

Abstract
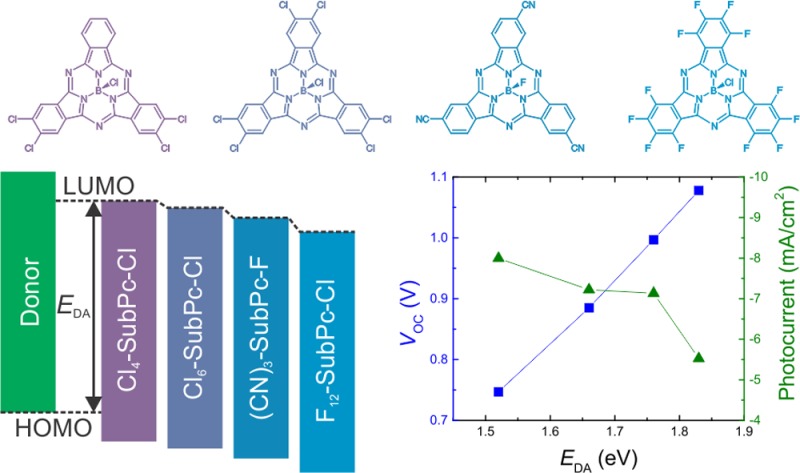
The use of non-fullerene acceptors in organic photovoltaic (OPV) devices could lead to enhanced efficiencies due to increased open-circuit voltage (VOC) and improved absorption of solar light. Here we systematically investigate planar heterojunction devices comprising peripherally substituted subphthalocyanines as acceptors and correlate the device performance with the heterojunction energetics. As a result of a balance between VOC and the photocurrent, tuning of the interface energy gap is necessary to optimize the power conversion efficiency in these devices. In addition, we explore the role of the charge transport layers in the device architecture. It is found that non-fullerene acceptors require adjusted buffer layers with aligned electron transport levels to enable efficient charge extraction, while the insertion of an exciton-blocking layer at the anode interface further boosts photocurrent generation. These adjustments result in a planar-heterojunction OPV device with an efficiency of 6.9% and a VOC above 1 V.
Introduction
Solar energy conversion in organic photovoltaic (OPV) devices generally relies on the dissociation of photogenerated excitons at the heterojunction between an electron-donating and an electron-accepting material. The free energy driving this charge transfer (CT) process is related to the energetic offset between the excitonic state and the charge-separated state. Transient absorption studies have shown that a reduction of this energetic driving force decreases the quantum yield of free charge carriers.1,2 As a consequence, efficient photocurrent generation is observed only when sufficient band energy offsets are present at the heterojunction.3,4 On the other hand, the open-circuit voltage (VOC) of OPV devices has been shown to scale with the interface energy gap EDA, which is the energy difference between the highest occupied molecular orbital (HOMO) of the donor and the lowest unoccupied molecular orbital (LUMO) of the acceptor.5,6 The photocurrent and VOC are thus both governed by the heterojunction energetics. Energy level engineering of the active materials will therefore be crucial to further enhance the power conversion efficiency (PCE) of OPV devices.
The use of fullerenes as acceptors in OPV devices generally enables efficient charge transfer and electron transport. However, fullerene molecules possess a low absorption intensity at longer visible wavelengths, and their frontier molecular orbital energy levels cannot easily be tuned. As a consequence, performance enhancement of OPV devices has mainly been achieved by the development of new donor materials.7,8 Chemical functionalization of small-molecule or polymer donor materials either adjusts the band gap to enhance the light absorption and photocurrent or shifts the HOMO level to increase the energy of the CT state and VOC. However, VOC of efficient fullerene-based OPV devices is generally limited to below 1 V because of the small optical band gap of fullerene acceptors.3 Therefore, it has been suggested that non-fullerene acceptors could enable an increased VOC in OPV devices without sacrificing photocurrent generation. Furthermore, non-fullerene acceptors offer additional advantages compared with fullerenes, such as efficient absorption of solar light and easy tuning of the band energies by chemical modification.
Boron subphthalocyanine chloride (SubPc) is an organic semiconducting molecule commonly used as an electron-donor material in OPV devices.9,10 In combination with fullerene acceptors, power conversion efficiencies (PCEs) above 5% have been reported.11,12 However, SubPc can also be employed as an acceptor material, provided that the selected donor material supplies sufficient energy offsets to enable efficient exciton dissociation at the heterojunction.13,14 The electronic properties of SubPc molecules can easily be tuned by the introduction of peripheral substituents.9 Similar to halogenation of phthalocyanines,15 peripheral halogenation of SubPc results in a shift of the molecular orbital energy levels, enabling the use of these SubPc derivatives as acceptors in combination with common donor materials. In contrast, axial substitution of the chlorine atom has shown little effect on the molecular orbital energy levels.10 Because neither peripheral nor axial substitutions alter the conjugated system of the molecule, the optical band gap and absorption spectrum of SubPc derivatives remain largely unchanged. As a consequence, the class of SubPc molecules is highly suited for use in investigating the influence of the acceptor’s energetic position on OPV device performance. Moreover, the easy tunability of their energy levels simplifies the optimization of interface energetics in donor–acceptor heterojunctions.
Both peripherally and axially substituted SubPc molecules have been applied as electron-acceptor materials in previous reports, resulting in high-voltage devices with VOC values of up to 1.3 V.16−18 However, low quantum efficiencies and the overlapping absorption profiles of the donor and acceptor materials resulted in low photocurrent generation. Selecting donor materials with absorption profiles complementary to those of the SubPc acceptors has resulted in efficiencies over 6%.19 Partial or full halogenation of the SubPc periphery (namely, F12-SubPc-Cl and Cl6-SubPc-Cl) yielded the most promising results. As a consequence, these compounds have been incorporated extensively in OPV devices in the past few years. Further efficiency enhancement could be achieved by fine-tuning the HOMO level and especially the LUMO level of the acceptor compound in the corresponding active layer.
In this study, we combined four small-band-gap donor materials with four peripherally substituted SubPc derivatives as acceptors in vacuum-evaporated planar-heterojunction OPV devices. Specifically, we explored for the first time an unsymmetrically substituted SubPc bearing two chlorine atoms at two isoindole units, Cl4-SubPc-Cl, as a compound with a slight increase in the LUMO energy relative to Cl6-SubPc-Cl. Moreover, cyano groups were introduced as novel electron-accepting peripheral substituents, giving access to the new evaporable electron acceptor (CN)3-SubPc-F bearing π-conjugated substituents and an electron-withdrawing character intermediate between those of Cl6-SubPc-Cl and F12-SubPc-Cl. As a result of the alteration of the peripheral substituent groups on the SubPc molecule, the LUMO energy of these non-fullerene acceptors could be tuned in this particular case by over 0.3 eV. However, the LUMO level of SubPc derivatives can be further lowered by introducing appropriate electron-withdrawing peripheral substituents. Thus, with nonsubstituted SubPc as a reference,9 the LUMO tunability range can formally increase to even 1 eV.
The facile energy level tuning of SubPc acceptors enabled us to systematically study the effect of the heterojunction energetics on the device performance, while the complementary absorption profiles of the donor and acceptor materials resulted in high photocurrent generation. The highest PCE was obtained for the combination of boron subnaphthalocyanine chloride (SubNc) as the donor and hexachlorinated SubPc (Cl6-SubPc-Cl) as the acceptor. The device performance was further improved by replacing the molybdenum oxide (MoO3) hole transport layer (HTL) by a diindenoperylene exciton-blocking layer, resulting in a planar-heterojunction OPV device with an efficiency of 6.9% and a VOC above 1 V.
Results and Discussion
Four peripherally substituted SubPc derivatives, shown in Figure 1a, were used in this study: two partially chlorinated SubPc molecules (Cl4-SubPc-Cl and Cl6-SubPc-Cl), a fully fluorinated SubPc molecule (F12-SubPc-Cl), and a tricyanated SubPc molecule ((CN)3-SubPc-F);20 the last of these contains a fluoride as the axial ligand instead of a chloride. The four SubPc derivatives have very similar absorption spectra in solution (Figure S1 in the Supporting Information), peaking at wavelengths around 570 nm. The electron affinities of these molecules, which are indicative of their LUMO energies, were estimated from the reduction potentials obtained by cyclic voltammetry measurements (Figure S2) and are schematically depicted in Figure 1c. The electron-withdrawing character of the peripheral substituents lowers the frontier orbital energies of the SubPc molecule, resulting in LUMO energies ranging from −3.54 to −3.85 eV. We note that caution should be used when comparing these LUMO levels with molecular energy levels in the literature.21 First of all, cyclic voltammetry measurements yield approximate values of the electron affinity (and LUMO energy). Second, the literature employs many different approaches for measuring and reporting molecular energy levels, and the experimental errors are often large. Therefore, the indicated LUMO levels and interface band gap energies in this work serve only to reflect the relative positions of the transport energy levels for the different SubPc derivatives rather than representing the absolute values of these energy levels.
Figure 1.
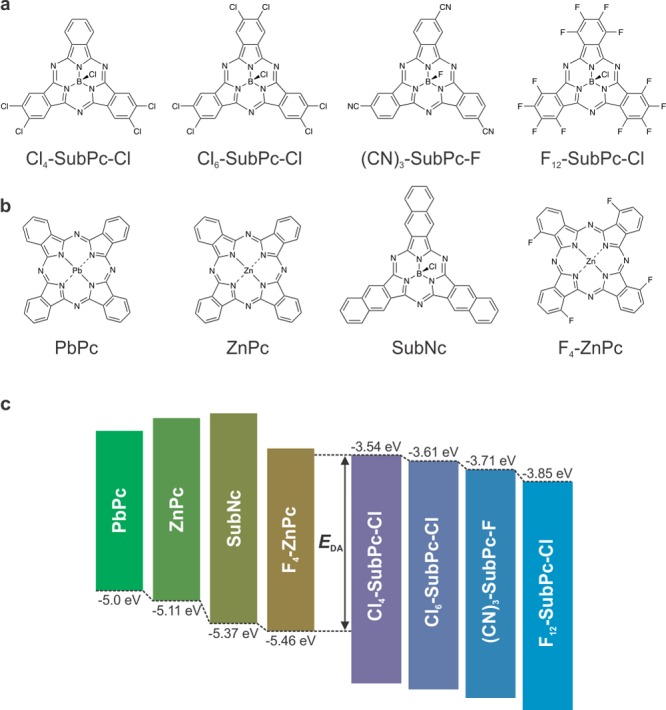
Structural and energetic properties of the active organic molecules used in this work. (a) Molecular structures of the SubPc acceptors. (b) Molecular structures of the donor materials. (c) Schematic representation of the LUMO energy levels of the SubPc acceptors, the HOMO energy levels of the donor materials, and the interface band gap EDA.
Planar-heterojunction devices containing the SubPc derivatives as acceptors were fabricated. These non-fullerene acceptors were combined with the small-band-gap donor materials shown in Figure 1b: lead phthalocyanine (PbPc), zinc phthalocyanine (ZnPc), SubNc, and zinc tetrafluorophthalocyanine (F4-ZnPc). These donor molecules were selected to span a range of HOMO energy levels (−5.0, −5.11, −5.37, and −5.46 eV, respectively).22−24 Because of their smaller band gaps, all of the donor materials have absorption spectra complementary to those of the SubPc acceptors (Figure S3). A 14 nm thick donor layer was deposited for SubNc, while a thickness of 40 nm was chosen for the other phthalocyanine donors. The thickness of the acceptor layer was set at 8 nm, irrespective of which SubPc derivative was used. Insertion of 5 nm of MoO3 at the anode interface ensured good hole extraction due to an improved energy level alignment with the donor materials.25 A 50 nm thick 1:1 blend of bathocuproine (BCP) and fullerene C60 was used as the electron transport layer (ETL). The low optical absorption and high conductivity of this layer ensured sufficient optical spacer thickness without compromising electron extraction.26 It has been shown that the presence of fullerene in the ETL does not actively contribute to the photocurrent generation in devices with a SubPc derivative as the acceptor.19,27 The effect of the SubPc acceptor on device performance was studied by current density–voltage (J–V) measurements under simulated solar illumination of 100 mW/cm2 (Figure S4) and by external quantum efficiency (EQE) measurements (Figure S5). The combination of each SubPc derivative with several donor materials resulted in a large set of data (Table S1), enabling us to assess the impact of the heterojunction energetics on the VOC, fill factor (FF), and photocurrent.
In this set of bilayer heterojunction devices, the VOC ranges from 0.3 to 1.1 V. For a single donor material, the VOC can be tuned by 0.3 V depending on the choice of the SubPc acceptor. Figure 2a shows that VOC increases linearly with the interface energy gap EDA. This corresponds to the well-known trend previously reported in the literature5,6 correlating VOC with the interface energetics. The upper limit for VOC is determined by the energy of the CT state,28 which is closely related to the interface gap EDA. However, the measured VOC in organic heterojunction devices never reaches this upper limit because of energy losses resulting from charge carrier recombination. The linear increase in VOC with EDA thus suggests that the different recombination processes occurring in these planar-heterojunction devices generally do not influence VOC. Only devices with a F4-ZnPc donor yielded a reduced VOC compared with the other donor materials, possibly resulting from significantly increased recombination losses. However, the reduced VOC could also be a related to dipole formation at the F4-ZnPc interface, as was suggested after similar observations for fullerene-based devices.29 Also considering the large uncertainty in the reported frontier orbital energies in the literature, both the interface energetics and the recombination dynamics at non-fullerene heterojunctions are therefore subject to further investigation. The remarkably low VOC for the device with the largest interface gap, i.e., the combination of F4-ZnPc and Cl4-SubPc-Cl, is a consequence of the lack of photocurrent generation at this heterojunction. With no free charge carriers available, no considerable photovoltage can be generated in this device structure. Disregarding the F4-ZnPc-based devices, the average difference between EDA and q·VOC is 0.8 eV. This value corresponds to those in previous literature reports,5,6 considering that the estimated interface energy gap EDA exceeds the energy of the CT state by the CT-state binding energy. The advantage of the SubPc acceptors hence lies in the facile tuning of EDA rather than reducing the energy losses at the active heterojunction.
Figure 2.

Relation of OPV performance parameters to the heterojunction energetics. (a) VOC scales linearly with the interface band gap energy EDA. (b) The photocurrent at reverse bias generally decreases with EDA. (c) The trade-off between photocurrent and VOC limits the PCE of organic heterojunction devices. The contour lines represent PCEs calculated assuming a 65% FF and a voltage-independent photocurrent. The arrow indicates a device with an exciton-blocking hole transport layer, which increases the photocurrent and consequently the PCE. (d) The FF is related to the LUMO energy of the acceptor.
The lack of photocurrent generation in the F4-ZnPc/Cl4-SubPc-Cl device warranted further investigation of the dependence of the photocurrent on the heterojunction energetics. Dissociation of bound excitons into free charge carriers is assumed to proceed through an intermediate CT state. A high quantum yield of free charge carriers is obtained only if this CT process is energetically favorable. As the interface energy gap EDA is closely related to the energy of the CT state, a correlation of EDA with photocurrent generation is expected. Figure 2b shows the photocurrents for all of the studied donor–acceptor heterojunctions in relation to their interface gaps. The photocurrent generation differs significantly for devices with different donor materials, depending on the absorption and spectral response of the specific donor material (see the EQE curves in Figure S5). In contrast, the different SubPc derivatives all have similar absorption spectra, and the variation in photocurrent density upon changing the acceptor material in these devices is therefore a good indication of their internal quantum efficiencies. We evaluated the photocurrent density at reverse bias (−2 V), as the photocurrent under short-circuit conditions is reduced by charge extraction issues in some device structures (see below). Figure 2b reveals a general trend toward low photocurrent generation at large EDA for all of the donor materials. Similar to previous reports,1−4 the reduced photocurrent is a consequence of the smaller free energy driving the CT process. The maximal interface gap where efficient CT still occurs differs for every donor material and depends on the optical band gap of the donor. For example, devices with SubNc as the donor, which has the largest band gap of the studied donor materials, generated reasonable photocurrents with all of the SubPc acceptors. In contrast, for devices containing PbPc, the donor with the smallest band gap in our study, the photocurrent was significantly reduced as EDA increased. In case of the F4-ZnPc/Cl4-SubPc-Cl heterojunction, EDA is too large and the energy of the CT state equals or exceeds the donor exciton energy. As a consequence, no free charges were generated at the heterojunction, and nearly no photocurrent was produced in this device structure. These observations confirm the correlation between the heterojunction energetics and the charge generation kinetics at the donor–acceptor interface. Besides the heterojunction energetics, other material properties determine the total photocurrent extraction in these devices, such as charge mobility, bulk recombination, layer morphology, and crystallinity. However, the degree to which these parameters affect the photocurrent requires further material characterization and is therefore a subject for future research.
Figure 2a,b makes clear that VOC and the photocurrent exhibit opposite trends with respect to the interface energy gap EDA: while VOC linearly increases with EDA, the photocurrent generally decreases for large EDA. As a result of this trade-off between VOC and photocurrent, efficiency enhancement of a heterojunction OPV device relies on optimization of the interface energy gap. Both EDA and VOC should be maximized without substantial reduction of the photocurrent. Figure 2c illustrates this trade-off for the presented planar-heterojunction devices. With the contour lines indicating PCEs calculated assuming a 65% FF and a voltage-independent photocurrent, we expect the highest PCE for the combination of a SubNc donor with a Cl6-SubPc-Cl acceptor. Disregarding possible variations in FF, all of the remaining donor–acceptor pairs yield either a reduced VOC or photocurrent and consequently cannot obtain higher efficiencies. For a specific donor material, however, the PCE can easily be maximized by selecting the most appropriate acceptor material. The LUMO level tunability of the SubPc derivatives thus offers an additional advantage in PCE enhancement of heterojunction OPV devices.
Figure 2d plots the measured FF as a function of the LUMO energy of the SubPc acceptor. While the Cl6-SubPc-Cl acceptor yields FFs of up to 66%, severe S-kinks in the J–V curve reduce the FF below 25% for the F12-SubPc-Cl acceptor. Low fill factors are often attributed to limited charge extraction by low charge carrier mobility or high bulk recombination in the device structure. However, the series resistances extracted from the measured J–V curves are similar for all of the donor–acceptor combinations in our study, indicating that potential mobility or bulk recombination effects are not the main cause of the reduced FF. The observed S-kinks are known to occur in planar-heterojunction devices when injection or extraction barriers are present at the interface between the active layer and the charge transport layer.30 Because the FF is mainly determined by the choice of acceptor material in our device set, we conclude that the energy alignment at the acceptor–ETL interface is critical to avoid S-kink manifestation. As electron extraction in the BCP:C60 ETL likely occurs through percolating conductive pathways of fullerene molecules, this blend functions as an excellent ETL for fullerene acceptors.26 However, a BCP:C60 ETL likely gives rise to charge injection or extraction barriers in combination with most non-fullerene acceptors, and energy level alignment between the SubPc acceptor and C60 is required to achieve high FFs. Clearly, Cl6-SubPc-Cl fulfills this requirement, which explains the highly efficient devices obtained in previous work.19 The remaining acceptor materials either form an extraction barrier (F12-SubPc-Cl and (CN)3-SubPc-F) or an injection barrier (Cl4-SubPc-Cl) with the BCP:C60 ETL, resulting in reduced FFs. (While the C60 LUMO energy is often reported to be around 3.9 eV,5 we repeat that the reported Cl6-SubPc-Cl LUMO energy of 3.61 eV is only approximate, and the claimed energy alignment is thus within experimental uncertainty.)
With an average FF of 65.3%, the SubNc/Cl6-SubPc-Cl heterojunction indeed yields the highest PCE in this systematic study, as expected from Figure 2c. As a result of the voltage-dependent photocurrent, the average PCE is yet limited to 3.9% in the general device structure, comprising MoO3 as the HTL and BCP:C60 as the ETL. However, specific modification of these transport layers can further enhance the performance of these planar-heterojunction devices. Figure 3 shows the J–V curves for the following device structures with adjusted electron and hole transport layers:
-
A:
ITO/MoO3 (5 nm)/SubNc (14 nm)/F12-SubPc-Cl (8 nm)/BCP:C60 (1:1, 50 nm)/Ag
-
B:
ITO/MoO3 (5 nm)/SubNc (14 nm)/F12-SubPc-Cl (8 nm)/BCP:Yb (5%, 50 nm)/Ag
-
C:
ITO/PEDOT:PSS/DIP (5 nm)/SubNc (14 nm)/F12-SubPc-Cl (8 nm)/BCP:Yb (5%, 50 nm)/Ag
-
D:
ITO/MoO3 (5 nm)/SubNc (14 nm)/Cl6-SubPc-Cl (8 nm)/BCP:C60 (1:1, 50 nm)/Ag
-
E:
ITO/PEDOT:PSS/DIP (5 nm)/SubNc (14 nm)/Cl6-SubPc-Cl (8 nm)/BCP:C60 (1:1, 50 nm)/Ag
Table 1 summarizes the performance parameters of the best-performing cell for each device structure.
Figure 3.
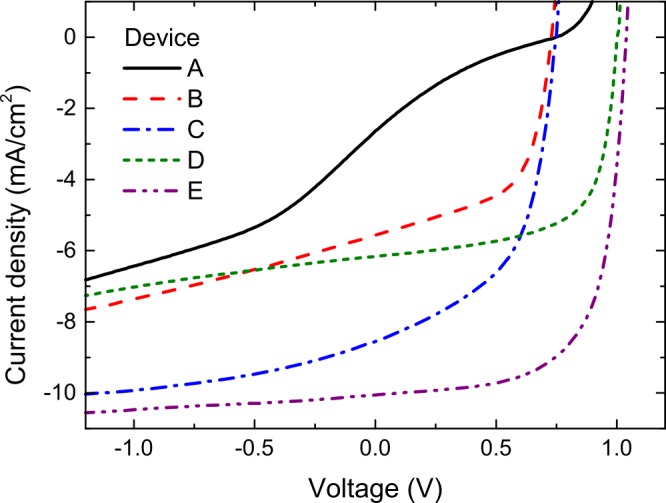
Current density–voltage measurements under simulated solar illumination for planar heterojunction devices with different electron and hole transport layers: (A) MoO3/SubNc/F12-SubPc-Cl/BCP:C60; (B) MoO3/SubNc/F12-SubPc-Cl/BCP:Yb; (C) PEDOT:PSS/DIP/SubNc/F12-SubPc-Cl/BCP:Yb; (D) MoO3/SubNc/Cl6-SubPc-Cl/BCP:C60; and (E) PEDOT:PSS/DIP/SubNc/Cl6-SubPc-Cl/BCP:C60.
Table 1. Solar Cell Performance Parameters for Planar-Heterojunction Devices with Different Electron and Hole Transport Layersa.
| device structure | VOC (V) | JSC (mA/cm2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| A: MoO3/SubNc/F12-SubPc-Cl/BCP:C60 | 0.75 | 2.64 | 17.4 | 0.34 |
| B: MoO3/SubNc/F12-SubPc-Cl/BCP:Yb | 0.73 | 5.57 | 57.6 | 2.25 |
| C: PEDOT:PSS/DIP/SubNc/F12-SubPc-Cl/BCP:Yb | 0.75 | 8.55 | 53.4 | 3.31 |
| D: MoO3/SubNc/Cl6-SubPc-Cl/BCP:C60 | 1.00 | 6.17 | 65.9 | 3.96 |
| E: PEDOT:PSS/DIP/SubNc/Cl6-SubPc-Cl/BCP:C60 | 1.04 | 10.1 | 66.6 | 6.86 |
For each device structure, the open-circuit voltage (VOC), short-circuit current density (JSC), fill factor (FF), and power conversion efficiency (PCE) of the best-performing cell are given.
In device structures with a F12-SubPc-Cl acceptor layer, we replaced the BCP:C60 ETL with a Yb-doped BCP layer. The high conductivity of this BCP:Yb layer ensures good electron extraction even at the high thickness needed in our device structures.31Figure 3 illustrates that the severe S-kink in the J–V curve for device A is no longer present for the BCP:Yb ETL in device B, confirming that BCP:C60 indeed impedes electron extraction from the F12-SubPc-Cl acceptor. We note, however, that a parasitic photoshunt effect in these devices limits the FF to 57%. We observed that the photocurrent slope at reverse bias does not depend on the choice of the electron transport layer. The origin of the photoshunt is therefore likely related to bulk effects in the active layer of the device.32,33 In Cl6-SubPc-Cl-based devices, for which S-kinks were not present, replacement of the ETL did not significantly affect the FF. However, the photocurrent was slightly reduced with BCP:Yb as the ETL (see Figure S6 and Table S2), which could result from exciton quenching by Yb clusters in the ETL or slight changes in the optical interference pattern.
At the anode interface of the device structure, a MoO3 HTL was used to ensure efficient hole extraction from the donor layers.25 Unfortunately MoO3 also significantly reduces the photocurrent as a result of quenching of donor excitons at its interface.34,35 The introduction of exciton-blocking12,36 or exciton-dissociating HTLs24 has been shown to significantly improve photocurrent generation in planar-heterojunction devices. Diindenoperylene (DIP) was recently introduced as an exciton-blocking HTL for SubNc-based devices.19 Here a spin-coated poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) layer was needed to provide a smooth surface, ensuring that DIP forms a closed layer during evaporation. Indeed, the replacement of MoO3 by DIP increased the short-circuit current density (JSC) by 54% in device C. Also VOC slightly increased upon insertion of the exciton-blocking DIP layer. With both the ETL and HTL replaced in the SubNc/F12-SubPc-Cl heterojunction device, the PCE of 3.31% was increased nearly 10-fold relative to that of the original device structure. Also, for the champion heterojunction, comprising a SubNc donor and Cl6-SubPc-Cl acceptor (device D), the photocurrent was further boosted by introducing an exciton-blocking DIP layer at the anode (device E). The increased JSC of 10.1 mA/cm2 led to further efficiency enhancement for this heterojunction, as illustrated in Figure 2c. Moreover, with a VOC above 1 V and a FF of 67%, the resulting PCE of 6.9% is among the highest reported efficiencies for bilayer OPV devices with a non-fullerene acceptor.
Conclusion
In conclusion, we have demonstrated the efficient use of SubPc derivatives as non-fullerene acceptors in bilayer heterojunction devices. In a systematic study, we explored how the device performance depends on the LUMO energy level of these acceptors. As a result of a trade-off between VOC and the photocurrent, the maximal PCE is obtained for heterojunctions with an optimized interface energy gap. Peripheral substitution of the SubPc acceptors enables this interface gap optimization, while the selection of the electron and hole transport layers is crucial to achieve high photocurrent generation and extraction. This study thus shows that the use of non-fullerene acceptors is a successful method to enhance the performance of organic solar cells. Further performance enhancement could result from advanced device structures, such as the insertion of a bulk-heterojunction layer, or the extension to cascade architectures.
Experimental Section
F12-SubPc-Cl and Cl6-SubPc-Cl were synthesized as described in the literature.19,37 Cl4-SubPc-Cl was prepared by cross-condensation of 4,5-dichlorophthalonitrile38 and phthalonitrile following the standard procedure. (CN)3-SubPc-F was prepared by palladium-mediated cyanation of triiodo-SubPc-OPhtBu39 and subsequent phenoxy group to fluorine axial exchange.40 Further details about the synthesis of these SubPc derivatives are given in the Supporting Information.
Electrochemical measurements were performed with an Autolab PGStat 30 system using a three-electrode configuration. The measurements were carried out using tetrahydrofuran solutions containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6). A glassy carbon electrode (3 mm diameter) was used as the working electrode, and a platinum wire and a Ag/AgNO3 (in CH3CN) electrode were employed as the counter and reference electrodes, respectively. Ferrocene (Fc) was added as an internal reference, and all of the potentials are given relative to the Fc/Fc+ couple. The scan rate was 100 mV/s.
OPV devices were fabricated on prepatterned indium tin oxide-coated glass substrates. Detergent and solvent cleaning of all substrates was followed by a 5 min oxygen plasma treatment to remove remaining carbon residue. PEDOT:PSS was spin-coated at 5000 rpm and this was followed by a bake-out at 130 °C in N2. All of the organic materials were purified by thermal gradient sublimation before being loaded into a high-vacuum evaporation chamber. All of the materials were deposited at an evaporation rate of 1 Å/s. The 120 nm thick Ag cathode was evaporated through a shadow mask defining an active area of 13.4 mm2.
Current density–voltage characteristics were measured under simulated solar illumination using a Keithley 2602 measurement unit and an Abet solar simulator calibrated with a Fraunhofer certified photovoltaic cell to yield a 100 mW/cm2 AM1.5G spectrum.
Acknowledgments
The research leading to these results received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under Grant Agreement 287818 of the X10D Project and from the European Community’s ERC Advanced Grant 320680 (EPOS CRYSTALLI). This work was also supported by the Spanish MINECO (CTQ-2014-52869-P) and Comunidad de Madrid (S2013/MIT-2841, FOTOCARBON). The authors thank Erwin Vandenplas for processing support.
Supporting Information Available
Synthetic procedures, absorption spectra, cyclic voltammograms, extinction coefficients, J–V curves measured under simulated solar illumination, and EQE spectra. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b02808.
The authors declare no competing financial interest.
Supplementary Material
References
- Ohkita H.; Cook S.; Astuti Y.; Duffy W.; Tierney S.; Zhang W.; Heeney M.; Mcculloch I.; Nelson J.; Bradley D. D. C.; Durrant J. R. J. Am. Chem. Soc. 2008, 130, 3030. [DOI] [PubMed] [Google Scholar]
- Shoaee S.; Clarke T. M.; Huang C.; Barlow S.; Marder S. R.; Heeney M.; Mcculloch I.; Durrant J. R. J. Am. Chem. Soc. 2010, 132, 12919. [DOI] [PubMed] [Google Scholar]
- Hoke E. T.; Vandewal K.; Bartelt J. A.; Mateker W. R.; Douglas J. D.; Noriega R.; Graham K. R.; Fréchet J. M. J.; Salleo A.; McGehee M. D. Adv. Energy Mater. 2012, 3, 220. [Google Scholar]
- Vandewal K.; Ma Z.; Bergqvist J.; Tang Z.; Wang E.; Henriksson P.; Tvingstedt K.; Andersson M. R.; Zhang F.; Inganäs O. Adv. Funct. Mater. 2012, 22, 3480. [Google Scholar]
- Wilke A.; Endres J.; Hörmann U.; Niederhausen J.; Schlesinger R.; Frisch J.; Amsalem P.; Wagner J.; Gruber M.; Opitz A.; Vollmer A.; Brütting W.; Kahn A.; Koch N. Appl. Phys. Lett. 2012, 101, 233301. [Google Scholar]
- Graham K. R.; Erwin P.; Nordlund D.; Vandewal K.; Li R.; Ngongang Ndjawa G. O.; Hoke E. T.; Salleo A.; Thompson M. E.; McGehee M. D.; Amassian A. Adv. Mater. 2013, 25, 6076. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Li Y.; Zhan X. Chem. Soc. Rev. 2012, 41, 4245. [DOI] [PubMed] [Google Scholar]
- Mishra A.; Bäuerle P. Angew. Chem., Int. Ed. 2012, 51, 2020. [DOI] [PubMed] [Google Scholar]
- Claessens C. G.; González-Rodríguez D.; Rodríguez-Morgade M. S.; Medina A.; Torres T. Chem. Rev. 2014, 114, 2192. [DOI] [PubMed] [Google Scholar]
- Morse G. E.; Bender T. P. ACS Appl. Mater. Interfaces 2012, 4, 5055. [DOI] [PubMed] [Google Scholar]
- Pandey R.; Zou Y.; Holmes R. J. Appl. Phys. Lett. 2012, 101, 033308. [Google Scholar]
- Lin C.-F.; Nichols V. M.; Cheng Y.-C.; Bardeen C. J.; Wei M.-K.; Liu S.-W.; Lee C.-C.; Su W.-C.; Chiu T.-L.; Han H.-C.; Chen L.-C.; Chen C.-T.; Lee J.-H. Sol. Energy Mater. Sol. Cells 2014, 122, 264. [Google Scholar]
- Beaumont N.; Cho S. W.; Sullivan P.; Newby D.; Smith K. E.; Jones T. S. Adv. Funct. Mater. 2012, 22, 561. [Google Scholar]
- Cnops K.; Rand B. P.; Cheyns D.; Verreet B.; Empl M. A.; Heremans P. Nat. Commun. 2014, 5, 3406. [DOI] [PubMed] [Google Scholar]
- Brinkmann H.; Kelting C.; Makarov S.; Tsaryova O.; Schnurpfeil G.; Wöhrle D.; Schlettwein D. Phys. Status Solidi A 2008, 205, 409. [Google Scholar]
- Gommans H.; Aernouts T.; Verreet B.; Heremans P.; Medina A.; Claessens C. G.; Torres T. Adv. Funct. Mater. 2009, 19, 3435. [Google Scholar]
- Sullivan P.; Duraud A.; Hancox I.; Beaumont N.; Mirri G.; Tucker J. H. R.; Hatton R. A.; Shipman M.; Jones T. S. Adv. Energy Mater. 2011, 1, 352. [Google Scholar]
- Morse G. E.; Gantz J. L.; Steirer K. X.; Armstrong N. R.; Bender T. P. ACS Appl. Mater. Interfaces 2014, 6, 1515. [DOI] [PubMed] [Google Scholar]
- Verreet B.; Cnops K.; Cheyns D.; Heremans P.; Stesmans A.; Zango G.; Claessens C. G.; Torres T.; Rand B. P. Adv. Energy Mater. 2014, 4, 1301413. [Google Scholar]
- This compound is in fact a 1:3 mixture of C3 and C1 regioisomers.
- Bredas J.-L. Mater. Horiz. 2014, 1, 17. [Google Scholar]
- Ikushima A. J.; Kanno T.; Yoshida S.; Maeda A. Thin Solid Films 1996, 273, 35. [Google Scholar]
- Riede M. K.; Uhrich C.; Widmer J.; Timmreck R.; Wynands D.; Schwartz G.; Gnehr W.-M.; Hildebrandt D.; Weiß A.; Hwang J.; Sundarraj S.; Erk P.; Pfeiffer M.; Leo K. Adv. Funct. Mater. 2011, 21, 3019. [Google Scholar]
- Barito A.; Sykes M. E.; Huang B.; Bilby D.; Frieberg B.; Kim J.; Green P. F.; Shtein M. Adv. Energy Mater. 2014, 4, 1400216. [Google Scholar]
- Hancox I.; Sullivan P.; Chauhan K. V.; Beaumont N.; Rochford L. A.; Hatton R. A.; Jones T. S. Org. Electron. 2010, 11, 2019. [Google Scholar]
- Bartynski A. N.; Trinh C.; Panda A.; Bergemann K. J.; Lassiter B. E.; Zimmerman J. D.; Forrest S. R.; Thompson M. E. Nano Lett. 2013, 13, 3315. [DOI] [PubMed] [Google Scholar]
- Verreet B.; Rand B. P.; Cheyns D.; Hadipour A.; Aernouts T.; Heremans P.; Medina A.; Claessens C. G.; Torres T. Adv. Energy Mater. 2011, 1, 565. [Google Scholar]
- Vandewal K.; Tvingstedt K.; Gadisa A.; Inganäs O.; Manca J. V. Phys. Rev. B 2010, 81, 125204. [Google Scholar]
- Meiss J.; Merten A.; Hein M.; Schuenemann C.; Schäfer S.; Tietze M.; Uhrich C.; Pfeiffer M.; Leo K.; Riede M. K. Adv. Funct. Mater. 2012, 22, 405. [Google Scholar]
- Tress W.; Leo K.; Riede M. K. Adv. Funct. Mater. 2011, 21, 2140. [Google Scholar]
- Mityashin A.; Cheyns D.; Rand B. P.; Heremans P. Appl. Phys. Lett. 2012, 100, 053305. [Google Scholar]
- Renshaw C. K.; Zimmerman J. D.; Lassiter B. E.; Forrest S. R. Phys. Rev. B 2012, 86, 085324. [Google Scholar]
- Jeong W.-I.; Lee Y. E.; Shim H.-S.; Kim T.-M.; Kim S.-Y.; Kim J.-J. Adv. Funct. Mater. 2012, 22, 3089. [Google Scholar]
- Xiao X.; Zimmerman J. D.; Lassiter B. E.; Bergemann K. J.; Forrest S. R. Appl. Phys. Lett. 2013, 102, 073302. [Google Scholar]
- Barito A.; Sykes M. E.; Bilby D.; Amonoo J.; Jin Y.; Morris S. E.; Green P. F.; Kim J.; Shtein M. J. Appl. Phys. 2013, 113, 203110. [Google Scholar]
- Hirade M.; Adachi C. Appl. Phys. Lett. 2011, 99, 153302. [Google Scholar]
- Claessens C. G.; González-Rodríguez D.; del Rey B.; Torres T.; Mark G.; Schuchmann H.-P.; von Sonntag C.; MacDonald J. G.; Nohr R. S. Eur. J. Org. Chem. 2003, 2547. [Google Scholar]
- Wöhrle D.; Eskes M.; Shigehara K.; Yamada A. Synthesis 1993, 194. [Google Scholar]
- Claessens C. G.; Torres T. Tetrahedron Lett. 2000, 41, 6361. [Google Scholar]
- Rodríguez-Morgade M. S.; Claessens C. G.; Medina A.; González-Rodríguez D.; Gutiérrez-Puebla E.; Monge A.; Alkorta I.; Elguero J.; Torres T. Chem.—Eur. J. 2008, 14, 1342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.