

Abstract
Liver fibrosis is a major pathological feature of chronic liver diseases and there is no effective therapy program at present. Schisandrin B (Sch B) is the major bioactive ingredient of Schisandra chinensis, with antioxidative, anti-inflammatory, antitumor, and hepatoprotective properties. This study aimed to investigate the protective effect and related molecular mechanism of Sch B against carbon tetrachloride (CCl4)-induced liver fibrosis in rats. The in vivo therapeutic effect of Sch B on liver fibrosis induced by CCl4 was examined in rats. In vitro, rat hepatic stellate cells (HSC-T6) were used to assess the effect of Sch B on the activation of HSCs. Sch B effectively attenuated liver damage and progression of liver fibrosis in rats, as evidenced by improved liver function and decreased collagen deposition. The effects of Sch B were associated with attenuating oxidative stress by activating nuclear factor-erythroid 2-related factor 2 (Nrf2)-mediated antioxidant signaling and suppressing HSC activation by inhibiting the transforming growth factor-β (TGF-β)/Smad signaling pathway. In an in vitro study, it was shown that Sch B inhibited TGF-β-induced HSC activation. Finally, Sch B significantly inhibited TGF-β1-stimulated phosphorylation of Smad and signaling of mitogen-activated protein kinases. This study demonstrates that Sch B prevents the progression of liver fibrosis by the regulation of Nrf2-ARE and TGF-β/Smad signaling pathways, and indicates that Sch B can be used for the treatment of liver fibrosis.
Keywords: schisandrin B, liver fibrosis, hepatic stellate cell activation, Nrf2, TGF-β/Smad
Introduction
Liver fibrosis, a chronic liver injury, is characterized by the excessive accumulation of extracellular matrix (ECM) and the distortion of normal hepatic architecture.1,2 With a persistent chronic liver injury, liver fibrosis can progress to liver cirrhosis, hepatocellular carcinoma, or even death.3,4 So liver fibrosis plays a crucial role in the development and progression of other chronic liver diseases. Therefore, it is necessary to elucidate the possible molecular mechanism underlying liver fibrosis for the sake of developing effective drugs.
It is widely recognized that activated hepatic stellate cells (HSCs) play a crucial role in the development of liver fibrosis.5–7 HSCs are normally quiescent, but after they are activated in response to liver damage, they become proliferative and get transformed into myofibroblasts, and subsequently into synthesis and secretion of ECM.8 In addition, activated HSCs and myofibroblasts also secrete large amounts of profibrotic cytokines that exacerbate the fibrotic process through autocrine and paracrine effects.9 Although the precise mechanism of HSC activation has not been fully elucidated, current research has revealed that transforming growth factor-β (TGF-β) is the most potent profibrotic cytokine.10 TGF-β stimulates HSC activation by activating Smad and mitogen-activated protein kinases (MAPK) signaling pathways in HSCs.11–14 Several studies have showed that the inhibition of TGF-β signaling pathways displays antifibrotic effects in experimental models.15–18 However, an effective therapy against liver fibrosis is not yet available in clinical practice. It is well known that hepatocyte damage is the precipitating event in the progression of liver fibrosis.19 Hepatocyte damage promotes the secretion of inflammatory and profibrogenic cytokines and directly promotes HSC activation.9 Given that hepatocyte damage and HSC activation are two important factors in the development of liver fibrosis, a combination therapy approach targeting them may be more effective. Thus, we searched for a suitable pharmacology to protect against hepatocyte damage and inhibit HSC activation for the treatment of liver fibrosis.
Schisandrin B (Sch B, Figure 1A), the main active ingredient of Schisandra chinensis, possesses diverse pharmacological activities such as antioxidative, anti-inflammatory, antitumor, and hepatoprotective properties.20–23 Some studies have demonstrated that the extract of S. chinensis protected hepatocytes against CCl4- and acetaminophen-induced liver injury by the inhibition of CYP-mediated bioactivation and the regulation of the Nrf2-ARE, antioxidant response element (ARE) pathway.24–27 Nuclear factor-erythroid 2-related factor 2 (Nrf2), a transcription factor that activates antioxidant response elements, protects a variety of tissues and cells against oxidative stress.28,29 In addition, other studies showed that Sch B inhibits TGF-β-mediated fibrotic signaling in A7r5 vascular smooth muscle cells and AML12 cells.30,31 All these findings implied that Sch B may protect hepatocyte against damage and suppress TGF-β-induced HSC activation, and could be used to treat liver fibrosis. However, the effect of Sch B on liver fibrosis has not been reported yet. Thus, the purpose of the present study was to investigate the protective effect and related molecular mechanism of Sch B against carbon tetrachloride (CCl4)-induced liver fibrosis in rats.
Figure 1.

Sch B protects against CCl4-induced liver injury.
Notes: (A) The structure of Schisandrin B (Sch B). (B) The experimental design scheme. (C) Serum ALT and AST activities. (D) Liver weight to body weight ratio. (E) H&E staining of liver sections (Scale bar: 100 and 50 μm; original magnification, ×50 and ×100). Data are expressed as the mean ± SEM (n=8). ##P<0.01 versus control group. *P<0.05 and **P<0.01 versus model group.
Abbreviations: CCl4, carbon tetrachloride; d, days; w, weeks; ALT, alanine transaminase; AST, aspartate transaminase.
Materials and methods
Chemicals and reagents
Sch B was purchased from the China pharmaceutical biological products analysis institute (Shanghai, China). CCl4 was purchased from Jiangsu Qiangsheng Chemical (Jiangsu, China). Monoclonal anti-p-Smad2/3 and Smad2/3 rabbit antibody were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Extracellular regulated protein kinases (ERK), p-ERK, P38, p-P38, c-Jun N-terminal kinase (JNK), p-JNK, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Monoclonal antibody against Nrf2 was purchased from Sangon Biotechnology (Shanghai, China). Secondary antibodies used in Western blot were IRDye®800CW antirabbit IgG (H+L) (LI-COR Biosciences, Lincoln, NE, USA) and IRDye800CW anti-mouse IgG (H+L) (LI-COR Biosciences).
Animals and experimental design
Male Wistar rats weighing 150–200 g (Shanghai SLAC Laboratory Animal Center, Shanghai, China) were housed under appropriate conditions (25°C±2°C and 12-h light/dark cycle) with free access to water and food. All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Animal Experimental Ethics Committee of the Second Military Medical University (Shanghai, China).
Experimental design was outlined in Figure 1B. The rats were randomly distributed into four groups (n=8 per group) as follows: 1) Sham control group, 2) Model group, 3) 25 mg/kg Sch B group (CCl4 + Sch B 25 mg/kg), and 4) 50 mg/kg Sch B group (CCl4 + Sch B 50 mg/kg). The model group was treated with CCl4 (50% CCl4/olive oil; 2 mL/kg) thrice a week for 8 weeks to induce liver fibrosis. In Sch B-treated group, the rats were administered CCl4 for 4 weeks and then administered Sch B at a dose of 25 or 50 mg/kg daily, respectively, together with CCl4 gavage for another 4 weeks. Equal volume of olive oil instead of CCl4 was given to rats by gavage as sham control.
At the end of the experiments, the animals were sacrificed. Serum was prepared and stored at −80°C until biochemical assay and the liver tissues were used for biochemistry analysis, real-time polymerase chain reaction (PCR), and Western blot.
ALT/AST assessment
Serum alanine transaminase (ALT) and aspartate transaminase (AST) were determined using a clinical automatic analyzer (Hitachi, Tokyo, Japan) and a commercial reagent kit (Roche Diagnostic, Mannheim, Germany) according to the manufacturer’s protocol.
Hydroxyproline content determination
Hepatic hydroxyproline content was measured with a hydroxyproline detection kit (JianCheng Institute of Biotechnology, NanJing, China) according to the manufacturer’s protocol.
Histological assays
Liver tissues were fixed in 10% formalin, paraffin-embedded, and sectioned. Liver sections were stained with hematoxylin/eosin (H&E) for histological examination. In addition, liver sections were stained with Sirius Red and Masson’s trichrome stains to estimate liver fibrosis. All sections were imaged by microscope (Olympus, Tokyo, Japan). The degree of liver fibrosis was quantified by Image-Pro Plus 6.0 software.
Immunohistological staining of α-smooth muscle actin and p-Smad2/3
Immunohistological examinations were carried out to detect the expression of α-smooth muscle actin (α-SMA) and p-Smad2/3. Briefly, liver sections were deparaffinized and treated with 3% H2O2 to block endogenous peroxidase activity. Antigen retrieval was performed in citrate buffer. After cooling, sections were treated with 5% bovine serum albumin (BSA) to block nonspecific protein binding. The sections were incubated with α-SMA and p-Smad2/3 primary antibody overnight at 4°C. Finally, the sections were washed with phosphate-buffered saline, incubated with a biotinylated secondary antibody followed by an avidin–biotin–peroxidase complex, and finally stained with 3, 3′-diaminobenzidine (DAB).
Cell culture and treatment
Immortalized rat HSC lines (HSC-T6) were purchased from the Fuxiang Biological Co. Ltd (Shanghai, China). The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific; Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone; Logan, UT, USA), 100 U/mL of penicillin (Thermo Fisher Scientific), and 100 μg/mL of streptomycin (Thermo Fisher Scientific) at 37°C with 5% CO2.
To evaluate the effect of Sch B on HSC proliferation, HSC-T6 cells were seeded in a 96-well plate with 100 μL (3×104 cells/mL) per well and incubated for 24 hours before the addition of the stimulus. Cells were incubated for 12, 24, or 48 hours and treated with different concentrations of Sch B (5, 10, and 30 μM). The effects of Sch B on HSC viability were tested by CCK8 (CK04; Dojindo; Kumamoto, Japan) assay.
To evaluate the effect of Sch B on HSC activation, cells were serum-starved overnight and then treated with TGF-β (2 ng/mL) in the presence or absence of Sch B for 24 hours. At the end of cell culture, cells were collected for Western blot analysis.
Real-time PCR analysis
Total RNA was extracted from the liver tissue using the TRI-zol reagent (15596026; Invitrogen) according to the standard protocol. The first-strand cDNA was synthesized by the PrimeScript RT reagent kit (#6210A; Takara; Kusatsu, Japan), and the total RNA (2 μg) was used as a template. The target mRNA expression was quantified with the SYBER Green PCR Master Mix (#RR420Q; Takara) using Step One Real-Time PCR System (Applied Biosystems, Warrington, UK). The conditions of real-time PCR analysis were as follows: 1) Holding stage: 95°C, 30 s; 2) Cycling Stage: 95°C, 5 s, 60°C, 34 s; 40 cycles; 3) Melt Curve Stage: 95°C, 15 s; 60°C, 1 min; 95°C, 15 s. GAPDH was amplified as reference genes. The primer sequences used in PCR are shown in Table 1. The expression levels were measured in terms of the cycle threshold (Ct) and then normalized to GAPDH expression using the 2−ΔΔCt method.
Table 1.
Primer sequences for real-time PCR assay
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| Col1A1 | GAGTGAGGCCACGCATGA | AGCCGGAGGTCCACAAAG |
| Col3A1 | TTTGGCACAGCAGTCCAATGTA | GACAGATCCCGAGTCGCAGA |
| TNF-α | TCAGTTCCATGGCCCAGAC | GTTGTCTTTGAGATCCATGCCATT |
| IL1-β | CCCTGAACTCAACTGTGAAATAGCA | CCCAAGTCAAGGGCTTGGAA |
| IL-6 | ATTGTATGAACAGCGATGATGCAC | CCAGGTAGAAACGGAACTCCAGA |
| TGF-β1 | ATTCCTGGCGTTACCTTGG | AGCCCTGTATTCCGTCTCCT |
| GCLc | GGGGTGACGAGGTGGAGTA | AAGCCGAGAATGCTGAGTTCA |
| HO-1 | AAGCCGAGAATGCTGAGTTCA | GCCGTGTAGATATGGTACAAGGA |
| NQO-1 | ATGGGAGGTGGTCGAATCTGA | GCCTTCCTTATACGCCAGAGATG |
| GAPDH | GACAACTTTGGCATCGTGGA | ATGCAGGGATGATGTTCTGG |
Abbreviation: PCR, polymerase chain reaction.
Western blot analysis
Protein was extracted and quantified. Then, Western blotting assays were performed as previously described. The samples were then separated by sodium dodecyl sulfate (SDS)-polyacrylamide gels (10%) and then transferred onto nitrocellulose membranes (HATF00010; Merck Millipore; Darmstadt, Germany). After being blocked with 5% skim milk (232100; Becton, Dickinson and Company; Franklin Lakes, NJ, USA) for 2 hours, the membranes were incubated with primary antibodies at 4°C overnight and then with the secondary antibodies for 2 hours at room temperature. Protein expression was imaged by the Odyssey Infrared Imaging System (LI-COR Biosciences).
Statistical analysis
Data are presented as the mean ± SEM. Differences between groups were determined by a two-tailed Student’s t-test in GraphPad Prism 5. P<0.05 was considered statistically significant.
Results
Sch B protects against CCl4-induced liver injury
In this study, the protective effect of Sch B against long-term CCl4-induced liver injury in rats was examined. Severe liver injury was induced by CCl4 in rats, as indicated by the elevation of ALT/AST activities and histopathological analysis. As shown in Figure 1C, CCl4 significantly elevated ALT and AST to 197.50±26.21 U/L and 239.70±20.25 U/L, respectively. On the other hand, Sch B treatment significantly reduced the upregulation of ALT and AST levels as compared to the model group. In addition, liver weight to body weight ratio was slightly increased in the model group; however, it was decreased nearly to the normal value in the Sch B-treated groups (Figure 1D). Furthermore, histological examination by H&E staining was performed to confirm the protective effect of Sch B. In agreement with serum ALT/AST activities, the livers of CCl4-treated rats displayed a large area of inflammatory cell infiltration, hepatocyte ballooning degeneration, and necrosis. However, Sch B treatment effectively ameliorated pathological lesions induced by CCl4 (Figure 1E). Taken together, these results indicate that Sch B has a protective effect against long-term CCl4-induced liver injury in rats.
Sch B ameliorates CCl4-induced liver fibrosis in rats
The collagen content of livers was detected to evaluate the effect of Sch B on liver fibrosis induced by CCl4 in rats. First, liver sections were stained by Sirius Red, which stains collagen fibers red. It was shown that the staining of collagen fiber in the CCl4-treated rats increased by 30-fold when compared with control rats. Interestingly, Sch B treatment markedly reduced collagen accumulation in the liver, with a 43% decrease in the 25 mg/kg group and a 58% decrease in the 50 mg/kg group. Similar results were also confirmed by Masson’s trichrome staining (Figure 2A and B). Furthermore, the mRNA levels of COL1A1 (encoding collagen-I) and COL3A1 (encoding collagen-III) were markedly increased in CCl4-treated rats, but were downregulated in Sch B-treated rats (Figure 2C). Additionally, the expression of collagen-I was increased after CCl4 treatment, but treatment with Sch B decreased its expression (Figure 2D and E). Finally, hydroxyproline, the major component of collagen protein, was also decreased in the Sch B-treated rats (Figure 2F). All these results suggest that Sch B could ameliorate CCl4-induced liver fibrosis in vivo.
Figure 2.

Sch B ameliorates CCl4-induced liver fibrosis in rats.
Notes: (A, B) Liver fibrosis assessed by Sirius Red and Masson. (C) The mRNA levels of COL1A1 and COL3A1. (D) Western blot analysis of collagen-I. (E) Densitometric analysis of Western blots. (F) Hydroxyproline content in rat livers. Data are expressed as the mean ± SEM (n=8). ###P<0.001 and ##P<0.01 versus control group. *P<0.05 and **P<0.01 versus model group.
Abbreviations: CCl4, carbon tetrachloride; Sch B, schisandrin B; Hyp, hydroxyproline.
Sch B regulates CCl4-induced hepatic oxidative stress and Nrf2-ARE signaling pathway in vivo
To investigate the effect of Sch B on liver oxidative stress caused by CCl4, the activities of malondialdehyde (MDA), superoxide dismutase (SOD), glutathione (GSH) and glutathione peroxidase (GSH-Px) were detected. After CCl4 treatment, the content of MDA was significantly elevated by 26.3% as compared to control rats, while the activities of GSH-Px and GSH were markedly reduced by 24.3% and 28.6%, respectively. Interestingly, these aberrant changes were partly reversed by Sch B treatment, suggesting that Sch B could block CCl4-induced oxidative liver damage (Figure 3A–D).
Figure 3.

Sch B regulates CCl4-induced hepatic oxidative stress and Nrf2-ARE signaling pathway in vivo.
Notes: (A) Lipid peroxidation MDA. (B) Liver GSH-Px activities. (C) Antioxidant enzyme SOD. (D) Liver GSH levels. (E) Western blot analysis of Nrf2 levels in the liver. (F) Densitometric analysis of Western blots. (G) The mRNA levels of GCLc, HO-1, and NQO1. Data are expressed as the mean ± SEM. #P<0.05 and ##P<0.01 versus control group. *P<0.05 and **P<0.01 versus model group.
Abbreviations: ARE, antioxidant response element; CCl4, carbon tetrachloride; Nrf2, nuclear factor-erythroid 2-related factor 2; Sch B, schisandrin B; MDA, malondialdehyde; SOD, superoxide dismutase; GSH, glutathione; GSH-Px, glutathione peroxidase.
Since the Nrf2-ARE signaling pathway plays an important role in oxidative stress, we investigated whether Nrf2 signaling was activated when rats were treated with Sch B. The results showed that the expression level of nuclear Nrf2 was slightly increased in the CCl4-treated rats, but was markedly increased after Sch B treatment (Figure 3E and F). Furthermore, the expressions of Nrf2-related antioxidant genes (GCLC, HO-1, and NQO1) were detected by real-time PCR. Compared with the model group, the expressions of hepatic GCLC, HO-1, and NQO1 were significantly elevated in the Sch B-treated group (Figure 3G). These results revealed that Sch B could suppress CCl4-induced oxidative stress perhaps by regulating the Nrf2-ARE signaling pathway.
Sch B alleviates CCl4-induced inflammation
Liver fibrosis is associated with inflammation response, so that the effect of Sch B on the expression of inflammatory cytokines induced by CCl4 was evaluated. Compared with the sham control group, the expressions of hepatic tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6 mRNA were significantly elevated after CCl4 treatment (P<0.01), whereas these inflammatory cytokines were reduced after 25 or 50 mg/kg Sch B treatment (Figure 4A–C). In addition, the level of TGF-β1 was increased by more than 3-fold after CCl4 treatment in the model group, but Sch B treatment blocked this trend especially at the dose of 50 mg/kg in TGF-β1 level (Figure 4D).
Figure 4.

Sch B alleviates CCl4-induced inflammatory response.
Notes: (A) Relative mRNA levels of TNF-α. (B) Relative mRNA levels of IL-1β. (C) Relative mRNA levels of IL-6. (D) Relative mRNA levels of TGF-β1. Data are expressed as the mean ± SEM (n=6). ##P<0.01 versus control group. *P<0.05 and **P<0.01 versus model group.
Abbreviations: CCl4, carbon tetrachloride; IL-6, interleukin-6; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α; Sch B, schisandrin B.
Sch B suppresses TGF-β/Smad-mediated HSC activation to inhibit liver fibrosis
To investigate the effect of Sch B on HSC activation, the expression level of α-SMA was measured to determine whether Sch B could inhibit HSC activation against liver fibrosis. As shown in Figure 5A, CCl4 treatment significantly increased α-SMA-positive cells in the liver as compared to the sham control group, confirming that HSCs were activated in the CCl4-induced liver fibrosis model. In contrast, α-SMA immunoreactive cells were largely decreased by treatment with Sch B. Furthermore, the expression of α-SMA in the liver was also detected by Western blot analysis. In agreement with immunohistochemical results, Sch B remarkably reduced the expression of α-SMA, suggesting that Sch B inhibited HSC activation in vivo.
Figure 5.

Sch B suppresses HSC activation and TGF-β/Smad signaling in CCl4-induced liver fibrosis.
Notes: (A) Immunohistochemistry staining of α-SMA and p-Smad2/3 in the liver tissues, the sections were photographed at Scale bar: 100 μm; original magnification: ×50; (B) Effects of Sch B on the expression of α-SMA, p-Smad2/3, and Smad2/3 in the liver tissues were measured by Western blot analysis; (C) Densitometric analysis of Western Blots. Data are expressed as the mean ± SEM. ##P<0.01 versus control group. *P<0.05 and **P<0.01 versus model group.
Abbreviations: CCl4, carbon tetrachloride; HSC, hepatic stellate cell; TGF-β, transforming growth factor β; Sch B, schisandrin B.
After coming to know that the TGF-β/Smad signaling pathway plays an important role in HSC activation and liver fibrosis, the phosphorylation-Smad2/3 was investigated to explore whether Sch B could block the TGF-β/Smad signaling pathway to inhibit HSC activation in rats after CCl4-induced liver fibrosis. The results showed that the expression level of phosphorylation-Smad2/3 was significantly increased after treatment with CCl4. Moreover, the increase of p-Smad2/3 in CCl4-treated rats was attenuated by Sch B treatment (Figure 5B and C). These findings indicated that Sch B treatment might prevent HSC activation during CCl4-induced liver fibrosis by inhibiting the TGF-β/Smad signaling pathway.
Sch B inhibits the activation of HSCs in vitro
As Sch B treatment ameliorates liver fibrosis induced by CCl4 in rats, the ability of Sch B on HSC proliferation and activation was further assessed in vitro. HSC-T6 cells were treated with various concentrations of Sch B for 12, 24, and 48 hours, and cell viability was measured by CCK8. As shown in Figure 6A and B, the results suggested that Sch B did not markedly inhibit HSC-T6 cell proliferation.
Figure 6.

Sch B inhibits the TGF-β1-induced HSC activation in vitro.
Notes: (A, B) Effects of Sch B on HSC-T6 cells viability. Cell viability was assessed by CCK8 assay; (C) Effects of Sch B on TGF-β1-induced HSC activation as assessed by α-SMA and collagen I expression; (D) Densitometric analysis of Western Blots. Data are expressed as the mean ± SEM. #P<0.05 and ##P<0.01 versus control group. *P<0.05 and **P<0.01 versus model group.
Abbreviations: α-SMA, α-smooth muscle actin; HSC, hepatic stellate cell; TGF-β, transforming growth factor-β; Sch B, schisandrin B.
Activation of HSCs is responsible for the collagen synthesis and deposition in the liver, so that the effect of Sch B on HSC activation was examined. HSC-T6 cells were treated with TGF-β1 for 24 hours after serum starvation. Consistent with previous studies, TGF-β1 (2 ng/mL) significantly increased HSC activation, as indicated by an increase in the expression of α-SMA and collagen I. However, the levels of α-SMA and collagen I were decreased after Sch B treatment, suggesting that Sch B could inhibit TGF-β1-induced HSC activation (Figure 6C and D).
Sch B inhibits TGF-β-mediated Smad and MAPK signaling molecules
To further explore the intracellular molecular mechanism, the phosphorylation level of Smad2/3 was examined. As shown in Figure 7A and B, TGF-β induced a strong increase of Smad2/3 phosphorylation in HSC-T6 cells. In contrast, Sch B decreased the expression of phosphorylated Smad2/3 induced by TGF-β. In addition, the ratio of p-Smad/total Smad confirmed that the induction of Smad2/3 phosphorylation by TGF-β was attenuated by Sch B treatment. Due to the fact that TGF-β also activates MAPK signaling, we investigated whether Sch B could inhibit TGF-β-mediated MAPK signaling. As shown in Figure 7C and D, TGF-β stimulated the phosphorylation of ERK, p38, and JNK in HSC-T6 cells. Interestingly, Sch B inhibited the phosphorylation of p38, ERK, and JNK. These results indicated that Sch B effectively blocked TGF-β signaling via the inhibition of Smad and MAPK signaling molecules.
Figure 7.

Sch B inhibits TGF-β-mediated Smad and MAPK signaling molecules in vitro.
Notes: (A, B) The expression of p-Smad2 and p-Smad3 was assessed by Western blot, and the densitometric analysis of Western blots. (C, D) Western blot analysis of p-p38, p38, p-ERK, ERK, p-JNK, and JNK in HSCs, and the densitometric analysis of Western blots. Data are expressed as the mean ± SEM. #P<0.05 and ##P<0.01 versus control group. *P<0.05 and **P<0.01 versus model group.
Abbreviations: HSCs, hepatic stellate cells; MAPK, mitogen-activated protein kinases; TGF-β, transforming growth factor-β; Sch B, schisandrin B.
Discussion
In this study, we demonstrated that Sch B could effectively ameliorate CCl4-induced liver fibrosis in rats and inhibit TGF-β-induced HSC activation in vitro. To assess the effect of Sch B on liver fibrosis, the collagen content in the liver was detected by various methods. First, Sirius Red and Masson staining results suggested that severe liver fibrosis was induced by CCl4 in rats, as revealed by extensive bridging fibrosis with substantial collagen deposition in the CCl4-treated rats as compared to sham control rats. However, fibrosis area was reduced in the Sch B-treated group, suggesting that Sch B could attenuate CCl4-induced liver fibrosis in rats. Furthermore, the expression level of collagen I protein and the mRNA levels of collagen I and collagen III were measured by Western blotting and real-time PCR, respectively. The results showed that Sch B suppressed CCl4-induced accumulation of collagen in rats, suggesting that Sch B has the ability to attenuate liver fibrosis. Finally, total collagen was measured by hydroxyproline content, and the results showed that Sch B markedly reduced the hydroxyproline content. Taken together, all results suggest that Sch B ameliorates CCl4-induced liver fibrosis in vivo.
Hepatocyte, the main cell type in the liver, is critical for the maintenance of liver homeostasis.32 Hepatocyte damage promotes the secretion of inflammatory and profibrogenic cytokines and directly promotes HSC activation, so hepatocyte damage is also the initial factor in the development of liver fibrosis.33,34 Many studies have demonstrated that Sch B could protect against CCl4-induced acute liver injury.26,35,36 In this study, the effect of Sch B on liver function was also examined. Consistent with previous studies, Sch B improves liver function, as evidenced by lower-serum ALT/AST levels and histopathological analysis. These results indicate that Sch B strongly protects hepatocytes, which offers a primary therapeutic advantage of Sch B, since it could improve hepatocyte integrity. It is well known that oxidative stress is a major contributor to hepatocyte death in the progression of liver fibrosis.37–39 Many studies have demonstrated that Sch B exerts its beneficial effects through attenuating the oxidative stress.26,40,41 In this study, CCl4 treatment significantly increased lipid peroxides as confirmed by elevation in MDA levels, accompanied by a significant depletion in liver GSH levels and GSH-Px activity. In agreement with previous studies, our results also demonstrated that Sch B effectively inhibited lipid peroxidation and enhanced the antioxidant capacity as evidenced by decreased MDA, and increased GSH, GSH-Px activity. These results indicated that Sch B could inhibit oxidative stress and protect against hepatocyte damage induced by CCl4.
How does Sch B inhibit CCl4-induced oxidative stress? Oxidative stress has been defined as an imbalance between reactive oxygen species generation and its clearance by antioxidants. CCl4 is known to be metabolized to trichloromethyl free radicals (CCl3) by cytochrome P450 enzymes and these free radicals may attack intracellular nucleic acid, protein, and lipid, ultimately leading to hepatocyte oxidative damage and death.42 Several studies showed that Sch B could inhibit oxidative stress-mediated liver injury by inhibiting CYP450 enzymes.43–45 Nrf2 is famous for a transcription factor that regulates the expression of various antioxidant genes in the cellular defense against oxidative stress. Upon stimulation, Nrf2 translocates from cytosol to nucleus, binds to ARE, induces the expression of antioxidant genes, and protects against oxidative damage. Importantly, recent studies have found that Nrf2 activators dramatically inhibited liver fibrosis, and Nrf2-null mice were more susceptible to liver fibrosis as compared to wild-type mice, suggesting that Nrf2 is a potential target to treat liver fibrosis.46–49 Interestingly, several studies have revealed that Sch B exhibited anti-inflammatory and antioxidant activities by activating Nrf2 pathways.50,51 In addition, recent reports also showed that S. chinensis extract and Sch B could activate Nrf2 reporter gene and induce the expression of HO-1 and NQO1.52 However, whether Sch B activates Nrf2-ARE signaling against liver fibrosis has not been studied. In the current study, Sch B markedly increased the expression level of nuclear Nrf2 as compared to the model group. Furthermore, the expression of Nrf2-target genes GCLc, HO-1, and NQO1 was increased in the Sch B-treated group. These results indicated that Sch B protects against liver fibrosis perhaps by activating the Nrf2-ARE pathway to inhibit oxidative stress-mediated hepatocyte damage in the rats with liver fibrosis.
Hepatocyte damage and death accompany inflammatory response. In our study, CCl4 increased the number of infiltrating inflammatory cells in the liver, and this was blocked by Sch B treatment. Furthermore, inflammatory cytokines were detected to assess the effect of Sch B on inflammation by real-time PCR. Our results also demonstrated that the mRNA levels of inflammatory cytokines TNF-α, IL-1β, and IL-6 were significantly increased after CCl4 treatment, which were attenuated by Sch B treatment in rats with CCl4-induced liver fibrosis. These results have shown that Sch B exerts antioxidant and anti-inflammation effects on CCl4-induced liver injury in the liver. But further studies are still necessary to explore the antifibrotic mechanism of Sch B with a focus on HSCs.
Activated HSCs are the major producers of ECM in liver fibrosis, and have been considered as an attractive target for antifibrotic therapy.5,53 In the healthy liver, HSCs are quiescent and located in the space of Disse. Liver injuries sensitize HSCs to paracrine stimuli, which ultimately activate HSCs and trans-differentiate into myofibroblasts.54 In the present study, our results clearly demonstrated that Sch B treatment significantly reduced the number of hepatic myofibroblasts, as shown by decreased α-SMA-positive cells in the CCl4-treated rats. A similar result was also confirmed by Western blot. These results suggest that Sch B inhibits HSC activation. HSCs are activated by several cytokines, and TGF-β is considered as the most potent profibrogenic cytokine in the development of liver fibrosis. Furthermore, we studied the effect of Sch B on the activation of HSCs in vitro. Consistent with previous studies, HSCs was activated by 2 ng/mL TGF-β1, as evidenced by increasing the expression of α-SMA and collagen-I proteins. Furthermore, in line with the results of animal experimental, these changes induced by TGF-β1 were reduced by Sch B. The results revealed that Sch B could inhibit HSC activation in vivo and in vitro, suggesting that Sch B prevents CCl4-induced liver fibrosis perhaps by inhibiting HSC activation.
TGF-β is a pleiotropic cytokine with key roles in cell proliferation and differentiation, which has three isoforms (TGF-β1, TGF-β2, and TGF-β3).55 Among them, TGF-β1 is the principal isoform involved in the development of liver fibrosis.10 TGFβ1 binds to the cognate receptor on the cell membrane and subsequently recruits Smad2 and Smad3. Smad complexes translocate from the cytoplasm to the nucleus, where they act as transcription factors and regulate target gene expression.12,56 In the study, phosphorylation-Smad2/3 was significantly increased after treatment with CCl4 and TGF-β1. Importantly, Sch B inhibits Smad2/3 phosphorylation in vivo and in vitro. In addition, TGF-β also activates other signaling pathways in HSC, such as MAPK pathways. It is reported that the p38 MAPK signaling pathway is involved in HSC activation and collagen synthesis.13,29 Our results also showed that Sch B reduced the expression of p-ERK, p-p38, and p-JNK. These data suggest that the antifibrotic effect of Sch B is associated with the inhibition of TGF-β signaling pathways.
Conclusion
In conclusion, this study clearly demonstrated that Sch B attenuated the development of liver fibrosis through multiple mechanisms (Figure 8). First, Sch B activates Nrf2-ARE pathway to inhibit oxidative stress-mediated hepatocyte damage and death. Furthermore, it is reasonable to assume that Sch B suppressed the TGF-β/Smad signaling pathway to inhibit HSC activation. These results suggest that Sch B has the potential for the effective treatment of liver fibrosis and provide a novel cellular mechanism for its antifibrotic effects. However, the effects of Sch B on liver fibrosis induced by a different in the future.
Figure 8.
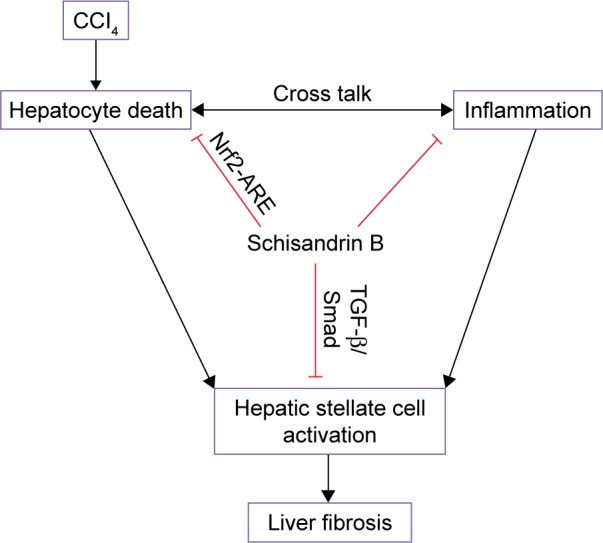
A diagram shows the possible mechanisms of Sch B against CCl4-induced liver fibrosis in rats.
Abbreviations: ARE, antioxidant response element; CCl4, carbon tetrachloride; Nrf2, nuclear factor-erythroid 2-related factor 2; Sch B, schisandrin B.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (81303192).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Ann Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 3.Zhang DY, Friedman SL. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology. 2012;56(2):769–775. doi: 10.1002/hep.25670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123(5):1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwaisako K, Jiang C, Zhang M, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U S A. 2014;111(32):E3297–E3305. doi: 10.1073/pnas.1400062111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reeves HL, Friedman SL. Activation of hepatic stellate cells–a key issue in liver fibrosis. Front Biosci. 2002;7:d808–d826. doi: 10.2741/reeves. [DOI] [PubMed] [Google Scholar]
- 8.Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123(5):1902–1910. doi: 10.1172/JCI66369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61(3):1066–1079. doi: 10.1002/hep.27332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inagaki Y, Okazaki I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56(2):284–292. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dooley S, Delvoux B, Streckert M, et al. Transforming growth factor beta signal transduction in hepatic stellate cells via Smad2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. TGFbeta signal transduction during transdifferentiation of hepatic stellate cells. FEBS Lett. 2001;502(1–2):4–10. doi: 10.1016/s0014-5793(01)02656-4. [DOI] [PubMed] [Google Scholar]
- 12.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8(12):970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 13.Tsukada S, Westwick JK, Ikejima K, Sato N, Rippe RA. SMAD and p38 MAPK signaling pathways independently regulate alpha1(I) collagen gene expression in unstimulated and transforming growth factor-beta-stimulated hepatic stellate cells. J Biol Chem. 2005;280(11):10055–10064. doi: 10.1074/jbc.M409381200. [DOI] [PubMed] [Google Scholar]
- 14.Xu P, Zhang Y, Liu Y, et al. Fibroblast growth factor 21 attenuates hepatic fibrogenesis through TGF-beta/smad2/3 and NF-kappaB signaling pathways. Toxicol Appl Pharmacol. 2016;290:43–53. doi: 10.1016/j.taap.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 15.Li JJ, Chen K, Li SN, et al. Protective effect of fucoidan from Fucus vesiculosus on liver fibrosis via the TGF-beta 1/Smad pathway-mediated inhibition of extracellular matrix and autophagy. Drug Des Dev Ther. 2016;10:619–630. doi: 10.2147/DDDT.S98740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dooley S, Delvoux B, Lahme B, Mangasser-Stephan K, Gressner AM. Modulation of transforming growth factor beta response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology. 2000;31(5):1094–1106. doi: 10.1053/he.2000.6126. [DOI] [PubMed] [Google Scholar]
- 17.Kim MJ, Park SA, Kim CH, et al. TGF-beta type I receptor kinase inhibitor EW-7197 suppresses cholestatic liver fibrosis by inhibiting HIF1alpha-induced epithelial mesenchymal transition. Cell Physiol Biochem. 2016;38(2):571–588. doi: 10.1159/000438651. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Wen XM, Lui EL, et al. Therapeutic targeting of the PDGF and TGF-beta-signaling pathways in hepatic stellate cells by PTK787/ZK22258. Lab Invest. 2009;89(10):1152–1160. doi: 10.1038/labinvest.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tu T, Calabro SR, Lee A, et al. Hepatocytes in liver injury: Victim, bystander, or accomplice in progressive fibrosis? J Gastroenterol Hepatol. 2015;30(12):1696–1704. doi: 10.1111/jgh.13065. [DOI] [PubMed] [Google Scholar]
- 20.Chun JN, Cho M, So I, Jeon JH. The protective effects of Schisandra chinensis fruit extract and its lignans against cardiovascular disease: a review of the molecular mechanisms. Fitoterapia. 2014;97:224–233. doi: 10.1016/j.fitote.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 21.Lu Y, Chen DF. Analysis of Schisandra chinensis and Schisandra sphenanthera. J Chromatogr A. 2009;1216(11):1980–1990. doi: 10.1016/j.chroma.2008.09.070. [DOI] [PubMed] [Google Scholar]
- 22.Panossian A, Wikman G. Pharmacology of Schisandra chinensis Bail: an overview of Russian research and uses in medicine. J Ethnopharmacol. 2008;118(2):183–212. doi: 10.1016/j.jep.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 23.He JL, Zhou ZW, Yin JJ, He CQ, Zhou SF, Yu Y. Schisandra chinensis regulates drug metabolizing enzymes and drug transporters via activation of Nrf2-mediated signaling pathway. Drug Des Devel Ther. 2015;9:127–146. doi: 10.2147/DDDT.S68501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan X, Chen P, Jiang Y, et al. Therapeutic efficacy of Wuzhi tablet (Schisandra sphenanthera Extract) on acetaminophen-induced hepatotoxicity through a mechanism distinct from N-acetylcysteine. Drug Metab Dispos. 2015;43(3):317–324. doi: 10.1124/dmd.114.062067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan X, Jiang Y, Wang Y, et al. Wuzhi tablet (Schisandra Sphenanthera extract) protects against acetaminophen-induced hepatotoxicity by inhibition of CYP-mediated bioactivation and regulation of NRF2-ARE and p53/p21 pathways. Drug Metab Dispos. 2014;42(12):1982–1990. doi: 10.1124/dmd.114.059535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie Y, Hao H, Wang H, Guo C, Kang A, Wang G. Reversing effects of lignans on CCl4-induced hepatic CYP450 down regulation by attenuating oxidative stress. J Ethnopharmacol. 2014;155(1):213–221. doi: 10.1016/j.jep.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 27.Cheng N, Ren N, Gao H, Lei X, Zheng J, Cao W. Antioxidant and hepatoprotective effects of Schisandra chinensis pollen extract on CCl4-induced acute liver damage in mice. Food Chem Toxicol. 2013;55:234–240. doi: 10.1016/j.fct.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284(20):13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang F, Ni C, Kong D, et al. Ligustrazine attenuates oxidative stress-induced activation of hepatic stellate cells by interrupting platelet-derived growth factor-beta receptor-mediated ERK and p38 pathways. Toxicol Appl Pharmacol. 2012;265(1):51–60. doi: 10.1016/j.taap.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 30.Park EJ, Chun JN, Kim SH, et al. Schisandrin B suppresses TGFbeta1 signaling by inhibiting Smad2/3 and MAPK pathways. Biochem Pharmacol. 2012;83(3):378–384. doi: 10.1016/j.bcp.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Park JH, Yoon J. Schizandrin inhibits fibrosis and epithelial-mesenchymal transition in transforming growth factor-beta1-stimulated AML12 cells. Int Immunopharmacol. 2015;25(2):276–284. doi: 10.1016/j.intimp.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 32.Qian H, Deng X, Huang ZW, et al. An HNF1alpha-regulated feedback circuit modulates hepatic fibrogenesis via the crosstalk between hepatocytes and hepatic stellate cells. Cell Res. 2015;25(8):930–945. doi: 10.1038/cr.2015.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147(4):765–783.e4. doi: 10.1053/j.gastro.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59(3):583–594. doi: 10.1016/j.jhep.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 35.Ip SP, Yiu HY, Ko KM. Differential effect of schisandrin B and dimethyl diphenyl bicarboxylate (DDB) on hepatic mitochondrial glutathione redox status in carbon tetrachloride intoxicated mice. Mol Cell Biochem. 2000;205(1–2):111–114. doi: 10.1023/a:1007065917665. [DOI] [PubMed] [Google Scholar]
- 36.Ip SP, Poon MK, Che CT, Ng KH, Kong YC, Ko KM. Schisandrin B protects against carbon tetrachloride toxicity by enhancing the mitochondrial glutathione redox status in mouse liver. Free Radic Biol Med. 1996;21(5):709–712. doi: 10.1016/0891-5849(96)00179-7. [DOI] [PubMed] [Google Scholar]
- 37.Torok NJ. Dysregulation of redox pathways in liver fibrosis. Am J Physiol Gastroint Liver Physiol. 2016;311(4):G667–G674. doi: 10.1152/ajpgi.00050.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35(2):297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- 39.Poli G. Pathogenesis of liver fibrosis: role of oxidative stress. Mol Aspects Med. 2000;21(3):49–98. doi: 10.1016/s0098-2997(00)00004-2. [DOI] [PubMed] [Google Scholar]
- 40.Giridharan VV, Thandavarayan RA, Arumugam S, et al. Schisandrin B ameliorates ICV-infused amyloid beta induced oxidative stress and neuronal dysfunction through inhibiting RAGE/NF-kappaB/MAPK and Up-Regulating HSP/Beclin expression. PLoS One. 2015;10(11):e0142483. doi: 10.1371/journal.pone.0142483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thandavarayan RA, Giridharan VV, Arumugam S, et al. Schisandrin B prevents doxorubicin induced cardiac dysfunction by modulation of DNA damage, oxidative stress and inflammation through inhibition of MAPK/p53 signaling. PLoS One. 2015;10(3):e0119214. doi: 10.1371/journal.pone.0119214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33(2):105–136. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]
- 43.Jiang Y, Fan X, Wang Y, et al. Hepatoprotective effects of six schisandra lignans on acetaminophen-induced liver injury are partially associated with the inhibition of CYP-mediated bioactivation. Chem Biol Interact. 2015;231:83–89. doi: 10.1016/j.cbi.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 44.Li WL, Xin HW, Yu AR, Wu XC. In vivo effect of Schisandrin B on cytochrome P450 enzyme activity. Phytomedicine. 2013;20(8–9):760–765. doi: 10.1016/j.phymed.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 45.Chiu PY, Leung HY, Poon MK, Lee SS, Ko KM. Schisandrin B induced antioxidant response is partly mediated by cytochrome P-4502E1 catalyzed reaction in mouse liver. Mol Cell Biochem. 2006;293(1–2):87–92. doi: 10.1007/s11010-006-2957-3. [DOI] [PubMed] [Google Scholar]
- 46.Kohler UA, Bohm F, Rolfs F, et al. NF-kappaB/RelA and Nrf2 cooperate to maintain hepatocyte integrity and to prevent development of hepatocellular adenoma. J Hepatol. 2016;64(1):94–102. doi: 10.1016/j.jhep.2015.08.033. [DOI] [PubMed] [Google Scholar]
- 47.Yang JJ, Tao H, Huang C, Li J. Nuclear erythroid 2-related factor 2: a novel potential therapeutic target for liver fibrosis. Food Chem Toxicol. 2013;59:421–427. doi: 10.1016/j.fct.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 48.Shimozono R, Asaoka Y, Yoshizawa Y, et al. Nrf2 activators attenuate the progression of nonalcoholic steatohepatitis-related fibrosis in a dietary rat model. Mol Pharmacol. 2013;84(1):62–70. doi: 10.1124/mol.112.084269. [DOI] [PubMed] [Google Scholar]
- 49.Xu W, Hellerbrand C, Kohler UA, et al. The Nrf2 transcription factor protects from toxin-induced liver injury and fibrosis. Lab Invest. 2008;88(10):1068–1078. doi: 10.1038/labinvest.2008.75. [DOI] [PubMed] [Google Scholar]
- 50.Leong PK, Ko KM. Schisandrin B induces an Nrf2-mediated thioredoxin expression and suppresses the activation of inflammasome in vitro and in vivo. Biofactors. 2015;41(5):314–323. doi: 10.1002/biof.1224. [DOI] [PubMed] [Google Scholar]
- 51.Checker R, Patwardhan RS, Sharma D, et al. Schisandrin B exhibits anti-inflammatory activity through modulation of the redox-sensitive transcription factors Nrf2 and NF-kappaB. Free Radic Biol Med. 2012;53(7):1421–1430. doi: 10.1016/j.freeradbiomed.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 52.He JL, Zhou ZW, Yin JJ, He CQ, Zhou SF, Yu Y. Schisandra chinensis regulates drug metabolizing enzymes and drug transporters via activation of Nrf2-mediated signaling pathway. Drug Des Devel Ther. 2015;9:127–146. doi: 10.2147/DDDT.S68501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50(4):1294–1306. doi: 10.1002/hep.23123. [DOI] [PubMed] [Google Scholar]
- 54.Ray K. Liver: hepatic stellate cells hold the key to liver fibrosis. Nat Rev Gastroenterol Hepatol. 2014;11(2):74. doi: 10.1038/nrgastro.2013.244. [DOI] [PubMed] [Google Scholar]
- 55.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Disco. 2012;11(10):790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]