

Abstract
ERdj3/DNAJB11 is an endoplasmic reticulum (ER)‐targeted HSP40 co‐chaperone that performs multifaceted functions involved in coordinating ER and extracellular proteostasis. Here, we show that ERdj3 assembles into a native tetramer that is distinct from the dimeric structure observed for other HSP40 co‐chaperones. An electron microscopy structural model of full‐length ERdj3 shows that these tetramers are arranged as a dimer of dimers formed by distinct inter‐subunit interactions involving ERdj3 domain II and domain III. Targeted deletion of residues 175‐190 within domain II renders ERdj3 a stable dimer that is folded and efficiently secreted from mammalian cells. This dimeric ERdj3 shows impaired substrate binding both in the ER and extracellular environments and reduced interactions with the ER HSP70 chaperone BiP. Furthermore, we show that overexpression of dimeric ERdj3 exacerbates ER stress‐dependent reductions in the secretion of a destabilized, aggregation‐prone protein and increases its accumulation as soluble oligomers in extracellular environments. These results reveal ERdj3 tetramerization as an important structural framework for ERdj3 functions involved in coordinating ER and extracellular proteostasis in the presence and absence of ER stress.
Keywords: ERdj3/DNAJB11, extracellular chaperone, HSP40, J‐protein co‐chaperone, secretory proteostasis
Subject Categories: Membrane & Intracellular Transport, Protein Biosynthesis & Quality Control, Structural Biology
Introduction
The HSP70 chaperoning pathway functions to regulate proteostasis through a highly conserved mechanism involving ATP‐dependent regulation of HSP70 binding to protein substrates (Kampinga & Craig, 2010; Kim et al, 2013; Cyr & Ramos, 2015; Nillegoda & Bukau, 2015). Mammals encode 11 HSP70 chaperones that localize to distinct intracellular environments including the cytosol, nucleus, mitochondria, and the endoplasmic reticulum (ER). These HSP70 chaperones are involved in diverse proteostasis functions including protein folding, protein degradation, protein disaggregation, translocation of polypeptides across membranes, and the regulation of stress‐responsive signaling pathways (Kampinga & Craig, 2010; Kim et al, 2013; Cyr & Ramos, 2015; Nillegoda & Bukau, 2015). This functional diversity is primarily mediated through a network of HSP40 co‐chaperones similarly localized throughout mammalian cells. HSP40s predominantly function by engaging misfolded protein substrates and delivering them to HSP70 for ATP‐dependent chaperoning (Kampinga & Craig, 2010; Kim et al, 2013; Cyr & Ramos, 2015; Nillegoda & Bukau, 2015). Through this basic mechanism, HSP40 co‐chaperones can influence many aspects of HSP70 function including client specificity, disaggregase activity, localization, and ATPase activity.
HSP40 co‐chaperones are classified into three categories based on similarity to the domain architecture of the E. coli HSP40 DNAJ (Kampinga & Craig, 2010). Type I HSP40 co‐chaperones have a conserved structure containing an N‐terminal J‐domain, a Gly/Phe flexible region, a bifurcated substrate binding domain separated by a zinc finger domain, and a C‐terminal dimerization domain. Type II HSP40 co‐chaperones are similar to Type I, although the zinc finger domain is replaced with a variable domain II. In contrast, Type III HSP40 co‐chaperones are highly diverse, but contain a J‐domain. Type I and Type II HSP40s generally assemble and function as dimers formed by inter‐subunit interactions between the C‐terminal dimerization domains (Kampinga & Craig, 2010; Alderson et al, 2016). Dimerization has previously been shown to be important for critical HSP40 functions including binding to misfolded protein substrates (Wu et al, 2005; Otero et al, 2014). However, these HSP40 dimers can vary in structure despite having similar domain organizations, potentially providing unique structural frameworks to allow for the many functions of HSP40 co‐chaperones (e.g., dictating HSP70 client specificity; Sha et al, 2000; Borges et al, 2005; Wu et al, 2005; Hu et al, 2008; Ramos et al, 2008). HSP40 dimers can also transiently assemble into higher order oligomers in the presence of protein aggregates to promote disaggregation (Nillegoda et al, 2015).
Seven different HSP40 co‐chaperones localize to the ER and regulate the folding, trafficking, or degradation of the secretory proteome (Kampinga & Craig, 2010; Otero et al, 2010; Melnyk et al, 2015). Of these ER HSP40s, one of the most abundant is the soluble Type II HSP40 co‐chaperone ERdj3/DNAJB11. ERdj3 has diverse functions involved in coordinating ER and extracellular proteostasis. In the ER, ERdj3 functions as a canonical HSP40 co‐chaperone, binding to misfolded proteins and delivering them to the ER HSP70 BiP for ATP‐dependent chaperoning (Shen et al, 2002; Shen & Hendershot, 2005; Jin et al, 2009; Marcinowski et al, 2011; Guo & Snapp, 2013; Behnke et al, 2016). Through this mechanism, ERdj3 functions to regulate the folding or degradation of secretory proteins including immunoglobulin light and heavy chains, the epithelial sodium channel, and glucocerebrosidase (Shen & Hendershot, 2005; Jin et al, 2009; Buck et al, 2010; Tan et al, 2014). In addition, ERdj3 is involved in regulating ER permeability and protein translocation into the ER through interaction with the Sec61 translocon (Dejgaard et al, 2010; Guo & Snapp, 2013; Schorr et al, 2015). However, in response to ER stress, ERdj3 is secreted to the extracellular space where it functions as an ATP‐independent extracellular chaperone to prevent the misfolding and/or aggregation of secreted proteins (Genereux et al, 2015). ERdj3 can also preemptively protect extracellular proteostasis through co‐secretion in a complex with destabilized, aggregation‐prone proteins in a process regulated by BiP availability (Genereux et al, 2015). Through these mechanisms, ERdj3 functions to protect the extracellular environment from misfolded, aggregation‐prone proteins that can be secreted during ER stress (Genereux & Wiseman, 2015). Thus, ERdj3 functions in both ATP‐dependent HSP70 chaperoning and ATP‐independent extracellular chaperoning to coordinate regulation between ER and extracellular proteostasis environments. ERdj3 can also function as an extracellular signaling molecule to regulate integrin signaling, indicating additional functions for secreted ERdj3 (Wang et al, 2013; Lee et al, 2016).
A key question is what are the structural features of ERdj3 required for its role in coordinating ER and extracellular proteostasis? The domain architecture of ERdj3 is similar to other Type I and Type II HSP40 co‐chaperones (Jin et al, 2009; Kampinga & Craig, 2010) and includes an N‐terminal J‐domain, a Gly/Phe‐rich linker, a bifurcated substrate binding domain separated by a Cys‐rich domain II containing two disulfide bonds, and a C‐terminal dimerization domain III (Fig 1A). ERdj3 is predicted to adopt a dimeric structure formed by inter‐subunit interactions between the ERdj3 dimerization domain III—a structure similar to that observed in other HSP40 co‐chaperones (Kampinga & Craig, 2010; Alderson et al, 2016). Mutations within ERdj3 domain III can render ERdj3 monomeric and impair substrate‐binding activity (Jin et al, 2009; Otero et al, 2014). Substrate binding can also be impaired through mutations in the substrate‐binding domain (Jin et al, 2009). In contrast, the ERdj3 J‐domain is required for functional interactions with BiP, but is not required for ERdj3 substrate binding or ATP‐independent chaperoning activity (Otero et al, 2014). Interestingly, the Cys‐rich ERdj3 domain II appears to have important roles for dictating both ERdj3 substrate binding and stability. Disruption of the domain II disulfide bonds reduces its binding to substrates and is predicted to destabilize ERdj3 (Marcus et al, 2007). Similarly, complete deletion of ERdj3 domain II inhibits ERdj3 chaperoning activity and decreases the intracellular half‐life of ERdj3 (Jin et al, 2009).
Figure 1. ERdj3WT assembles as a native tetramer.
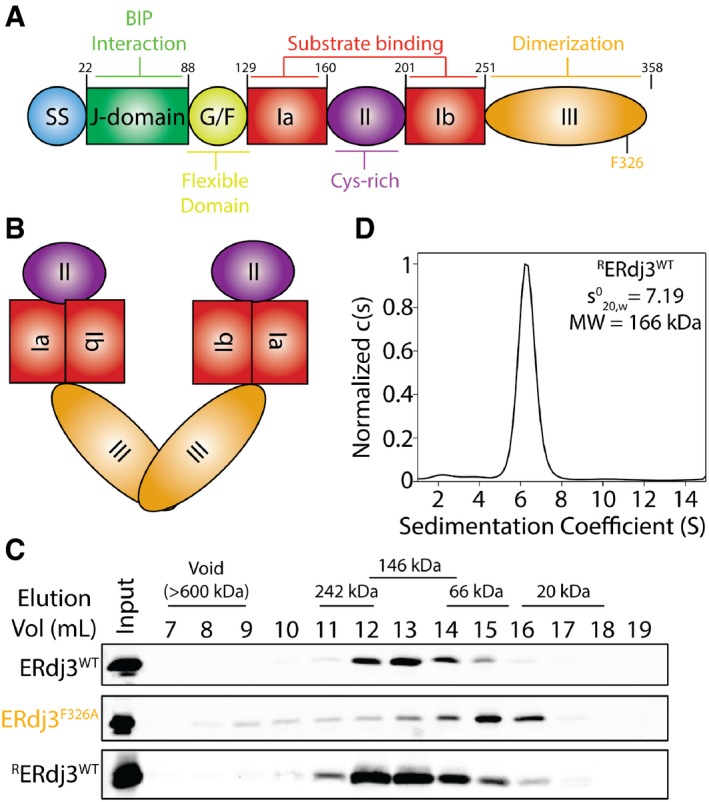
- Image showing the domain architecture of ERdj3 including the cleavable N‐terminal signal sequence (ss), J‐domain, flexible Gly/Phe‐rich domain (G/F), bifurcated substrate binding domain I (Ia and Ib), Cys‐rich domain II, and C‐terminal dimerization domain III.
- Illustration of the predicted dimeric structure of ERdj3/DNAJB11 based on comparisons with other dimeric Type I and Type II HSP40 co‐chaperones (e.g., DNAJB1). The colors reflect the domains shown in (A).
- Immunoblot of Superdex 200 gel filtration fractions of conditioned media collected from HEK293T cells expressing ERdj3WT or ERdj3F326A. Gel filtration fractions of conditioned media prepared on mock‐transfected HEK293T cells supplemented with recombinant ERdj3WT (RERdj3WT) are also shown. The migration of molecular weight standards is shown above.
- Analytical ultracentrifugation (AUC) sedimentation velocity c(S) distribution for RERdj3WT (1.12 mg/ml). The inset shows the extrapolated value for RERdj3WT and the predicted molecular weight calculated using a frictional coefficient of 1.5.
Source data are available online for this figure.
Here, we employ biochemical and structural approaches to define the ERdj3 quaternary structure. Surprisingly, we show that ERdj3 assembles into a native tetramer stabilized by distinct inter‐subunit interactions involving its dimerization domain III and the Cys‐rich domain II. As previously shown, disruptions in domain III inhibit ERdj3 oligomerization, rendering the protein monomeric. However, targeted deletions within domain II that maintain the disulfides renders ERdj3 a stable dimer. This dimeric ERdj3 shows impaired functional interactions with the ER chaperone BiP and reduced capacity for binding substrates both within the ER and in the extracellular space. Furthermore, we show that overexpression of dimeric ERdj3 enhances ER stress‐dependent reductions in the secretion of destabilized FTTTRA25T and exacerbates ER stress‐dependent accumulation of this protein as soluble oligomers in conditioned media. These results identify ERdj3 as the first canonical HSP40 co‐chaperone to assemble into a stable, native tetramer and demonstrates that this tetrameric assembly is important for the role of ERdj3 in coordinating secretory proteostasis during conditions of ER stress.
Results
Gel filtration and analytical ultracentrifugation show that endogenous and recombinant ERdj3WT assemble into tetramers
ERdj3 is a multi‐domain protein with a well‐defined domain architecture (Fig 1A). ERdj3 is homologous to other canonical Type II HSP40 co‐chaperones such as DNAJB1/Hdj1 (Appendix Fig S1A). Crystal structures of DNAJB1 and other HSP40 co‐chaperones lacking their N‐terminal J‐domain have revealed that these proteins dimerize through inter‐subunit contacts between their dimerization domain III (Appendix Fig S1B; Sha et al, 2000; Borges et al, 2005; Wu et al, 2005; Hu et al, 2008; Ramos et al, 2008). This led to speculation that ERdj3 also assembles into a dimer through its dimerization domain III (Fig 1B).
In order to test this prediction, we used gel filtration chromatography to determine the size of overexpressed wild‐type ERdj3 (ERdj3WT) secreted from transfected HEK293T cells. Surprisingly, secreted ERdj3WT eluted with fractions that corresponded to a molecular weight of ~146 Da, which is significantly higher than the predicted 76 kDa dimer (Fig 1C). Instead, this is more consistent with ERdj3 being a stable tetramer that does not dissociate during the high dilution associated with gel filtration chromatography. Recombinant wild‐type ERdj3 (RERdj3WT) added to media conditioned on mock‐transfected HEK293 cells eluted in the same fractions as cell‐secreted ERdj3WT (Fig 1C). This shows that RERdj3WT adopts the same oligomeric state as the cell‐secreted protein. However, secreted ERdj3 mutants harboring the mutation F326A in the dimerization domain III (ERdj3F326A) eluted in fractions corresponding to the 38 kDa monomer. This is consistent with previous results showing that mutation at F326 disrupts ERdj3 oligomerization, rendering ERdj3 a monomer (Jin et al, 2009; Otero et al, 2014).
We next probed the oligomeric state of RERdj3WT using an in vitro crosslinking assay. Increasing concentrations of three different lysine crosslinking reagents (DSP, DSS, and EGS) show stabilization of an ERdj3 oligomer of ~150 kDa, which is consistent with the 153 kDa ERdj3 tetramer (Appendix Fig S1C). These oligomers were further defined using analytical ultracentrifugation (AUC). Sedimentation velocity experiments show that RERdj3WT is monodisperse and has a sedimentation coefficient () of 7.19 ± 0.3 (Fig 1D, Appendix Fig S1D). Using a frictional coefficient of 1.5, which is similar to that previously calculated for the HSP40 co‐chaperone Sis1 and takes into account the elongated structure of HSP40 co‐chaperones (Silva et al, 2011), our AUC results reveal a molecular weight for RERdj3WT of 166 kDa. This molecular weight is close to the predicted 153 kDa ERdj3 tetramer and supports the hypothesis that RERdj3WT assembles into a tetramer. While AUC experiments cannot be performed using secreted ERdj3WT due to the low concentrations of secreted ERdj3 and the complexity of cell culture media, the similar gel filtration elution of RERdj3WT and cell‐secreted ERdj3WT indicates that secreted ERdj3WT also assembles into a tetramer.
Electron microscopy (EM) reveals a 19 Å structural model for full‐length RERdj3WT
Structure determination of canonical full‐length HSP40 co‐chaperones is challenged by the conformational flexibility observed for this class of protein. For some type I and type II HSP40 co‐chaperones, this conformational flexibility can be reduced using recombinant protein lacking the J‐domain, allowing structure determination by X‐ray crystallography (Sha et al, 2000; Borges et al, 2005; Wu et al, 2005; Hu et al, 2008; Ramos et al, 2008). We attempted to crystallize full‐length RERdj3WT and an RERdj3WT lacking the J‐domain using 380 crystallizing conditions at two temperatures. However, none of the crystals obtained showed diffraction sufficient for structure determination.
Fortunately, the assembly of full‐length RERdj3WT into a tetramer increases the protein size to 153 kDa, which is amenable to structure determination by electron microscopy (EM). We performed reference‐free 2D analysis of negative stained RERdj3WT particles, which revealed that the complex adopts a diamond‐shaped morphology (Figs 2A and EV1A). Subsequent 3D reconstruction of these data yielded a 19 Å resolution structure, which reveals ERdj3 as a tetramer organized as a “dimer of dimers” (Figs 2B and EV1B and C). The structure comprises 4 tube‐like densities of similar length, with two distinct types of vertices responsible for connecting these 4 densities (Fig 2B and C). Four globular densities localize above and below the diamond, which appear to be flexible domains that are independent of the core dimer or dimers structure. Unfortunately, the high degree of conformational flexibility of RERdj3WT, which hinders structure determination by crystallography, also prevents higher resolution structure determination by EM. In an attempt to reduce this conformational flexibility, we performed an analogous EM analysis of RERdj3WT in the presence of a substrate: the monomeric variant of transthyretin (M‐TTR). We confirmed that RERdj3WT binds M‐TTR by co‐immunopurification (Fig EV1D). However, in the presence of M‐TTR, RERdj3WT remained in a flexible tetrameric conformation, demonstrating that the presence of substrate does not influence ERdj3 tetramerization or reduce conformational flexibility (Fig EV1E).
Figure 2. Negative stain EM structure of RERdj3WT reveals the native tetramer.
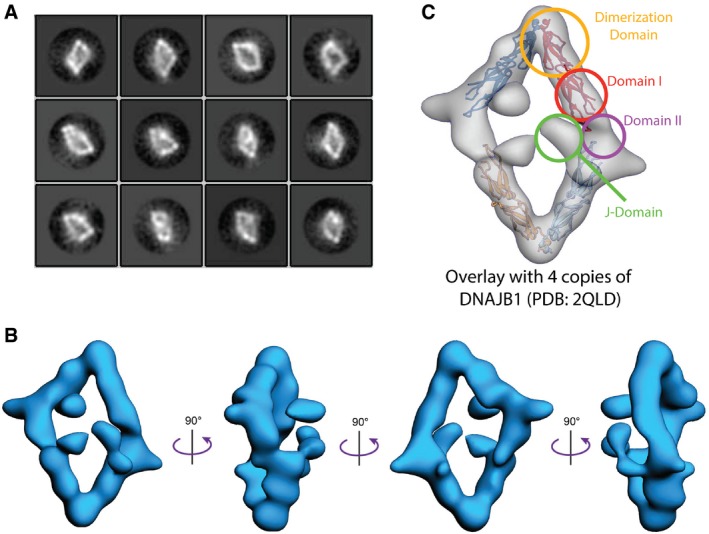
- 2D reference‐free class averages of negative stained RERdj3WT complexes show a diamond‐like shape.
- A 19 Å resolution structure of the RERdj3 tetramer is shown in 4 orthogonal views, displaying the flat, diamond‐shaped organization of the tetramer. The EM map is deposited in the EM data bank (EMD‐8707).
- A crystal structure of the ERdj3 homolog DNAJB1 (PDB: 2QLD; Hu et al, 2008) is docked into the RERdj3 EM density to depict the domain organization.
Figure EV1. Negative stain EM structure of RERdj3WT reveals the native tetramer (related to Fig 2).
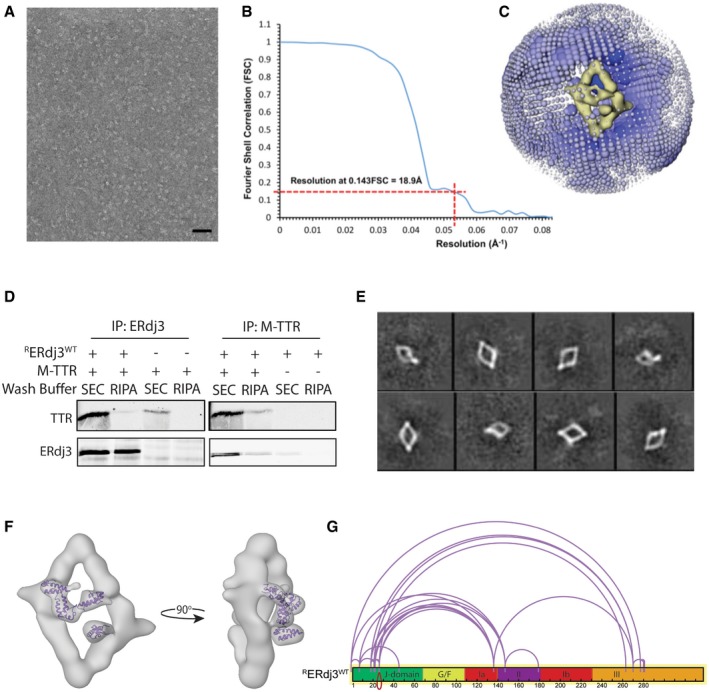
- A negative stain micrograph of recombinant ERdj3 (RERdj3; scale bar, 70 nm).
- Gold standard Fourier Shell Correlation (FSC) plot reporting the resolution of the final map to be 18.9 Å (at 0.143 FSC).
- Angular distribution of particles contributing to the 3D reconstruction, with the diameter of each sphere related to the number of particles at that Euler angle.
- Immunoblot of RERdj3 or monomeric transthyretin (M‐TTR) immunopurifications (IP) of solutions containing mixtures of RERdj3WT and M‐TTR, as indicated. The resulting IPs were washed with either the mild buffer used for size‐exclusion chromatography (SEC) or high‐detergent RIPA buffer, as indicated.
- 2D reference‐free class averages of negative stained RERdj3WT prepared in the presence of M‐TTR.
- EM image showing the NMR structure of DNAJB1 J‐domain (1HDJ; Qian et al, 1996) docked into the density localized above the ERdj3 tetramer reconstruction.
- Illustration showing the domain relationship for crosslinked peptides of RERdj3WT incubated in the presence of the amine‐reactive crosslinker DSS (10 μM). Crosslinked peptides were identified by liquid chromatography/mass spectrometry and mapped onto the RERdj3WT sequence using the xiNET crosslink viewer (Combe et al, 2015).
Source data are available online for this figure.
In order to determine the domain organization of RERdj3WT, we docked the crystal structure of four DNAJB1 subunits lacking their J‐domain (DNAJB1ΔJ; PDB: 2QLD; Hu et al, 2008) into our negative stain EM reconstruction (Fig 2C). In the DNAJB1ΔJ crystal structure, DNAJB1 forms a dimer through inter‐subunit interactions between the DNAJB1 dimerization domain III. Interestingly, two of these dimeric DNAJB1ΔJ units efficiently dock into our EM model, with the DNAJB1 dimerization domain interface corresponding to the acute angle vertices of RERdj3WT. In contrast, the obtuse angle vertices of our RERdj3WT reconstruction represent a second interface responsible for dimer‐to‐dimer contacts that extends beyond the DNAJB1ΔJ substrate‐binding domain—a site that would correspond with localization of ERdj3 domain II. Interestingly, DNAJB1 has a substantially shorter domain II as compared to RERdj3WT (Appendix Fig S1A), suggesting that this second interface may be mediated through inter‐subunit contacts between the elongated RERdj3WT domain II.
Our EM structure was determined using full‐length RERdj3WT, which includes the J‐domains that are normally removed for structure determination. This allows us to define the organization of the J‐domains within the ERdj3 tetramer. Based on the docking of DNAJB1ΔJ into our RERdj3WT model, the globular structures localized above the core ERdj3 diamond likely represent the J‐domain of this full‐length protein (Fig 2C). The size of these globular structures is consistent with the size of crystallized J‐domains (Fig EV1F). Furthermore, a crosslinking‐mass spectrometric analysis of RERdj3WT using a low concentration of the lysine reactive crosslinker DSS reveals multiple crosslinks between peptides comprising the J‐domain and other ERdj3 domains including the substrate binding domain and the dimerization domain, indicating that the J‐domain is available to interact with multiple parts of the protein (Fig EV1G). These results indicate that the J‐domain is highly flexible and localizes above (or below) the core ERdj3 tetramer, providing a dynamic surface to form functional interactions with the ER HSP70 chaperone BiP.
Targeted deletion in ERdj3 domain II disrupts formation of the native tetramer
Our EM reconstruction indicates that the unique tetramerization interface of ERdj3 is mediated through domain II inter‐subunit interactions. This domain contains 4 Cys residues that form stabilizing disulfide bonds important for ERdj3 stability and function (Marcus et al, 2007). Non‐reducing SDS–PAGE/immunoblotting shows that these disulfides are intramolecular (Marcus et al, 2007), indicating that the tetramerization interface predicted for domain II is not mediated through inter‐subunit disulfide bond formation. Interestingly, secondary structure prediction software including PSIPRED (Jones, 1999; Buchan et al, 2013) identified two potential β‐sheets separated by a turn that localize between the two pairs of cysteine residues within ERdj3 domain II (Fig 3A). These same β‐sheets were predicted in previous structural models of ERdj3 domain II (Jin et al, 2009). The presence of this structural motif suggests it may be important for dictating ERdj3 tetramerization through domain II inter‐subunit contacts identified by EM.
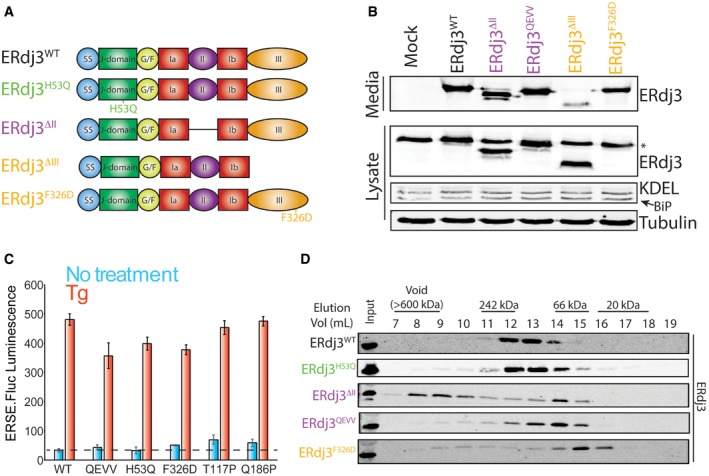
Figure 3. Targeted mutations/deletions within domain II disrupts tetramerization of secreted ERdj3.
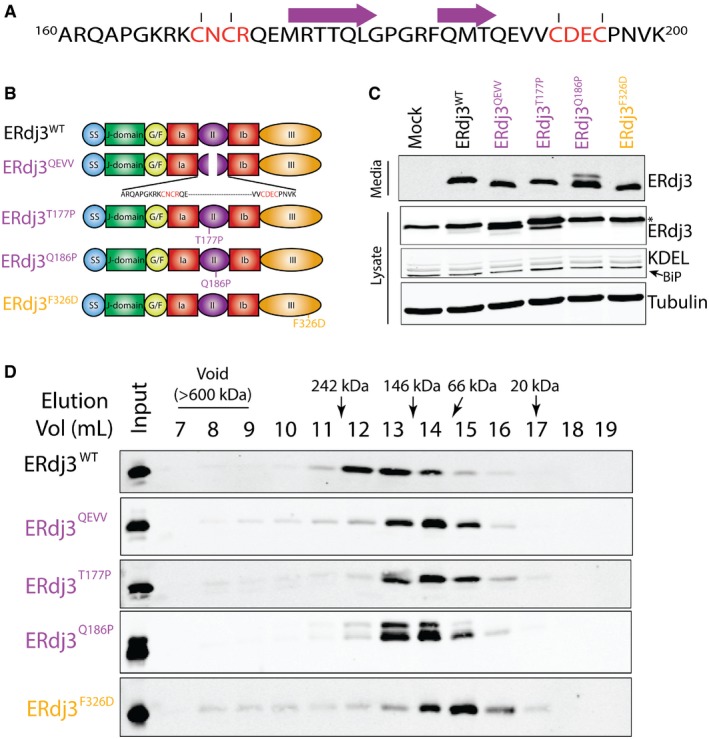
- Protein sequence for the ERdj3 domain II (residues 160‐200) including a sheet‐turn‐sheet structural motif within this sequence predicted using the PSIPRED algorithm (Jones, 1999; Buchan et al, 2013). The two predicted β sheets are shown by the purples arrows. The Cys residues that form stabilizing disulfides are also shown (red).
- Illustration showing the domain organization of ERdj3 mutants that contain mutations/deletions within domain II (ERdj3WT, ERdj3QEVV, ERdj3T177P, and ERdj3Q186P) or domain III (ERdj3F326D).
- Immunoblot of media and lysates prepared from HEK293T cells overexpressing ERdj3WT, ERdj3QEVV, ERdj3T177P, ERdj3Q186P, or ERdj3F326D. The arrow indicates BiP in the KDEL immunoblot. The asterisk indicates the migration of endogenous ERdj3WT in cell lysates.
- Immunoblot of Superdex 200 gel filtration fractions of conditioned media collected from HEK293T cells overexpressing ERdj3WT, ERdj3QEVV, ERdj3T177P, ERdj3Q186P, or ERdj3F326D. The migration of molecular weight standards is shown above.
Source data are available online for this figure.
We defined the importance for domain II in ERdj3 tetramerization by monitoring the oligomeric state of ERdj3 mutants secreted from mammalian cells using gel filtration. We collected conditioned media from HEK293T cells transfected with ERdj3 mutants including the J‐domain mutant ERdj3H53Q [which disrupts functional interactions between ERdj3 and BiP (Shen & Hendershot, 2005)], ERdj3ΔII (which completely removes the ERdj3 domain II), ERdj3QEVV (a targeted deletion of residues 175–190 that constitute the two predicted β‐sheets shown in Fig 3A), ERdj3ΔIII (a mutant lacking the entirety of domain III), and ERdj3F326D [a mutant harboring a point mutation within domain III that disrupts ERdj3 oligomerization (Jin et al, 2009; Otero et al, 2014)] (Figs 3B and EV2A). We also overexpressed two ERdj3 point mutants, ERdj3T177P and ERdj3Q186P, containing mutations predicted to disrupt the β‐sheets within domain II. Initially, we confirmed that these ERdj3 variants are secreted to conditioned media using immunoblotting. We previously showed that ERdj3H53Q is efficiently secreted from mammalian cells (Genereux et al, 2015). All of the other ERdj3 variants also accumulate in conditioned media, reflecting efficient secretion from cells (Figs 3C and EV2B). However, ERdj3ΔIII shows lower protein levels in conditioned media, suggesting that deletion of domain III destabilizes this variant and attenuates its secretion. Overexpression of ERdj3Q186P also reproducibly shows the presence of a band with reduced mobility on SDS–PAGE, suggesting that this mutant leads to alternative ERdj3 posttranslational processing.
Figure EV2. Targeted mutations/deletions within domain II disrupts tetramerization of secreted ERdj3 (related to Fig 3).
- Illustration showing the domain organization of ERdj3 mutants that contain disruptions within the J‐domain (ERdj3H53Q), domain II (ERdj3ΔII), or domain III (ERdj3ΔIII or ERdj3F326D).
- Immunoblot of media and lysates prepared from HEK293T cells overexpressing ERdj3WT, ERdj3ΔII, ERdj3QEVV, ERdj3ΔIII, or ERdj3F326D. The arrow indicates BiP in the KDEL immunoblot. The asterisk (*) indicates the migration of endogenous ERdj3WT in cell lysates.
- Graph showing activation of the ERSE.FLuc unfolded protein response (UPR) reporter in stably transfected HEK293T cells overexpressing the indicated ERdj3 variant. The ER stressor thapsigargin (Tg; 0.5 μM) was added where indicated. Error bars show SEM for n = 3. The dashed line shows reporter activation in vehicle‐treated HEK293T cells overexpressing ERdj3WT.
- Immunoblot of Superdex 200 gel filtration fractions of conditioned media collected from HEK293T cells expressing ERdj3WT, ERdj3H53Q, ERdj3ΔII, ERdj3QEVV, or ERdj3F326D. The migration of molecular weight standards is shown above.
Source data are available online for this figure.
We next evaluated the impact of overexpressing these ERdj3 variants on global ER proteostasis by monitoring activation of an ER stress‐responsive luciferase reporter (ERSE.Fluc). Overexpression of ERdj3WT, ERdj3H53Q, ERdj3QEVV, or ERdj3F326D does not significantly activate this reporter in the absence of ER stress (Fig EV2C). Similarly, overexpression of these mutants does not increase intracellular levels of the ER stress‐responsive chaperone BiP (Figs 3C and EV2B). However, overexpression of ERdj3T177P and ERdj3Q186 shows a two‐fold increase in ERSE.Fluc activation and increased BiP protein in cell lysates (Figs 3C and EV2C). This shows that overexpression of these ERdj3 variants induces a moderate amount of ER stress, likely through the presence of the unstructured β‐sheet in domain II (Fig 3A). However, none of the ERdj3 mutants influence ERSE.FLuc activation in the presence of the ER stressor thapsigargin (Tg), indicating that these ERdj3 variants do not disrupt ER stress induced UPR activation.
We used gel filtration chromatography to compare the size of secreted ERdj3 variants in conditioned media. The secreted J‐domain mutant ERdj3H53Q elutes at an identical size to that observed for ERdj3WT, indicating that this mutation does not influence ERdj3 oligomerization (Fig EV2D). In contrast, secreted ERdj3F326D elutes at much later fractions that correspond to the monomeric molecular weight of 38 kDa (Fig EV2D). This is consistent with previous work showing this point mutation ablates ERdj3 oligomerization (Jin et al, 2009; Otero et al, 2014) and confirms that domain III interactions are necessary for ERdj3 oligomerization. Interestingly, ERdj3ΔII partially elutes in intermediate fractions between the peak fractions of tetrameric ERdj3WT and monomeric ERdj3F326D (Fig EV2D). This suggests that secreted ERdj3ΔII migrates as a dimer. However, a significant population of secreted ERdj3ΔII also elutes as high molecular weight aggregates (> 600 kDa), reflecting that complete deletion of ERdj3 domain II destabilizes the protein (Jin et al, 2009). ERdj3 mutants that selectively disrupt the β‐sheet motifs within ERdj3 (ERdj3QEVV, ERdj3T177P, and ERdj3Q186P) also show migrations that correspond to dimeric ERdj3 (Fig 3D). However, we do not observe high molecular weight aggregates for these variants. These results indicate that the β‐sheets within ERdj3 domain II are important for tetramerization.
We further tested the importance for ERdj3 domain II for tetramerization using RERdj3ΔII and RERdj3QEVV—two recombinant ERdj3 mutants that contain targeted disruptions within this domain (Fig 4A). We were unable to purify RERdj3T177P due to aggregation, likely reflecting the presence of unstructured ERdj3 domain II β‐sheets that also led to UPR activation in cells (Fig EV2C). Importantly, RERdj3QEVV and RERdj3ΔII have nearly identical circular dichroism spectra to that obtained for RERdj3WT, indicating that these mutants do not globally disrupt the ERdj3 secondary structure (Appendix Fig S2A). Gel filtration of RERdj3ΔII and RERdj3QEVV shows that these proteins elute with a peak fraction consistent with dimeric ERdj3 (Fig 4B) and identical to those observed for secreted ERdj3ΔII and ERdj3QEVV (Fig 3D). AUC sedimentation velocity experiments showed that RERdj3ΔII and RERdj3QEVV are monodisperse species that migrate with sedimentation coefficients () of 4.23 and 3.79, respectively (Fig 4C and D, and Appendix Fig S2B and C). Using the same frictional coefficient employed for RERdj3WT, these results show that these proteins assemble into oligomers of 76 or 64 kDa, respectively. These molecular weights are similar to those predicted for dimers of these mutants (67 and 73 kDa). The AUC peak for RERdj3ΔII is broader than that observed for the other ERdj3 domain II mutants, suggesting that RERdj3ΔII exhibits more structural heterogeneity as compared to RERdj3QEVV. This is consistent with previous results showing that complete deletion of domain II destabilizes ERdj3 in the ER (Jin et al, 2009) and our results showing that secreted RERdj3ΔII aggregates into high molecular weight oligomers (Fig EV2D). We further demonstrated the importance of domain II for ERdj3 tetramerization by crosslinking RERdj3QEVV with different lysine crosslinking reagents, all of which predominantly result in formation of ERdj3 dimers (Appendix Fig S2D).
Figure 4. Targeted deletion of the domain II β‐sheets renders ERdj3 a stable dimer.
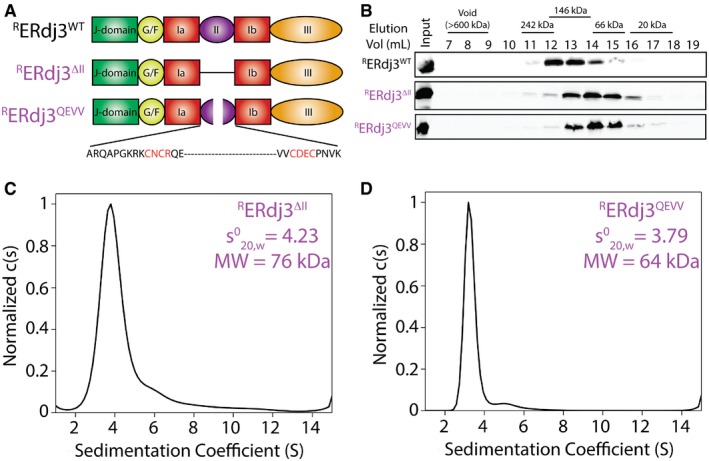
- Illustration showing the domain architecture for RERdj3WT, RERdj3ΔII, or RERdj3QEVV.
- Immunoblot of Superdex 200 gel filtration fractions of RERdj3WT, RERdj3ΔII, or RERdj3QEVV is shown. The migration of molecular weight standards is shown above.
- Analytical ultracentrifugation (AUC) sedimentation velocity c(S) distribution of RERdj3ΔII (0.8 mg/ml). The inset shows the extrapolated value for RERdj3ΔII and the predicted molecular weight calculated using a frictional coefficient of 1.5.
- Analytical ultracentrifugation (AUC) sedimentation velocity c(S) distribution of RERdj3QEVV (0.87 mg/ml). The inset shows the extrapolated value for RERdj3QEVV and the predicted molecular weight calculated using a frictional coefficient of 1.5.
Source data are available online for this figure.
The results shown in Figs 3 and 4 demonstrate that ERdj3 tetramerization requires inter‐subunit interactions between domain II. Interestingly, domain II is highly variable across other human Type I and II HSP40 co‐chaperones (Kampinga & Craig, 2010), suggesting that other HSP40 co‐chaperones are unlikely to form the same tetramerization interface used by ERdj3. However, the ERdj3 domain II is conserved across multicellular organisms, although it is divergent from the cytosolic yeast homolog YDJ1 (Appendix Fig S2E). This suggests that ERdj3 tetramerization may have evolved to perform chaperoning activities important for multicellular organisms, such as the coordination of ER and extracellular proteostasis environments.
Targeted disruption of domain II decreases ERdj3 substrate binding
ERdj3 regulates proteostasis throughout the secretory pathway (Genereux & Wiseman, 2015; Genereux et al, 2015). In the ER, ERdj3 functions as a canonical HSP40 co‐chaperone by binding misfolded proteins and delivering these proteins to BiP for ATP‐dependent chaperoning. However, in response to ER stress, ERdj3 can be secreted to the extracellular space either alone or in complex with non‐native substrates to protect the extracellular environment from destabilized, aggregation‐prone proteins that can aggregate into toxic oligomers and amyloid fibrils (Genereux & Wiseman, 2015; Genereux et al, 2015). Here, we define how targeted deletion within domain II influences ERdj3 functions important for regulating proteostasis throughout the secretory pathway.
We used an immunopurification (IP) approach to define how targeted deletion within ERdj3 domain II influences functional interactions with BiP (Kampinga & Craig, 2010; Genereux et al, 2015). We co‐expressed FLAG‐tagged BiP (FTBiP) and ERdj3WT, the J‐domain mutant ERdj3H53Q, the dimeric ERdj3QEVV, or the monomeric ERdj3F326D in HEK293T cells. We then treated these cells with the cell‐permeable crosslinker DSP (Shoulders et al, 2013; Cooley et al, 2014; Genereux et al, 2015) and quantified interactions between ERdj3 variants and FTBiP by measuring the recovery of ERdj3 in anti‐FLAG IPs from these crosslinked lysates. ERdj3WT efficiently co‐IPs with FTBiP (Fig 5A and B). However, the ERdj3 H53Q mutation disrupts functional interactions with BiP, reflected by the decreased recovery of ERdj3H53Q in FTBiP IPs. This is consistent with previous results (Shen & Hendershot, 2005). ERdj3QEVV also shows reduced recovery in FTBiP IPs, indicating that this mutant also decreases functional interactions with BiP. In contrast, ERdj3F326D does not demonstrate reduced association with BiP, potentially reflecting that BiP recognizes this monomeric mutant as a client (Fig 5A and B).
Figure 5. Stable, dimeric ERdj3QEVV shows reduced binding to BiP and destabilized substrates.
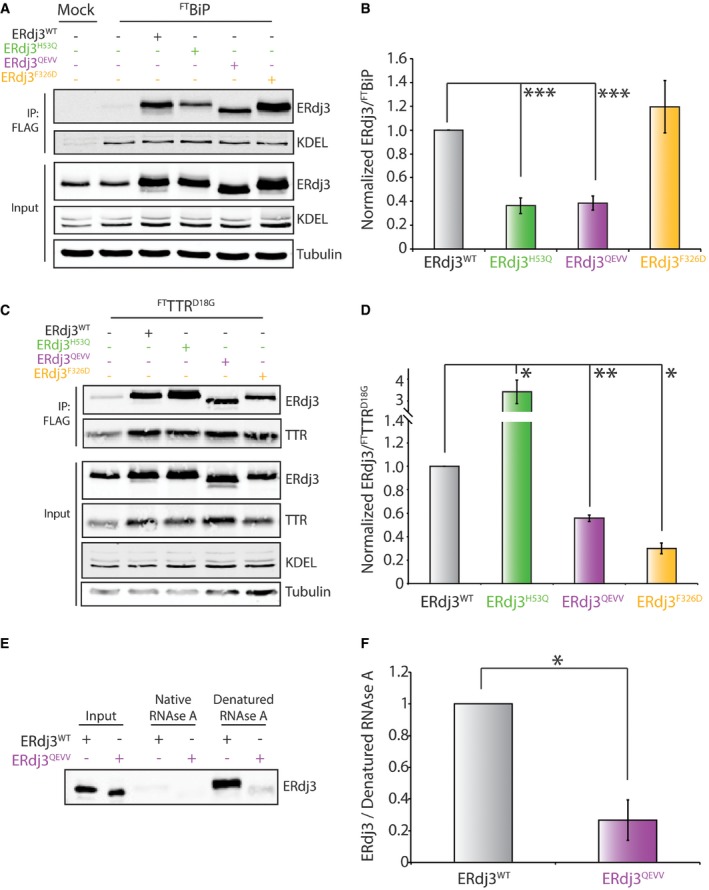
- Representative immunoblot for inputs and anti‐FLAG immunoprecipitations (IPs) of lysates prepared from HEK293T cells overexpressing FTBiP and the indicated ERdj3 mutant. Mock‐transfected HEK293T cells are included as a control. Cells were treated with the cell‐permeable, reversible crosslinker DSP prior to lysis to immortalize transient FTBiP‐ERdj3 interactions.
- Bar graph depicting quantification of ERdj3 mutants co‐immunopurifying with FTBiP from immunoblots as shown in (A). Data are shown normalized to ERdj3WT. Error bars represent standard error for n = 3. ***P < 0.005 from a paired t‐test.
- Representative immunoblots for inputs and anti‐FLAG immunoprecipitations (IPs) of lysates prepared from HEK293T cells co‐overexpressing FTTTRD18G and the indicated ERdj3 mutant. Mock‐transfected HEK293T cells are included as a control. Cells were treated with the cell‐permeable, reversible crosslinker DSP prior to lysis to immortalize transient FTTTRD18G‐ERdj3 interactions.
- Bar graph depicting quantification of ERdj3 mutants co‐immunopurifying with FTTTRD18G from immunoblots as shown in (C). Data are shown normalized to ERdj3WT. Error bars represent standard error for n = 3. *P < 0.05. **P < 0.01 from a paired t‐test.
- Representative immunoblot of secreted ERdj3WT or ERdj3QEVV from conditioned media collected from transfected HEK293T cells purified using an affinity Sepharose resin conjugated to native or denatured RNase A. Conditioned media containing ERdj3WT or ERdj3QEVV are shown as input.
- Bar graph depicting quantification of secreted ERdj3WT and ERdj3QEVV mutants associated with denatured RNase A beads as in (E). Data are normalized to ERdj3WT. Error bars represent standard error for n = 3. *P < 0.05 from a paired t‐test.
Source data are available online for this figure.
Next, we defined how targeted deletion within ERdj3 influences intracellular binding to substrates such as the non‐secreted, destabilized D18G TTR mutant (TTRD18G). We co‐overexpressed ERdj3 mutants and FLAG‐tagged TTRD18G (FTTTRD18G) in HEK293T cells and performed an identical co‐IP approach to that described above. Overexpressed ERdj3WT is efficiently recovered in FTTTRD18G IPs, confirming this protein as an ERdj3 substrate (Fig 5C and D). Overexpression of the J‐domain mutant ERdj3H53Q further increases recovery of ERdj3 in FTTTRD18G IPs. This is consistent with previous reports showing that genetic disruption of ERdj3‐BiP interactions can increase intracellular interactions between ERdj3 and destabilized substrates (Shen & Hendershot, 2005). In contrast, the recovery of overexpressed ERdj3QEVV in FTTTRD18G IPs is reduced 50% relative to ERdj3WT. This is consistent with previous results showing that complete deletion of ERdj3 domain II or selective disruption of disulfides within domain II reduces substrate binding (Marcus et al, 2007; Jin et al, 2009). Overexpressed ERdj3F326D also shows reduced recovery in FTTTRD18G IPs, which could reflect the destabilization of this variant afforded by disruption of the dimerization domain. Similar results were observed using the secreted, destabilized ERjd3 substrate FTALLC (Fig EV3A and B). These results indicate that disruption of the ERdj3 tetramer through targeted disruption of domain II or mutation in the dimerization domain decreases ERdj3 substrate binding.
Figure EV3. Stable, dimeric ERdj3QEVV shows reduced binding to BiP and destabilized substrates (related to Fig 5).
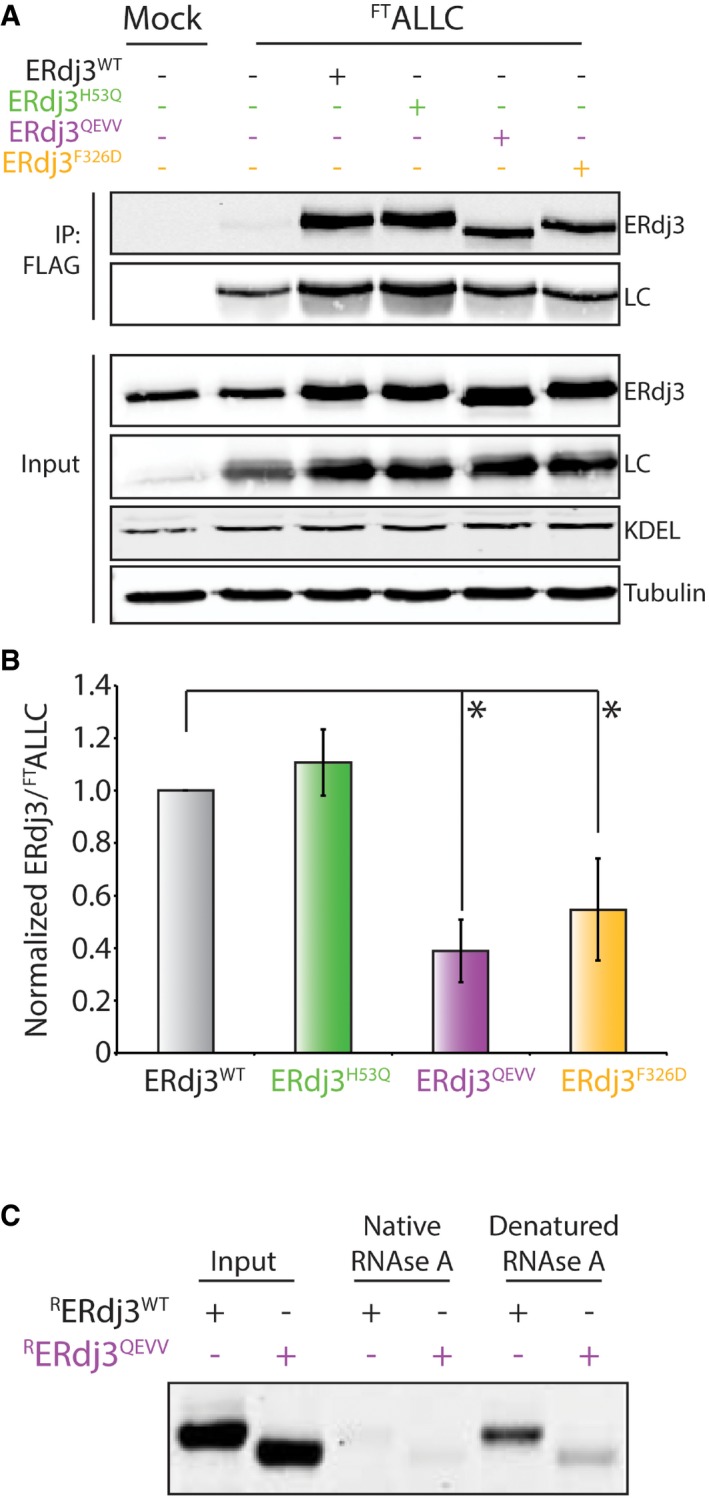
- Representative immunoblot of input and anti‐FLAG immunoprecipitations (IPs) from lysates prepared from HEK293T cells co‐overexpressing FTALLC and the indicated ERdj3 mutant. Cells were treated with the cell‐permeable, reversible crosslinker DSP prior to lysis to immortalize transient FTALLC‐ERdj3 interactions.
- Quantification of the indicated ERdj3 mutant co‐immunoprecipitating with FTALLC as described in (A). The data are normalized to ERdj3WT. Error bars show standard error for n = 3. *P < 0.05 from a paired t‐test.
- Representative Coomassie stain gel of RERdj3WT or RERdj3QEVV purified using an affinity sepharose resin conjugated to either native or denatured RNase A. RERdj3WT and RERdj3QEVV inputs are also shown.
Source data are available online for this figure.
We next sought to determine how targeted deletion within domain II influences the binding of secreted ERdj3 to non‐native protein conformations in extracellular environments (Genereux et al, 2015). For these experiments, we employed Sepharose beads conjugated to native or denatured RNase A. We then incubated these beads with either recombinant or cell‐secreted ERdj3QEVV and monitored interactions by co‐purification and immunoblotting. We previously used this exact assay to show that secreted ERdj3WT and ERdj3H53Q selectively bind non‐native protein substrates (Genereux et al, 2015). Both recombinant and cell‐secreted ERdj3QEVV show reduced binding to denatured RNase A, relative to ERdj3WT, demonstrating that targeted disruption of ERdj3 domain II also impairs substrate binding in the extracellular space (Figs 5E and F, and EV3C).
Overexpression of ERdj3QEVV exacerbates ER stress‐dependent imbalances in FTTTRA25T secretory proteostasis
The reduced interactions between ERdj3QEVV and both destabilized substrates and BiP indicate that this mutant should impair the capacity for ERdj3 to regulate secretory proteostasis of destabilized proteins. We tested this prediction using the destabilized A25T TTR variant (TTRA25T). ERdj3 binds to FTTTRA25T in the ER and extracellular environments (Shoulders et al, 2013; Genereux et al, 2015). In addition, FTTTRA25T secretory proteostasis is highly sensitive to ER stress (Chen et al, 2016). Tg‐induced ER stress decreases secretion of total FTTTRA25T (Shoulders et al, 2013) and increases the extracellular population of FTTTRA25T that accumulates as soluble aggregates commonly associated with human disease (Chen et al, 2016). Thus, we defined how overexpression of ERdj3QEVV and other ERdj3 variants influence FTTTRA25T secretory proteostasis in the presence or absence of ER stress.
We transfected HEK293T cells with FTTTRA25T and ERdj3WT, ERdj3QEVV, ERdj3F326D, or ERdj3H53Q. We then collected conditioned media from these cells in the absence or presence of Tg. All conditioned media was prepared in the presence of the non‐cell‐permeable small molecule Tafamidis‐Sulfonate (Taf‐S). Taf‐S is a kinetic stabilizer of the native FTTTRA25T tetramer that selectively stabilizes tetrameric TTR only upon secretion to the extracellular space (Chen et al, 2016). The addition of Taf‐S during conditioning allows quantification of the population of FTTTRA25T secreted in non‐native conformations that accumulate in the extracellular space as soluble aggregates (Chen et al, 2016).
Initially, we quantified total FTTTRA25T (including stabilized tetramers and soluble aggregates) that accumulate in these conditioned media using SDS–PAGE/immunoblotting (Fig 6A). In the absence of Tg, we do not observe significant differences in the amounts of total FTTTRA25T in conditioned media of cells overexpressing dimeric ERdj3QEVV or ERdj3F326D, as compared to cells overexpressing ERdj3WT (Figs 6B and EV4A). This indicates that overexpression of these ERdj3 mutants does not globally influence FTTTRA25T secretion. The addition of Tg significantly decreases secretion of FTTTRA25T in all conditions (Figs 6B and C, and EV4A). This is consistent with previous results showing Tg‐dependent reduction in FTTTRA25T secretion (Shoulders et al, 2013). Interestingly, overexpression of ERdj3QEVV enhanced the Tg‐dependent reduction in FTTTRA25T secretion, as compared to ERdj3WT (Figs 6C and EV4A). Similar results were observed when secreted FTTTRA25T was measured by metabolic labeling (Fig EV4B). In contrast, overexpression of ERdj3F326D or ERdj3H53Q does not significantly influence the Tg‐dependent reduction in secreted FTTTRA25T (Figs 6C and EV4A).
Figure 6. Overexpression of ERdj3QEVV exacerbates Tg‐induced alterations in secretory proteostasis for the destabilized, aggregation‐prone FTTTRA25T .
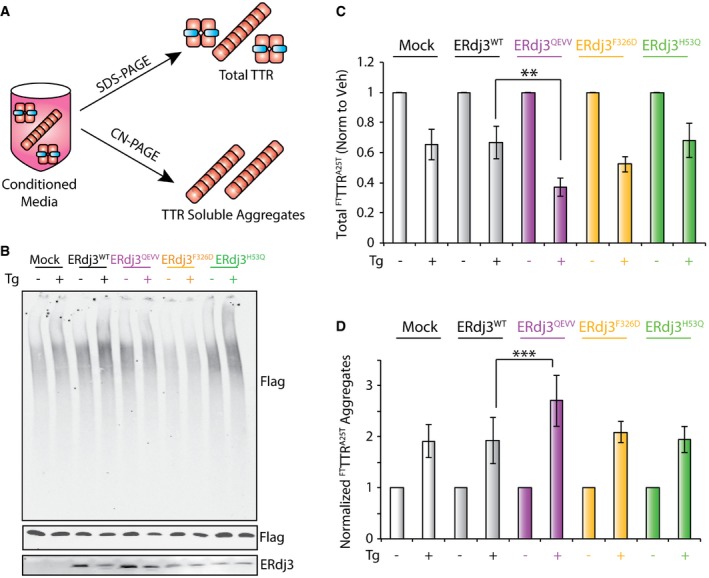
- Illustration showing the experimental approach to probe total and aggregate FTTTRA25T in conditioned media prepared on cells overexpressing FTTTRA25T and ERdj3 variants. Conditioned media was prepared in the presence of 10 μM Taf‐S (blue) to stabilize FTTTRA25T tetramers secreted to the extracellular space. Total FTTTRA25T (including stabilized tetramers and soluble aggregates) is quantified by SDS–PAGE/immunoblotting, and soluble FTTTRA25T aggregates is quantified by Clear‐Native (CN)–PAGE/immunoblotting.
- Representative CN–PAGE and SDS–PAGE immunoblots of conditioned media prepared on HEK293T cells overexpressing FTTTRA25T and ERdj3WT, ERdj3QEVV, ERdj3F326D, or ERdj3H53Q. Media was conditioned for 16 h in the presence of Taf‐S (10 μM). Tg (500 nM) was added during conditioning, as indicated.
- Quantification of total FTTTRA25T in conditioned media prepared as described in (B). The levels of FTTTRA25T are normalized to vehicle‐treated cells for each transfection. Error bars show SEM for n = 5. **P < 0.01 from a paired t‐test.
- Quantification of normalized FTTTRA25T soluble aggregates in conditioned media prepared as described in (B). Normalized aggregates were calculated as previously described (Chen et al, 2016) using the formula: normalized FTTTRA25T aggregates for condition X = [aggregate FTTTRA25T condition X/aggregate FTTTRA25T veh‐treated mock‐transfected cells]/[total FTTTRA25T condition X/total FTTTRA25T in veh‐treated mock‐transfected cells]. The data are shown relative to vehicle‐treated cells for each transfection. Error bars show SEM for n = 5. ***P < 0.005 from a paired t‐test.
Source data are available online for this figure.
Figure EV4. Overexpression of ERdj3QEVV exacerbates Tg‐induced alterations in secretory proteostasis regulation for the destabilized, aggregation‐prone FTTTRA25T (related to Fig 6).
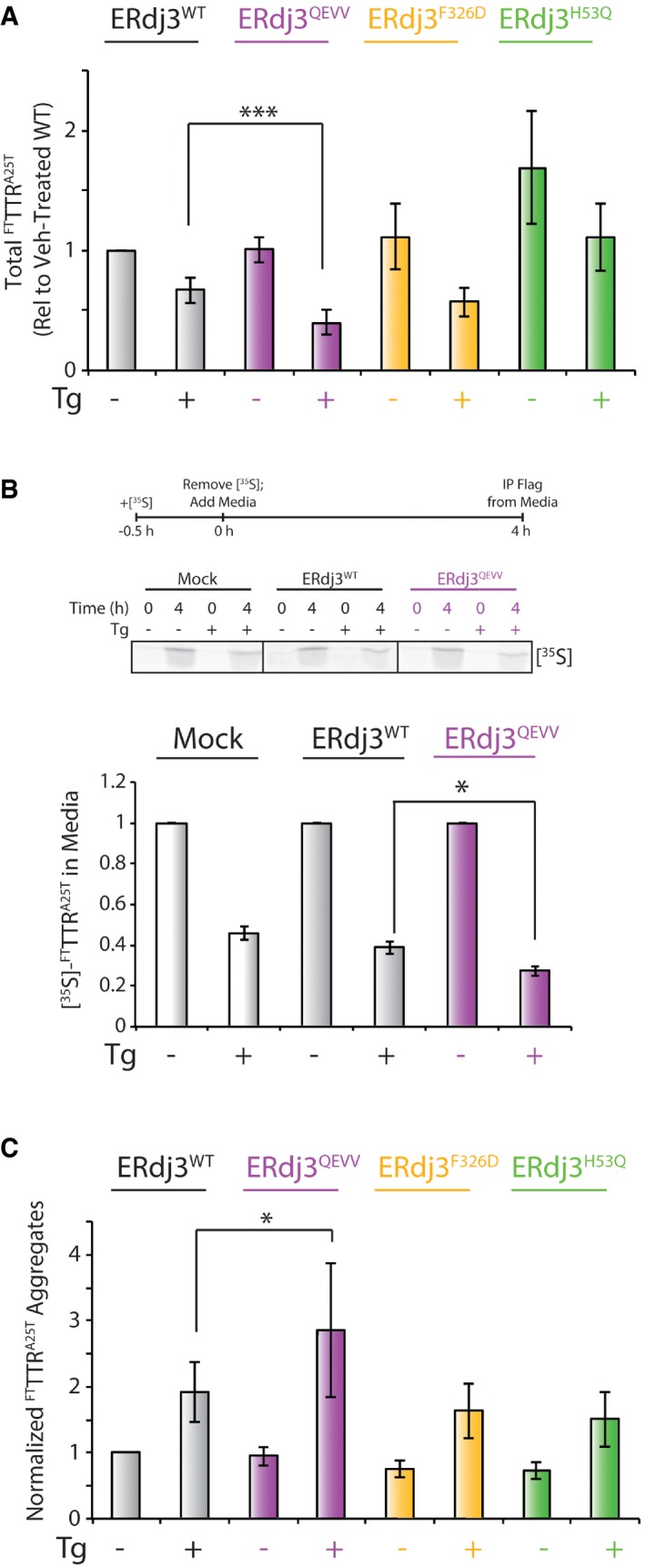
- Quantification of total FTTTRA25T in conditioned media measured by SDS–PAGE/immunoblotting. The levels of FTTTRA25T in conditioned media are shown relative to vehicle‐treated cells overexpressing ERdj3WT. Error bars show SEM for n = 5. ***P < 0.005 from a paired t‐test. A representative immunoblot for this quantification is shown in Fig 6B.
- Representative autoradiogram and quantification of HEK293T [35S]‐labeled FTTTRA25T in conditioned media prepared on HEK293 cells transfected with FTTTRA25T and ERdj3WT or ERdj3QEVV, as indicated. The [35S] metabolic labeling protocol is shown above. Error bars show SEM for n = 4. *P < 0.05 from a paired t‐test.
- Quantification of normalized FTTTRA25T aggregates in conditioned media measured by CN–PAGE/immunoblotting. Normalized FTTTRA25T aggregates were calculated by the following formula: normalized FTTTRA25T aggregates for a given condition X = [aggregate FTTTRA25T condition X/aggregate FTTTRA25T veh‐treated mock‐transfected cells]/[total FTTTRA25T condition X/total FTTTRA25T in veh‐treated mock‐transfected cells]. The data are shown relative vehicle‐treated cells transfected with ERdj3WT. Error bars show SEM for n = 5. *P < 0.05 from a paired t‐test.
We next quantified soluble aggregates of FTTTRA25T in conditioned media by Clear‐Native (CN)–PAGE/immunoblotting, as previously described (Chen et al, 2014; Fig 6A). In this analysis, we divided the relative aggregate signal determined by CN–PAGE by the total amount of FTTTRA25T in conditioned media quantified by SDS–PAGE/immunoblotting. This reflects the normalized population of FTTTRA25T that accumulates as soluble aggregates in conditioned media (Chen et al, 2016). In the absence of Tg, overexpression of ERdj3QEVV, ERdj3F326D, or ERdj3H53Q does not significantly influence normalized FTTTRA25T aggregates, as compared to ERdj3WT (Figs 6A and EV4C). The addition of Tg significantly increases normalized FTTTRA25T aggregates in conditioned media prepared on all cellular conditions (Figs 6D and EV4C). This is consistent with previous results showing Tg increases the population of secreted FTTTRA25T that accumulates as soluble aggregates (Chen et al, 2016). Interestingly, overexpression of ERdj3QEVV exacerbated the Tg‐dependent increase in normalized FTTTRA25T aggregates (Figs 6D and EV4C). In contrast, overexpression of ERdj3F326D or ERdj3H53Q did not significantly influence normalized FTTTRA25T aggregates in conditioned media. These results show that overexpression of ERdj3QEVV increases the population of FTTTRA25T that accumulates as soluble aggregates in conditioned media during ER stress.
Collectively, our results show that overexpression of the dimeric ERdj3QEVV enhances Tg‐dependent reductions in FTTTRA25T secretion and exacerbates Tg‐dependent increases in the population of FTTTRA25T that accumulate as soluble aggregates. The inability for monomeric ERdj3F326D to similarly disrupt FTTTRA25T secretory proteostasis during ER stress likely reflects the inability for this mutant to heterooligomerize with endogenous ERdj3WT, preventing its ability to function as a dominant negative to suppress endogenous ERdj3 activity during ER stress. Regardless, our results show that disruption of the ERdj3 tetramer through targeted deletions within domain II significantly decreases the capacity for ERdj3 to regulate secretory proteostasis in response to ER stress, highlighting the importance of domain II‐dependent ERdj3 tetramerization for maintaining the integrity of the secretory proteome during conditions of ER stress.
Discussion
Here, we show that ERdj3 assembles as a native tetramer formed by distinct inter‐subunit interactions involving domain II and domain III. We demonstrate that targeted deletion of ERdj3 domain II residues 175–190 renders ERdj3 a stable dimer. These stable dimers show impaired interactions with the ER HSP70 BiP and reduced binding to substrates in the ER and extracellular space. Furthermore, dimeric ERdj3 attenuates cellular capacity to regulate secretory proteostasis of destabilized FTTTRA25T in response to acute ER stress. Collectively, these results show that ERdj3 tetramerization is a key structural feature important for ERdj3‐dependent coordination of proteostasis throughout the secretory pathway.
Our identification of ERdj3 as a homo‐tetramer is supported by previous biochemical observations indicating that ERdj3 behaves as a protein substantially larger than the predicted dimer. Native gel electrophoresis previously showed that ERdj3 migrates as an oligomer with a molecular weight of ~200 kDa, which is substantially larger than the predicted dimer and more consistent with an ERdj3 tetramer (Guo & Snapp, 2013). Furthermore, fluorescence recovery after photobleaching experiments shows that ERdj3 diffuses in the ER at rates slower than those predicted for the ERdj3 dimer (Guo & Snapp, 2013). This slower diffusion likely reflects interactions between ERdj3 and ER proteostasis factors including Sec61 and an ER‐localized multi‐protein chaperoning complex (Meunier et al, 2002; Dejgaard et al, 2010; Guo & Snapp, 2013). However, our results suggest that ERdj3 tetramerization could also contribute to this slower diffusion within the ER.
We show that the dimeric ERdj3QEVV decreases substrate binding in the ER and extracellular environments and disrupts functional interactions with BiP. This indicates that disruption of the ERdj3 tetramer impairs ERdj3‐dependent regulation of secretory proteostasis. Consistent with this, overexpression of ERdj3QEVV exacerbates ER stress‐dependent alterations in secretory proteostasis for destabilized, aggregation‐prone FTTTRA25T. This disruption in FTTTRA25T secretory proteostasis likely reflects the decreased capacity for ERdj3QEVV to bind misfolded substrates, but not the reduced BiP binding, since similar results were not observed upon overexpression of ERdj3H53Q—an ERdj3 mutant that disrupts BiP interactions, but not substrate binding. These results strongly indicate that ERdj3 tetramerization offers a unique structural framework to coordinate ER and extracellular proteostasis of destabilized, aggregation‐prone protein in response to acute ER stress.
ERdj3 tetramerization offers many advantages that could facilitate its role in regulating in secretory proteostasis in the absence and presence of ER stress. Tetrameric ERdj3 contains four substrate‐binding domains that are all available to engage hydrophobic regions within misfolded protein substrates. The organization of these four binding sites into a single complex could promote substrate binding through increased avidity. This is consistent with our results showing that dimeric ERdj3QEVV binds substrates less efficiently than ERdj3WT. However, we cannot rule out the possibility that this reduced substrate binding results from alterations in the structure or presentation of the substrate binding domains, afforded by deletion of residues 175–190 of domain II, that could also influence interactions with misfolded substrates.
Tetramerization of ERdj3 may also provide a template to differentially influence the folding, degradation, or aggregation of structurally distinct client proteins in the ER or extracellular environments. Interactions with ERdj3 have been shown to distinctly impact the fate of ER or extracellular substrates. In the ER, ERdj3 promotes BiP‐dependent folding, inhibits the aggregation, and/or reduces the degradation of ER‐targeted proteins such as immunoglobulin heavy and light chain (Shen & Hendershot, 2005; Kampinga & Craig, 2010; Behnke et al, 2016). However, ERdj3 has also been proposed to promote the degradation of proteins such as the epithelial sodium channel and destabilized glucocerebrosidase variants (Buck et al, 2010; Tan et al, 2014). The presence of four substrate‐binding sites in a single ERdj3 tetramer could allow for multiple interactions with distinct hydrophobic regions of a client protein or complex. These types of multivalent interactions between ERdj3 and substrates could potentially stabilize a specific conformation that promote targeting to folding or degradation pathways (e.g., BiP chaperoning pathway and ER‐associated degradation, respectively) or inhibit aggregation into toxic oligomeric conformations. Currently, the ability for an HSP40 co‐chaperone to influence targeting of a substrate toward folding or degradation through this type of mechanism has not been demonstrated. However, the ATP‐independent secreted chaperones clusterin can stabilize small oligomeric conformations of amyloidogenic proteins such as Aβ, preventing their further aggregation into toxic oligomers or amyloid fibrils (Narayan et al, 2012). ERdj3 tetramers may similarly stabilize small oligomeric conformations of amyloidogenic proteins through multiple interactions with its four substrate binding domains, providing a mechanism to explain the substoichiometric inhibition of protein aggregation observed for ERdj3 (Genereux et al, 2015).
ERdj3 tetramerization could also influence substrate fate by providing a structural template to modulate functional interactions between ERdj3 and other proteostasis pathways. In the ER, ERdj3 forms functional interactions with Sec61 and the ER HSP70 BiP (Dejgaard et al, 2010; Guo & Snapp, 2013; Otero et al, 2014; Schorr et al, 2015). In addition, ERdj3 has also been shown to incorporate into a multi‐chaperone complex including many other ER proteostasis factors including GRP94, HYOU1, PDI, and UDP‐glucosyltransferase in the presence of substrates such as immunoglobulin heavy chain (Meunier et al, 2002). ERdj3 tetramers increase binding sites on this co‐chaperone, which could increase the capacity to form interactions with one or several of these interaction partners. Specific ERdj3 domains important for interacting with many of these ER proteostasis factors remain currently undefined. However, our results show that ERdj3 interactions with the ER HSP70 chaperone BiP are impaired in the dimeric ERjd3QEVV mutant, suggesting tetramerization may be important for interactions with functional partners. ERdj3 tetramerization could also serve an important role for dictating non‐proteostasis interactions between ERdj3 and cellular factors involved in other functions such as integrin signaling (Wang et al, 2013; Lee et al, 2016). The importance of ERdj3 tetramerization for integrin signaling could potentially reflect the emergence of domain II sequence conservation in multi‐cellular organisms where this type of signaling is observed.
To our knowledge, our results identify ERdj3 as the first type I or type II HSP40 co‐chaperone to assemble into a native tetramer, as opposed to the dimeric structure observed for most HSP40 co‐chaperones of these types. As discussed above, the formation of the tetramer provides a unique structural framework to define how ERdj3 coordinates its diverse functions involved in regulating ER and extracellular proteostasis. In addition, our identification of residues 175–190 of domain II as the key determinants in ERdj3 tetramerization provides new insights into the importance of domain II for ERdj3 function and reveals potential mechanisms to regulate ERdj3 substrate binding and proteostasis regulation through posttranslational alterations targeting this domain.
Materials and Methods
Mammalian cell culture, immunoprecipitation, and quantitative immunoblotting
HEK293T cells were cultured in DMEM (Mediatech) media with 10% FBS supplemented with pen/strep and glutamine (Gibco). ERdj3 mutants and FTTTRD18G, FTTTRA25T, FTALLC, or FTBiP plasmids were transfected using the calcium phosphate transfection method. The transfected cells were incubated overnight before harvesting. Lysate was prepared in the lysis buffer (20 mM HEPES pH 7.0, 150 mM NaCl, 1% Triton X‐100, 5 mM EDTA) plus cOmplete® protease inhibitor cocktail (Roche).
For immunoprecipitation with the FLAG‐tagged constructs following crosslinking, the harvested cells were resuspended in DPBS (Gibco), and 1 mM DSP was added to the suspension from 100 mM stock in DMSO. The cells were incubated at ambient temperature for 30 min, and 100 mM Tris pH 8.0 was added to quench the reaction for another 15 min. The cells were then lysed as described above for immunoprecipitation with M2 anti‐FLAG magnetic beads (Sigma) overnight at 4°C. The beads were washed four times in RIPA buffer before elution by boiling in 3× SDS loading buffer without DTT. 100 mM DTT was added to the eluents, and the sample was boiled again before SDS–PAGE and Western blot. After overnight primary antibody incubation at 4°C, the blots were incubated with IR Dye conjugated secondary antibodies (LICOR) and the quantified with the Odyssey system (LICOR).
Quantification of total and aggregate FTTTRA25T in conditioned media
HEK293T cells were transfected with FTTTRA25T and the indicated ERdj3 variant using calcium phosphate. Cells were then cultured overnight in cell culture media containing 10 μM Tafamidis‐Sulfonate (Taf‐S). Thapsigargin (Tg; 500 nM) was added as indicated. Conditioned media was then collected and analyzed by SDS–PAGE/immunoblotting and Clear‐Native–PAGE/immunoblotting, as previously described (Chen et al, 2014, 2016). The relative amounts of total or aggregate FTTTRA25T were quantified with the Odyssey system (LICOR).
Conditioned media analytical gel filtration chromatography
Conditioned media sample was first passed through 0.22 μm filter to remove insoluble species. Then, 500 μl of sample was injected onto a self‐packed Superdex 200 (GE Healthcare) analytical column (10 × 300 mm) using the ÄKTA FPLC system at flow rate of 0.4 ml/min. 1× DPBS (Gibco) was used as the mobile phase at ambient temperature. The column was calibrated with NativeMark™ Unstained Protein Standard (Life Technology). The fractions were collected with interval of 1 ml and analyzed by SDS–PAGE and Western blot.
RERdj3 purification and characterization
6xHis Smt3 tagged ERdj3 mutants were transformed into SHuffle® T7 Express competent E. coli (NEB) and incubated at 30°C. The overnight culture grown at 30°C in LB media was inoculated in LB media at ratio of 1:1,000 to grow at 30°C till O.D. reaches ~0.6, and then, 0.1 mM IPTG was used to induce overnight expression at 25°C. The harvested cells were sonicated in Ni A buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 20 mM imidazole) with cOmplete™ Ultra EDTA free protease inhibitor (Roche). The lysate was centrifuged and then filtered through 0.45 μm filter before loading onto HisTrap FF column (GE Healthcare) using ÄKTA FPLC system (GE Healthcare). The column was washed with 5% Ni B buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 250 mM imidazole) before eluted with 100% Ni B buffer. The eluents was dialyzed overnight at 4°C in Ni A buffer with ULP1 protease added to cleave the 6xHis Smt3 tag. The sample was then loaded onto HisTrap column to collect untagged RERdj3 in the flow through. Next, the concentrated sample was loaded onto Superdex 200 prep grade column (GE Healthcare, self‐packed) in HN buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol). The collected fractions were concentrated and flash‐frozen to be stored at −80°C. For circular dichroism analysis, the proteins were dialyzed overnight in 50 mM Na Pi pH 7.5, 300 mM NaCl, 10% glycerol at 4°C. The dialyzed sample was diluted to 6 μM, and CD measurements were made with an Aviv 62A DS Circular Dichroism spectrometer, using quartz cuvettes with a 0.2 cm path length. CD spectra were obtained by monitoring molar ellipticity from 260 to 200 nm, with 10‐s averaging times.
Analytical ultracentrifugation and velocity data analysis
Recombinant ERdj3WT and mutants were dialyzed overnight at 4°C in 50 mM HEPES pH 7.5, 300 mM NaCl to remove glycerol. The dialyzed samples were then diluted to final concentrations and run at 201,600 g with Beckman Optima XL‐I Analytical Ultracentrifuge. Sedimentation velocity data were collected using UV absorbance at 280 nm every 10 min for up to 600 cycles. The buffer density (1.0144 g/ml), viscosity (1.0611E‐2), and Vbar for each protein (RERdj3WT: 0.7319, RERdj3QEVV: 0.7328, RERdj3Δ2: 0.7346) were calculated using the Sednterp program. The sedimentation velocity data were fit using continuous distribution C(s) model with SedFit. The apparent s values of each run were obtained from the integration of the C(s) peak and corrected it to standard condition s20,w of using SedFit. The s distribution and residue maps were plotted using the GUSSI program (Brautigam, 2015) by importing the data from SedFit. The s20,w data were extrapolated linearly to concentration of 0 mg/ml to obtain reported in the text, and the error represents the standard deviation of peak value. The molecular weights of each mutant were calculated using SedFit with s value at 0 mg/ml and f/f 0 ratio of 1.5.
RNase A beads assay
RNase A‐conjugated beads were incubated in 20 mM Tris, pH 8.8, 6 M guanidinium hydrochloride, and 100 mM dithiothreitol overnight to disrupt the disulfide bonds in RNase A. The denatured and reduced RNase A beads were then incubated in 100 mM Tris, pH 8, and 37.5 mM iodoacetamide to block free thiols, thus preventing RNase A from adopting its native structure. To examine the affinity of secreted ERdj3 for either native or denatured RNase A beads, culture medium of HEK293T cells overexpressing either ERdj3WT or ERdj3QEVV was collected and their relative levels were quantified using immunoblotting. Similar amount of ERdj3WT and ERdj3QEVV were incubated with either native or denatured RNase A beads overnight at 4°C. The beads were washed with buffer A (50 mM HEPES, 300 mM NaCl, 10% glycerol, pH 7.5) containing 0.1% Triton X‐100. Proteins bound to the beads were then eluted with reducing Laemmli buffer and analyzed by immunoblotting. To measure the interaction of RERdj3 with either native or denatured RNase A beads, 30 μg recombinant ERdj3 was incubated with buffer A overnight at 4°C then washed with buffer A containing 0.5% Triton X‐100. Protein bound to the beads was analyzed using the protocol described above.
Additional Materials and Methods are included in the Appendix Supplementary Methods.
Data accession
The final EM map showing the 19 Å structure of RERdj3WT was deposited in EM data bank with the accession number EMD‐8707. Additional information discussing the negative stain electron microscopy data collection and image processing used to prepare this model is described in the Appendix Supplementary Methods.
Author contributions
K‐CC, SQ, SC, GCL, and RLW conceived of the project. K‐CC, SQ, ICN, SC, JDS, and LP performed the experiments. K‐CC, SQ, SC, ICN, JDS, LP, ETP, JWK, GCL, and RLW analyzed the data. K‐CC, SQ, SC, GCL, and RLW wrote the manuscript. K‐CC, SQ, SC, ICN, JDS, LP, ETP, JWK, GCL, and RLW edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Dennis Wolan (TSRI) and Joseph Genereux (UC‐Riverside) for helpful discussions and experimental support. This work was supported by the National Institutes of Health (NS092829, DK107604, DK102635 to RLW; DP2 EB020402 to GCL; DK046335 to JWK), the Amyloidosis Foundation (RLW), The Pew Scholar Program (GCL), and the Searle Scholar Program (GCL). LP is funded by a Leukemia and Lymphoma Society Postdoctoral Fellowship.
The EMBO Journal (2017) 36: 2296–2309
References
- Alderson TR, Kim JH, Markley JL (2016) Dynamical structures of Hsp70 and Hsp70‐Hsp40 complexes. Structure 24: 1014–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke J, Mann MJ, Scruggs FL, Feige MJ, Hendershot LM (2016) Members of the Hsp70 family recognize distinct types of sequences to execute ER quality control. Mol Cell 63: 739–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges JC, Fischer H, Craievich AF, Ramos CH (2005) Low resolution structural study of two human HSP40 chaperones in solution. DJA1 from subfamily A and DJB4 from subfamily B have different quaternary structures. J Biol Chem 280: 13671–13681 [DOI] [PubMed] [Google Scholar]
- Brautigam CA (2015) Calculations and publication‐quality illustrations for analytical ultracentrifugation data. Methods Enzymol 562: 109–133 [DOI] [PubMed] [Google Scholar]
- Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT (2013) Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res 41: W349–W357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck TM, Kolb AR, Boyd CR, Kleyman TR, Brodsky JL (2010) The endoplasmic reticulum‐associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol Biol Cell 21: 1047–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Genereux JC, Qu S, Hulleman JD, Shoulders MD, Wiseman RL (2014) ATF6 activation reduces the secretion and extracellular aggregation of destabilized variants of an amyloidogenic protein. Chem Biol 21: 1564–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Genereux JC, Suh EH, Vartabedian VF, Rius B, Qu S, Dendle MT, Kelly JW, Wiseman RL (2016) Endoplasmic reticulum proteostasis influences the oligomeric state of an amyloidogenic protein secreted from mammalian cells. Cell Chem Biol 23: 1282–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combe CW, Fischer L, Rappsilber J (2015) xiNET: cross‐link network maps with residue resolution. Mol Cell Proteomics 14: 1137–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley CB, Ryno LM, Plate L, Morgan GJ, Hulleman JD, Kelly JW, Wiseman RL (2014) Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc Natl Acad Sci USA 111: 13046–13051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr DM, Ramos CH (2015) Specification of Hsp70 function by type I and type II Hsp40. Subcell Biochem 78: 91–102 [DOI] [PubMed] [Google Scholar]
- Dejgaard K, Theberge JF, Heath‐Engel H, Chevet E, Tremblay ML, Thomas DY (2010) Organization of the Sec61 translocon, studied by high resolution native electrophoresis. J Proteome Res 9: 1763–1771 [DOI] [PubMed] [Google Scholar]
- Genereux JC, Qu S, Zhou M, Ryno LM, Wang S, Shoulders MD, Kaufman RJ, Lasmezas CI, Kelly JW, Wiseman RL (2015) Unfolded protein response‐induced ERdj3 secretion links ER stress to extracellular proteostasis. EMBO J 34: 4–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereux JC, Wiseman RL (2015) Regulating extracellular proteostasis capacity through the unfolded protein response. Prion 9: 10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Snapp EL (2013) ERdj3 regulates BiP occupancy in living cells. J Cell Sci 126: 1429–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Wu Y, Li J, Qian X, Fu Z, Sha B (2008) The crystal structure of the putative peptide‐binding fragment from the human Hsp40 protein Hdj1. BMC Struct Biol 8: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Zhuang M, Hendershot LM (2009) ERdj3, a luminal ER DnaJ homologue, binds directly to unfolded proteins in the mammalian ER: identification of critical residues. Biochemistry 48: 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT (1999) Protein secondary structure prediction based on position‐specific scoring matrices. J Mol Biol 292: 195–202 [DOI] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11: 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer‐Hartl M, Hartl FU (2013) Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82: 323–355 [DOI] [PubMed] [Google Scholar]
- Lee JY, Chen JY, Shaw JL, Chang KT (2016) Maintenance of stem cell niche integrity by a novel activator of integrin signaling. PLoS Genet 12: e1006043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinowski M, Holler M, Feige MJ, Baerend D, Lamb DC, Buchner J (2011) Substrate discrimination of the chaperone BiP by autonomous and cochaperone‐regulated conformational transitions. Nat Struct Mol Biol 18: 150–158 [DOI] [PubMed] [Google Scholar]
- Marcus NY, Marcus RA, Schmidt BZ, Haslam DB (2007) Contribution of the HEDJ/ERdj3 cysteine‐rich domain to substrate interactions. Arch Biochem Biophys 468: 147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnyk A, Rieger H, Zimmermann R (2015) Co‐chaperones of the mammalian endoplasmic reticulum. Subcell Biochem 78: 179–200 [DOI] [PubMed] [Google Scholar]
- Meunier L, Usherwood YK, Chung KT, Hendershot LM (2002) A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol Biol Cell 13: 4456–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan P, Orte A, Clarke RW, Bolognesi B, Hook S, Ganzinger KA, Meehan S, Wilson MR, Dobson CM, Klenerman D (2012) The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid‐beta(1‐40) peptide. Nat Struct Mol Biol 19: 79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Bukau B (2015) Metazoan Hsp70‐based protein disaggregases: emergence and mechanisms. Front Mol Biosci 2: 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, Arnsburg K, Gao X, Scior A, Aebersold R, Guilbride DL, Wade RC, Morimoto RI, Mayer MP, Bukau B (2015) Crucial HSP70 co‐chaperone complex unlocks metazoan protein disaggregation. Nature 524: 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero JH, Lizak B, Hendershot LM (2010) Life and death of a BiP substrate. Semin Cell Dev Biol 21: 472–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero JH, Lizak B, Feige MJ, Hendershot LM (2014) Dissection of structural and functional requirements that underlie the interaction of ERdj3 protein with substrates in the endoplasmic reticulum. J Biol Chem 289: 27504–27512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian YQ, Patel D, Hartl FU, McColl DJ (1996) Nuclear magnetic resonance solution structure of the human Hsp40 (HDJ‐1) J‐domain. J Mol Biol 260: 224–235 [DOI] [PubMed] [Google Scholar]
- Ramos CH, Oliveira CL, Fan CY, Torriani IL, Cyr DM (2008) Conserved central domains control the quaternary structure of type I and type II Hsp40 molecular chaperones. J Mol Biol 383: 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorr S, Klein MC, Gamayun I, Melnyk A, Jung M, Schauble N, Wang Q, Hemmis B, Bochen F, Greiner M, Lampel P, Urban SK, Hassdenteufel S, Dudek J, Chen XZ, Wagner R, Cavalie A, Zimmermann R (2015) Co‐chaperone specificity in gating of the polypeptide conducting channel in the membrane of the human endoplasmic reticulum. J Biol Chem 290: 18621–18635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha B, Lee S, Cyr DM (2000) The crystal structure of the peptide‐binding fragment from the yeast Hsp40 protein Sis1. Structure 8: 799–807 [DOI] [PubMed] [Google Scholar]
- Shen Y, Meunier L, Hendershot LM (2002) Identification and characterization of a novel endoplasmic reticulum (ER) DnaJ homologue, which stimulates ATPase activity of BiP in vitro and is induced by ER stress. J Biol Chem 277: 15947–15956 [DOI] [PubMed] [Google Scholar]
- Shen Y, Hendershot LM (2005) ERdj3, a stress‐inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP's interactions with unfolded substrates. Mol Biol Cell 16: 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR III, Su AI, Kelly JW, Wiseman RL (2013) Stress‐independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 3: 1279–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JC, Borges JC, Cyr DM, Ramos CH, Torriani IL (2011) Central domain deletions affect the SAXS solution structure and function of yeast Hsp40 proteins Sis1 and Ydj1. BMC Struct Biol 11: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YL, Genereux JC, Pankow S, Aerts JM, Yates JR III, Kelly JW (2014) ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher's disease. Chem Biol 21: 967–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Juan HC, Wong YH, Kuo WC, Lu YL, Lin SF, Lu CJ, Fann MJ (2013) Protogenin prevents premature apoptosis of rostral cephalic neural crest cells by activating the alpha5beta1‐integrin. Cell Death Dis 4: e651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Li J, Jin Z, Fu Z, Sha B (2005) The crystal structure of the C‐terminal fragment of yeast Hsp40 Ydj1 reveals novel dimerization motif for Hsp40. J Mol Biol 346: 1005–1011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6