

Abstract
Repetitive DNA is prone to replication fork stalling, which can lead to genome instability. Here, we find that replication fork stalling at telomeres leads to the formation of t‐circle‐tails, a new extrachromosomal structure that consists of circular telomeric DNA with a single‐stranded tail. Structurally, the t‐circle‐tail resembles cyclized leading or lagging replication intermediates that are excised from the genome by topoisomerase II‐mediated cleavage. We also show that the DNA damage repair machinery NHEJ is required for the formation of t‐circle‐tails and for the resolution of stalled replication forks, suggesting that NHEJ, which is normally constitutively suppressed at telomeres, is activated in the context of replication stress. Inhibition of NHEJ or knockout of DNA‐PKcs impairs telomere replication, leading to multiple‐telomere sites (MTS) and telomere shortening. Collectively, our results support a “looping‐out” mechanism, in which the stalled replication fork is cut out and cyclized to form t‐circle‐tails, and broken DNA is religated. The telomere loss induced by replication stress may serve as a new factor that drives replicative senescence and cell aging.
Keywords: looping‐out, NHEJ, replication fork stalling, telomeres, topoisomerase II
Subject Categories: DNA Replication, Repair & Recombination
Introduction
In vertebrate species, chromosome ends termed telomeres are composed of tandem repeats of short DNA sequences (TTAGGG/CCCTAA)n 1 that are associated with specialized shelterin proteins 2, 3. Telomeres protect against deleterious repair activities by preventing chromosome ends from being recognized as broken DNA by the DNA repair machinery. Progressive telomere attrition occurs during cell division due to the “end replication problem” 4 and processing events. Eventually, extremely short telomeres result in “uncapping” of chromosome ends that triggers cellular senescence and apoptosis 5, 6.
The maintenance of telomere structure and function relies on efficient replication of telomeric DNA. The telomere DNA is duplicated by conventional semi‐conservative DNA replication, replication fork can initiate from origins within telomere or subtelomere 7, a G/C‐rich region adjacent to telomeres. However, the heterochromatic nature of telomeres/subtelomeres, the long G‐rich repetitive tracts prone to form G‐quadruplex secondary structure, and possible T‐loop configuration all cause significant replicative stress and may block the progression of the DNA replication fork 8. Replication forks stalled at telomeres or subtelomeres may be restarted with the aid of shelterin protein TRF1/TRF2, the human RecQ helicases such as WRN, BLM, or RTEL‐1, as well as replication accessory proteins like CST complex (CTC1, STN1, and TEN1) 9, 10. The failure in resolution of stalled replication fork leads to the generation of replication‐dependent abnormal structures termed fragile telomeres that is often associated with genome instability 11.
Long telomeres are particularly prone to replicative stress 9, 12, 13, 14. The stochastic “telomere trimming”, a deletion of large segments of telomeric DNA, has been observed in cells with relatively long telomeres, including normal lymphocytes, mature sperm, hiPSCs (human‐induced pluripotent stem cells), hESCs (human embryonic stem cells), and cancer cells with overexpressed hTR 13, 14, 15. In addition, replication stress‐induced “microdeletions” have also been observed at common fragile sites in the genome 16, 17. Furthermore, aphidicolin‐induced replication stress in normal human cells leads to large submicroscopic deletions of DNA segments that are linked to the change of gene copy number 18. Although the mechanism underlying “DNA deletion” under high replication stress is not clear, these observations reveal an intriguing correlation between replication stress and “intralocus DNA (telomere) deletion”.
DNA damage repair machineries including homologue recombination and non‐homologous end‐joining (NHEJ) are associated with telomeric DNA 19, 20, 21. Given that NHEJ activity is constitutively suppressed by TRF2 at chromosome ends, the function of NHEJ at telomeres remains elusive. Interestingly, the inhibition/deficiency of NHEJ in mouse and human cells resulted in phenomena connected to the replication defect of telomeric DNA 22, 23, 24, 25, raising the possibility that NHEJ machinery may be required for successful telomere replication. Especially, it has been hypothesized that telomere replication might be stalled during S phase (phase I), restart and completion of synthesis occurs in late S/G2 (phase II) 19, 20, 21. In this scenario, DNA damage response (DDR) is activated 26, and NHEJ may participate in resolution of stalled replication forks at telomeres.
Here, we analyzed telomeric DNA structures and identified a novel, extrachromosomal, double‐stranded circular telomeric molecule with a long single‐stranded C‐rich tail (t‐circle‐tail). These “t‐circle‐tail” molecules are present in almost all tested human cells and are derived from stalled replication forks at telomeres. The process that generates t‐circle‐tail requires topoisomerase II‐mediated cleavage and NHEJ‐mediated ligation. Inhibition of NHEJ in human cells or knockout of DNA‐PKcs in mouse cells impairs the formation of t‐circle‐tail, resulting in the defect of telomere replication. Based on these findings, we propose a “looping‐out” model in which stalled telomeric replication fork is excised from genome, the released DNA is looped out to form the t‐circle‐tail, and the broken DNA is ligated to initiate new replication fork. This mechanism allows cells to resolve stalled replication forks at telomeric DNA.
Results
Identification of extrachromosomal t‐circle with C‐rich single‐stranded tail
We investigated unusual telomeric DNA structures using two‐dimensional agarose gel electrophoresis (2D gel; Fig 1A). Genomic DNA from HTC75 cells was first digested with HinfI and RsaI and then subjected to 2D gel analysis and “in‐gel” hybridization with a telomere‐specific probe under native and denaturing conditions. As expected, the vast majority of telomere‐hybridizing DNA consisted of linear dsDNA of variable length with G‐rich ssDNA overhangs (e.g., C‐probe hybridization under native and denatured conditions; Fig 1B). A minor fraction of the telomeric DNA signal appeared on the 2D gel as a smear spreading outward from the loading well (tending toward faster mobility/smaller size). This fraction was described previously by Deng et al 27 and designated “slow‐mobility structures”. Unexpectedly, these slow‐mobility structures can only be detected by G‐rich probe under native conditions in HTC75 cells (Fig 1B) and in HeLa cells (Appendix Fig S1A). These results indicate that the structures contain both double‐stranded telomeric DNA and single‐stranded C‐rich DNA.
Figure 1. Identification and characterization of t‐circle‐tail.
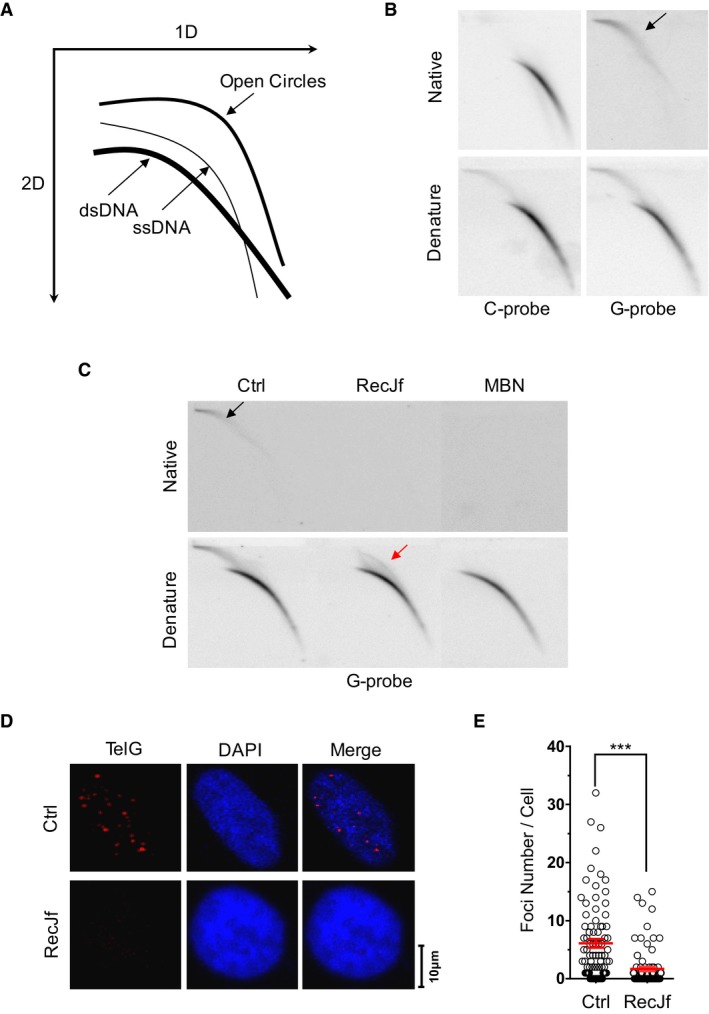
- Schematic of 2D gel and in‐gel hybridization.
- HinfI‐ and RsaI‐digested HTC75 genomic DNA was hybridized to C‐rich telomeric probe or G‐rich telomeric probe under native or denaturing conditions, as indicated. The arrow indicates t‐circle‐tail.
- Genomic DNA was hybridized with G‐rich probe as in (B), except that DNA was digested with RecJf or mung bean nuclease (MBN) prior to 2D gel electrophoresis. The black arrow indicates t‐circle‐tail. The red arrow indicates nicked circular DNA.
- Native FISH hybridization with telomeric G‐rich‐Cy3 probe. HTC75 cells were treated with or without (Ctrl) RecJf prior to FISH.
- Quantification of (D). Each dot represents the number of foci in a single cell. Data represent the mean ± SEM of three independent experiments. P‐value was obtained from two‐tailed unpaired Student's t‐test. ***P < 0.001.
When we treated the genomic DNA with RecJf, a 5′ to 3′ exonuclease with a high specificity for ssDNA, prior to 2D gel analysis, we found that “slow‐mobility structures” were converted into closed‐circular telomeric DNA (e.g., t‐circle DNA; Fig 1C, hybridization under denaturing conditions). Moreover, these “slow‐mobility structures” were completely degraded by mung bean nuclease, an endonuclease specific for single‐stranded DNA or single‐stranded regions on dsDNA (Fig 1C), suggesting that the t‐circle DNA is partially single‐stranded. Taken together, these observations indicate that the slow‐migrating telomere‐homologous signal includes partially single‐stranded t‐circles with 5′ C‐rich tails. We refer to this fraction of telomeric DNA as “t‐circle‐tail”.
We further verified the structure of the t‐circle‐tail in a series of experiments. We found that t‐circle‐tail is insensitive to RNase H (Appendix Fig S1B), indicating that there is no RNA constituent in the structure. T‐circle‐tail was converted to closed‐circular DNA when treated with Plasmid‐safe™ ATP‐dependent DNase, which exonucleolytically digests linear DNA, but not circular DNA (Appendix Fig S1C), further demonstrating the presence of closed‐circular DNA in the structure. Finally, when we generated closed‐circular DNA by treating t‐circle‐tail with RecJf, we found that t‐circle‐tail could be regenerated by the highly processive Φ29 DNA polymerase, which catalyzes rolling circle DNA synthesis and generates long single‐stranded DNA with a 5′ free end 28 (Appendix Fig S1D).
Next, we looked for in vivo evidence of t‐circle‐tail. We performed in situ hybridization of normal HCT75 cells with a fluorescence‐labeled G‐rich telomeric probe under native conditions, with or without prior treatment with RecJf (Fig 1D and E). Whereas distinct nuclear foci were detected in HTC75 cells, they were barely observed in cells pre‐treated with RecJf (Fig 1D and E). In addition, we treated cells with methyl‐methanesulfonate (MMS), a mutagen that specifically introduces DNA lesions at cytosine residues 29, 30. We observed a decreased amount of t‐circle‐tail DNA and a corresponding increase in closed‐circular DNA in MMS‐treated cells (Appendix Fig S1E). These results demonstrate that t‐circle‐tail exists in vivo.
Replication stress induces the formation of t‐circle‐tail
We detected t‐circle‐tail structures in variety of human cell lines, including normal human BJ fibroblasts, hF2, hEF, PBMC, T cells, immortalized 293T cells and many other human cancer cell lines (Appendix Fig S2A). The relative abundance of t‐circle‐tail, calculated as [(intensity of signal in the t‐circle‐tail)/(total intensity of signal) × 100%], varied from 0.11 to 22.63% (Appendix Fig S2A and B). The greater abundance of t‐circle‐tail is correlated with longer telomere length as measured by the TRF assay (P = 0.0039; Fig 2A and Appendix Fig S2C). The same correlation was observed in HeLa cells with different telomere length (HeLa, HeLa 229 and HeLa 1.2.11; Appendix Fig S2D and E). Moreover, when DNA was pre‐treated with RecJf that converts t‐circle‐tail to telomeric circle by removing 5′ single‐stranded tail, we observed that HeLa cells with longer telomere harbored bigger telomeric circle (Appendix Fig S2E), demonstrating that in addition to higher abundance, bigger size of t‐circle‐tail is also correlated with longer telomeres.
Figure 2. T‐circle‐tail is formed in cells experiencing replicative stress.
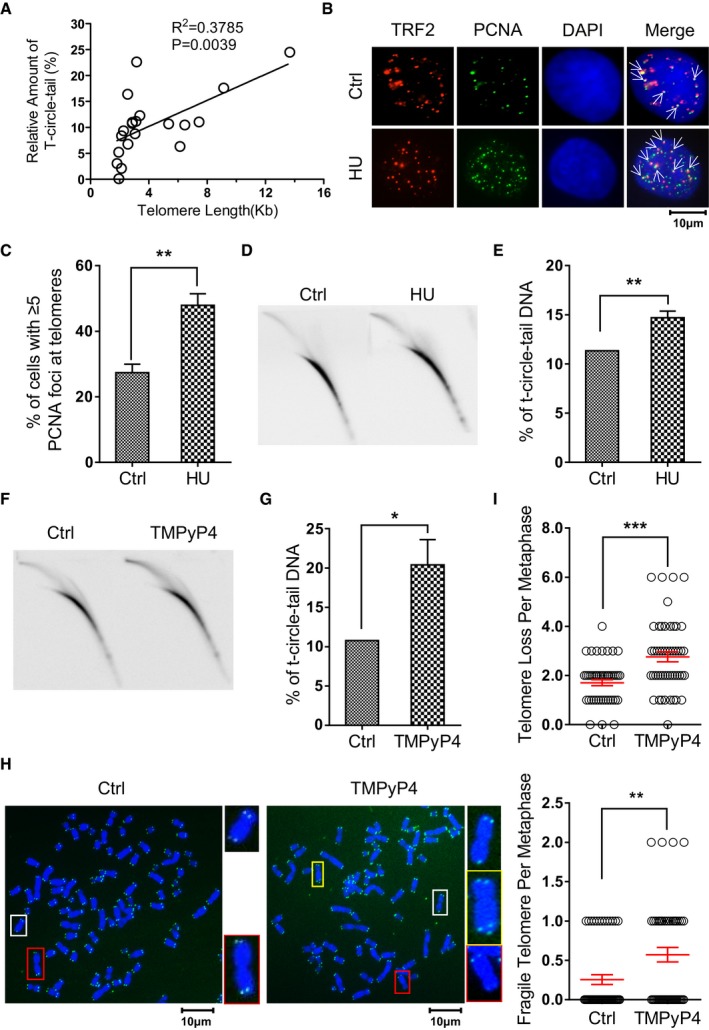
- T‐circle‐tail was quantified in cells with telomeres of different lengths. Raw data are shown in Appendix Fig S2. Linear regression analysis yielded a correlation constant and P‐value of R 2 = 0.3785 and P = 0.0039, respectively.
- HeLa cells were treated with or without HU and incubated for 24 h. Cells were fixed and analyzed by DAPI staining and IF for PCNA or TRF2, as indicated. Colocalized foci are indicated by white arrowheads.
- Quantification of (B). The percent of cells with ≥ 5 co‐localized foci is shown. > 100 cells were counted for each group. Bars represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to obtain P‐values. **P < 0.01.
- Genomic DNA from PBS (Ctrl) or HU‐treated HeLa cells was analyzed by 2D gel electrophoresis and hybridized to G‐rich telomeric probe under denaturing conditions.
- Quantification of (D). Bars represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to obtain P‐values. **P < 0.01.
- 2D gel analysis was performed as in (D), except that cells were treated with or without TMPyP4.
- Quantification of (F). Bars represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to obtain P‐values. *P < 0.05.
- Metaphase spread and FISH showing telomere‐free ends and fragile telomeres in control and TMPyP4‐treated HeLa cells.
- Quantification of (H). Data represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to obtain P‐values. **P < 0.01, ***P < 0.001.
Since long telomeres are prone to replication stress 9, 12, 13, we hypothesized that high replication stress might promote formation of t‐circle‐tail. To test this, we exposed cells to HU, which depletes dNTP pools and induces replication fork stalling on the genome 31. As expected, HU treatment led to an increase in PCNA foci, a marker for stalled replication forks 32, at telomeres and throughout the genome (Fig 2B and C). 2D gel electrophoresis showed that the abundance of t‐circle‐tail increased in HU‐treated cells (Fig 2D and E). Increased t‐circle‐tail was also observed in cells treated with aphidicolin (Appendix Fig S3A and B), which induces replication fork stalling by inhibiting DNA polymerase α and δ, and in cells with knockdown of TRF1 (Appendix Fig S3C–E), a component of shelterin that ensures efficient replication of telomeres 11. These results revealed that replication fork stalling in telomeric DNA induces the production of t‐circle‐tail. To test whether telomere dysfunction induces the generation of t‐circle‐tail, TRF2 was knocked out in HeLa cells by inducible CRISPR/Cas9 system. We observed significant increase of telomere dysfunction induced foci (TIFs) in TRF2‐deficient cells (Dox) that is not correlated with increase in t‐circle‐tail (Appendix Fig S4A–D), suggesting that the formation of t‐circle‐tail is not caused by dysfunctional telomeres. It has been reported that p53 participated in chromosome end protection 33, we then explored a potential function of p53 in t‐circle‐tail formation. Our data showed that depletion of p53 in HTC116 cells (with wild‐type p53) resulted in no change of t‐circle‐tail abundance (Appendix Fig S4E–G), indicating that p53 is not involved in production of t‐circle‐tail.
To explore the cellular consequence associated with the formation of t‐circle‐tail, cells were treated with TMPyP4, a ligand that induces severe fork stalling at G‐rich sequence by promoting the formation of “G‐quadruplex” structure during replication 34. As expected, a significant increase in t‐circle‐tail in TMPyP4‐treated cells was observed (Fig 2F and G). In addition, increased frequency of fragile telomere as evidenced by multi‐telomeric signals (MTS) at chromosome end was detected (Fig 2H), indicating the failure in replicating some telomeres 11. Interestingly, we also observed a significant increase in telomere‐free ends (the chromosome ends with undetectable telomere signal by FISH; Fig 2H and I), suggesting that rapid telomeric DNA deletion occurs.
The t‐circle‐tail structure resembles a cyclized leading or lagging strand during replication
The specific correlation between t‐circle‐tail formation and loss of telomeric DNA raised the possibility that the t‐circle‐tail might be derived from stalled replication forks. To investigate the mechanism underlying the production of t‐circle‐tail, we first confirmed a link between the generation of t‐circle‐tail and telomere replication. HeLa cells were synchronized at G1/S, released into S phase, and then harvested at early, middle, or late S phase and G2 (Appendix Fig S5A). Genomic DNA was isolated and analyzed to determine the abundance of t‐circle‐tail (Appendix Fig S5B). Our results showed that the percentage of t‐circle‐tail was proportional to total cellular DNA throughout the cell cycle (Appendix Fig S5C), suggesting that nascent t‐circle‐tail is generated when the DNA content doubles during S phase. Next, we synchronized HeLa cells at G1/S, released them into S phase for 3 h, and pulse‐labeled with BrdU for 1 h during mid‐S, and immediately harvested (Fig 3A). Incorporation of BrdU into nascent t‐circle‐tail and newly synthesized telomeres (leading and lagging daughter) imparts a higher density, allowing separation and isolation by CsCl density gradient centrifugation (Appendix Fig S5D) 35. We analyzed newly synthesized telomeres (leading and lagging daughter) by 2D gel electrophoresis and found that during 1 h of exposure to BrdU, the production of nascent t‐circle‐tail coincided with telomere replication (Fig 3B), demonstrating that the generation of t‐circle‐tail is closely associated with telomere replication.
Figure 3. T‐circle‐tail consists of newly synthesized leading and lagging daughter DNA .
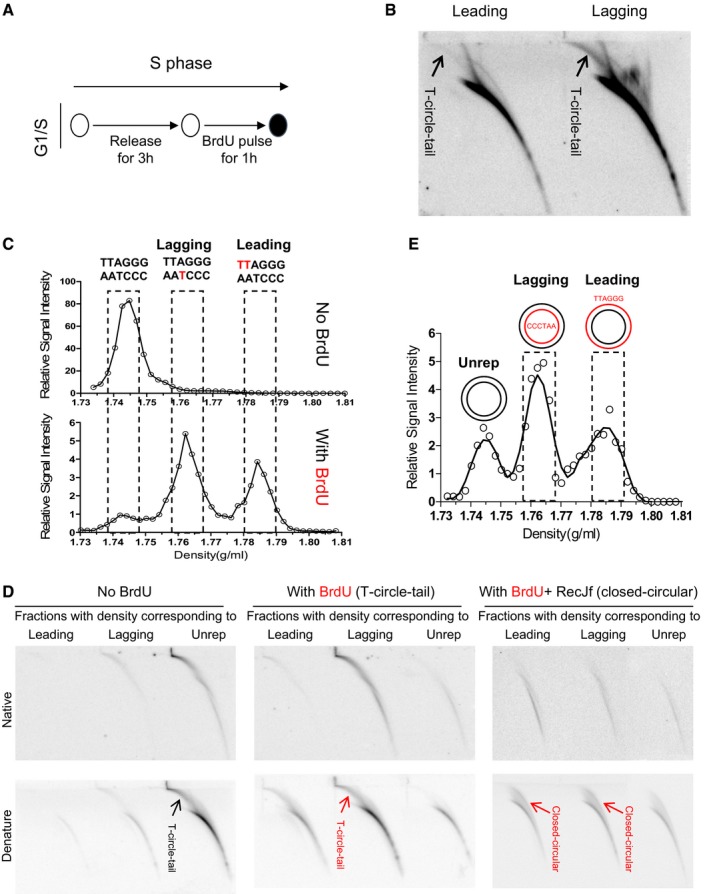
- The experimental protocol used to pulse label cells synchronized at G1/S.
- Cells were pulse‐labeled with BrdU for 1 h, and replicated DNA (leading and lagging daughter) was isolated from a CsCl gradient and analyzed by 2D gel electrophoresis.
- CsCl density gradient centrifugation analysis of BrdU‐labeled telomeric DNA (see Materials and Methods for details). Peaks corresponding to leading and lagging strand replicative intermediates and unreplicated DNA are indicated.
- Telomeric DNA in fractions containing leading, lagging, or unreplicated DNA were analyzed by 2D gel electrophoresis and hybridized with a G‐rich probe. Hybridization conditions are indicated. In the right panel, BrdU‐labeled DNA was digested with RecJf prior to CsCl gradient centrifugation.
- CsCl density gradient centrifugation analysis of the closed‐circular moiety of t‐circle‐tail after RecJf treatment. The fractions corresponding to leading, lagging, or unreplicated DNA are indicated.
We then explored the formation of t‐circle‐tail during S phase by determining its strand synthesis. For this purpose, we released G1/S synchronized HeLa cells into S phase for 12 h in the presence of BrdU. We fractionated total cellular DNA by CsCl gradient centrifugation (Fig 3C). The fractions with a density corresponding to leading, lagging, or unreplicated DNA were analyzed by 2D gel electrophoresis. We found that the majority of newly generated BrdU‐labeled t‐circle‐tail DNA, which shifts to a higher density, was enriched in the fractions corresponding to the lagging strand telomeric DNA in which the C‐rich strand (CCCTAA) is newly synthesized (Fig 3D, left and middle figure). This suggests that new synthesis of t‐circle‐tail involves addition of a long C‐rich strand and is consistent with the long single‐stranded C‐rich strand of t‐circle‐tail. Similar results were obtained in telomerase‐negative human normal BJ fibroblasts and MRC5 cells (Appendix Fig S5E). When genomic DNA was digested with RecJf prior to CsCl gradient centrifugation (Fig 3E), the t‐circle‐tail was converted to closed‐circular DNA, as expected (Fig 3D, middle and right figure). The closed‐circular DNA co‐fractionated with leading and lagging daughter telomeres (Fig 3D, right and Fig 3E), indicating that newly generated closed‐circular moiety of the t‐circle‐tail contains a nascent G‐rich (leading daughter) or C‐rich strand (lagging daughter). The same amount of closed‐circular DNA was detected in leading and lagging fractions (Fig 3D, right), suggesting that cells adopt a mechanism that equally utilizes leading and lagging strand DNA to produce t‐circle‐tail. Altogether, these results demonstrated that nascent t‐circle‐tail consists of newly synthesized leading or lagging DNA in closed‐circular moiety and newly produced long C‐rich single‐stranded tail.
T‐circle‐tail is generated by Topo II‐dependent cleavage of replication forks
The tension generated by DNA replication‐associated unwinding of the DNA duplex is alleviated by DNA Topoisomerase II (Topo II), which is localized to both sides of the replication fork 36. Topo II facilitates progression of the replication fork by creating double‐stranded breaks (DSB) that release superhelical tension 36, 37, 38. Interestingly, Topo II is enriched at telomeres in cells treated with HU (Fig 4A and B), implying that Topo II might be involved in the processing of stalled replication forks. To test whether Topo II is required for the formation of t‐circle‐tail, we treated cells with ICRF‐187, a specific inhibitor of the cleavage activity of Topo II 36. We observed that the abundance of t‐circle‐tail decreased in ICRF‐187‐treated cells (Fig 4C and D), demonstrating that the cleavage activity of Topo II is indispensable for generation of t‐circle‐tail. A similar result was obtained when merbarone, another specific inhibitor of Topo II's cleavage activity 39, was used (Appendix Fig S6A and B).
Figure 4. Effect of topoisomerase II inhibitors on the abundance of t‐circle‐tail.
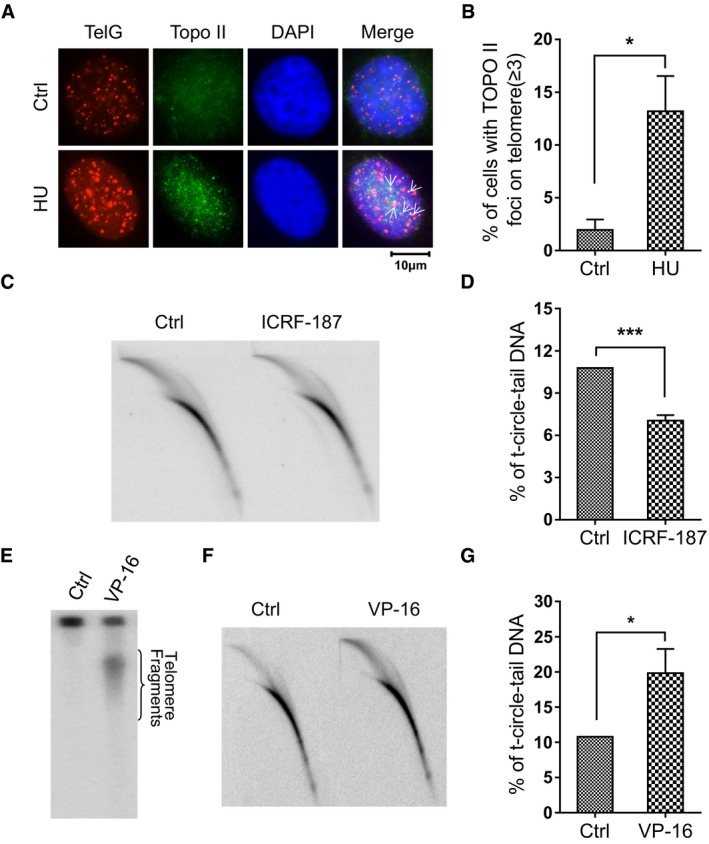
- IF (anti‐Topo II) and FISH (telomeres) were performed on HU‐treated or untreated HeLa cells. Colocalized foci are indicated by white arrowheads.
- Quantification of (A). Cells containing ≥ 3 co‐stained foci were scored. > 100 cells were counted for each group. Bars represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to calculate P‐values. *P < 0.05.
- Genomic DNA was purified from ICRF‐187‐treated and ICRF‐187‐untreated HeLa cells and subjected to 2D gel electrophoresis and hybridization under denaturing conditions using a G‐rich telomeric probe.
- Quantification of (C). Bars represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to calculate P‐values. ***P < 0.001.
- VP‐16‐treated HeLa cells were embedded in an agarose plug, digested (see Materials and Methods) and analyzed by agarose gel (i.e., plug assay, see Materials and Methods).
- DNA was isolated from VP‐16‐treated or control HeLa cells and analyzed by 2D gel for t‐circle‐tail using G‐rich telomeric probe hybridization under denaturing condition.
- Quantification of (F). Bars represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to calculate P‐values. *P < 0.05.
It is thus likely that t‐circle‐tail is generated by Topo II‐dependent cleavage of stalled replication fork. If so, it is speculated that increased “cleavage” by Topo II would generate more t‐circle‐tail. To test this, we treated cells with etoposide (VP‐16), a small molecule drug that traps Topo II‐DNA complex and prevents the religation of cleaved DNA 36, 40, 41. As expected, VP‐16 treatment induced replication fork stalling at telomeres as evidenced by increased telomeric PCNA foci (Appendix Fig S6C and D). In association with this, constant‐field gel electrophoresis (CFGE) 42 assay detected an increased amount of telomeric DNA segments released from genome in VP‐16‐treated cells (Fig 4E). Accordingly, we observed a significant increase of t‐circle‐tail in VP‐16‐treated cells as compared to control cells (Fig 4F and G). Together, these results support the conclusion that t‐circle‐tail is cut from replicating telomeres in a Topo II‐dependent manner.
This conclusion was further confirmed by the presence of subtelomeric sequence in t‐circle‐tail. Given the fact that replication fork can be initiated from the origins within telomeres and subtelomeres 7, it is speculated that t‐circle‐tail that is derived from stalled replication fork may consist of both telomeric and subtelomeric sequences. To test this, PCR amplification of purified t‐circle‐tail DNA was performed using primers targeting subtelomeric sequence on specific chromosomes 43. Our results demonstrated the presence of subtelomeric in t‐circle‐tail DNA (Appendix Fig S7).
NHEJ machinery is required for the formation of t‐circle‐tail
DNA replication fork stalling activates DNA damage response (DDR) signaling 26. Indeed, treatment with HU caused accumulation of 53BP1 foci, an indicator of DDR 44, throughout the genome and at telomeres (Fig 5A and B). We examined cells for evidence of DDR activity, which can take place through recruitment of NHEJ or homologous recombination (HR) machinery. Interestingly, both phosphorylated DNA‐PKcs (pDNA‐PKcs), an NHEJ factor, and the HR protein Rad51 were enriched at telomeres in HU‐treated cells (Fig 5C and D, and Appendix Fig S8A and B), indicating that both NHEJ and HR pathways are activated when replication forks are stalled at telomeres. Although a role for HR in resolving stalled replication forks is not novel 45, 46, 47, a role for NHEJ in this process in mammalian cells has not been previously identified.
Figure 5. Localization of DNA‐PKcs at telomeres and the involvement of NHEJ in the formation of t‐circle‐tail.
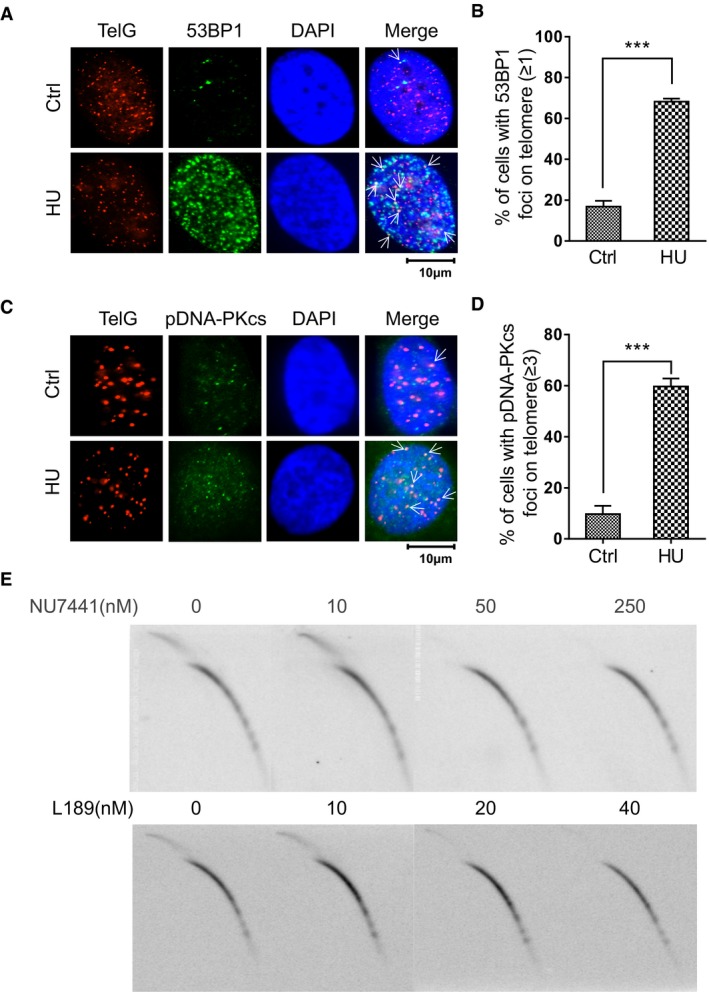
- Cells were treated with HU and fixed, then analyzed by IF using anti‐53BP1 and hybridized to telomeric probe, as indicated. Colocalized foci are indicated by white arrowheads.
- Quantification of (A). Cells with more than 1 co‐localized 53BP1, and telomere signals were scored. > 100 cells were counted for each group. Bars represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to calculate P‐values. ***P < 0.001.
- HeLa cells were treated with HU, fixed and analyzed by IF‐FISH using anti‐pDNA‐PKcs (S2056) and telomere probe, as indicated. Colocalized foci are indicated by white arrowheads.
- Quantification of (C). Cells with ≥ 3 co‐localized signals for pDNA‐PKcs and telomeric DNA were scored. > 100 cells were counted for each group. Bars represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to calculate P‐values. ***P < 0.001.
- HTC75 cells were treated with the indicated concentration of NU7441 or L189 for 24 h and analyzed by 2D gel electrophoresis and hybridization using G‐rich telomeric probe under denaturing conditions.
To determine the impact of the repair pathways on t‐circle‐tail formation, we used NU7441 to inhibit DNA‐PKcs 48, 49, L189 to inhibit Ligase IV 50, or B02 to inhibit Rad51 51. Strikingly, NU7441 and L189 inhibited formation of t‐circle‐tail in a dose‐dependent manner and completely eliminated t‐circle‐tail at higher concentration (Fig 5E), whereas B02 had no effect (Appendix Fig S8C and D). In addition to canonical NHEJ, alternative NHEJ (alt‐NHEJ), whose activity is dependent upon PARP1 protein 52, was also tested. We thus used olaparib, a specific inhibitor of PARP1, to treat the cells and found that it has no/limited effect on the generation of t‐circle‐tail (Appendix Fig S8E and F). These results indicated that canonical NHEJ machinery, but not HR/Rad51 or alt‐NHEJ/PARP1, is required for the formation of t‐circle‐tail.
Given the fact that telomerase is recruited to stalled replication fork in ATM‐/ATR‐dependent manner 11, 53, we suspected that telomerase expression might suppress the function of NHEJ in t‐circle‐tail formation. To test it, telomerase (hTERT) was overexpressed in 293T cells and the relative abundance of t‐circle‐tail was determined by 2D gel electrophoresis. We observed that overexpression of hTERT induced 30% decrease of t‐circle‐tail (Appendix Fig S9), suggesting that recruitment of telomerase suppresses the formation of t‐circle‐tail. Furthermore, when L189 or NU7441 was used to treat hTERT overexpressed cells, no change of t‐circle‐tail was observed, indicating that telomerase expression alleviates the effect of NHEJ inhibition (Appendix Fig S9).
Topo II and NHEJ facilitates the telomere replication
The results above revealed that Topo II‐mediated cleavage and NHEJ machinery are required for generation of t‐circle‐tail when replication forks are stalled at telomeres. We then asked whether inhibition of Topo II or NHEJ affects the resolution of stalled replication forks. For this purpose, we inhibited Topo II or DNA‐PKcs using ICRF‐187 or NU7441, respectively, and visualized stalled replication forks at telomeres by co‐staining for PCNA and TRF2. We observed that the abundance of PCNA foci at telomeres increased in cells treated with ICRF‐187 or NU7441 (Fig 6A and B), indicating the requirement of Topo II or NHEJ in resolving stalled replication forks at telomeres. In addition, additive inhibitory effect was observed when combining usage of ICRF‐187 and NU7441 to treat the cells, suggesting that Topo II and DNA‐PKcs might work in synergy to resolve replication fork stalling at telomeres (Fig 6A and B). This result also suggests that NHEJ/DNA‐PKcs might be required for the religation of broken telomeric DNA after stalled forks are cut off by Topo II. In supporting this hypothesis, when L189 was used to inhibit Ligase IV, we also observed increased frequency of PCNA foci at telomeres (Appendix Fig S10A and B), implying that ligation step is essential for completion of resolving stalled forks.
Figure 6. Topo II and DNA‐PKcs are required for telomere replication.
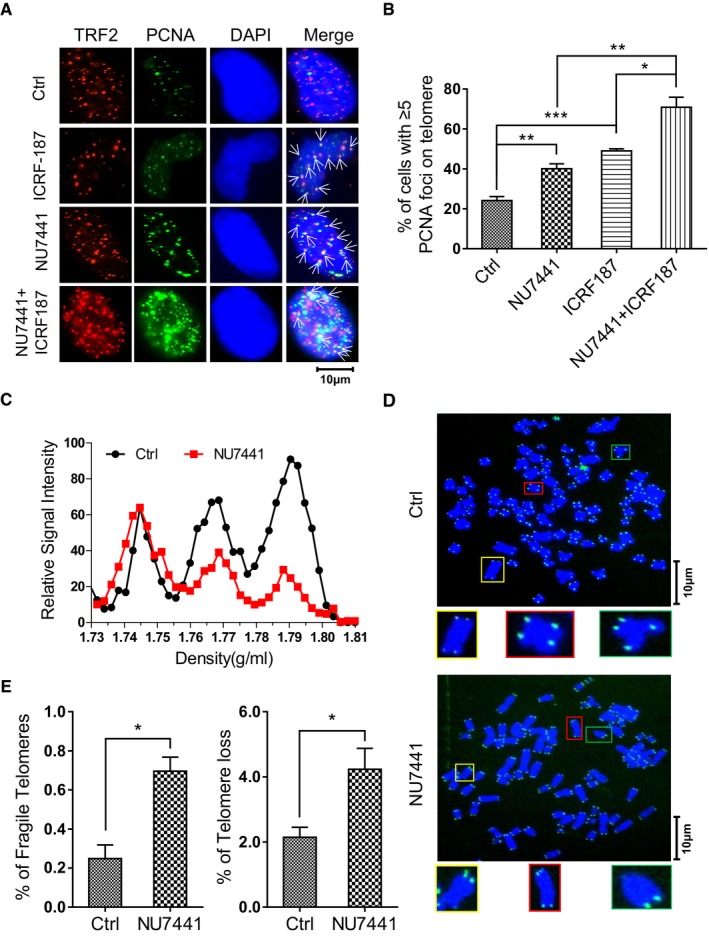
- ICRF‐187‐ and NU7441‐treated HeLa cells were analyzed by IF using anti‐PCNA or anti‐TRF2, as indicated. Colocalized foci are indicated by white arrowheads.
- Quantification of (A). Cells with ≥ 5 co‐stained foci were scored. > 100 cells were counted for each group. Bars represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to calculate P‐values. *P < 0.05, **P < 0.01, ***P < 0.001.
- G1/S synchronized HeLa cells were released into S phase for 9 h in the presence or absence (Ctrl) of NU7441. Genomic DNA was purified and subjected to CsCl gradient centrifugation and slot blot analysis using telomere‐specific probes.
- Metaphase spread and FISH showing telomere‐free ends and fragile telomeres in control and NU7441‐treated HeLa cells.
- Quantification of (D). For quantification, > 3,000 chromosomes were counted for each condition. Bars indicate the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to calculate P‐values. *P < 0.05.
We further examined how inhibition of NHEJ affects telomere replication and the fragility of telomeres. We used CsCl gradient centrifugation to separate replicated telomeres from unreplicated based on the density change resulting from the incorporation of BrdU 54. We found that inhibition of DNA‐PKcs by NU7441 decreased the amount of replicated telomeric DNA (leading and lagging daughters) during S phase (9 h from G1/S), indicating that telomere replication is inhibited by NU7441 (Fig 6C). NU7441 does not affect the progression of cell cycle (Appendix Fig S10C) and the replication of global genomic DNA (Appendix Fig S10D), suggesting a telomere‐specific requirement for DNA‐PKcs in DNA replication. NU7441 treatment also significantly increased fragile telomeres and telomere‐free chromosome ends (Fig 6D and E), consistent with previous work showing that telomere fragility is often associated with loss of telomeres due to replication failure 55.
DNA‐PKcs deficiency leads to telomere replication defects in mice
To further verify the role of NHEJ in telomere replication, we knocked out the DNA‐PKcs gene in mice using a self‐excision gene‐targeting approach 25, 56, 57. We confirmed the complete depletion of DNA‐PKcs in mouse embryonic fibroblast (MEF) cells by Western blot (Appendix Fig S10E). Mouse cells contain extremely long telomeres and the presence of an unidentified high molecular weight telomere‐homologous signal interfered with the detection of t‐circle‐tail. However, we still observed significant reduction in telomeric signal at the expected t‐circle‐tail position in DNA‐PKcs‐deficient cells (Appendix Fig S10F). Accordingly, there were more fragile telomeres in DNA‐PKcs‐deficient cells (KO‐9) compared to wild‐type cells (TA2‐9; Fig 7A and B). Also, DNA‐PKcs‐deficient cells displayed much shorter telomeres than wild‐type cells after long‐term culture (Fig 7C), supporting that DNA‐PKcs is required for successful telomere replication and is essential for the maintenance of telomere length and integrity.
Figure 7. Effect of DNA‐PKcs deficiency on telomere stability/length in mouse cells.
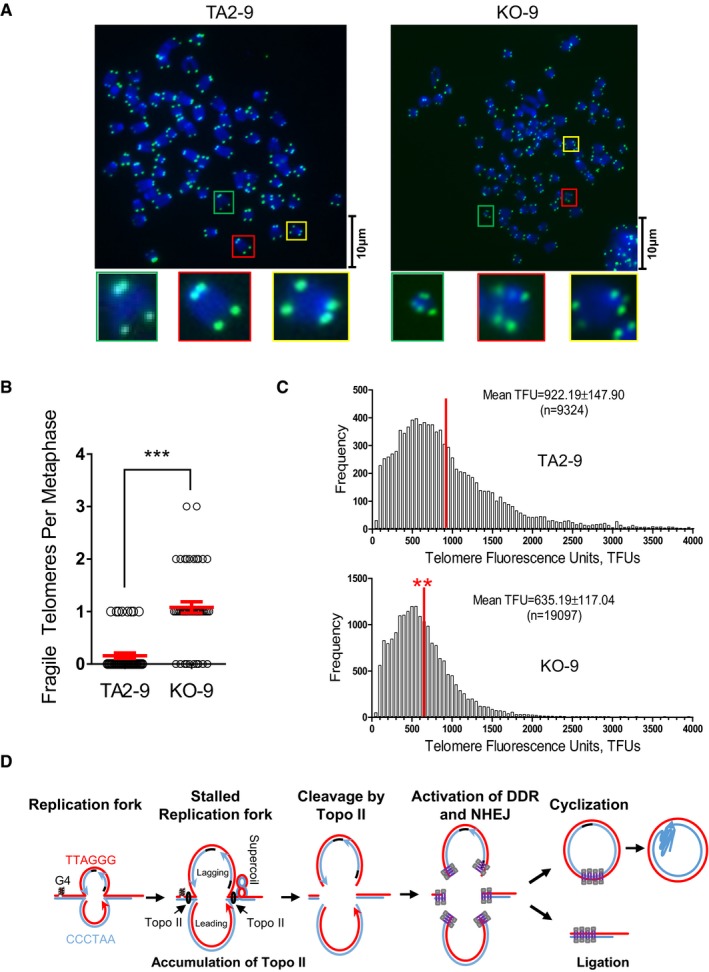
- Control (TA2‐9) and DNA‐PKcs‐deficient (KO‐9) MEF cells were analyzed by Q‐FISH using a telomere‐specific probe.
- Quantification of (A). The number of fragile telomeres in each metaphase cell was scored. > 50 cells were counted for each group. Data represent the mean ± SEM of three independent experiments. Two‐tailed unpaired Student's t‐test was used to calculate P‐values. ***P < 0.001.
- Telomere length distribution in wild‐type and DNA‐PKcs‐deficient MEF cells as determined by Q‐FISH. Red vertical lines indicate the mean of telomere length (TFUs). Mean TFUs ± SEM was provided in the image. Two‐tailed unpaired Student's t‐test was used to calculate P‐values. **P < 0.01.
- Model for the resolution of a telomere‐localized stalled replication fork and the release of an extrachromosomal t‐circle‐tail molecule (see text for details).
Discussion
Here, we have characterized a structure called a t‐circle‐tail, which is present in all examined human cell types. T‐circle‐tail is different from previously identified T‐complex 58 in many respects: (i) t‐circle‐tail appears as a smear on 2D gel spreading outward from the loading well (Fig 1B and Appendix Fig S1A) Versus T‐complex DNA stacks in first dimension gel and cannot migrate into second dimension gel, (ii) only single‐stranded C‐rich DNA is present in t‐circle‐tail (Fig 1B and Appendix Fig S1A) Versus both single‐stranded G‐rich and C‐rich DNA are present in T‐complex, (iii) t‐circle‐tail is sensitive to RecJf (5′ to 3′ exonuclease; Fig 1C and Appendix Fig S2E) Versus T‐complex is resistant. Therefore, T‐complex is proposed to be a highly branched telomeric DNA with internal single‐stranded G‐ and C‐rich DNA, which is different from t‐circle‐tail that consists of double‐stranded telomeric circle and single‐stranded C‐rich tail. The circle‐with‐tail structure was also observed in yeast by 2D gel electrophoresis or electron microscope 59, 60. We find that t‐circle‐tail is induced by stalled replication forks at telomeres. Thus, the discovery of t‐circle‐tail provides a useful tool to study telomere replication. Through investigating the formation of t‐circle‐tail, we revealed an unexpected role for NHEJ machinery in resolving stalled replication forks.
T‐circle‐tail and looping‐out mechanism for resolving stalled replication forks
Telomeric DNA consists of highly G/C‐rich repetitive sequences that are difficult to replicate 8, 11. Although many factors are recruited to telomeres to facilitate replication or to restart stalled replication forks 61, it is inevitable that replication stress due to severe DNA damage or structural obstacles will prevent at least some collapsed replication forks from being restarted. To mimic this situation, we induced artificial replication fork stalling on the genome and/or at telomeres by treating cells with HU, aphidicolin, TMPyP4 or siRNA targeting TRF1. All treatments yielded a similar phenotype, that is, the generation of t‐circle‐tail, suggesting that t‐circle‐tail may derive from stalled replication forks (Fig 2 and Appendix Fig S3). Moreover, DNA synthesis analysis of t‐circle‐tail formation revealed that the closed‐circular moiety of t‐circle‐tail derives from either nascent G‐rich (leading daughter) or C‐rich (lagging daughter) strands of replicating telomeres (Fig 3D and E). In support of this, increased amount of telomeric DNA segments excised from replicating telomeres by Topo II corresponds to increased abundance of t‐circle‐tail in cells treated with VP‐16 (Fig 4E, F and G). Conversely, ICRF‐187 or merbarone suppressed the cleavage activity of Topo II and decreased the abundance of t‐circle‐tail (Fig 4C and D, and Appendix Fig S6A and B). Based on these observations, we conclude that the leading and lagging replication intermediates in stalled replication forks are cut out from the genome by a Topo II‐mediated process and then cyclized to form the t‐circle‐tail (Fig 7D).
NHEJ machinery is localized to stalled replication forks and is indispensable for the formation of t‐circle‐tail (Fig 5). The complete resolution of stalled replication forks requires removal of the replication fork by Topo II followed by religation of 5′‐end and 3′‐end telomeric DNA, thus allowing a new replication fork to be initiated. The inhibition of DNA‐PKcs or Ligase IV, and direct knockout of the DNA‐PKcs gene caused accumulation of stalled replication forks at telomeres (Fig 6A and B, and Appendix Fig S10A and B), resulting in fragile telomeres and telomere loss (Figs 6D and E, and 7A and B). These results indicate that NHEJ is required for complete resolution of stalled replication forks.
Taken together, we propose a looping‐out mechanism for resolving stalled replication forks at telomeres (Fig 7D): (i) Topo II accumulates on both sides of fork, cleaves and releases stalled replication fork (both leading and lagging daughter DNA), (ii) the induced DSB activates DDR including HR and NHEJ, (iii) NHEJ ligates the released leading and lagging DNA, as well as broken telomeres, (iv) leading and lagging DNA are cyclized to form the closed‐circular moiety of the t‐circle‐tail, respectively, and (v) replication is re‐started at newly ligated telomeres (Fig 7D). In this model, the generation of t‐circle‐tail is a key process in resolving stalled replication forks. A prediction of this model would be that inhibiting t‐circle‐tail formation would impair the resolution process, leading to fragile sites at telomeres. Indeed, we found that inhibition of NHEJ in human cells or knockout of DNA‐PKcs gene in mice results in telomere replication defects that are evidenced by an increase of fragile telomeres (Figs 6 and 7).
We found that the tail of t‐circle‐tail could be regenerated by the highly processive Φ29 DNA polymerase, which catalyzes rolling circle DNA synthesis and generates long single‐stranded DNA with 5′ free end (Appendix Fig S1D). While the exact mechanism underlying the formation of tail remains to be demonstrated, we speculated that the manner similar to rolling circle replication might occur in cells. T‐circle‐tail serves as a byproduct in resolving stalled replication forks at telomeres, it may play a role during telomere lengthening. It has been proposed that telomeric circular DNA may be involved in the extension of telomeres in ALT cells 62. In this case, the t‐circle‐tail with its long single‐stranded C‐rich strand could provide an appropriate template for synthesis and elongation of the G‐rich overhang of telomeres.
T‐circle‐tail and telomere length regulation
A direct consequence of the looping‐out mechanism would be the deletion of a DNA sequence to form a corresponding circle of DNA. Extrachromosomal circular DNA (eccDNA) is ubiquitous in eukaryotic organisms, and most eccDNA consists of chromosomal tandem repeats that may bear high replication stress 63. It is likely that stalled replication fork resolution by looping‐out may represent a common mechanism for replication of fragile DNA in the genome.
The looping‐out mechanism could also contribute to telomere length regulation. “Telomere trimming” is characterized by the deletion of large segments of telomeric DNA that is often observed in cells with very long telomeres such as lymphocytes, iPSCs, hESCs, germ line cells and cells with overexpressed telomerase 12, 13, 14, 15. Extremely long telomeres may not be beneficial to cells due to their high replication difficulty. Our results show that increased generation of t‐circle‐tail by TMPyP4 treatment results in a rapid deletion of telomeric DNA (Fig 2F–I), suggesting that “telomere trimming” may result from replication stress. In addition, “telomere trimming” is often associated with the formation of t‐circle DNA 13, 14. Given the structural similarity between the t‐circle‐tail and the t‐circle, these two molecules are interconvertible. In this context, the generation of t‐circle‐tail by the looping‐out mechanism represents a new approach allowing cells to shorten those extremely long telomeres to relieve high replication stress.
In normal human somatic cells, telomerase is absent and human telomeres lose 50–300 base pairs during each cell division 64, 65. It is widely accepted that telomere shortening is attributed to the unidirectional nature of DNA polymerase and additional processing events. However, this does not fully explain the large variation in telomere shortening rates between different cell lines. Our results showed that t‐circle‐tail is present in human normal fibroblast, PBMC, and T cells (Appendix Fig S2), indicating the occurrence of spontaneous “telomere trimming” during DNA replication that contributes to telomere shortening in these cells. Therefore, replication stress represents a new factor that could induce telomere shortening.
NHEJ machinery and telomere replication
The looping‐out model proposed here is similar to the mechanism of immunoglobulin heavy‐chain class switching (V(D)J recombination). The latter involves RAG‐mediated excision of the immunoglobulin gene followed by NHEJ‐mediated ligation, generating an extrachromosomal circle DNA (eccDNA) 66, 67.
Although the accumulated evidence indicates that NHEJ is involved in promoting DNA replication, a mechanistic understanding is lacking. It has been previously reported that NHEJ‐deficient cells display a high incidence of fragile sites 68 and that replication fork progression is reduced in the absence of NHEJ 69. Moreover, cells carrying the DNA‐PKcs3A/3A mutant allele demonstrate impaired replication of leading daughter strands at telomeres 25. However, it remains unclear whether NHEJ functions through indirect (signaling) roles and/or direct roles involving DNA end‐joining 70. Here, our data show that NHEJ is required for resolving stalled replication forks: First, DNA‐PKcs is activated (phosphorylated) and recruited to stalled telomeres; and second, both DNA‐PKcs and Ligase IV are responsible for the formation of t‐circle‐tail and for preventing the formation of fragile telomeres. These results support a direct role for NHEJ (ligation) in telomere replication and provide a feasible explanation for previous observations 22, 23, 24, 25.
It is widely believed that NHEJ is constitutively suppressed in telomeres by TRF2, thus preventing end fusion between chromosomes 71. Our results reveal that NHEJ is activated at telomeric DNA in the context of replication fork arrest. The regulatory mechanism underlying switch between suppression and activation is an intriguing topic for future study.
Materials and Methods
Cell culture and treatment
HeLa, HeLa 229, and HeLa 1.2.11 cells were cultured at 37°C and 5% CO2 in Glutamax‐DMEM (Life) containing 10% newborn calf serum (PAA), 100 U/ml penicillin and 1% streptomycin (Hyclone). Other cell lines used in this study were cultured in Glutamax‐DMEM with 10% fetal bovine serum (Life). HeLa, HeLa 229 and majority of cell lines in Appendix Fig S2 were purchased from Chinese Academy of Sciences of Type Culture Collection. HeLa 1.2.11 was a kind gift from Carolyn M. Price. HTC75 was a kind gift from Zhou Songyang. Cell lines were treated with 2 mM hydroxyurea (HU, Sigma), 5 μM TMPyP4 (Sigma), methyl‐methanesulfonate (MMS, Sigma), 50 μg/ml ICRF‐187 (Selleck), 10 μM etoposide (VP‐16, Sigma), NU7441 (Selleck) or L189 (Santa Cruz) or 27.4 μM B02 (EMD Millipore) or 100 μM Olaparib (MCE). The following siRNA was used to transfect the cells during knockdown experiments: siTRF1 sense‐UUUAACAUGACACUUGUCCdTdT, antisense‐GGACAAGUGUCAUGUUAAAdTdT; sip53‐1 sense‐GCAUGAACCGGAGGCCCAUdTdT, antisense‐ AUGGGCCUCCGGUUCAUGCdTdT;sip53‐2 sense‐GACUCCAGUGGUAAUCUACdTdT, antisense‐GUAGAUUACCACUGGAGUCdTdT.
Cell cycle synchronization and BrdU labeling
HeLa cells were synchronized using the “double thymidine” approach, as previously described 35. Briefly, 2 mM thymidine (Sigma) was added to exponentially growing cells for 19 h, cells were then washed three times with prewarmed 1 × PBS and released into fresh medium for 10 h, followed by addition of 2 mM thymidine and incubation for 14 h. After washing three times with prewarmed 1 × PBS, cells were incubated with fresh medium containing 100 μM 5‐bromo‐2‐deoxyuridine (BrdU, Sigma) for a variable length of time. Alternatively, for pulse labeling, cells were released into fresh medium, incubated for 3 h, and then transferred and incubated in culture medium containing 100 μM BrdU for 1 h. For BJ fibroblast and MRC5 cell labeling, cells were cultured in medium containing 100 μM BrdU for 48 h. FACS analysis was carried out as previously described 35.
Knockout of TRF2 by inducible CRISPR/Cas9 system
HeLa cells were transfected with lentivirus carrying inducible‐Cas9 (pHAGE‐TRE‐Cas9) and selected with neomycin for 10 days. Two lentivirus (p‐Lenti‐puro‐sgTRF2‐1, p‐Lenti‐BSD‐sgTRF2‐2) with individual sgRNA (sgTRF2‐1‐F CACCGCCTTTCGGGGTAGCCGGTA, sgTRF2‐1‐R AAACTACCGGCTACCCCGAAAGGC, sgTRF2‐2‐F CACCGAACCC GCAGCAATCGGGACA, sgTRF2‐2‐R AAACTGTCCCGATTGCTGCGGGTTC) were used to sequentially transfect selected cells. After two rounds of selection with puromycin (2 μg/ml) and blasticidin (10 μg/ml), the clones were selected and doxycycline (1 μg/ml) was then added into culture medium to induce Cas9 expression. After 5 days, cells were harvested for experiments.
Genomic DNA purification and enzyme treatment
Genomic DNA was extracted with AxyPrep Blood Genomic DNA Miniprep Kit (Axygen) and dissolved in TE buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0). 10 μg genomic DNA was digested overnight with 10 U HinfI (Thermo), 10 U RsaI (Thermo), and 2 μg/ml RNase A (Takara) at 37°C. The reaction was terminated with EDTA, and DNA was subjected to 2D gel electrophoresis. 30 U RecJf (NEB) or 20 U Mungbean Nuclease (NEB) was used to digest genomic DNA in combination with HinfI and RsaI. 20 U Plasmid‐Safe™ ATP‐Dependent DNase (Epicentre) was used to digest DNA samples digested with HinfI and RsaI.
Immunofluorescence (IF) and immunofluorescence in situ hybridization (IF‐FISH)
IF and IF‐FISH were carried out as previously described 72. Briefly, cells grown on glass coverslips were washed in chilled PBS and fixed with 4% paraformaldehyde in PBS, permeabilized with 0.5% Triton X‐100 and blocked with 5% goat serum. Fixed cells were incubated with primary antibody at 4°C overnight and incubated with secondary antibody at room temperature for 2 h. Coverslips were counterstained with DAPI for observation under a Zeiss Axioplan II microscope.
For IF‐FISH assays, following secondary antibody incubation, coverslips were fixed again with 4% paraformaldehyde and sequentially dehydrated with 75, 95, and 100% ethanol. The coverslips were then incubated with hybridization mix containing Cy3‐(TTAGGG)3 (Panagene, F1006‐5). DNA was heat‐denatured at 85°C for 5 min, and the slides were incubated at 37°C for 2 h protected from light. The coverslips were washed, dehydrated as described above, and counterstained with DAPI. Antibodies used in this assay are as follows: anti‐TRF2 (EMD Millipore, 05‐521), anti‐53BP1 (Novus Biologicals, NB100‐304), anti‐PCNA (Gene Tex, GTX100539), anti‐Topo II (Abcam, ab52934), anti‐pDNA‐PKcs (S2056; Abcam, ab18192), anti‐Rad51 (Santa Cruz, SC‐8349), DyLight 488‐conjugated goat anti‐mouse IgG H&L (Multisciences, 4120353), DyLight 488‐conjugated goat anti‐rabbit IgG H&L (Multisciences, 4220431), and DyLight 549‐conjugated goat anti‐mouse IgG H&L (Multisciences, 4120816).
Native FISH
Native FISH was performed as described 73 with a minor modification. Briefly, coverslips were fixed in 2% paraformaldehyde at room temperature for 10 min, permeabilized for 10 min in KCM buffer [0.1% Triton X‐100, 10 mM Tris–HCl (pH 7.5), 120 mM KCl, and 20 mM NaCl] and treated with RNase A and RecJf (20 U) at 37°C for at least 2 h. Cells not treated with RecJf were used as a control. Hybridization was performed in hybridization buffer containing 10 mM Tris–HCl (pH 7.5), 85.6 mM KCl, 0.5% blocking reagent (Roche, 11096176001), 70% formamide, and 40 nM Cy3‐(TTAGGG)3 (Panagene, F1006‐5). Slides were incubated at room temperature for 2 h, washed, and counterstained with DAPI. Images were acquired using 100× objective on a Leica TCS SP5 microscope.
Two‐dimensional gel electrophoresis
2D agarose gel electrophoresis was performed as described 74, 75. Briefly, 10 μg genomic DNA was digested with RsaI and HinfI (Fermentas, Thermo Scientific) and loaded onto a 0.4% agarose gel. Electrophoresis was carried out in 1 × TBE at 1 V/cm for 12 h at room temperature. The lane containing DNA was excised from the gel and the gel buffer was exchanged with 1 × TBE with 0.3 μg/ml ethidium bromide (EB; Sigma). The gel slice was transferred and cast with 1% agarose gel in 1 × TBE containing 0.3 μg/ml EB. The gel was run at 4°C for 6 h at 3 V/cm.
Detection of subtelomeric DNA
HinfI‐ and RsaI‐digested HeLa DNA was subjected to 2D agarose gel as described above. The gel with t‐circle‐tail signal was excised and DNA in gel was purified with QIAquick Gel Extraction Kit (QIAGEN). Purified DNA was used as a template for PCR with primers targeting subtelomeric sequence of specific chromosomes. The primer pairs were referred to that previously described 43.
Telomere restricted fragment (TRF) assay
The telomere length assay was performed as previously described 35.
Plug assay/Constant‐field gel electrophoresis of embedded cells
The plug assay was performed as previously described 76. Briefly, 1 × 106 cells were rinsed twice with 1 × PBS, embedded in 50 μl 0.7% prewarmed agarose (45°C), and solidified in a 1 ml decapitated injector. Agarose plugs were incubated with digestion buffer (10 mM Tris–HCl pH8.0, 10 mg/ml Protease K, 0.5% SDS, 40 mg/ml RNase A, 100 mM EDTA) at 37°C for 20 h. Plugs were placed into the wells of a 0.7% agarose gel and sealed with 0.7% agarose. Electrophoresis was carried out at 1 V/cm at room temperature for 8 h.
Native/denatured in‐gel hybridization
In‐gel hybridization analysis of telomeric DNA was performed as described 77 with a minor modification: Agarose gels were pumped dry at room temperature. For native in‐gel hybridization, gels were hybridized in Denhart's hybridization buffer with 32P‐labeled C‐/G‐probe after prehybridization. The C‐/G‐probes were generated as previously described. Gels were washed three times with 2 × SSC + 0.5% SDS twice, and 2 × SSC + 0.1% SDS once. For denatured in‐gel hybridization, gels were denatured with 0.5 M NaOH and neutralized with 1 M Tris–HCl (pH 8.0) prior to following the procedure for native hybridization. The gels were exposed to a PhosphorImager screen (GE Healthcare) and scanned on a Typhoon imager (GE Healthcare). Calculations of the relative abundance of t‐circle‐tail were made using Image Quant software.
Western blot analysis
Western blotting was performed according to standard protocols. Antibodies used are as follows: anti‐TRF2 (EMD Millipore, 05‐521), anti‐TRF1 (Gene Tex, GTX77605), anti‐DNA‐PKcs (Thermo Scientific, Ab‐4), anti‐β‐actin (Proteintech, 60008‐1‐Ig) and anti‐GAPDH (Proteintech, 60004‐1‐Ig), HRP‐conjugated goat anti‐mouse IgG (H+L; Proteintech, SA00001‐1), HRP‐conjugated goat anti‐mouse IgG (H+L; Proteintech, SA00001‐2).
Author contributions
YZ and TPZ designed the study, analyzed data, and wrote the manuscript. TPZ performed majority of experiments. ZPZ, QH, FL, and SCZ were involved in experiments. HYL, MFT, WBM, JJH, ZSY, YKR, and BPCC offered ideas and helped analyzing data. YZ supervised the project. All authors read and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
We thank all the members in Dr. Zhao's laboratory for insightful scientific discussion. This work was supported by National Natural Science Foundation of China Grants [31271472; 31322033; 31301110], National Basic Research Program of China [2014CB964703], the Science and Technology Planning Project of Guangdong Province (2015B020228002), and Pearl River Scholar Funded Scheme (2013).
EMBO Reports (2017) 18: 1412–1428
References
- 1. Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, Meyne J, Ratliff RL, Wu JR (1988) A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci USA 85: 6622–6626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xin H, Liu D, Songyang Z (2008) The telosome/shelterin complex and its functions. Genome Biol 9: 232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19: 2100–2110 [DOI] [PubMed] [Google Scholar]
- 4. Watson JD (1972) Origin of concatemeric T7 DNA. Nat New Biol 239: 197–201 [DOI] [PubMed] [Google Scholar]
- 5. Smogorzewska A, de Lange T (2004) Regulation of telomerase by telomeric proteins. Annu Rev Biochem 73: 177–208 [DOI] [PubMed] [Google Scholar]
- 6. Allsopp RC, Chang E, Kashefi‐Aazam M, Rogaev EI, Piatyszek MA, Shay JW, Harley CB (1995) Telomere shortening is associated with cell division in vitro and in vivo . Exp Cell Res 220: 194–200 [DOI] [PubMed] [Google Scholar]
- 7. Drosopoulos WC, Kosiyatrakul ST, Yan Z, Calderano SG, Schildkraut CL (2012) Human telomeres replicate using chromosome‐specific, rather than universal, replication programs. J Cell Biol 197: 253–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gilson E, Geli V (2007) How telomeres are replicated. Nat Rev Mol Cell Biol 8: 825–838 [DOI] [PubMed] [Google Scholar]
- 9. Stewart JA, Wang F, Chaiken MF, Kasbek C, Chastain PD II, Wright WE, Price CM (2012) Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J 31: 3537–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leon‐Ortiz AM, Svendsen J, Boulton SJ (2014) Metabolism of DNA secondary structures at the eukaryotic replication fork. DNA Repair (Amst) 19: 152–162 [DOI] [PubMed] [Google Scholar]
- 11. Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138: 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pickett HA, Reddel RR (2012) The role of telomere trimming in normal telomere length dynamics. Cell Cycle 11: 1309–1315 [DOI] [PubMed] [Google Scholar]
- 13. Pickett HA, Cesare AJ, Johnston RL, Neumann AA, Reddel RR (2009) Control of telomere length by a trimming mechanism that involves generation of t‐circles. EMBO J 28: 799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rivera T, Haggblom C, Cosconati S, Karlseder J (2017) A balance between elongation and trimming regulates telomere stability in stem cells. Nat Struct Mol Biol 24: 30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pickett HA, Henson JD, Au AY, Neumann AA, Reddel RR (2011) Normal mammalian cells negatively regulate telomere length by telomere trimming. Hum Mol Genet 20: 4684–4692 [DOI] [PubMed] [Google Scholar]
- 16. Durkin SG, Ragland RL, Arlt MF, Mulle JG, Warren ST, Glover TW (2008) Replication stress induces tumor‐like microdeletions in FHIT/FRA3B. Proc Natl Acad Sci USA 105: 246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Durkin SG, Glover TW (2007) Chromosome fragile sites. Annu Rev Genet 41: 169–192 [DOI] [PubMed] [Google Scholar]
- 18. Arlt MF, Mulle JG, Schaibley VM, Ragland RL, Durkin SG, Warren ST, Glover TW (2009) Replication stress induces genome‐wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am J Hum Genet 84: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Verdun RE, Karlseder J (2007) Replication and protection of telomeres. Nature 447: 924–931 [DOI] [PubMed] [Google Scholar]
- 20. Verdun RE, Karlseder J (2006) The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 127: 709–720 [DOI] [PubMed] [Google Scholar]
- 21. Verdun RE, Crabbe L, Haggblom C, Karlseder J (2005) Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell 20: 551–561 [DOI] [PubMed] [Google Scholar]
- 22. Ruis BL, Fattah KR, Hendrickson EA (2008) The catalytic subunit of DNA‐dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol Cell Biol 28: 6182–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Espejel S, Martin M, Klatt P, Martin‐Caballero J, Flores JM, Blasco MA (2004) Shorter telomeres, accelerated ageing and increased lymphoma in DNA‐PKcs‐deficient mice. EMBO Rep 5: 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Espejel S, Franco S, Sgura A, Gae D, Bailey SM, Taccioli GE, Blasco MA (2002) Functional interaction between DNA‐PKcs and telomerase in telomere length maintenance. EMBO J 21: 6275–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang S, Matsunaga S, Lin YF, Sishc B, Shang Z, Sui J, Shih HY, Zhao Y, Foreman O, Story MD et al (2015) Spontaneous tumor development in bone marrow‐rescued DNA‐PKcs mice due to dysfunction of telomere leading strand deprotection. Oncogene 35: 3909–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deng Z, Dheekollu J, Broccoli D, Dutta A, Lieberman PM (2007) The origin recognition complex localizes to telomere repeats and prevents telomere‐circle formation. Curr Biol 17: 1989–1995 [DOI] [PubMed] [Google Scholar]
- 28. Henson JD, Cao Y, Huschtscha LI, Chang AC, Au AY, Pickett HA, Reddel RR (2009) DNA C‐circles are specific and quantifiable markers of alternative‐lengthening‐of‐telomeres activity. Nat Biotechnol 27: 1181–1185 [DOI] [PubMed] [Google Scholar]
- 29. Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA et al (2012) Clustered mutations in yeast and in human cancers can arise from damaged long single‐strand DNA regions. Mol Cell 46: 424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A (2013) Migrating bubble during break‐induced replication drives conservative DNA synthesis. Nature 502: 389–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Elia AE, Wang DC, Willis NA, Boardman AP, Hajdu I, Adeyemi RO, Lowry E, Gygi SP, Scully R, Elledge SJ (2015) RFWD3‐dependent ubiquitination of RPA regulates repair at stalled replication forks. Mol Cell 60: 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mailand N, Gibbs‐Seymour I, Bekker‐Jensen S (2013) Regulation of PCNA‐protein interactions for genome stability. Nat Rev Mol Cell Biol 14: 269–282 [DOI] [PubMed] [Google Scholar]
- 33. Tutton S, Azzam GA, Stong N, Vladimirova O, Wiedmer A, Monteith JA, Beishline K, Wang Z, Deng Z, Riethman H et al (2016) Subtelomeric p53 binding prevents accumulation of DNA damage at human telomeres. EMBO J 35: 193–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vannier JB, Sandhu S, Petalcorin MI, Wu X, Nabi Z, Ding H, Boulton SJ (2013) RTEL1 is a replisome‐associated helicase that promotes telomere and genome‐wide replication. Science 342: 239–242 [DOI] [PubMed] [Google Scholar]
- 35. Zhao Y, Sfeir AJ, Zou Y, Buseman CM, Chow TT, Shay JW, Wright WE (2009) Telomere extension occurs at most chromosome ends and is uncoupled from fill‐in in human cancer cells. Cell 138: 463–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nitiss JL (2009) DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 9: 327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Germe T, Miller K, Cooper JP (2009) A non‐canonical function of topoisomerase II in disentangling dysfunctional telomeres. EMBO J 28: 2803–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ye J, Lenain C, Bauwens S, Rizzo A, Saint‐Leger A, Poulet A, Benarroch D, Magdinier F, Morere J, Amiard S et al (2010) TRF2 and apollo cooperate with topoisomerase 2alpha to protect human telomeres from replicative damage. Cell 142: 230–242 [DOI] [PubMed] [Google Scholar]
- 39. Fortune JM, Osheroff N (1998) Merbarone inhibits the catalytic activity of human topoisomerase IIalpha by blocking DNA cleavage. J Biol Chem 273: 17643–17650 [DOI] [PubMed] [Google Scholar]
- 40. Yeo CQ, Alexander I, Lin Z, Lim S, Aning OA, Kumar R, Sangthongpitag K, Pendharkar V, Ho VH, Cheok CF (2016) p53 Maintains genomic stability by preventing interference between transcription and replication. Cell Rep 15: 132–146 [DOI] [PubMed] [Google Scholar]
- 41. Aparicio T, Baer R, Gottesman M, Gautier J (2016) MRN, CtIP, and BRCA1 mediate repair of topoisomerase II‐DNA adducts. J Cell Biol 212: 399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mao P, Liu J, Zhang Z, Zhang H, Liu H, Gao S, Rong YS, Zhao Y (2016) Homologous recombination‐dependent repair of telomeric DSBs in proliferating human cells. Nat Commun 7: 12154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Deng Z, Wang Z, Stong N, Plasschaert R, Moczan A, Chen HS, Hu S, Wikramasinghe P, Davuluri RV, Bartolomei MS et al (2012) A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J 31: 4165–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Panier S, Boulton SJ (2014) Double‐strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15: 7–18 [DOI] [PubMed] [Google Scholar]
- 45. Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T (2010) Hydroxyurea‐stalled replication forks become progressively inactivated and require two different RAD51‐mediated pathways for restart and repair. Mol Cell 37: 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nagaraju G, Scully R (2007) Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair 6: 1018–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Anand RP, Lovett ST, Haber JE (2013) Break‐induced DNA replication. Cold Spring Harb Perspect Biol 5: a010397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, Smith GC (2004) Identification of a highly potent and selective DNA‐dependent protein kinase (DNA‐PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett 14: 6083–6087 [DOI] [PubMed] [Google Scholar]
- 49. Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, Calvert AH, Newell DR, Smith GC, Curtin NJ (2006) Preclinical evaluation of a potent novel DNA‐dependent protein kinase inhibitor NU7441. Cancer Res 66: 5354–5362 [DOI] [PubMed] [Google Scholar]
- 50. Chen X, Zhong S, Zhu X, Dziegielewska B, Ellenberger T, Wilson GM, MacKerell AD Jr, Tomkinson AE (2008) Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Can Res 68: 3169–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV (2011) Identification of specific inhibitors of human RAD51 recombinase using high‐throughput screening. ACS Chem Biol 6: 628–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Doksani Y, de Lange T (2016) Telomere‐internal double‐strand breaks are repaired by homologous recombination and PARP1/Lig3‐dependent end‐joining. Cell Rep 17: 1646–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tong AS, Stern JL, Sfeir A, Kartawinata M, de Lange T, Zhu XD, Bryan TM (2015) ATM and ATR signaling regulate the recruitment of human telomerase to telomeres. Cell Rep 13: 1633–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huang C, Dai X, Chai W (2012) Human Stn1 protects telomere integrity by promoting efficient lagging‐strand synthesis at telomeres and mediating C‐strand fill‐in. Cell Res 22: 1681–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vannier JB, Pavicic‐Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ (2012) RTEL1 dismantles T loops and counteracts telomeric G4‐DNA to maintain telomere integrity. Cell 149: 795–806 [DOI] [PubMed] [Google Scholar]
- 56. Bunting M, Bernstein KE, Greer JM, Capecchi MR, Thomas KR (1999) Targeting genes for self‐excision in the germ line. Genes Dev 13: 1524–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang S, Yajima H, Huynh H, Zheng J, Callen E, Chen HT, Wong N, Bunting S, Lin YF, Li M et al (2011) Congenital bone marrow failure in DNA‐PKcs mutant mice associated with deficiencies in DNA repair. J Cell Biol 193: 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grolimund L, Aeby E, Hamelin R, Armand F, Chiappe D, Moniatte M, Lingner J (2013) A quantitative telomeric chromatin isolation protocol identifies different telomeric states. Nat Commun 4: 2848 [DOI] [PubMed] [Google Scholar]
- 59. Groff‐Vindman C, Cesare AJ, Natarajan S, Griffith JD, McEachern MJ (2005) Recombination at long mutant telomeres produces tiny single‐ and double‐stranded telomeric circles. Mol Cell Biol 25: 4406–4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nosek J, Rycovska A, Makhov AM, Griffith JD, Tomaska L (2005) Amplification of telomeric arrays via rolling‐circle mechanism. J Biol Chem 280: 10840–10845 [DOI] [PubMed] [Google Scholar]
- 61. Pfeiffer V, Lingner J (2013) Replication of telomeres and the regulation of telomerase. Cold Spring Harb Perspect Biol 5: a010405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cesare AJ, Reddel RR (2010) Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 11: 319–330 [DOI] [PubMed] [Google Scholar]
- 63. Cohen S, Segal D (2009) Extrachromosomal circular DNA in eukaryotes: possible involvement in the plasticity of tandem repeats. Cytogenet Genome Res 124: 327–338 [DOI] [PubMed] [Google Scholar]
- 64. Harley CB, Futcher AB, Greider CW (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345: 458–460 [DOI] [PubMed] [Google Scholar]
- 65. Huffman KE, Levene SD, Tesmer VM, Shay JW, Wright WE (2000) Telomere shortening is proportional to the size of the G‐rich telomeric 3′‐overhang. J Biol Chem 275: 19719–19722 [DOI] [PubMed] [Google Scholar]
- 66. von Schwedler U, Jack HM, Wabl M (1990) Circular DNA is a product of the immunoglobulin class switch rearrangement. Nature 345: 452–456 [DOI] [PubMed] [Google Scholar]
- 67. Lieber MR, Ma Y, Pannicke U, Schwarz K (2004) The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair 3: 817–826 [DOI] [PubMed] [Google Scholar]
- 68. Schwartz M, Zlotorynski E, Goldberg M, Ozeri E, Rahat A, le Sage C, Chen BP, Chen DJ, Agami R, Kerem B (2005) Homologous recombination and nonhomologous end‐joining repair pathways regulate fragile site stability. Genes Dev 19: 2715–2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Alexander JL, Barrasa MI, Orr‐Weaver TL (2015) Replication fork progression during re‐replication requires the DNA damage checkpoint and double‐strand break repair. Curr Biol 25: 1654–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Allen C, Ashley AK, Hromas R, Nickoloff JA (2011) More forks on the road to replication stress recovery. J Mol Cell Biol 3: 4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sfeir A, de Lange T (2012) Removal of shelterin reveals the telomere end‐protection problem. Science 336: 593–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen Y, Deng Z, Jiang S, Hu Q, Liu H, Songyang Z, Ma W, Chen S, Zhao Y (2015) Human cells lacking coilin and Cajal bodies are proficient in telomerase assembly, trafficking and telomere maintenance. Nucleic Acids Res 43: 385–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. O'Sullivan RJ, Arnoult N, Lackner DH, Oganesian L, Haggblom C, Corpet A, Almouzni G, Karlseder J (2014) Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat Struct Mol Biol 21: 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Brewer BJ, Fangman WL (1988) A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell 55: 637–643 [DOI] [PubMed] [Google Scholar]
- 75. Friedman KL, Brewer BJ (1995) Analysis of replication intermediates by two‐dimensional agarose gel electrophoresis. Methods Enzymol 262: 613–627 [DOI] [PubMed] [Google Scholar]
- 76. Dahm‐Daphi J, Dikomey E (1996) Rejoining of DNA double‐strand breaks in X‐irradiated CHO cells studied by constant‐ and graded‐field gel electrophoresis. Int J Radiat Biol 69: 615–621 [DOI] [PubMed] [Google Scholar]
- 77. Ouellette MM, Liao M, Herbert BS, Johnson M, Holt SE, Liss HS, Shay JW, Wright WE (2000) Subsenescent telomere lengths in fibroblasts immortalized by limiting amounts of telomerase. J Biol Chem 275: 10072–10076 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File