for 24 h, before the medium was replaced with 100 μl medium containing 1.0 mg/ml SAA1, 0.2 mg/ml biotinylated SAA1 and HDL. After a 24‐h incubation period, the medium was removed and the extracellular proteins were removed by trypsinization. Hundred microlitres medium supplemented with 1.0 mg/ml SAA1 and HDL were added and exchanged after 48 h altogether with SAA1 and HDL. After 96 h, the medium was removed and the samples were incubated for 20 min at RT with 100 μl 1% (w/v) BSA solution in PBS. The solution was replaced with 100 μl blocking solution supplemented with 5% (v/v) gold‐conjugated streptavidin (10 nm; Thermo Fisher Scientific) and incubated at RT for 30 min. The samples were washed three times with 100 μl PBS for 5 min and incubated at RT for 3 h with 100 μl 0.1 M sodium phosphate buffer pH 7.3 supplemented with 2.5% (w/v) of glutaraldehyde and 1% (w/v) sucrose. Samples were washed for 2 min with 100 μl PBS and dehydrated by plunging them into a series of flasks containing 100 ml 30% (v/v), 50% (v/v), 70% (v/v) propanol for 5 min, 90% (v/v) propanol for 2 min and 100% (v/v) propanol for 10 min each. The dehydrated samples were critical point dried using a CPD BalTec 030 Critical Point Dryer (Leica) and coated by electron beam evaporation with a 20‐nm carbon layer using a Balzers BAF 300 (Bal‐Tec). Samples were analysed using a Hitachi S‐5200 scanning electron microscope (Hitachi) and an yttrium–aluminium–garnet‐backscattered electron detector (Hitachi).
In other experiments (see Appendix Fig S6), the samples were sputtered with gold/palladium (20 nm thickness) in a Sputter Coater (MED010, Balzers) and analysed using a Zeiss 962 SEM (Zeiss) at 20‐kV acceleration voltage and a working distance of 11 mm.
Circular dichroism (CD) measurements
Lyophilised protein was dissolved in water at a concentration of 10 mg/ml (see Appendix Fig S2A) or of 1 mg/ml (see Appendix Fig S2B). Protein concentration was determined by absorption at 280 nm 68 and diluted to a final concentration of 0.5 mg/ml (see Appendix Fig S2A) or 0.3 mg/ml (see Appendix Fig S2B) with Tris buffer (pH 8). All spectra were acquired with a J‐810 CD spectrometer (Jasco Analytical Instruments) and a 0.1‐mm quartz cuvette (SUPRASIL® 106‐QS) or a 1‐mm quartz cuvette (SUPRASIL® 110‐QS). We used a band width of 1 nm and a scanning speed of 100 nm/min. Resulting spectra represent the average of 20 accumulations. The mean residue weight ellipticity [θ]MRW was calculated using the measured ellipticity θ, the mean residue weight MRW, cuvette path length d and the protein concentration c according to formula (7). The MRW is the protein molecular weight divided by the number of peptide bonds.
| (7) |
Secondary structural composition was analysed using the programs CDSSTR, ContinLL and Selcon3 (http://sites.bmb.colostate.edu/sreeram/CDPro/). Results of all three programs were averaged and represented.
Congo red (CR) absorbance spectroscopy
All measurements were carried out with a Lambda 35 UV/VIS spectrometer (Perkin Elmer) in a SUPRASIL® Ultra‐Micro quartz UV/VIS cuvette (Type No.: 105.201‐QS, Hellma) at RT. The absorbance spectra were recorded from 200 to 700 nm, using a slit width of 1 nm and a scan speed of 480 nm/min with 1‐nm intervals. Each sample had a final volume of 200 μl and contained 0.5 mg/ml freshly dissolved (soluble) SAA1 protein or 0.25 mg/ml SAA1 fibrils, 10 μM CR and 10 mM Tris buffer (pH 8), as indicated. We used half the fibril concentration compared with soluble SAA1 to not exceed the detector limit.
CR green birefringence
Cells were grown on glass cover slips in medium containing 0.5 mg/ml SAA1 and HDL for up to 6 days, as indicated. At the end of the experiment, the medium was removed. After washing each well with 100 μl PBS, we added 100 μl ice‐cold methanol to each well and incubated the samples for 10 min at 4°C. The methanol was replaced with 100 μl CR solution which consisted of 80% (v/v) ethanol, 3% (w/v) NaCl and 0.6% (w/v) CR (Carl Roth). The plate was kept for 45 min on an orbital platform shaker (Heidolph Rotamax 120) that was constantly agitated at 75 rpm. The CR solution was removed, and each well was washed thrice with 100 μl water. Subsequently, the samples were stained for 2 min at RT with 100 μl Mayer's Hemalaun solution (Roth) and washed with 100 μl 70% (v/v) ethanol and thrice with 100 μl water. The glass cover slips were then removed from the wells and plunged thrice into 90% (v/v) ethanol, thrice into 100% (v/v) ethanol and twice into 100% (v/v) xylol (1 sec each). The glass cover slips were mounted on microscopic slides using Roti®‐Histokitt (Carl Roth) and examined with an Eclipse 80i polarizing microscope (Nikon). While CR green birefringence does not intrinsically discriminate intra‐ from extracellular amyloid species, only extracellular deposits are usually large enough to give a strong signal within this method.
Thioflavin T (ThT) fluorescence spectroscopy
All fluorescence spectra at 450 nm were recorded between 460 and 700 nm using a LS 55 fluorescence spectrometer (Perkin Elmer). All samples had a volume of 200 μl and contained 0.5 mg/ml freshly dissolved (soluble) SAA1 protein or 0.25 mg/ml SAA1 fibrils, 20 μM ThT and 10 mM Tris buffer (pH 8) or cell culture medium, as indicated. We used half the fibril concentration compared with soluble SAA1 to not exceed the detector limit. Samples were measured in a SUPRASIL® Micro quartz fluorescence cuvette (Type No.: 105.253‐QS, Hellma), accumulating three scans per sample and using a scan speed of 100 nm/min. The EX and EM slits were both set to 7 nm, and all spectra were recorded at RT.
ThT fibrillation kinetics measurements
Measurements were carried out in a black 96‐well plate (Greiner Bio‐One) at 37°C using a FLUOstar OMEGA plate reader (BMG Labtech). A 10 mg/ml SAA1 stock solution was obtained by dissolving recombinant SAA1 in water. The final sample volume in each well was 100 μl and samples contained 10 mM Tris buffer pH 8, 50 μM SAA1, 20 μM ThT and 52 μg/ml HDL as indicated in the experiment. Every 20 min the sample was agitated by orbital shaking for 10 s at 100 rpm and the fluorescence was measured in each well (EX/EM: 450/490 nm) over a period of 140 h.
ThT aggregation kinetics of SAA1101Cys‐AF594
Samples consisted of 49 μM SAA1, 1 μM SAA1101Cys‐AF594 and 10 μM ThT in 10 mM Tris buffer pH 8.0 and had a volume of 100 μl. They were incubated in a black 96‐well plate (Greiner Bio‐One) that was analysed for 120 h at 37°C under continuous agitation (100 rpm) in a FLUOstar OMEGA plate reader (BMG Labtech). The fluorescence was measured every 30 min using the EX/EM settings: 450/490 nm (ThT fluorescence) and 450/620 nm (FRET). The lag time was determined as described elsewhere 69.
Size exclusion chromatography (SEC)
SEC was performed using an Äkta Purifier system (GE Healthcare) and a Superdex 200 5/150 GL column (GE Healthcare) equilibrated and run with 10 mM sodium phosphate, 150 mM NaCl (pH 7.4). The column was calibrated with blue dextran and Gel Filtration Low Molecular Weight and High Molecular Weight Kits (GE Healthcare) that we used according to the manufacturer. 30 μl of sample was injected and detected by absorption at 215 nm and 280 nm using a flow rate of 0.3 ml/min.
Western blot
Proteins were separated on NuPAGE® 4–12% (w/v) Bis–Tris gradient gels (Thermo Fisher Scientific) using NuPAGE® MES LDS running buffer (Thermo Fisher Scientific). Samples were mixed with 4× NuPAGE® LDS sample buffer (Thermo Fisher Scientific) and denatured by heating for 10 min at 95°C. Subsequently, proteins were blotted onto 0.45‐μm Whatman® Protran® BA 85 nitrocellulose membrane (GE Healthcare) using a Trans‐Blot® Semi‐Dry transfer system (Bio Rad) (35 min, 20 V). The membrane was equilibrated with transfer buffer (5 ml 20× NuPAGE® transfer buffer, 20 ml methanol, 75 ml H2O). Afterwards, the membrane was blocked for 1 h with 5% (w/v) milk in PBS at RT. Primary anti‐mSAA1 antibody (1:400; Pineda) was applied in PBST containing 5% (w/v) milk powder and 0.1% (v/v) Tween and incubated at RT for 1 h. The secondary antibody (anti‐rabbit‐HRP, 1:1,000; Dako) was applied in PBST containing 5% (w/v) milk and 0.1% (v/v) Tween and incubated at RT for 1 h. After each antibody incubation step, the membrane was washed trice with PBST containing 0.1% (v/v) Tween for 10 min. Detection was performed using the SuperSignal® West‐Kit (Thermo Scientific).
Statistical analysis
Error bars represent the standard deviation (SD), and results were analysed by the Student's t‐test (unpaired, unequal variances).
Author contributions
SC, KM, TA, IP‐G, JL and CH performed the research. SC, KM, TA, IP‐G, CH, TSy, TSi, PW and MF analysed the data. CH, TSy, TSi and PW contributed reagents and techniques. MF designed the research. SC, KM and MF wrote the manuscript. All authors commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
The work was supported by grants from the Deutsche Forschungsgemeinschaft to M.F. and C.H. (FA 456/15‐1 and HA 7138/2‐1). S.C. acknowledges a stipend from the Carl Zeiss Foundation. I.P.‐G. was supported by the International Graduate School in Molecular Medicine Ulm. The authors thank Anastasia Weber and Beate Garbers for helping in protein purification, Dr. Angelika Schierhorn (Martin‐Luther‐Universität Halle‐Wittenberg) for mass spectrometry and the Core Facility Konfokale und Multiphotonen Mikroskopie (Ulm University) for technical advice.
EMBO Reports (2017) 18: 1352–1366
Notes
Correction added on 1 August 2017, after first online publication: the detection method was corrected.
Correction added on 1 August 2017, after first online publication: “EX filter” was changed to “EM filter”.
References
- 1. Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75: 333–366 [DOI] [PubMed] [Google Scholar]
- 2. Merlini G, Bellotti V (2003) Molecular mechanisms of amyloidosis. N Engl J Med 349: 583–596 [DOI] [PubMed] [Google Scholar]
- 3. Eisenberg D, Jucker M (2012) The amyloid state of proteins in human diseases. Cell 148: 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xue WF, Homans SW, Radford SE (2008) Systematic analysis of nucleation‐dependent polymerization reveals new insights into the mechanism of amyloid self‐assembly. Proc Natl Acad Sci USA 105: 8926–8931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15: 384–396 [DOI] [PubMed] [Google Scholar]
- 6. Westermark G, Fändrich M, Westermark P (2015) AA amyloidosis: pathogenesis and targeted therapy. Annu Rev Pathol Mech Dis 10: 321–344 [DOI] [PubMed] [Google Scholar]
- 7. Hazenberg BP, van Gameren II, Bijzet J, Jager PL, van Rijswijk MH (2004) Diagnostic and therapeutic approach of systemic amyloidosis. Neth J Med 62: 121–128 [PubMed] [Google Scholar]
- 8. Pepys MB (2006) Amyloidosis. Annu Rev Med 57: 223–241 [DOI] [PubMed] [Google Scholar]
- 9. Caughey B, Lansbury PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26: 267–298 [DOI] [PubMed] [Google Scholar]
- 10. Röcken C, Shakespeare A (2002) Pathology, diagnosis and pathogenesis of AA amyloidosis. Virchows Arch 440: 111–122 [DOI] [PubMed] [Google Scholar]
- 11. Uhlar CM, Whitehead AS (1999) Serum amyloid A, the major vertebrate acute‐phase reactant. Eur J Biochem 265: 501–523 [DOI] [PubMed] [Google Scholar]
- 12. Röcken C, Kisilevsky R (1998) Comparison of the binding and endocytosis of high‐density lipoprotein from healthy (HDL) and inflamed (HDL(SAA)) donors by murine macrophages of four different mouse strains. Virchows Arch 432: 547–555 [DOI] [PubMed] [Google Scholar]
- 13. Ye RD, Sun L (2015) Emerging functions of serum amyloid A in inflammation. J Leukoc Biol 98: 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patke S, Srinivasan S, Maheshwari R, Srivastava SK, Aguilera JJ, Colón W, Kane RS (2013) Characterization of the oligomerization and aggregation of human Serum Amyloid A. PLoS ONE 8: e64974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jayaraman S, Haupt C, Gursky O (2015) Thermal transitions in serum amyloid A in solution and on the lipid: implications for structure and stability of acute‐phase HDL. J Lipid Res 56: 1531–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCubbin WD, Kay CM, Narindrasorasak S, Kisilevsky R (1988) Circular‐dichroism studies on two murine serum amyloid A proteins. Biochem J 256: 775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smetana H (1927) The relation of the reticulo‐endothelial system to the formation of amyloid. J Exp Med 45: 619–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takahashi M, Yokota T, Kawano H, Gondo T, Ishihara T, Uchino F (1989) Ultrastructural evidence for intracellular formation of amyloid fibrils in macrophages. Virchows Arch A Pathol Anat Histopathol 415: 411–419 [DOI] [PubMed] [Google Scholar]
- 19. Shirahama T, Cohen AS (1975) Intralysosomal formation of amyloid fibrils. Am J Pathol 81: 101–116 [PMC free article] [PubMed] [Google Scholar]
- 20. Shirahama T, Miura K, Ju ST, Kisilevsky R, Gruys E, Cohen AS (1990) Amyloid enhancing factor‐loaded macrophages in amyloid fibril formation. Lab Invest 62: 61–68 [PubMed] [Google Scholar]
- 21. Lundmark K, Vahdat Shariatpanahi A, Westermark GT (2013) Depletion of spleen macrophages delays AA amyloid development: a study performed in the rapid mouse model of AA amyloidosis. PLoS ONE 8: e79104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kennel SJ, Macy S, Wooliver C, Huang Y, Richey T, Heidel E, Wall JS (2014) Phagocyte depletion inhibits AA amyloid accumulation in AEF‐induced huIL‐6 transgenic mice. Amyloid 21: 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kluve‐Beckerman B, Liepnieks JJ, Wang L, Benson MD (1999) A cell culture system for the study of amyloid pathogenesis. Amyloid formation by peritoneal macrophages cultured with recombinant serum amyloid A. Am J Pathol 155: 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elimova E, Kisilevsky R, Szarek WA, Ancsin JB (2004) Amyloidogenesis recapitulated in cell culture: a peptide inhibitor provides direct evidence for the role of heparan sulfate and suggests a new treatment strategy. FASEB J 18: 1749–1751 [DOI] [PubMed] [Google Scholar]
- 25. Gellermann GP, Appel TR, Tannert A, Radestock A, Hortschansky P, Schroeckh V, Leisner C, Lütkepohl T, Shtrasburg S, Röcken C et al (2005) Raft lipids as common components of human extracellular amyloid fibrils. Proc Natl Acad Sci USA 102: 6297–6302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kollmer M, Meinhardt K, Haupt C, Liberta F, Wulff M, Linder J, Handl L, Heinrich L, Loos C, Schmidt M et al (2016) Electron tomography reveals the fibril structure and lipid interactions in amyloid deposits. Proc Natl Acad Sci USA 113: 5604–5609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Derebe MG, Zlatkov CM, Gattu S, Ruhn KA, Vaishnava S, Diehl GE, MacMillan JB, Williams NS, Hooper LV (2014) Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. eLife 3: e03206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu J, Yu Y, Zhu I, Cheng Y, Sun PD (2014) Structural mechanism of serum amyloid A‐mediated inflammatory amyloidosis. Proc Natl Acad Sci USA 111: 5189–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Westermark P (2014) Nomenclature 2014: amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 21: 221–224 [DOI] [PubMed] [Google Scholar]
- 30. Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA et al (2012) Direct observation of the interconversion of normal and toxic forms of α‐synuclein. Cell 149: 1048–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tosatto L, Horrocks MH, Dear AJ, Knowles TP, Dalla Serra M, Cremades N, Dobson CM, Klenerman D (2015) Single‐molecule FRET studies on alpha‐synuclein oligomerization of Parkinson's disease genetically related mutants. Sci Rep 5: 16696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mettlen M, Pucadyil T, Ramachandran R, Schmid SL (2009) Dissecting dynamin's role in clathrin‐mediated endocytosis. Biochem Soc Trans 37: 1022–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Claus S, Puscalau‐Girtu I, Walther P, Syrovets T, Simmet T, Haupt C, Fändrich M (2017) Cell‐to‐cell transfer of SAA1 protein in a cell culture model of systemic AA amyloidosis. Sci Rep 7: 45683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kluve‐Beckerman B, Manaloor J, Liepnieks JJ (2001) Binding, trafficking and accumulation of serum amyloid A in peritoneal macrophages. Scand J Immunol 53: 393–400 [DOI] [PubMed] [Google Scholar]
- 35. Röcken C, Menard R, Bühling F, Vöckler S, Raynes J, Stix B, Krüger S, Roessner A, Kähne T (2005) Proteolysis of serum amyloid A and AA amyloid proteins by cysteine proteases: cathepsin B generates AA amyloid proteins and cathepsin L may prevent their formation. Ann Rheum Dis 64: 808–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chronopoulos S, Laird DW, Ali‐Khan Z (1994) Immunolocalization of serum amyloid A and AA amyloid in lysosomes in murine monocytoid cells: confocal and immunogold electron microscopic studies. J Pathol 173: 361–369 [DOI] [PubMed] [Google Scholar]
- 37. Okoshi T, Yamaguchi I, Ozawa D, Hasegawa K, Naiki H (2015) Endocytosed 2‐microglobulin amyloid fibrils induce necrosis and apoptosis of rabbit synovial fibroblasts by disrupting endosomal/lysosomal membranes: a novel mechanism on the cytotoxicity of amyloid fibrils. PLoS ONE 10: e0139330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Monis GF, Schultz C, Ren R, Eberhard J, Costello C, Connors L, Skinner M, Trinkaus‐Randall V (2006) Role of endocytic inhibitory drugs on internalization of amyloidogenic light chains by cardiac fibroblasts. Am J Pathol 169: 1939–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, Hutchinson WL, Mangione PP, Gallimore JR, Millar DJ et al (2010) Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature 468: 93–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loos C, Syrovets T, Musyanovych A, Mailänder V, Landfester K, Simmet T (2014) Amino‐functionalized nanoparticles as inhibitors of mTOR and inducers of cell cycle arrest in leukemia cells. Biomaterials 35: 1944–1953 [DOI] [PubMed] [Google Scholar]
- 41. Hirakura Y, Carreras I, Sipe JD, Kagan BL (2002) Channel formation by serum amyloid A: a potential mechanism for amyloid pathogenesis and host defense. Amyloid 9: 13–23 [DOI] [PubMed] [Google Scholar]
- 42. Butterfield SM, Lashuel HA (2010) Amyloidogenic protein‐membrane interactions: mechanistic insight from model systems. Angew Chem Int Ed Engl 49: 5628–5654 [DOI] [PubMed] [Google Scholar]
- 43. Friedrich RP, Tepper K, Rönicke R, Soom M, Westermann M, Reymann K, Kaether C, Fändrich M (2010) Mechanism of amyloid plaque formation suggests an intracellular basis Aβ pathogenicity. Proc Natl Acad Sci USA 107: 1942–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harper JD, Lansbury PT Jr (1997) Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time‐dependent solubility of amyloid proteins. Annu Rev Biochem 66: 385–407 [DOI] [PubMed] [Google Scholar]
- 45. Kimura N, Yanagisawa K (2007) Endosomal accumulation of GM1 ganglioside‐bound amyloid beta‐protein in neurons of aged monkey brains. NeuroReport 18: 1669–1673 [DOI] [PubMed] [Google Scholar]
- 46. Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee JM (2009) Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid‐beta peptide. Proc Natl Acad Sci USA 106: 20324–20329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Axelrad MA, Kisilevsky R, Willmer J, Chen SJ, Skinner M (1982) Further characterization of amyloid‐enhancing factor. Lab Invest 47: 139–146 [PubMed] [Google Scholar]
- 48. Glenner GG (1975) Current concepts of the formation and composition of amyloid. Ann Clin Lab Sci 5: 257–263 [PubMed] [Google Scholar]
- 49. Keeling J, Teng J, Herrera GA (2004) AL‐amyloidosis and light‐chain deposition disease light chains induce divergent phenotypic transformations of human mesangial cells. Lab Invest 84: 1322–1338 [DOI] [PubMed] [Google Scholar]
- 50. Westermark P (1973) Fine structure of islets of Langerhans in insular amyloidosis. Virchows Arch A Pathol Pathol Anat 359: 1–18 [DOI] [PubMed] [Google Scholar]
- 51. Campistol JM, Solé M, Bombi JA, Rodriguez R, Mirapeix E, Muñoz‐Gomez J, Revert OW (1992) In vitro spontaneous synthesis of beta 2‐microglobulin amyloid fibrils in peripheral blood mononuclear cell culture. Am J Pathol 141: 241–247 [PMC free article] [PubMed] [Google Scholar]
- 52. Arciello A, De Marco N, Del Giudice R, Guglielmi F, Pucci P, Relini A, Monti DM, Piccoli R (2011) Insights into the fate of the N‐terminal amyloidogenic polypeptide of ApoA‐I in cultured target cells. J Cell Mol Med 15: 2652–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jeffrey M, McGovern G, Goodsir CM, Brown KL, Bruce ME (2000) Sites of prion protein accumulation in scrapie‐infected mouse spleen revealed by immuno‐electron microscopy. J Pathol 191: 323–332 [DOI] [PubMed] [Google Scholar]
- 54. Peters PJ, Mironov A Jr, Peretz D, van Donselaar E, Leclerc E, Erpel S, DeArmond SJ, Burton DR, Williamson RA, Vey M et al (2003) Trafficking of prion proteins through a caveolae‐mediated endosomal pathway. J Cell Biol 162: 703–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Glenner GG, Ein D, Eanes ED, Bladen HA, Terry W, Page DL (1971) Creation of “amyloid” fibrils from Bence Jones proteins in vitro . Science 174: 712–714 [DOI] [PubMed] [Google Scholar]
- 56. Lai Z, Colón W, Kelly JW (1996) The acid‐mediated denaturation pathway of transthyretin yields a conformational intermediate that can self‐assemble into amyloid. Biochemistry 35: 6470–6482 [DOI] [PubMed] [Google Scholar]
- 57. Magde D, Wong R, Seybold PG (2002) Fluorescence quantum yields and their relation to lifetimes of rhodamine 6G and fluorescein in nine solvents: improved absolute standards for quantum yields. Photochem Photobiol 75: 327–334 [DOI] [PubMed] [Google Scholar]
- 58. Wang LH, Rothberg KG, Anderson RG (1993) Mis‐assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol 123: 1107–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lunov O, Syrovets T, Loos C, Beil J, Delacher M, Tron K, Nienhaus GU, Musyanovych A, Mailänder V, Landfester K et al (2011) Differential uptake of functionalized polystyrene nanoparticles by human macrophages and a monocytic cell line. ACS Nano 5: 1657–1669 [DOI] [PubMed] [Google Scholar]
- 60. Band AH, Chitamber SD, Bhattacharya A, Talwar GP (1986) Mechanism of phagocytosis of mycobacteria by Schwann cells and their comparison with macrophages. Int J Lepr Other Mycobact Dis 54: 292–299 [PubMed] [Google Scholar]
- 61. Sandgren KJ, Wilkinson J, Miranda‐Saksena M, McInerney GM, Byth‐Wilson K, Robinson PJ, Cunningham AL (2010) A differential role for macropinocytosis in mediating entry of the two roms of vaccinia virus into dendritic cells. PLoS Pathog 6: e10000866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hill TA, Gordon CP, McGeachie AB, Venn‐Brown B, Odell LR, Chau N, Quan A, Mariana A, Sakoff JA, Chircop M et al (2009) Inhibition of dynamin mediated endocytosis by the dynoles‐synthesis and functional activity of a family of indoles. J Med Chem 52: 3762–3773 [DOI] [PubMed] [Google Scholar]
- 63. Aoki T, Nomura R, Fujimoto T (1999) Tyrosine phosphorylation of caveolin‐1 in the endothelium. Exp Cell Res 253: 629–636 [DOI] [PubMed] [Google Scholar]
- 64. Oliveira CA, Kashman Y, Mantovani B (1996) Effects of latrunculin A on immunological phagocytosis and macrophage spreading‐associated changes in the F‐actin/G‐actin content of the cells. Chem Biol Interact 100: 141–153 [DOI] [PubMed] [Google Scholar]
- 65. Schlegel R, Dickson RB, Willingham MC, Pastan IH (1982) Amantadine and dansylcadaverine inhibit vesicular stomatitis virus uptake and receptor‐mediated endocytosis of α2‐macroglobulin. Proc Natl Acad Sci USA 79: 2291–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sen D, Deerinck TJ, Ellisman MH, Parker I, Cahalan MD (2008) Quantum dots for tracking dendritic cells and priming an immune response in vitro and in vivo . PLoS ONE 3: e3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Walther P, Ziegler A (2002) Freeze substitution of high‐pressure frozen samples: the visibility of biological membranes is improved when the substitution medium contains water. J Microsc 208: 3–10 [DOI] [PubMed] [Google Scholar]
- 68. Gill SC, von Hippel PH (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem 182: 319–326 [DOI] [PubMed] [Google Scholar]
- 69. Christopeit T, Hortschansky P, Schroeckh V, Gührs K, Zandomeneghi G, Fändrich M (2005) Mutagenic analysis of the nucleation propensity of oxidized Alzheimer's beta‐amyloid peptide. Protein Sci 14: 2125–2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File