

Abstract
A distinctive feature of cancer cells is their elevated levels of reactive oxygen species (ROS), a trait that can cause cancer cells to be more sensitive to ROS-inducing agents than normal cells. ROS takes several forms, each with different reactivity and downstream consequence. Here we show that simultaneous generation of superoxide and hydrogen peroxide within cancer cells results in significant synergy, causing potent and selective cancer cell death. In these experiments superoxide is generated using the NAD(P)H quinone oxidoreductase 1 (NQO1) substrate deoxynyboquinone (DNQ), and hydrogen peroxide is generated using the lactate dehydrogenase A (LDH-A) inhibitor NHI-Glc-2. This combination reduces tumor burden and prolongs survival in a mouse model of lung cancer. These data suggest that simultaneous induction of superoxide and hydrogen peroxide can be a powerful and selective anticancer strategy.
INTRODUCTION
Cancer cells harbor increased levels of reactive oxygen species (ROS) relative to normal cells, and ROS plays an important role in signaling, tumorigenesis, proliferation, angiogenesis, and metastasis.1 Due to the elevated ROS levels in cancer, treatment with drugs that generate ROS (either directly or indirectly) has been suggested as an effective strategy for selectively pushing cancer cells over the ROS threshold and into cell death.2–7 Unfortunately, cancer cells also have adaptations that enable them to survive under increased oxidative stress, and these mechanisms can lead to reduced efficacy of ROS-inducing drugs, including in clinical studies.8, 9
Given the recognized potential for ROS induction as an anticancer strategy, various approaches have been attempted to enhance the efficacy of ROS generators, for example, the combination of a ROS inducer with an inhibitor of ROS scavenging.10–13 While such strategies can increase the quantity of general ROS, dramatic synergy is rarely observed.
An alternative approach is to combine two different ROS generators. Several forms of ROS can exist, including hydrogen peroxide (H2O2), superoxide (O2˙−), hydroxyl radical (˙OH), and peroxynitrite (ONOO−), with each having a different reactivity and function.14–16 Superoxide is typically converted to H2O2 by superoxide dismutase (SOD). H2O2 has relatively low reactivity, but it can generate highly reactive hydroxyl radical through the reaction with transition metals such as Fe2+ (Fenton reaction).17–19 We hypothesized that superoxide and H2O2 generators would potently synergize due to their ability to induce the superoxide-driven Fenton reaction. Specifically, the superoxide generator is predicted to drive the Haber-Weiss reaction (Fe3+ + O2˙− → Fe2+ +O2), providing the ferrous iron necessary to react with H2O2 in the Fenton reaction (Fe2+ + H2O2 → Fe3+ + OH− + ˙OH). The production of the highly reactive hydroxyl radical then is capable of damaging nearly all macromolecules found within the cell.8, 20 If superoxide and H2O2 could be generated selectively in cancer cells, they might induce significant synergy and selective cancer cell death.
Described herein we test this hypothesis through use of two experimental therapeutics, deoxynyboquinone (DNQ21–24) and NHI-Glc-225 (Figure 1). These well-characterized compounds operate through two distinct mechanisms, superoxide formation by DNQ through activation by NAD(P)H quinone oxidoreductase 1 (NQO1), and H2O2 formation by NHI-Glc-2 through lactate dehydrogenase A (LDH-A) inhibition. NQO1 is an NAD(P)H dependent two-electron reductase and typically acts as a detoxification enzyme. NQO1 is highly expressed in many solid tumors with minimal expression in normal tissues.26–28 Additionally, elevated levels of NQO1 correlate with poor prognosis in many cancers including lung cancer.29 Reduction of DNQ by NQO1 generates an unstable hydroquinone, which is rapidly and spontaneously oxidized back to the parent, forming superoxide in the process (Figure 1A).24 Greater than 60 mols of superoxide are generated by each mole of DNQ;22, 24 this burst of superoxide overwhelms the cellular capacity to convert it to hydrogen peroxide, thus DNQ is an outstanding compound for generation of rapid and persistent cellular superoxide. LDH-A catalyzes the conversion of pyruvate to lactate, and high LDH-A levels are frequently found in tumors and correlate with poor prognosis and low response to chemotherapy.30, 31 Inhibition of LDH-A results in cancer cell death in culture and in vivo by increased oxidative stress (Figure 1B).32, 33 Here, we show that the combination of DNQ and NHI-Glc-2 significantly increases cancer cell death in a mechanistically distinct fashion. This synergism was shown to be dependent upon NQO1 activity and is operational in vivo. These results suggest that the combination of therapeutics that generate different forms of ROS can potently synergize to induce selective cancer cell death.
Figure 1.
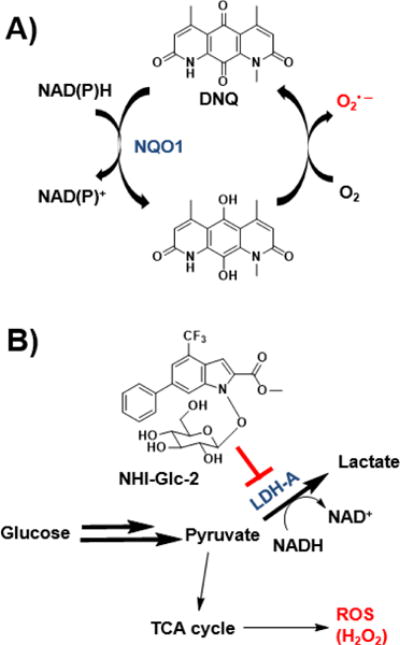
ROS generation by DNQ and NHI-Glc-2. A) DNQ is reduced by NQO1, and the resulting hydroquinone is unstable and spontaneously oxidizes to the parent in the presence of oxygen, producing superoxide. The regenerated DNQ can be used as a substrate for another round of NQO1-mediated redox cycling. B) Aerobic glycolysis is increased in cancer cells. Inhibition of LDH-A increases oxidative phosphorylation, ultimately generating H2O2.
RESULTS AND DISCUSSION
The combination of NHI-Glc-2 and DNQ synergistically induce death of cancer cells in culture
The ability of a combination treatment of NHI-Glc-2 and DNQ to induce cell death was examined in A549 non-small cell lung cancer (NSCLC) cells. These cells express high levels of both LDH-A and NQO1 (Supporting Figure S1A). A549 cell death induced by DNQ was dramatically increased by the addition of NHI-Glc-2 in a dose dependent manner (Figure 2A and Supporting Figure S2). For example, as shown in Figure 2A, single agent treatment with DNQ (50 nM) or NHI-Glc-2 (5 μM) has little effect, but the combination results in >90% cell death. To determine synergy, the combination index (CI) was calculated with the Chou and Tallay method using Compusyn software.34 Combinations describe synergistic interactions when CI value <1, and the lower the CI value, the stronger the synergy. The combination index shows that the combination of DNQ and NHI-Glc-2 in A549 NSCLC cells is markedly synergistic (Figure 2B).
Figure 2.
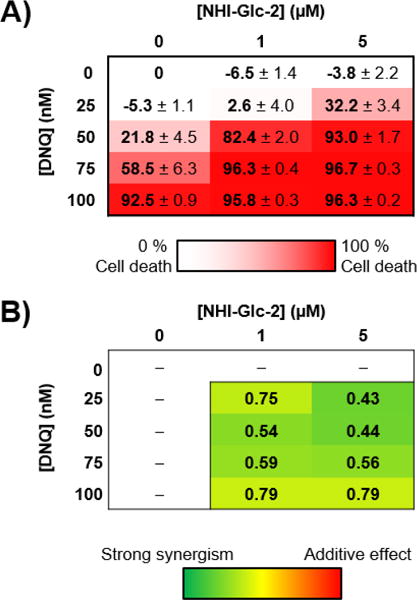
DNQ synergizes with NHI-Glc-2 to enhance cell death in culture. A) A549 NSCLC cells were treated with the indicated concentrations of DNQ and NHI-Glc-2 for 48 h. Percent cell death was assessed using the SRB assay.35 Error represents the s.e.m. of four replicates. B) Combination index values calculated for each combination with CompuSyn software (<1 indicates synergistic interaction).
The synergistic cell death induced by NHI-Glc-2 and DNQ is dependent on NQO1
DNQ is an excellent substrate for NQO1, and its anticancer activity is dependent on NQO1.24 The NQO1 dependency of the combination was investigated by comparing cell death induction in an isogenic cell line pair, MDA-MB-231 (human breast cancer cells) with or without NQO1 expression. In MDA-MB-231 cells that do not express NQO1 (Supporting Figure S1B), DNQ does not induce cell death (Figure 3A, left panel), and no synergy is observed. However, using MDA-MB-231 transfected to express NQO1,22 DNQ induces cell death in a dose dependent manner and synergy is observed upon co-treatment with NHI-Glc-2 (Figure 3A, right panel). Cellular NQO1 activity was also modulated by treating A549 cells with the NQO1 inhibitors ES936 or dicoumarol (DIC) prior to treatment with DNQ (50 nM) and/or NHI-Glc-2 (15 μM). Consistent with previous reports,22, 23 cell death induced by DNQ is reduced by co-treatment with ES936 or DIC, whereas the activity of NHI-Glc-2 is not changed by treatment with NQO1 inhibitors. In the absence of NQO1 inhibitors, dramatic increases of cell death are observed in the combination treatment with NHI-Glc-2 and DNQ. This dramatic increase is not observed when NQO1 is inhibited (Figure 3B). These experiments demonstrate that both the activity of DNQ and the synergy observed with DNQ in combination with NHI-Glc-2 are dependent on NQO1.
Figure 3.
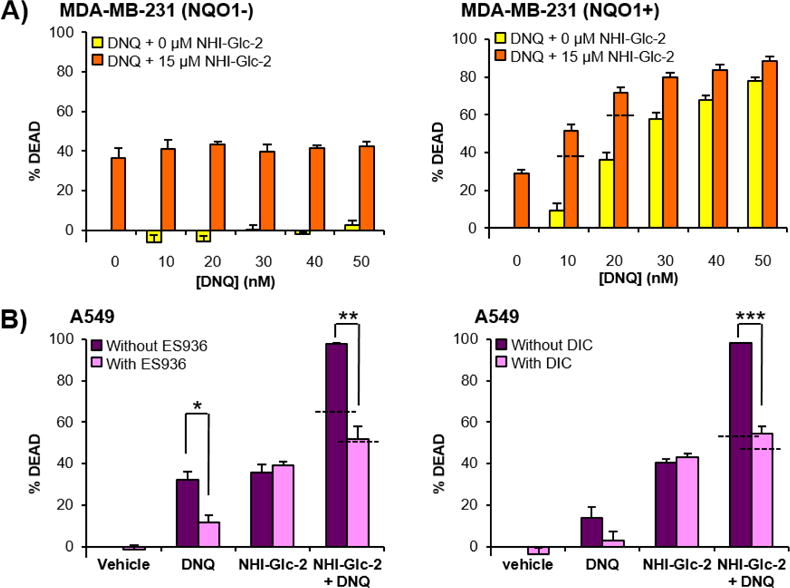
DNQ and NHI-Glc-2 synergize to induce cancer cell death in an NQO1 dependent manner. A) MDA-MB-231 cells transformed with empty plasmid (MDA-MB-231 NQO1-) and with a plasmid containing the gene for NQO1 (MDA-MB-231 NQO1+) were treated with the indicated concentrations of DNQ and NHI-Glc-2 for 48 h, and cell death was assessed using the SRB assay. B) The NQO1 inhibitor ES936 or DIC protects against and DNQ/NHI-Glc-2 mediated cell death in A549 cells. A549 cells were treated with ES936 (100 nM) or DIC (25 μM) prior to treatment with DNQ (50 nM) and/or NHI-Glc-2 (15 μM). Percent cell death observed after 48 h treatment of DNQ and NHI-Glc-2 in the presence and the absence of NQO1 inhibitor using the SRB assay. Error bars represent the s.e.m. of three replicates, and the dashed horizontal lines represent the additive effect of DNQ and NHI-Glc-2 for each drug combination. Statistical analysis was performed using an unpaired, two-tailed student’s t test. * 0.01 ≤ p < 0.05, ** 0.001 ≤ p < 0.01, *** p < 0.001.
NHI-Glc-2 and DNQ synergistically induce death in NQO1-expressing lung cancer cells
The generality of the synergy of NHI-Glc-2 and DNQ was examined in various human lung cancer cell lines (H460, H1993, HCC15, H1299) and normal lung fibroblast cells (IMR90). H460, H1993, and HCC15 cells show high expression of NOQ1, while H1299 and IMR90 cells show little-to-no expression of NQO1 (Supporting Figure S1A). Co-treatment of NHI-Glc-2 and DNQ in the cells with high NQO1 expression such as H460, HCC15, and H1993 dramatically induces increased cell death (Supporting Figure S3A). In particular, in H460 cells where single agent treatment with NHI-Glc-2 (5 μM) or DNQ (40 nM) induces less than 10% cell death, greater than 90% of the cells are killed by the co-treatment; the combination indices in H460 and H1993 cell show strong synergism (Supporting Figure S3B). In contrast, the combination in H1299, which has low NQO1 expression, demonstrates minimal increases in cell death and the combination indices indicate an additive effect or no synergism. IMR90 cells are normal lung fibroblast and do not express NQO1.23 Single agent treatment with DNQ in IMR90 cells is ineffective, and co-treatment with NHI-Glc-2 only reflects the activity of NHI-Glc-2 (Supporting Figure S3). These results are consistent with the experiments performed in the NQO1 +/− isogenic cell lines and with NQO1 inhibitors shown in Figure 3. Taken together, these experiments demonstrate the synergistic cell death induced by the combination of NHI-Glc-2 and DNQ is dependent on NQO1.
ROS generation increases substantially in A549 cells treated with NHI-Glc-2 and DNQ
NQO1-mediated bio-reduction of DNQ to the hydroquinone, and its subsequent oxidation to DNQ rapidly generates ROS in a catalytic fashion (Figure 1A). To observe ROS in cells, ROS detection after 1 hr was accomplished using the fluorescent probes MitoSox Red for superoxide detection, and carboxy-H2DCFDA for detection of general ROS including cytosolic hydrogen peroxide (H2O2); importantly, carboxy-H2DCFDA does not detect superoxide.36 In DNQ-treated A549 cells mitochondrial superoxide generation was markedly increased, while high levels of general ROS (presumably H2O2) were detected in NHI-Glc-2 treated cells; high levels of both superoxide and general ROS were detected in the combination treated cells (Figure 4A). This phenomenon was quantified by flow cytometric analysis, showing superoxide generation by DNQ and H2O2 generation by NHI-Glc-2 are dose dependent (Supporting Figure S4A), with high level of both forms of ROS observed in combination treated cells (Supporting Figure S4B). Importantly, while NHI-Glc-2-treated cells produce H2O2 within the 1 h duration of this experiment, DNQ-treated cells do not produce H2O2 in 1 h, but instead generate large quantities of superoxide during this timeframe (Supporting Figure 4C).
Figure 4.
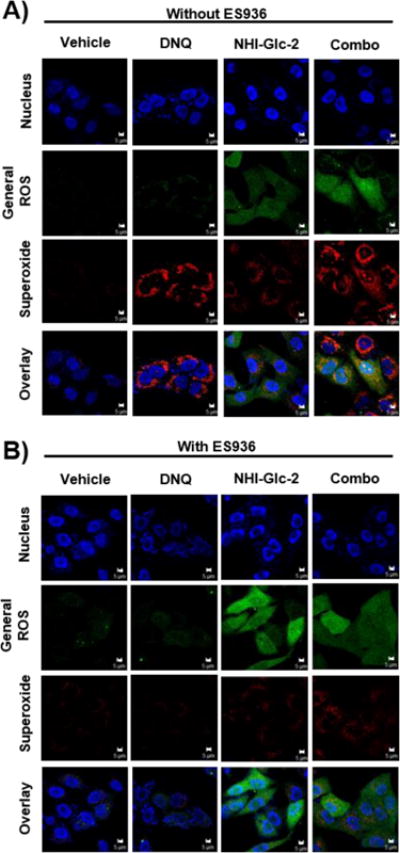
The combination of DNQ and NHI-Glc-2 induces ROS production in A549 cells. A) A549 cells were treated with DNQ (250 nM) and/or NHI-Glc-2 (15 μM) for 1 h. Superoxide and H2O2 production after each compound treatment were visualized using MitoSox Red and carboxy-H2DCFDA respectively. Fluorescence intensities of each ROS indicator were measured using LSM 700. B) The NQO1 inhibitor ES936 protects against DNQ or DNQ/NHI-Glc-2 -mediated ROS production in A549 cells. A549 cells were treated with DNQ (250 nM) and/or NHI-Glc-2 (15 μM) for 1 h after pretreatment of ES936 (100 nM) for 1h. Scale bar indicates 5 μm.
To investigate the role of ROS in the synergistic cell death, cells were treated in the absence and the presence of the reducing agent N-Acetyl Cysteine (NAC). The results show that cell death induced by NHI-Glc-2 (15 μM) was not affected by NAC, while cell death induced by DNQ (50 nM) was reduced by the pre-incubation with NAC (Supporting Figure S5). Additionally, cell death induced by the combination of DNQ (25 nM) and NHI-Glc-2 (15 μM) was significantly reduced by addition of NAC. These results suggest ROS plays an important role in DNQ and combination-mediated cell death.
Increased ROS generation is NQO1 dependent
Because the dramatic synergy observed with the combination of NHI-Glc-2 and DNQ is NQO1 dependent, an assessment was made of whether the ROS generation was also due to NQO1 activity. For this experiment, A549 cells were pretreated with the NQO1 inhibitor ES936 for 1 h. In the absence of ES936, DNQ treatment greatly increased production of superoxide in both the single agent and combination treatment as seen in Figure 4A. However, increased superoxide generation was not detected in DNQ and the combination treated cells after pre-treatment with ES936 (Figure 4B). ROS generation by NHI-Glc-2 was unchanged regardless of NQO1 inhibition. Combination treated cells only showed ROS produced by NHI-Glc-2 after the inhibition of NQO1.
The combination of NHI-Glc-2 with most other ROS-inducing small molecules does not synergistically induce cancer cell death
To test whether NHI-Glc-2 synergizes with other ROS-inducing compounds, A549 were co-treated with NHI-Glc-2 and elesclomol3 or menadione37 for 48h and biomass was assessed. Unlike DNQ, cell death induced by elesclomol or menadione did not synergize with co-treatment of NHI-Glc-2 (Supporting Figure S6A–D). Little to no increase in cell death was observed and combination indices showed additive effects or no synergism. As shown in Supporting Figure S6E, some cells treated with elesclomol or menadione show increased H2O2 generation but no superoxide formation; only DNQ-treated cells displayed a high level of superoxide. Thus, superoxide produced by DNQ is an important and distinctive factor for potentiation with NHI-Glc-2. 2-Methoxyestradiol (2-ME)38 and Embelin39 are reported to be superoxide generators by inhibition of SOD activity and expression respectively. However, cell death induced by 2-ME or Embelin was not increased by addition of NHI-Glc-2 and combination indices showed no synergism (Supporting Figure S7A–S7B). In contrast to the results with 2-ME and Embelin, the compound YM15540, 41 produces dramatic levels of superoxide similar to DNQ and causes synergistic cancer cell death when treated in combination with NHI-Glc-2 (Supporting Figure S7). As shown in Supporting Figure S8, YM155 is not a substrate for NQO1, and therefore YM155 produces superoxide via a different mechanism from DNQ. Regardless, generation of rapid and persistent cellular superoxide by DNQ or YM155 is needed to produce synergistic cytotoxicity with NHI-Glc-2 (Supporting Figure S7C and S7D), likely explaining the dramatic difference in the ability of these compounds to potentiate H2O2.
The combination of DNQ with hydrogen peroxide or tert-butyl hydroperoxide (TBHP) increases cell death
A549 cells were treated with DNQ and hydrogen peroxide (H2O2) or tert-butyl hydroperoxide (TBHP) for 48 h and cell death was assessed. Similar to NHI-Glc-2, H2O2 and TBHP potentiate DNQ-mediated cell death (Figure 5A and 5B), with treatment of DNQ (50 nM) and TBHP (10 μM) inducing <20% cell death, but with ~90% of cells killed by the combination (Figure 5B). To determine whether the superoxide-driven Fenton reaction is responsible for this synergy, cell death mediated by the combination of DNQ and NHI-Glc-2 or DNQ and TBHP were compared in the absence and the presence of the iron chelator deferoxamine (DFO).42 In A549 cells, cell death induced by TBHP (15 μM) was 35%, but it was reduced to 6% by the pretreatment of DFO (Figure 5C) suggesting that TBHP dependent cell death is primarily caused by highly reactive tert-butoxy radical formation via the Fenton reaction.19 As a single agent, NHI-Glc-2 and DNQ induced cell death were not significantly altered by DFO. However, DNQ + NHI-Glc-2, or DNQ + TBHP mediated cell death was protected by DFO (Figure 5C). These results suggest that the combination of DNQ + NHI-Glc-2 or DNQ + THBP cause synergistic cell death by their ability to induce the Haber-Weiss reaction followed by the Fenton reaction resulting in production of highly reactive and toxic radicals such as hydroxyl or alkoxyl radical.
Figure 5.
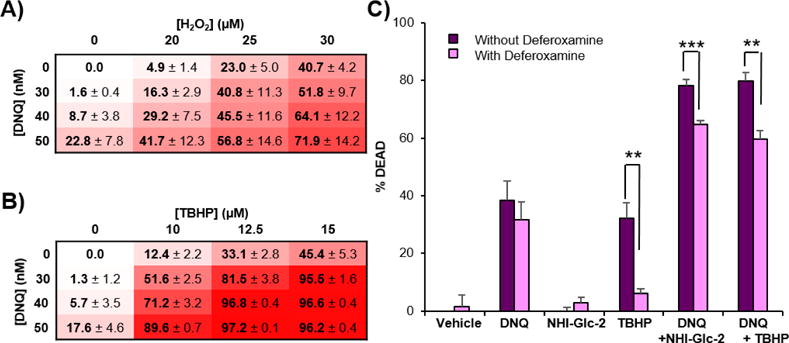
H2O2 or TBHP treatment potentiates DNQ-mediated cell death. A, B) A549 cells were treated with the indicated concentrations of DNQ and H2O2 (A) or TBHP (B) for 48 h. Percent cell death observed after 48 h using SRB assay. Error represents the s.e.m of three replicate. C) Treatment with the iron chelator deferoxamine (DFO) protects from TBHP, DNQ + NHI-Glc-2, or DNQ + TBHP− mediated cell death. A549 cells were pretreated with DFO (100 μM) for 1 h and then DNQ (100 nM), NHI-Glc-2 (10 μM), or TBHP (15 μM) were added and co-treated for 2 h. After washing, the cells were incubated for 48 h in drug free media. Percent cell death was assessed using SRB assay. Error bars represent the s.e.m of four replicates. Statistical analysis was performed using an unpaired, two-tailed student’s t test. ** 0.001 ≤ p < 0.01, *** p < 0.001.
ROS generation by DNQ increases glucose uptake into cells by inducing GLUT1 over-expression
ROS is closely correlated with carbohydrate metabolism,43, 44 inducing hypoxia inducible factor 1 (HIF1) activation, which alters the expression level of glucose transporters (GLUT) and glycolytic proteins including LDH-A.45 Previously, it was shown that GLUTs transcription level in U-937 cells is increased upon the treatment with DNQ.21 As NHI-Glc-2 is taken up by cells in a GLUT specific fashion,25 the manner in which DNQ-derived ROS affects GLUT1 expression and glucose uptake in synergy with NHI-Glc-2 was assessed. GLUT1 expression dramatically increased following DNQ treatment in A549, H460, and H1993 cells (Figure 6A and Supporting Figure S9A). GLUT1 expression was most dramatically increased by DNQ treatment in H460 cells and the synergism was greatest in this cell line, consistent with a strong relationship between ROS, glucose uptake, and synergistic cell death. To further explore this connection, uptake of a fluorescently-labeled glucose probe, 2-NBDG, was measured by flow cytometry. Cellular uptake of 2-NBDG was increased with increasing concentrations of DNQ, and inhibited by ES936 (Figure 6B and Supporting Figure S9B). In order to assess if ROS generation resulted in increased cellular NHI-Glc-2 uptake, cellular uptake of NHI-Glc-2 following DNQ treatment was measured (using HPLC) and found to be increased 1.3-fold (Figure 6C). Taken together, these results demonstrate that ROS generation by DNQ is NQO1 dependent and results in increased GLUT1 expression, leading to increased cellular uptake of glucose and NHI-Glc-2. In cells, NHI-Glc-2 reduces lactate production and aerobic glycolysis by inhibition of LDH-A.25 Therefore, these cells produce more ATP through oxidative phosphorylation, resulting in increased ROS (H2O2) production. DNQ-generated superoxide synergizes with the resulting H2O2 to generate highly reactive hydroxyl radical through the superoxide-driven Fenton reaction. A schematic of how ROS produced by the combination of NHI-Glc-2 and DNQ induces dramatic cancer cell death is shown in Figure 6D.
Figure 6.
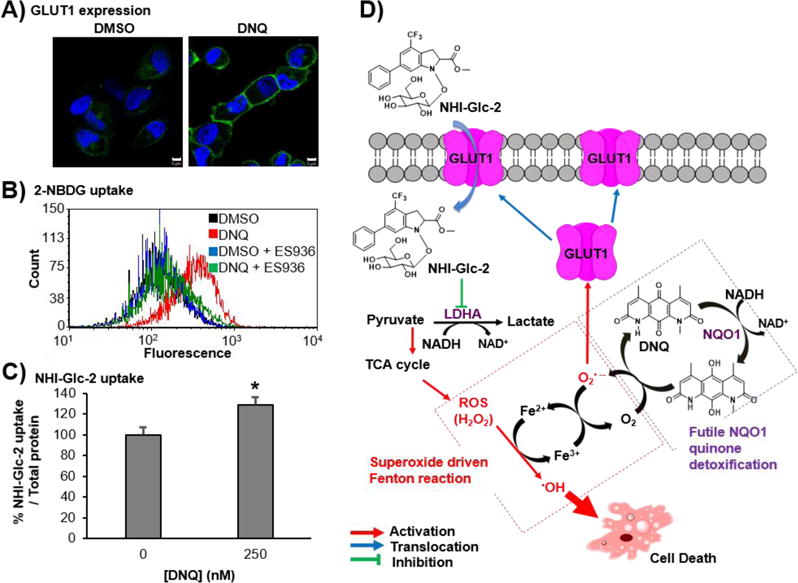
DNQ induces ROS production and glucose uptake in A549 cells. A) GLUT1 expression in A549 cells is increased by DNQ treatment. A549 cells were treated with DNQ (250 nM) for 4 h, and GLUT1 expression in plasma membrane was assessed using immunofluorescence. Scale bar indicates 5 μm. B) 2-NBDG cellular uptake is increased by DNQ treatment. A549 cells were treated with DNQ (100 nM) for 2 h in the absence and the presence of ES936 (100 nM) and then incubated with 2-NBDG (50 μM) for 30 min. 2-NBDG cellular uptake was analyzed using flow cytometry. C) Cellular uptake of NHI-Glc-2 is increased by DNQ treatment in A549 cells. A549 cells were treated with DNQ (250 nM) for 4 h and then incubated with NHI-Glc-2 (20 μM) for 30 min. NHI-Glc-2 cellular uptake was analyzed by HPLC after cell lysis. Error bars represent the s.e.m. of three replicates. Statistical analysis was performed using an unpaired, two-tailed student’s t test. * p < 0.05 relative to DMSO treatment. D) Proposed mechanism of action.
The combination of NHI-Glc-2 and IB-DNQ is active in a murine tumor model
In order to explore the antitumor activity of the combination in vivo, IB-DNQ, a derivative of DNQ that possess superior pharmacokinetic properties (Supporting Figure S10A and S10B) was used. Like DNQ, IB-DNQ-mediated cell death is potent and NQO1-dependent,23, 46 and IB-DNQ also powerfully synergizes with NHI-Glc-2 to enhance cancer cell death in H460 cells (Supporting Figure S10C and S10D). To determine the optimal timing and order of compound addition, a colony forming assay was used to assess multiple dose of NHI-Glc-2 and IB-DNQ. A549 cells were treated either simultaneously with IB-DNQ and NHI-Glc-2 (Supporting Figure S11A) or with NHI-Glc-2 being added 6 (Supporting Figure S11B) to 24 h (Supporting Figure S11C) after IB-DNQ treatment. At these concentrations, treatment of IB-DNQ or NHI-Glc-2 alone has little to no effect on inhibition of colony formation. Simultaneous treatment of compounds and NHI-Glc-2 addition 6 h post IB-DNQ treatment were equally effective. A span of 24 h between IB-DNQ and NHI-Glc-2 was less effective at reducing colony formation. Next, the order of compound addition was examined. A549 cells were treated with IB-DNQ and NHI-Glc-2 simultaneously (Supporting Figure S11D), NHI-Glc-2 was added 6 h after treatment with IB-DNQ (Supporting Figure S11E), or IB-DNQ was added 6h after treatment with NHI-Glc-2 (Supporting Figure S11F). The compound treatments were repeated every other day for four total treatments. More colonies were observed when cells were treated with NHI-Glc-2 prior to IB-DNQ addition. In contrast, the number of colonies was similar when NHI-Glc-2 was added simultaneously or 6 h after IB-DNQ. This trend was also observed with H460 cells (Supporting Figure S11G–S11L).
Based upon the results of the colony forming assays, dosing strategies were designed using oral gavage of IB-DNQ followed by oral gavage of NHI-Glc-2 six hours later. To assess efficacy in vivo, a H460 surgical intervention metastasis model was utilized, where tumor cells were implanted subcutaneously, allowed to reach 2 cm3, and then removed prior to drug treatment. The four groups of mice received either vehicle, IB-DNQ, NHI-Glc-2, or the combination of IB-DNQ and NHI-Glc-2. Kaplan-Meier survival curves showed an increased survival of mice treated with the combination of IB-DNQ and NHI-Glc-2 (Figure 7A). 60% of the combination treated mice were alive 60 days post resection, while all of vehicle treated mice were dead. In addition, tumor burden in the lungs (lung metastases) was significantly reduced in combination treated mice (Figure 7B–7D).
Figure 7.
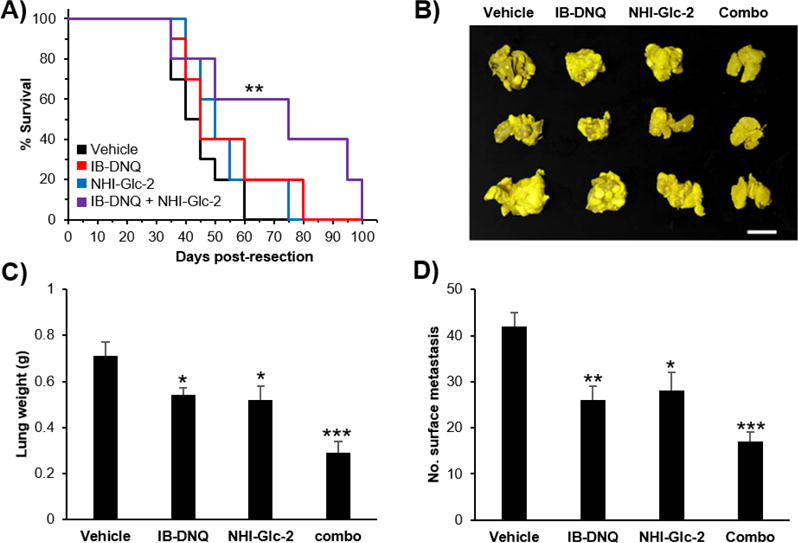
The combination of IB-DNQ and NHI-Glc-2 has an antitumor effect in vivo. H460 cells were inoculated subcutaneously into SCID mice. After 8 weeks the size of tumors were 2 cm3, and at this point the tumors were resected. The animals were split into four groups and treated with vehicle, IB-DNQ (15 mg/kg), NHI-Glc-2 (125 mg/kg), and IB-DNQ and NHI-Glc-2 (15 mg kg−1 and 125 mg kg−1, respectively). The drugs were given by oral gavage twice-a-week for 11 weeks. A) Kaplan-Meier survival curve shows prolonged survival of mice by treatment with IB-DNQ and/or NHI-Glc-2. Statistical analysis was performed using Log Rank Survival Test using Origin Pro 9. ** 0.001 ≤ p < 0.01 relative to vehicle treatment. B-D) Lung tumor burden is reduced by the combination treatment. Scale bar indicates 1 cm. Error bars show s.e.m. (n = 5─10 mice per group). Statistical analysis was performed using an unpaired, two-tailed student’s t test. * 0.01 ≤ p < 0.05, ** 0.001 ≤ p < 0.01, *** p < 0.001 relative to vehicle treatment.
CONCLUSION
Cancer cells often have elevated ROS compared to normal tissues thus making them, in principle, more sensitive to changes in ROS levels. In practice, the general toxicity of ROS-generating drugs has led to narrow therapeutic windows and diminished clinical impact.47–50 While the combination of ROS generators and cancer drugs with other mechanisms of action is an active area of investigation,1, 8 the synergistic combination of different forms of ROS remains underexplored. As ROS signaling is essential for normal cell function and extreme levels of ROS are toxic to normal tissues, selective production of ROS in cancer cells is critical. Described herein is a strategy whereby selective generation of superoxide in cancer cells synergizes with drug-generated hydrogen peroxide, resulting in potent and selective cancer cell death. This strategy capitalizes on mechanism-based synergy, with many aspects that are cancer cell specific: 1) Cancer cells have an enhanced antioxidant defense due to high levels of ROS, and NRF2 acts as the master antioxidant regulator by controlling the expression of the enzymes with antioxidant functions, including NOQ1.1, 29 As such the expression of NQO1 is dramatically increased in many cancer types compared to normal cells. 2) DNQ-derived superoxide is continuously generated by a futile NQO1-mediated process, driving the Fenton reaction. 3) Finally, the cellular uptake of glucose and aerobic glycolysis are also dramatically increased in many cancers via the Warburg effect. Thus, high LDH-A levels are frequently found in cancer cells.
The work described herein utilized the combination of the NQO1 substrate DNQ and LDH-A inhibitor NHI-Glc-2 as the superoxide and hydrogen peroxide generators, respectively. However, alternative small molecule sources of superoxide51, 52 and hydrogen peroxide can be envisioned; for example as shown herein DNQ strongly synergizes TBHP (Figure 5). As DNQ operates at the diffusion-controlled limit for NQO1 processing,23 this compound appears to be a particularly strong choice for rapid, potent, and selective superoxide generation in cancer cells.
The selectivity of this strategy, as shown by the minimal effects in normal lung fibroblast IMR-90 cells (or in cancer cells with low levels of NQO1), is worthy of note and suggests in vivo promise. In addition, while the work herein has focused on NSCLC, this drug combination is very likely to be effective in the many other cancer types with overexpression of NQO1,27, 28 including pancreatic,53 breast,54 and head-and-neck cancers.55 More generally, the potential of different ROS forms to synergize is intriguing and could be a general manner to exploit mechanism-based synergy to induce selective cancer cell death.
METHODS
See the Supporting Information for details.
Supplementary Material
Acknowledgments
We thank T. Fan (UIUC), L. Dirikolu (Louisiana State University), and A. Lee (UIUC) for the technical assistance of the MTD and pharmacokinetic analysis of IB-DNQ and NHI-Glc-2. We also thank R. Botham for helpful comments. We are grateful to the NIH grants R01-GM098453 (to PJH, FM, and PS), and R01-CA152330 (to PS), the University of Pisa, Beth Israel Deaconess Medical Center, and the University of Illinois for support of this work.
Footnotes
Supporting Information
The Supporting Information is available for free of charge on the ACS Publications website at DOI:
Experimental procedures and supporting figures S1 ─ S11
References
- 1.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 2.Griffith O. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem. 1982;257:13704–13712. [PubMed] [Google Scholar]
- 3.Kirshner JR, He S, Balasubramanyam V, Kepros J, Yang CY, Zhang M, Du Z, Barsoum J, Bertin J. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther. 2008;7:2319–2327. doi: 10.1158/1535-7163.MCT-08-0298. [DOI] [PubMed] [Google Scholar]
- 4.Miller WH, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 5.Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014;2:17. doi: 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by b-phenylethyl isothiocyanate. Cancer cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 7.Parkinson EI, Hergenrother PJ. Runaway ROS as a selective anticancer strategy. ChemMedChem. 2011;6:1957–1959. doi: 10.1002/cmdc.201100381. [DOI] [PubMed] [Google Scholar]
- 8.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 9.Wondrak GT. Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid Redox Signal. 2009;11:3013–3069. doi: 10.1089/ars.2009.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halasi M, Pandit B, Wang M, Nogueira V, Hay N, Gartel AL. Combination of oxidative stress and FOXM1 inhibitors induces apoptosis in cancer cells and inhibits xenograft tumor growth. Am J Pathol. 2013;183:257–265. doi: 10.1016/j.ajpath.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maeda H, Hori S, Ohizumi H, Segawa T, Kakehi Y, Ogawa O, Kakizuka A. Effective treatment of advanced solid tumors by the combination of arsenic trioxide and L-buthionine-sulfoximine. Cell Death Differ. 2004;11:737–746. doi: 10.1038/sj.cdd.4401389. [DOI] [PubMed] [Google Scholar]
- 12.Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, Keating MJ, Huang P. Inhibition of mitochondrial respiration a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem. 2003;278:37832–37839. doi: 10.1074/jbc.M301546200. [DOI] [PubMed] [Google Scholar]
- 13.Tomasetti M, Santarelli L, Alleva R, Dong LF, Neuzil J. Redox-active and redox-silent compounds: synergistic therapeutics in cancer. Curr Med Chem. 2015;22:552–568. doi: 10.2174/0929867321666140915142219. [DOI] [PubMed] [Google Scholar]
- 14.Auten RL, Davis JM. Oxygen toxicity and reactive oxygen species: the devil is in the details. Pediatr Res. 2009;66:121–127. doi: 10.1203/PDR.0b013e3181a9eafb. [DOI] [PubMed] [Google Scholar]
- 15.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 16.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 17.Indo HP, Yen HC, Nakanishi I, Matsumoto K-i, Tamura M, Nagano Y, Matsui H, Gusev O, Cornette R, Okuda T, Minamiyama Y, St Clair DK, Majima HJ. A mitochondrial superoxide theory for oxidative stress diseases and aging. J Clin Biochem Nutr. 2015;56:1–7. doi: 10.3164/jcbn.14-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat Res. 1996;145:532–541. [PubMed] [Google Scholar]
- 19.Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 20.Tong L, Chuang CC, Wu S, Zuo L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015;367:18–25. doi: 10.1016/j.canlet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 21.Bair JS, Palchaudhuri R, Hergenrother PJ. Chemistry and biology of deoxynyboquinone, a potent inducer of cancer cell death. J Am Chem Soc. 2010;132:5469–5478. doi: 10.1021/ja100610m. [DOI] [PubMed] [Google Scholar]
- 22.Huang X, Dong Y, Bey EA, Kilgore JA, Bair JS, Li LS, Patel M, Parkinson EI, Wang Y, Williams NS, Gao J, Hergenrother PJ, Boothman DA. An NQO1 substrate with potent antitumor activity that selectively kills by PARP1-induced programmed necrosis. Cancer Res. 2012;72:3038–3047. doi: 10.1158/0008-5472.CAN-11-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkinson EI, Bair JS, Cismesia M, Hergenrother PJ. Efficient NQO1 substrates are potent and selective anticancer agents. ACS Chem Biol. 2013;8:2173–2183. doi: 10.1021/cb4005832. [DOI] [PubMed] [Google Scholar]
- 24.Parkinson EI, Hergenrother PJ. Deoxynyboquinones as NQO1-Activated Cancer Therapeutics. Acc Chem Res. 2015;48:2715–2723. doi: 10.1021/acs.accounts.5b00365. [DOI] [PubMed] [Google Scholar]
- 25.Calvaresi EC, Granchi C, Tuccinardi T, Di Bussolo V, Huigens RW, III, Lee HY, Palchaudhuri R, Macchia M, Martinelli A, Minutolo F, Hergenrother PJ. Dual Targeting of the Warburg Effect with a Glucose-Conjugated Lactate Dehydrogenase Inhibitor. ChemBioChem. 2013;14:2263–2267. doi: 10.1002/cbic.201300562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malkinson AM, Siegel D, Forrest GL, Gazdar AF, Oie HK, Chan DC, Bunn PA, Mabry M, Dykes DJ, Harrison J, Steadman D, Ross D. Elevated DT-diaphorase activity and messenger RNA content in human non-small cell lung carcinoma: relationship to the response of lung tumor xenografts to mitomycin C. Cancer Res. 1992;52:4752–4757. [PubMed] [Google Scholar]
- 27.Siegel D, Ross D. Immunodetection of NAD (P) H: quinone oxidoreductase 1 (NQO1) in human tissues. Free Radic Biol Med. 2000;29:246–253. doi: 10.1016/s0891-5849(00)00310-5. [DOI] [PubMed] [Google Scholar]
- 28.Yang Y, Zhang Y, Wu Q, Cui X, Lin Z, Liu S, Chen L. Clinical implications of high NQO1 expression in breast cancers. J Exp Clin Cancer Res. 2014;33:14. doi: 10.1186/1756-9966-33-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Z, Zhang Y, Jin T, Men J, Lin Z, Qi P, Piao Y, Yan G. NQO1 protein expression predicts poor prognosis of non-small cell lung cancers. BMC cancer. 2015;15:207. doi: 10.1186/s12885-015-1227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koukourakis M, Giatromanolaki A, Sivridis E, Bougioukas G, Didilis V, Gatter K, Harris A. Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. Br J Cancer. 2003;89:877–885. doi: 10.1038/sj.bjc.6601205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 33.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA. 2010;107:2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 35.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 36.Zhu H, Bannenberg GL, Moldéus P, Shertzer HG. Oxidation pathways for the intracellular probe 2′, 7′-dichlorofluorescin. Arch Toxicol. 1994;68:582–587. doi: 10.1007/s002040050118. [DOI] [PubMed] [Google Scholar]
- 37.Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, Sutton R, Peterson OH. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem. 2006;281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- 38.Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407:390–395. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]
- 39.Allensworth JL, Aird KM, Aldrich AJ, Batinic-Haberle I, Devi GR. XIAP inhibition and generation of reactive oxygen species enhances TRAIL sensitivity in inflammatory breast cancer cells. Mol Cancer Ther. 2012;11:1518–1527. doi: 10.1158/1535-7163.MCT-11-0787. [DOI] [PubMed] [Google Scholar]
- 40.Nakahara T, Takeuchi M, Kinoyama I, Minematsu T, Shirasuna K, Matsuhisa A, Kita A, Tominaga F, Yamanaka K, Kudoh M, Sasamata M. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 41.Winter GE, Radic B, Mayor-Ruiz C, Blomen VA, Trefzer C, Kandasamy RK, Huber KV, Gridling M, Chen D, Klampfl T, Kralovics R, Kubicek S, Fernandez-Capetillo O, Brummelkamp TR, Superti-Furga G. The solute carrier SLC35F2 enables YM155-mediated DNA damage toxicity. Nat Chem Biol. 2014;10:768–773. doi: 10.1038/nchembio.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abe K, Saito H. Characterization of t-Butyl Hydroperoxide Toxicity in Cultured Rat Cortical Neurones and Astrocytes. Pharmacol Toxicol. 1998;83:40–46. doi: 10.1111/j.1600-0773.1998.tb01440.x. [DOI] [PubMed] [Google Scholar]
- 43.Kang SW, Lee S, Lee EK. ROS and energy metabolism in cancer cells: alliance for fast growth. Arch Pharm Res. 2015;38:338–345. doi: 10.1007/s12272-015-0550-6. [DOI] [PubMed] [Google Scholar]
- 44.Liemburg-Apers DC, Willems PH, Koopman WJ, Grefte S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch Toxicol. 2015;89:1209–1226. doi: 10.1007/s00204-015-1520-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dang CV, Kim J-w, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51–56. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- 46.Lundberg AP, Francis JM, Pajak M, Parkinson EI, Wycislo KL, Rosol TJ, Brown ME, London CA, Dirikolu L, Hergenrother PJ, Fan TM. Pharmacokinetics and derivation of an anticancer dosing regimen for the novel anti-cancer agent isobutyl-deoxynyboquinone (IB-DNQ), a NQO1 bioactivatable molecule, in the domestic felid species. Invest New Drugs. 2017;35:134–144. doi: 10.1007/s10637-016-0414-z. [DOI] [PubMed] [Google Scholar]
- 47.Glasauer A, Chandel NS. Targeting antioxidants for cancer therapy. Biochem Pharmacol. 2014;92:90–101. doi: 10.1016/j.bcp.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 48.Gupta SC, Hevia D, Patchva S, Park B, Koh W, Aggarwal BB. Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal. 2012;16:1295–1322. doi: 10.1089/ars.2011.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sborov DW, Haverkos BM, Harris PJ. Investigational cancer drugs targeting cell metabolism in clinical development. Expert Opin Investig Drugs. 2015;24:79–94. doi: 10.1517/13543784.2015.960077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foufelle F, Fromenty B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharma Res Per. 2016;4:e00211. doi: 10.1002/prp2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD (P) H: quinone oxidoreductase activity is the principal determinant of β-lapachone cytotoxicity. J Biol Chem. 2000;275:5416–5424. doi: 10.1074/jbc.275.8.5416. [DOI] [PubMed] [Google Scholar]
- 52.Tudor G, Gutierrez P, Aguilera-Gutierrez A, Sausville EA. Cytotoxicity and apoptosis of benzoquinones: redox cycling, cytochrome c release, and BAD protein expression. Biochem Pharmacol. 2003;65:1061–1075. doi: 10.1016/s0006-2952(03)00013-3. [DOI] [PubMed] [Google Scholar]
- 53.Li LS, Bey EA, Dong Y, Meng J, Patra B, Yan J, Xie XJ, Brekken RA, Barnett CC, Bornmann WG, Gao J, Boothman DA. Modulating endogenous NQO1 levels identifies key regulatory mechanisms of action of β-lapachone for pancreatic cancer therapy. Clin Cancer Res. 2011;17:275–285. doi: 10.1158/1078-0432.CCR-10-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bey EA, Reinicke KE, Srougi MC, Varnes M, Anderson VE, Pink JJ, Li LS, Patel M, Cao L, Moore Z, Rommel A, Boatman M, Lewis C, Euhus DM, Bornmann WG, Buchsbaum DJ, Spitz DR, Gao J, Boothman DA. Catalase Abrogates β-Lapachone-Induced PARP1 Hyperactivation-Directed Programmed Necrosis in NQO1-Positive Breast Cancers. Mol Cancer Ther. 2013;12:2110–2120. doi: 10.1158/1535-7163.MCT-12-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li LS, Reddy S, Lin ZH, Liu S, Park H, Chun SG, Bornmann WG, Thibodeaux J, Yan J, Chakrabarti G, Xie XJ, Sumer BD, Boothman DA, Yordy JS. NQO1-mediated tumor-selective lethality and radiosensitization for head and neck cancer. Mol Cancer Ther. 2016;15:1757–1767. doi: 10.1158/1535-7163.MCT-15-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.