

Abstract
O‐GlcNAcylation is an essential, nutrient‐sensitive post‐translational modification, but its biochemical and phenotypic effects remain incompletely understood. To address this question, we investigated the global transcriptional response to perturbations in O‐GlcNAcylation. Unexpectedly, many transcriptional effects of O‐GlcNAc transferase (OGT) inhibition were due to the activation of NRF2, the master regulator of redox stress tolerance. Moreover, we found that a signature of low OGT activity strongly correlates with NRF2 activation in multiple tumor expression datasets. Guided by this information, we identified KEAP1 (also known as KLHL19), the primary negative regulator of NRF2, as a direct substrate of OGT. We show that O‐GlcNAcylation of KEAP1 at serine 104 is required for the efficient ubiquitination and degradation of NRF2. Interestingly, O‐GlcNAc levels and NRF2 activation co‐vary in response to glucose fluctuations, indicating that KEAP1 O‐GlcNAcylation links nutrient sensing to downstream stress resistance. Our results reveal a novel regulatory connection between nutrient‐sensitive glycosylation and NRF2 signaling and provide a blueprint for future approaches to discover functionally important O‐GlcNAcylation events on other KLHL family proteins in various experimental and disease contexts.
Keywords: KEAP1, KLHL, NRF2, O‐GlcNAcylation, OGT
Subject Categories: Metabolism; Post-translational Modifications, Proteolysis & Proteomics
Introduction
O‐linked β‐N‐acetylglucosamine (O‐GlcNAc) is a dynamic post‐translational modification (PTM) that reversibly decorates serine and threonine residues of thousands of nuclear, cytoplasmic and mitochondrial proteins (Hart et al, 2011; Bond & Hanover, 2015). As with other intracellular PTMs, dedicated enzymes add or remove O‐GlcNAc on target proteins to control their functions in response to various physiological and metabolic cues. In mammals, O‐GlcNAc is added by O‐GlcNAc transferase (OGT) and removed by O‐GlcNAcase (OGA; Hart et al, 2011; Bond & Hanover, 2015). O‐GlcNAcylation of target proteins requires UDP‐GlcNAc, a nucleotide‐sugar derived from glucose and other metabolites via the hexosamine biosynthetic pathway (HBP), directly linking nutrient status to O‐GlcNAc signaling (Hart et al, 2011; Bond & Hanover, 2015). The genetic ablation of either OGT or OGA in mice is lethal, highlighting their essential role in embryonic development (Shafi et al, 2000; Yang et al, 2012; Keembiyehetty et al, 2015). Moreover, aberrant O‐GlcNAcylation is implicated in various human diseases, particularly cancer (Hart et al, 2011; Bond & Hanover, 2013; Ma & Vosseller, 2013; Ferrer & Reginato, 2014; Ferrer et al, 2016). For example, numerous oncoproteins (e.g., Myc, Akt) and tumor suppressors (e.g., p53, AMPK) are O‐GlcNAcylated, affecting oncogenic signaling and treatment responses (Chou et al, 1995; Yang et al, 2006; Luo et al, 2007; Kang et al, 2008; Slawson & Hart, 2011; Ma & Vosseller, 2014).
Despite its broad pathophysiological significance, major aspects of O‐GlcNAc signaling remain poorly understood. Significant challenges include identifying the specific glycoprotein substrates that are most functionally important for signal transduction, and elucidating the biochemical effects that O‐GlcNAc exerts on its substrates. Therefore, new strategies are needed to understand these aspects of O‐GlcNAc biology. We addressed these challenges through an interdisciplinary approach, combining genetics, biochemistry, and chemical biology. Because many chromatin proteins and transcription factors are O‐GlcNAcylated (Sakabe et al, 2010; Lewis & Hanover, 2014; Leturcq et al, 2017), we focused on the role of O‐GlcNAc cycling in gene regulation. In doing so, we discovered an unexpected connection between OGT and the nuclear factor‐erythroid 2‐related factor 2 (NRF2) pathway.
NRF2 is a basic leucine zipper transcription factor whose levels and activity are suppressed in unstressed cells by KEAP1 (also called KLHL19; Itoh et al, 1999), a substrate adaptor protein for the Cullin‐3 (CUL3)‐dependent E3 ubiquitin ligase complex. KEAP1–CUL3 represses NRF2 by mediating its ubiquitination and subsequent proteasomal degradation (Cullinan et al, 2004; Kobayashi et al, 2004; Zhang et al, 2004; Furukawa & Xiong, 2005). Oxidative stress or electrophiles modify cysteine residues in KEAP1, resulting in conformational changes and reduced ubiquitination of bound NRF2 (Zhang & Hannink, 2003; Wakabayashi et al, 2004; Zhang et al, 2004; Kobayashi et al, 2006), as illustrated by the “hinge and latch” model for KEAP1 action (Tong et al, 2006b). Therefore, under stressed conditions, newly translated NRF2 molecules remain free of KEAP1, allowing NRF2 accumulation and nuclear translocation (Padmanabhan et al, 2006; Tong et al, 2006a,b). In the nucleus, NRF2 binds promoters containing antioxidant response element (ARE) DNA sequences, activating the transcription of many genes involved in oxidative stress responses, such as heme oxygenase 1 (HO‐1), superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase 1 (GPX1), NAD(P)H:quinone oxidoreductase (NQO1), gamma‐glutamylcysteine synthetase (GCS), and glutathione reductase (Kansanen et al, 2013). NRF2‐regulated expression of ARE‐driven genes, especially those involved in glutathione (GSH) biosynthesis and recycling (Harvey et al, 2009), is critical for cell survival during oxidative stress in various disease models (Chan & Kwong, 2000; Lee et al, 2003; Sangokoya et al, 2010; Doss et al, 2016).
Several mechanisms for NRF2 pathway regulation have been described (Sykiotis & Bohmann, 2010). For example, oxidative stress or electrophilic xenobiotics can disrupt either the KEAP1–NRF2 (Levonen et al, 2004; Canning et al, 2015; Huerta et al, 2016) or KEAP1–CUL3 interaction (Gao et al, 2007; Rachakonda et al, 2008; Eggler et al, 2009; Cleasby et al, 2014), thereby reducing NRF2 ubiquitination and degradation, and activating downstream transcription of NRF2 targets. In addition, the NRF2 pathway is subject to regulation by PTMs, including succination of KEAP1 (Adam et al, 2011; Kinch et al, 2011) and phosphorylation of NRF2 itself (Alam et al, 2000; Huang et al, 2002; Cullinan & Diehl, 2004; Salazar et al, 2006). However, the impact of O‐GlcNAc cycling on NRF2 signaling remains poorly defined. Here, using the transcriptional response to OGT inhibition as guide, we show that O‐GlcNAcylation of KEAP1 at serine 104 is required to restrain the NRF2 pathway in unstressed cells. Interestingly, we also found that global O‐GlcNAcylation and NRF2 activation co‐vary in response to glucose changes in cultured cells. Therefore, our results reveal a new mechanism of NRF2 regulation through nutrient‐sensitive O‐GlcNAcylation and have important implications for antioxidant signaling in both normal and disease contexts.
Results
Global transcriptional responses to OGT and OGA inhibition
O‐GlcNAc transferase (OGT) dynamically glycosylates numerous chromatin components and transcription factors (Ozcan et al, 2010; Sakabe et al, 2010; Lewis & Hanover, 2014; Leturcq et al, 2017). However, it remains challenging to identify the particular O‐GlcNAc substrate(s) that are most functionally important in particular gene expression programs. To address this challenge, we profiled the global transcriptional responses to OGT or OGA inhibition to guide our downstream biochemical investigation. We analyzed the transcriptomes of MDA‐MB‐231 cells treated with DMSO (vehicle) or small molecular inhibitors of OGT (5SGlcNAc; Gloster et al, 2011) or OGA (Thiamet‐G; Yuzwa et al, 2008; Fig 1A). First, we confirmed that Thiamet‐G and 5SGlcNAc increased and decreased global O‐GlcNAcylation, respectively, as expected (Fig EV1A). Then, we collected RNA samples at multiple time points after DMSO, Thiamet‐G, or 5SGlcNAc treatment and analyzed them by Affymetrix U133A2 arrays (deposited into GEO as GSE81740). The array data were normalized by the robust multi‐array (RMA) method and zero‐transformed against the average expression of three DMSO samples (Keenan et al, 2015; Tang et al, 2015) to derive the transcriptional responses to OGA or OGT inhibition (Fig 1B). As early as 8 h, Thiamet‐G induced OGA mRNA and downregulated OGT mRNA, whereas 5SGlcNAc induced OGT and downregulated OGA mRNAs (Fig 1B). These changes reflect previously reported compensation responses to O‐GlcNAc perturbation (Zhang et al, 2014), confirming the expected effects of OGA and OGT inhibition on gene expression in our system. In addition to the OGA/OGT mRNA changes, we noted large clusters of altered gene expression after 24 h of OGT inhibition (Fig 1B). Many of the induced genes (Appendix Table S1) encode stress response proteins, including HO‐1, NQO1, GCLM, and SLC7A11, whereas several repressed genes (Appendix Table S2) are inflammatory pathway components, such as IL‐8 and Cox‐2. We verified the induction and repression of selected genes by real‐time RT–PCR (Fig EV1B).
Figure 1. OGT inhibition and NRF2 activation induce similar gene expression patterns.
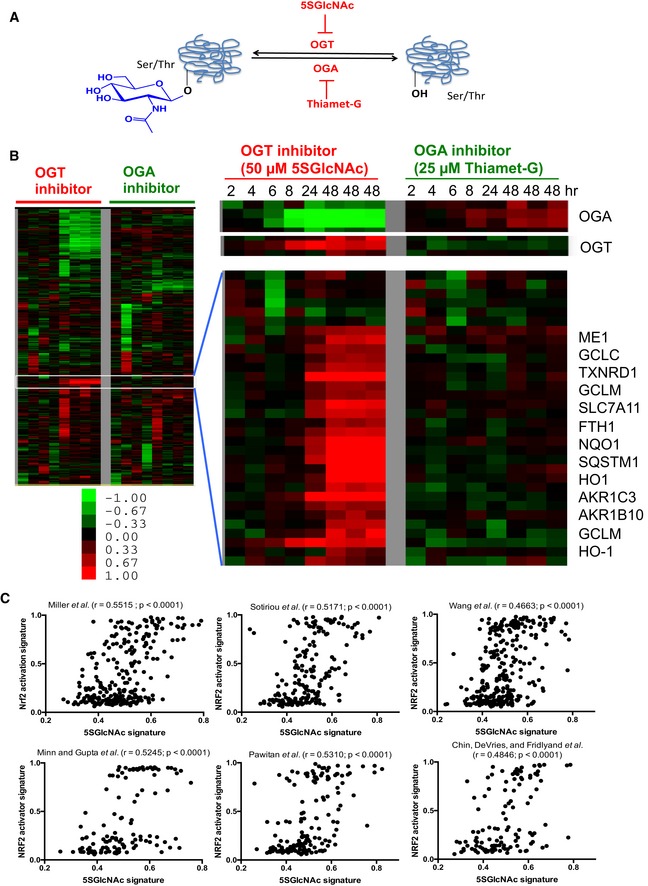
- O‐GlcNAc is added onto serine (shown) or threonine residues of intracellular proteins by the glycosyltransferase OGT and removed by the glycoside hydrolase OGA. OGT and OGA are inhibited by the specific small molecules 5SGlcNAc and Thiamet‐G, respectively.
- Heatmap of the global transcriptional response of MDA‐MB‐231 cells to the inhibition of OGT or OGA for the indicated time points. Heatmap of selected induced and repressed genes and gene clusters during OGT and OGA inhibitions are shown with several indicated gene names. The color bar indicates log2 (fold change); green: decreased expression (i.e., repression); red: increased expression (i.e., induction).
- The gene expression signature of low OGT activity (i.e., 5SGlcNAc) shows positive correlation with the NRF2 activation signature when projected into six different tumor datasets. r is the Pearson's correlation coefficient. The P‐values (two‐tailed) were derived from the Pearson's correlation coefficient as performed by GraphPad Prism 5.0.
Figure EV1. Transcriptional response to inhibitors of OGT and OGA (related to Fig 1).
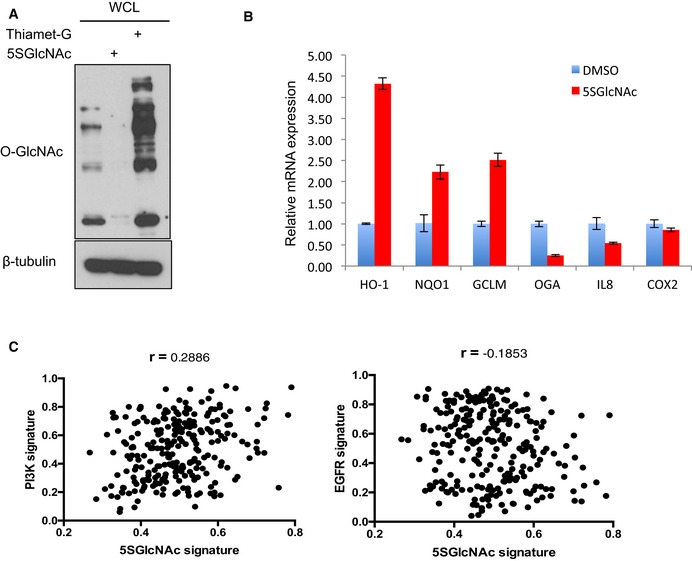
- OGT and OGA inhibitors reduced and increased, respectively, global O‐GlcNAc levels. MDA‐MB‐231 cells were treated with 50 μM 5SGlcNAc or 25 μM Thiamet‐G for 24 h and WCLs were analyzed by Western blots.
- qPCR validation of indicated up‐ and downregulated genes from microarray data. MDA‐MB‐231 cells were treated with DMSO vehicle or 50 μM 5SGlcNAc for 48 h and extracted mRNA was analyzed by qPCR (normalized to β‐actin). n = 3, error bars represent standard deviation.
- Lack of correlation between low‐OGT activity (i.e., 5SGlcNAc) and PI3K or EGFR signaling gene expression signatures when projected into the Miller breast tumor dataset. r = Pearson's correlation coefficient.
Source data are available online for this figure.
To elucidate the mechanistic basis of these transcriptional changes, we used tumor gene expression data to examine the in vivo relationship between the OGT inhibition response and other known signaling pathways. We defined a gene signature of low OGT activity (i.e., 5SGlcNAc treatment) based on our data and projected it into six different human breast tumor gene expression datasets (Sotiriou et al, 2003; Miller et al, 2005; Minn et al, 2005; Pawitan et al, 2005; Wang et al, 2005; Chin et al, 2006). We then examined several previously identified gene signatures of different signaling pathways and determined their correlations with the low‐OGT activity signature in the same datasets (Gatza et al, 2011). Interestingly, among all signaling pathways tested, the strongest correlation existed between the low‐OGT activity signature and NRF2 activation [i.e., treatment with sulforaphane (SFN) or tert‐butylhydroquinone (tBHQ); Pearson's correlation coefficient r ranges from 0.47 to 0.55 for six different datasets; P < 0.0001; Fig 1C], with weaker correlation with other pathways, including PI3K (r = 0.2886, Miller et al dataset as an example) and EGFR (r = −0.1853, Miller et al dataset as an example; Fig EV1C). Therefore, human tumors with the low‐OGT activity gene signature tend to have strong NRF2 activation signatures, suggesting an unexpected in vivo connection between these two pathways.
OGT inhibition activates a NRF2‐dependent transcriptional program
Because of the strong in vivo correlation between low OGT activity and NRF2 induction in human tumors, and because most of the 5SGlcNAc‐induced genes in our dataset are known NRF2 targets (Fig 1B), we tested whether OGT inhibition activates NRF2. Indeed, Western blots confirmed that 5SGlcNAc triggered the accumulation and nuclear translocation of NRF2 (Fig 2A). 5SGlcNAc also increased NRF2 transactivation in a well‐characterized NQO1‐ARE–luciferase reporter assay (Moehlenkamp & Johnson, 1999; Sangokoya et al, 2010; Appendix Fig S1A). In addition, live cell imaging using H1299 lung cancer cells tagged with YFP at the endogenous NQO1 locus (Sigal et al, 2006; Cohen et al, 2009) demonstrated increasing NRF2 expression and activity throughout 5SGlcNAc treatment (Fig 2B and Appendix Fig S1B). However, 5SGlcNAc did not induce the mRNA of NRF2 itself, or of the related transcription factors NFE2, NRF1, or NRF3 (Appendix Fig S1C). We concluded that OGT inhibition by 5SGlcNAc robustly activates NRF2 protein and activity through a post‐transcriptional mechanism.
Figure 2. OGT inhibition induces a NRF2‐dependent antioxidant response without causing oxidative stress.
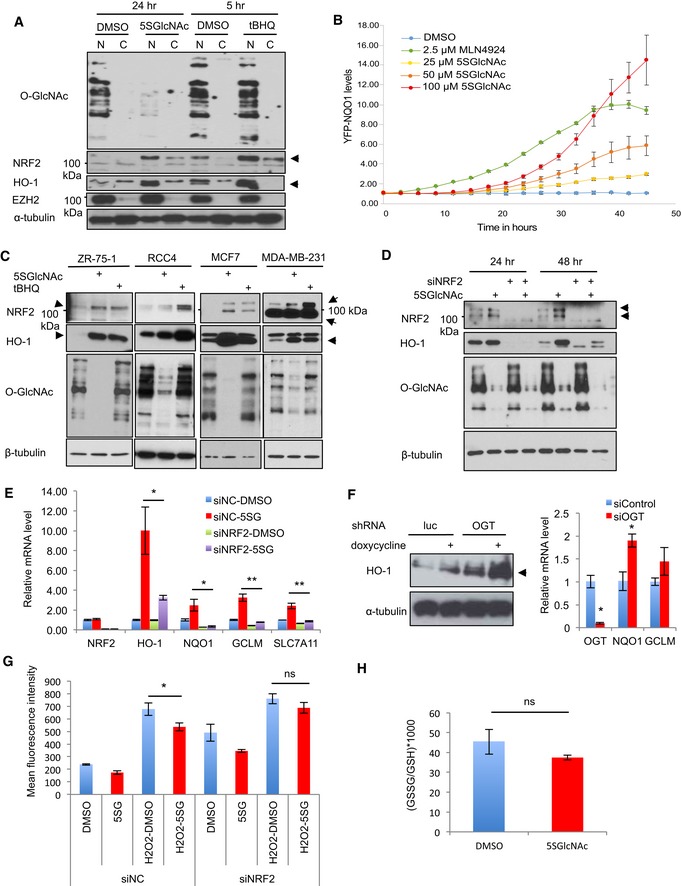
- 5SGlcNAc increased the nuclear level of NRF2 and HO‐1 proteins, similar to the known NRF2 activator tBHQ. MDA‐MB‐231 cells were treated with 50 μM 5SGlcNAc for 24 h or 25 μM tBHQ for 5 h. Lysates were separated into cytosolic and nuclear fractions for Western blot by indicated antibodies. EZH2 and α‐tubulin are loading controls for nuclear and cytosolic fractions, respectively. Arrows indicate the protein of interest.
- 5SGlcNAc treatment increased the levels of YFP‐NQO1. H1299 cells expressing YFP‐NQO1 were treated with 25, 50, or 100 μM 5SGlcNAc or 2.5 μM MLN4924 (NRF2 activator, positive control) and analyzed in real time for YFP levels using the IncuCyte platform. Data are from one experiment with five technical replicates and are presented as mean ± SD. Data were reproduced in a biological replicate with five technical replicates.
- 5SGlcNAc increased NRF2 and HO‐1 protein levels and reduced global O‐GlcNAcylation in ZR‐75‐1, RCC4, MCF7, and MDA‐MB‐231 cells. Indicated cell lines were treated with 50 μM 5SGlcNAc or tBHQ (50 μM for ZR‐75‐1 and RCC4; 25 μM for MCF7 and MDA‐MB‐231) for 48 h and harvested for Western blot analysis by indicated antibodies. Arrows indicate the induced protein of interest.
- Silencing NRF2 by siRNAs abolishes 5SGlcNAc‐triggered HO‐1 induction. MCF7 cells were treated with non‐targeting control siRNA or siNRF2 for 24 h and incubated with 50 μM 5SGlcNAc for additional 24 or 48 h before harvesting and Western blotting. Arrows indicate the protein of interest.
- MDA‐MB‐231 cells were transfected with either control (siNC) or siNRF2 siRNAs for 24 h and then treated with 50 μM 5SGlcNAc for additional 48 h. Relative gene expression levels were assessed by quantitative PCR (qPCR) and normalized to β‐actin mRNA and the DMSO/siNC group. n = 3, error bars represent standard deviations.
- Left panel: The level of HO‐1 protein in MDA‐MB‐231 before and after doxycycline‐induced shRNA‐mediated silencing of luciferase (control) or OGT. Arrows indicate the protein of interest. Right panel: OGT depletion by siRNA increases NQO1 and GCLM mRNA. qPCR measurement of the indicated mRNAs in MDA‐MB‐231 transfected with control or OGT‐targeting siRNA for 24 h is shown (normalized to β‐actin mRNA). n = 3, error bars represent standard deviations.
- MDA‐MB‐231 cells were transfected with either siNC or siNRF2 siRNAs for 24 h and then treated with 87.5 μM 5SGlcNAc (5SG, 48 h) or H2O2 (882 μM, 1 h). Then, cells were incubated with H2‐DCFDA dye and relative ROS levels were measured by flow cytometry. n = 3, error bars represent standard deviations.
- MDA‐MB‐231 cells were treated with DMSO or 50 μM 5SGlcNAc (48 h), and GSSG/GSH ratios were measured by MS. n = 3, error bars represent standard deviations.
We confirmed that 5SGlcNAc increased both endogenous NRF2 protein, which migrates at ~100 kDa in an SDS–PAGE gel (Lau et al, 2013), and its transcriptional target HO‐1 in ZR‐75‐1, RCC4, MCF7, and MDA‐MB‐231 cells (Fig 2C), as did the known NRF2 activator tBHQ (Li et al, 2005). This result indicates that the OGT/NRF2 connection is conserved across cells from diverse tissue types. The mRNAs of several NRF2 target genes were also induced by 5SGlcNAc in RCC4, ZR‐75‐1, and BT‐474 cells (Appendix Fig S1D–F). Furthermore, NRF2 is required for this transcriptional response to OGT inhibition, because silencing NRF2 by siRNA greatly reduced HO‐1 protein induction by 5SGlcNAc, without altering its effect on O‐GlcNAcylation (Fig 2D). Similarly, NRF2 silencing significantly reduced the 5SGlcNAc induction of HO‐1, NQO1, GCLM, and SLC7A11 mRNA (Fig 2E). In contrast, silencing the related transcription factor NRF1 only affected SLC7A11 induction by 5SGlcNAc (Appendix Fig S1G). Collectively, these results indicate that OGT inhibition specifically activates a NRF2‐dependent transcriptional program in diverse cell types.
OGT inhibition activates NRF2 through a specific signaling event
We next tested whether NRF2 pathway induction by OGT inhibition was due to a specific signaling event, versus off‐target effects or non‐specific stress. First, we generated MDA‐MB‐231 cells with inducible shRNAs that target either luciferase or OGT. We found that doxycycline‐induced silencing of OGT robustly upregulated HO‐1 (Fig 2F, left panel). In addition, we silenced OGT in MDA‐MB‐231 cells using siRNA targeting a different sequence and observed an induction of the NRF2 targets NQO1 and GCLM (Fig 2F, right panel). These results show that the genetic silencing of OGT activates NRF2, phenocopying our 5SGlcNAc results and arguing against off‐target effects of the small molecule. In addition, 5SGlcNAc did not increase intracellular reactive oxygen species (ROS; Fig 2G) or the ratio of oxidized/reduced glutathione (Fig 2H and Appendix Fig S1H). In fact, 5SGlcNAc decreased ROS levels in hydrogen peroxide‐treated cells, and this effect was abolished when NRF2 was silenced (Fig 2G). These data demonstrate that OGT inhibition induces a NRF2‐dependent antioxidant response but does not increase ROS, arguing that OGT inhibition does not trigger NRF2 activation by causing non‐specific stress.
To further confirm this hypothesis, we asked whether the link between O‐GlcNAc and NRF2 could be exploited to produce a predicted result in a cellular stress response. Several studies have shown that certain breast and renal cancer cell lines are cystine‐addicted and undergo non‐apoptotic death due to oxidative stress upon cystine deprivation (Timmerman et al, 2013; Tang et al, 2016a,b). Consistent with NRF2 induction and reduced oxidative stress, OGT inhibition rescued MDA‐MB‐231 cells from cystine‐deprived death (Appendix Fig S1I). Taken together, these data strongly suggest that OGT inhibition triggers NRF2 signaling through a specific O‐GlcNAc‐mediated signaling event, and not an off‐target effect or non‐specific oxidative stress. We concluded that O‐GlcNAcylation of a particular substrate or substrates is required to restrain the NRF2 pathway in unstressed cells.
OGT activity is required for optimal NRF2 ubiquitination
Since OGT inhibition did not affect NRF2 mRNA levels (Fig 2E and Appendix Fig S1C), we investigated how NRF2 protein is regulated by O‐GlcNAcylation. First, we blocked translation elongation with cycloheximide (Schneider‐Poetsch et al, 2010), which, as expected, depleted the majority of NRF2 protein because of its short half‐life (Stewart et al, 2003; Furukawa & Xiong, 2005; Fig 3A, lane 3). However, even in the presence of cycloheximide, we still observed detectable NRF2 induction after a short period of OGT inhibition (Fig 3A, lane 4). Similarly, rapamycin, an inhibitor of cap‐dependent translation initiation (Beretta et al, 1996), also failed to suppress 5SGlcNAc‐mediated NRF2 accumulation (Fig EV2A, lanes 1 and 2). These results indicate that OGT inhibition does not induce NRF2 protein through increased translation. Next, we tested the role of proteasome‐mediated degradation, thought to be the major mode of NRF2 regulation in most contexts (Kensler et al, 2007; Kansanen et al, 2013). Interestingly, we found that OGT inhibition reduced the amount of polyubiquitinated NRF2 in proteasome‐inhibited cells (Figs 3B and EV2A and B) without affecting global ubiquitination (Fig EV2C). In summary, OGT inhibition reduced NRF2 ubiquitination and led to NRF2 protein accumulation. These observations suggested that O‐GlcNAcylation might somehow promote the activity of an E3 ligase complex that ubiquitinates NRF2, and that OGT inhibition could disrupt this regulation to trigger NRF2 accumulation and activity.
Figure 3. O‐GlcNAcylation regulates NRF2 post‐translationally.
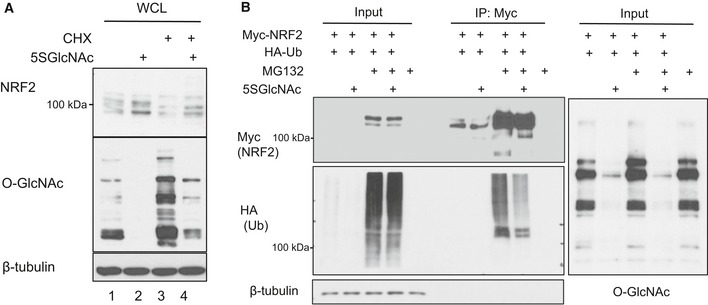
- MCF7 cells were treated with cycloheximide (CHX, 25 μg/ml) for 1 h to block translation and then 5SGlcNAc (50 μM) for additional 10 h. Whole‐cell lysates (WCLs) were analyzed for NRF2 protein and global O‐GlcNAcylation by Western blot.
- Polyubiquitination of NRF2 is decreased upon OGT inhibition. 293T cells were transfected with MYC‐NRF2 and/or HA‐ubiquitin (Ub) as indicated for 24 h and then treated with vehicle or 50 μM 5SGlcNAc for additional 48 h, with or without MG132 (10 μM, 3 h). Lysates were analyzed by IP and Western blot.
Figure EV2. O‐GlcNAcylation regulates NRF2 post‐translationally (related to Fig 3).
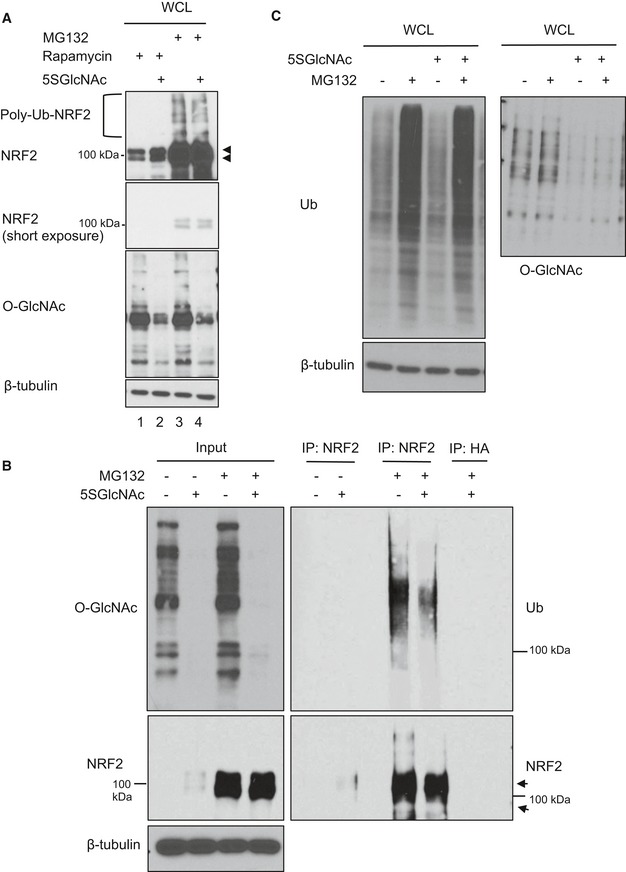
- MCF7 cells were treated with rapamycin (50 nM) for 1 h and then 5SGlcNAc (50 μM) for additional 8 h. Then, MG132 (10 μM) was added 2 h before harvesting (total experimental time: 9 h). WCLs were analyzed by Western blot. Arrows indicate NRF2.
- 5SGlcNAc reduced the ubiquitination of endogenous NRF2 in MCF7 cells. MCF7 cells were treated with 50 μM 5SGlcNAc (48 h) and 10 μM MG132 (6 h), and WCLs were analyzed by IP/Western blot. Arrows indicate NRF2.
- 5SGlcNAc does not affect global poly ubiquitination levels. MDA‐MB‐231 cells were treated with 25 μM 5SGlcNAc for 45 h and 10 μM MG132 for additional 3 h. WCLs were analyzed by Western blots.
Source data are available online for this figure.
KEAP1 is O‐GlcNAcylated by OGT
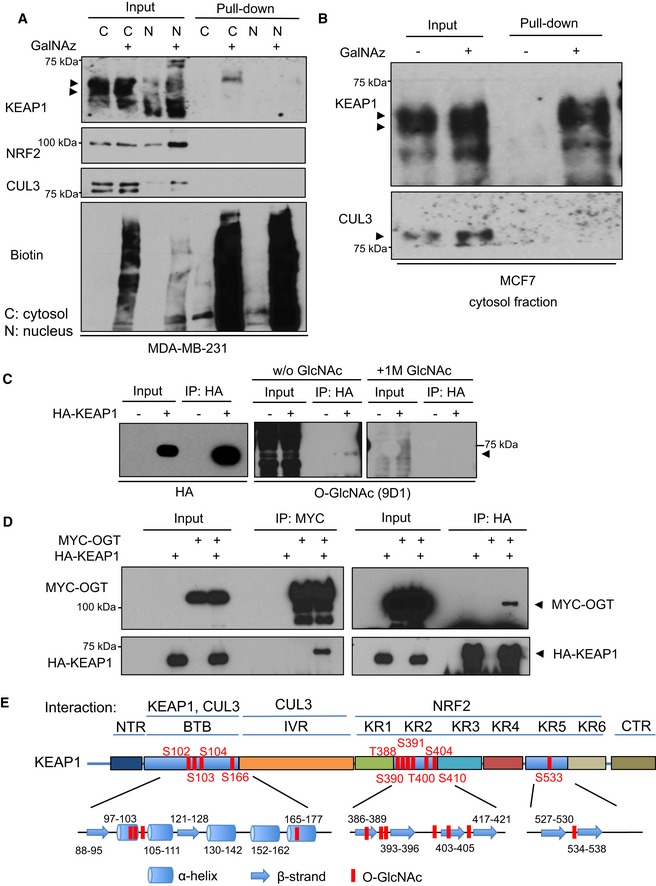
We next used a chemical biology approach that we developed previously (Boyce et al, 2011; Palaniappan et al, 2013) to identify O‐glycosylated proteins that might regulate NRF2 ubiquitination. MDA‐MB‐231 cells were incubated with peracetylated N‐azidoacetylgalactosamine (GalNAz; Boyce et al, 2011; Palaniappan et al, 2013). GalNAz is metabolized to a UDP‐GlcNAc nucleotide‐sugar analog and used by endogenous OGT to modify its native substrates with an azidosugar (Boyce et al, 2011; Palaniappan et al, 2013). Next, we used copper‐catalyzed azide‐alkyne cycloaddition (CuAAC, or “click chemistry”) to chemoselectively ligate a biotin probe to endogenous OGT substrates and then capture them by streptavidin affinity chromatography (Boyce et al, 2011; Palaniappan et al, 2013). The eluted proteins were separated by SDS–PAGE and probed with antibodies against known NRF2 regulators. Interestingly, we found that GalNAz labeled endogenous KEAP1 in MDA‐MB‐231 (Fig 4A) and MCF7 (Fig 4B), but not CUL3 or NRF2 itself.
Figure 4. KEAP1 is O‐GlcNAcylated by OGT at multiple residues.
-
A, BMDA‐MB‐231 cells (A) or MCF7 cells (B) were incubated with DMSO or 100 μM GalNAz for 24 h and then separated into cytosolic (C) and nuclear (N) fractions. Fractions were subjected to a CuAAC reaction with an alkyne‐biotin probe and precipitated to remove unreacted probe. Then, biotin‐labeled proteins were affinity‐purified via NeutrAvidin bead pull‐down and analyzed by Western blot. Arrows indicate the protein of interest.
-
CGlcNAc competition assay. 293T cells transfected with HA‐KEAP1 were lysed and analyzed by IP and Western blot. GlcNAc competition (a specificity control) was performed by pre‐incubating the O‐GlcNAc antibody with 1 M free GlcNAc before blotting. Blots were performed simultaneously with equal sample loading and chemiluminescence exposure time. Arrows indicate the protein of interest.
-
DOGT interacts with KEAP1 in human cells. 293T cells were transfected with MYC‐OGT and/or HA‐KEAP1 constructs as indicated for 48 h, and lysates were analyzed by IP/Western blot. Arrows indicate the protein of interest.
-
EEleven candidate O‐GlcNAcylated residues of KEAP1, identified by MS, are shown in the context of protein domains, secondary structure elements, and selected known interacting partners of KEAP1. NTR, N‐terminal region; BTB, broad complex/tramtrack/bric‐à‐brac domain; IVR, intervening region; KR, Kelch repeat domain; CTR, C‐terminal region.
These experiments suggested that OGT may restrain NRF2 through glycosylation of KEAP1, the major adaptor for the CUL3‐containing E3 ligase complex that regulates NRF2 protein stability (Cullinan et al, 2004; Kobayashi et al, 2004; Zhang et al, 2004; Furukawa & Xiong, 2005). Consistent with this hypothesis, we found that recognition of KEAP1 by an anti‐O‐GlcNAc monoclonal antibody was reduced by either treating cells with 5SGlcNAc (Fig EV3A) or GlcNAc competition (Fig 4C; Sakabe et al, 2010). Furthermore, KEAP1 co‐immunoprecipitated (co‐IP‐ed) with OGT (Figs 4D and EV3B), and this interaction was independent of the KEAP1 broad complex/tramtrack/bric‐à‐brac (BTB) domain (Fig EV3C). To confirm that the KEAP1/OGT interaction is direct, we purified recombinant GST‐KEAP1 and His‐OGT from Escherichia coli and performed in vitro protein–protein interaction assays. We did not observe a direct KEAP1/OGT interaction in this in vitro context (Fig EV3D), likely because additional proteins or PTMs are required for the observed in vivo interaction. Together, these data indicate that KEAP1 is O‐GlcNAcylated by OGT.
Figure EV3. KEAP1 is O‐GlcNAcylated and interacts with OGT in human cells (related to Fig 4).
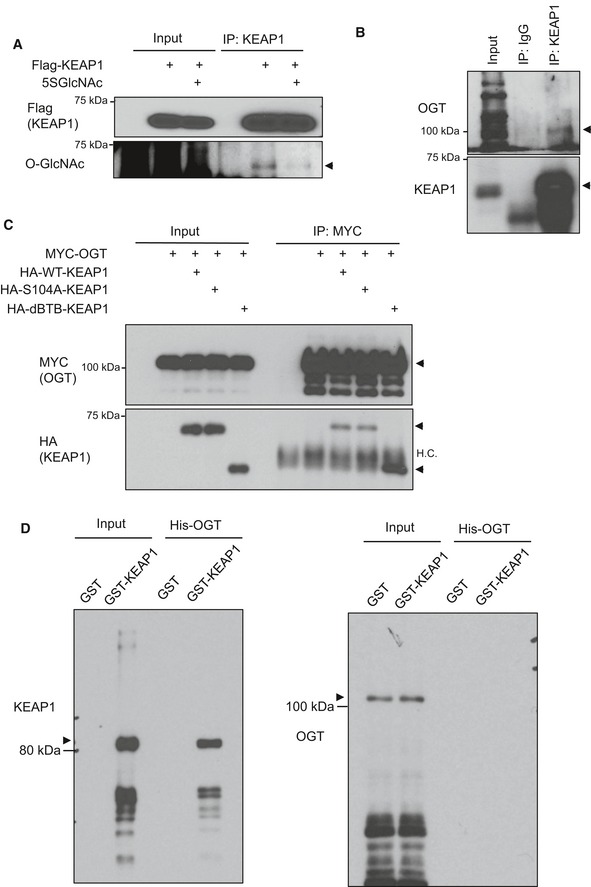
- O‐GlcNAcylation of KEAP1 is reduced upon 5SGlcNAc treatment. H838 cells were transfected with Flag‐KEAP1 and then treated with DMSO or 50 μM 5SGlcNAc for 48 h. WCLs were analyzed by IP/Western blot. Arrow indicates KEAP1.
- KEAP1 associates with OGT in human cells. Endogenous KEAP1 was IP‐ed from 293T lysates and blotted with antibodies against OGT and KEAP1 (arrows).
- Interaction between OGT and KEAP1 mutants. The indicated KEAP1 constructs and/or MYC‐OGT were transfected into 293T cells (48 h), and WCLs were analyzed by IP/Western blot. Arrows indicate the proteins of interest. HC: IP‐ing antibody heavy chain.
- Purified GST‐KEAP1 and His‐OGT do not interact in vitro. Recombinant GST‐KEAP1 and His‐OGT were purified individually from Escherichia coli, mixed, and analyzed by glutathione agarose pull‐down and Western blot. Arrows indicate the proteins of interest.
Source data are available online for this figure.
To elucidate the functional effects of KEAP1 glycosylation, we purified recombinant human KEAP1 from 293T cells because they transfect efficiently, allowing us to generate the required amount of material for site‐mapping experiments. Mass spectrometry (MS)‐based proteomic analysis of the purified KEAP1 identified 11 candidate O‐GlcNAcylation sites (Fig 4E). Four putative O‐GlcNAc sites (S102, S103, S104, and S166; Appendix Fig S2) lie within the α‐helices (The UniProt, 2017) of the BTB domain, which is required for KEAP1 homodimerization and CUL3 interaction (Zipper & Mulcahy, 2002; Zhang et al, 2004; Furukawa & Xiong, 2005). Six additional potential O‐GlcNAc sites (T388, S390, S391, T400, S404, and S410; Appendix Fig S2) are in the β‐strands of the second Kelch motif, while the final candidate site (S533; Appendix Fig S2) is in the fifth Kelch motif. The KEAP1 Kelch motifs interact with NRF2 to regulate NRF2 levels (Itoh et al, 1999; Kobayashi et al, 2002; Fig 4E). Taken together, these results suggest that OGT interacts with and constitutively glycosylates KEAP1 under unstressed conditions and that inhibition of OGT reduces KEAP1 O‐GlcNAcylation.
KEAP1 is required for O‐GlcNAc‐mediated NRF2 regulation and erastin‐induced cell death
To test the requirement for KEAP1 in the regulation of NRF2 by O‐GlcNAcylation, we took advantage of A549 and H838 cells, which harbor KEAP1 loss‐of‐function mutations (G333C and frameshift non‐sense mutation, respectively; Singh et al, 2006). These mutations make A549 and H838 cells appropriate KEAP1‐null systems for testing the functional role of expressed KEAP1 mutants, without confounding effects from endogenous KEAP1. 5SGlcNAc failed to induce NRF2 target genes in A549 and H838 cells, demonstrating that functional KEAP1 is required for NRF2 activation by OGT inhibition (Appendix Fig S3A and B). Next, we created unglycosylatable mutations in each of the 11 putative KEAP1 O‐GlcNAc sites by converting serine or threonine to alanine, and used H838 and A549 cells to test whether any individual unglycosylatable KEAP1 mutant lost its ability to destabilize NRF2 and repress NQO1‐ARE‐reporter activity. First, we confirmed that the expression levels of the KEAP1 mutants were comparable (Appendix Fig S3C and E). Then, we verified that wild‐type KEAP1 expression reduced NRF2 protein level (Appendix S3C) and NQO1‐ARE‐reporter activity (~90%) in both cell lines (Appendix Fig S3D and F). Among all point mutants tested, only S104A consistently exhibited reduced capacity to destabilize NRF2 or to repress the ARE‐reporter (Appendix Fig S3C–F). Consistent with this result, the S104A KEAP1 mutant, when compared to wild‐type KEAP1, also exhibited a significantly reduced ability to repress NRF2 protein levels in a proteasome‐dependent manner (Appendix Fig S3G).
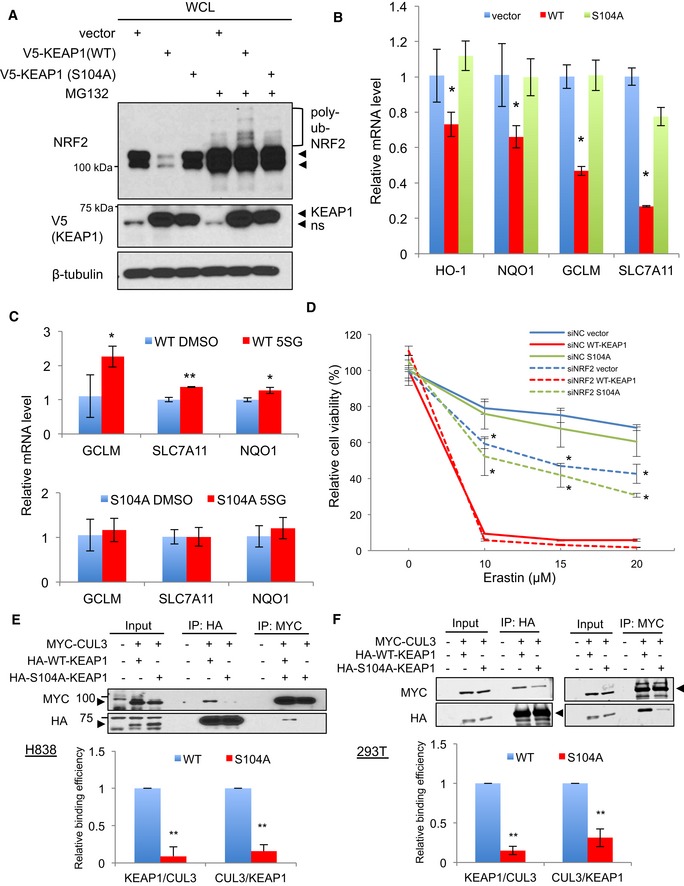
The KEAP1 S104A mutant also failed to repress NRF2 protein in stably transduced H838 cells (Fig 5A). In addition, the polyubiquitination of NRF2 in KEAP1 S104A‐expressing cells was similar to that of vector control cells, but significantly lower than that of cells expressing wild‐type KEAP1 (Fig 5A). Furthermore, we found that cells stably expressing the KEAP1 S104A mutant were unable to repress endogenous NRF2 target gene expression (Fig 5B). Next, we asked whether KEAP1 S104 is required for the induction of NRF2 target genes by 5SGlcNAc. We examined the expression of GCLM, SLC7A11, NQO1, and HO‐1 mRNA in wild‐type or S104A KEAP1‐reconstituted H838 cells after treatment with 5SGlcNAc. KEAP1 wild‐type‐expressing cells (Fig 5C, upper panel) but not S104A‐expressing cells (Fig 5C, lower panel) showed significant induction by 5SGlcNAc of most NRF2 target genes. HO‐1 mRNA was also induced by 5SGlcNAc in wild‐type KEAP1‐expressing cells but did not reach statistical significance at 95% confidence interval (P = 0.059, data not shown). Together, these data indicate that the induction of NRF2 signaling by 5SGlcNAc is due to reduced O‐GlcNAcylation of KEAP1 S104.
Figure 5. O‐GlcNAcylation of KEAP1 S104 regulates the KEAP1–CUL3 interaction and NRF2 activity.
-
AThe KEAP1 S104A mutant has reduced ability to target NRF2 for proteasome‐dependent destruction. WCLs from H838 cells stably expressing vector, wild‐type KEAP1, or the KEAP1 S104A mutant were treated with vehicle or 10 μM MG132 for 3 h, and lysates were analyzed by Western blot. ns, non‐specific band. The arrowheads indicate NRF2 protein doublet in the NRF2 blot. In the V5 blot, the upper arrowhead is KEAP1 protein, the lower arrowhead is a non‐specific band (ns).
-
BKEAP1 S104 is required to suppress NRF2 transcriptional activity. RNA collected from stable H838 cells was analyzed by qPCR (normalized to β‐actin mRNA). n = 3, error bars represent standard deviations; P‐values were calculated by Student's t‐test; *P < 0.05.
-
CReconstitution of H838 cells with wild‐type KEAP1, but not S104A KEAP1, restores induction of NRF2 targets by 5SGlcNAc. Stable H838 cells were treated with 50 μM 5SGlcNAc for 48 h, and mRNAs were analyzed by qPCR (normalized to β‐actin mRNA) and compared to DMSO treatment. n = 3 for wild type; n = 7 for S104A. Error bars represent standard deviations; P‐values were calculated by Student's t‐test, *P < 0.05, **P < 0.001.
-
DWild‐type, but not S104A, KEAP1 sensitizes H838 cells to erastin‐induced cell death in a NRF2‐dependent manner. Stable H838 cells were transfected with either siNC (non‐targeting control) or siNRF2 for 24 h and then treated with erastin as indicated for additional 48 h. Cell viability was measured by CellTiter‐Glo assay and normalized to the siNC/DMSO control of each stable cell line. n = 3, P‐values were calculated by Student's t‐test (siNC vector versus siNRF2 vector or siNC S104A versus siNRF2 S104A), *P < 0.05.
-
E, FThe KEAP1 S104A mutant exhibits reduced interaction with CUL3. H838 (E) or 293T (F) cells were transfected with the indicated constructs for 32 or 48 h, respectively, and lysates were analyzed by IP/fluorescent Western blot. Quantification of the KEAP1–CUL3 interaction was performed by the ImageJ (E) or LI‐COR (F) software platform and is given in the lower panels. n = 3, error bars represent standard deviation; P‐values were calculated by Student's t‐test, **P < 0.001.
In non‐small‐cell lung cancer, somatic mutations in KEAP1 often result in defective KEAP1 function and constitutive activation of NRF2 (Padmanabhan et al, 2006; Hast et al, 2014), promoting tumor growth and resistance to therapies (Singh et al, 2006). For example, NRF2 induction confers resistance to erastin‐induced cell death (Yagoda et al, 2007; Dixon et al, 2012; Sun et al, 2016). Our model predicts that KEAP1 S104A‐expressing cells would be resistant to erastin, relative to wild‐type KEAP1‐expressing cells, due to constitutively higher NRF2 activation. Indeed, we found that S104A stable cells were resistant to erastin, consistent with their higher NRF2 pathway activation (Fig 5D). In contrast, cells stably expressing wild‐type KEAP1 are more susceptible to erastin (Fig 5D). Importantly, depletion of NRF2 by siRNA significantly sensitized the vector control and S104A‐expressing cells to erastin, as determined by two orthogonal cell viability assays (Fig 5D and Appendix Fig S3H). These results indicate that S104A KEAP1, unlike wild‐type, cannot fully repress NRF2 or sensitize H838 to erastin‐mediated cell death. Together, our data indicate that O‐GlcNAcylation of KEAP1 at S104 is required for its ability to mediate NRF2 ubiquitination and proteasomal destruction, and unglycosylatable S104A mutant KEAP1 confers NRF2‐dependent resistance to erastin‐induced cell death.
O‐GlcNAcylation of KEAP1 promotes its productive interaction with CUL3
Because O‐GlcNAcylation regulates NRF2 ubiquitination (Figs 3 and EV2A and B), we hypothesized that O‐GlcNAcylation of KEAP1 might affect the interactions between components of the NRF2–KEAP1–CUL3 E3 ligase complex. We found that neither OGT nor OGA inhibition affected the interaction of NRF2 with KEAP1 (Appendix Fig S3I). In contrast, OGT inhibition markedly reduced the interaction between CUL3 and endogenous (Fig EV4A) or transfected (Fig EV4B) KEAP1. Furthermore, we observed that the KEAP1 S104A mutant exhibited a > 70% reduction in its interaction with CUL3 in H838 and 293T cells (Fig 5E and F). Based on recent structural and modeling experiments (PDB: 5NLB), S104 likely does not participate directly in KEAP1 dimerization in KEAP1–CUL3 complexes (Fig EV5A). However, prior studies are conflicting about whether S104 is required for KEAP1 dimerization (Zipper & Mulcahy, 2002; McMahon et al, 2006). We tested the effects of OGT inhibition and S104 mutation on KEAP1 dimerization and found that neither OGT nor OGA inhibition affected KEAP1 dimerization (Appendix Fig S3J). Furthermore, the S104A KEAP1 mutant retained its ability to interact with wild‐type KEAP1 (Appendix Fig S3K). These results indicate that S104 glycosylation is not required for KEAP1 dimerization.
Figure EV4. O‐GlcNAcylation of KEAP1 regulates CUL3 interaction (related to Fig 5).
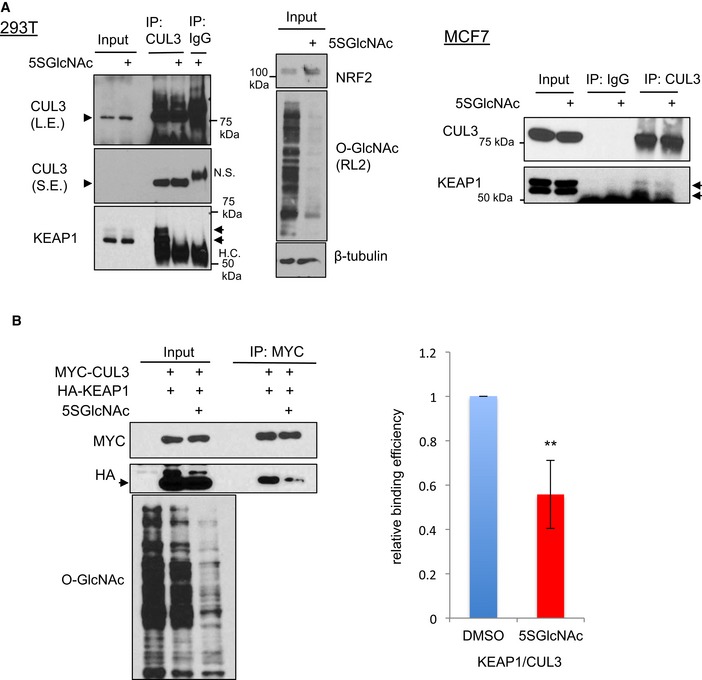
- 5SGlcNAc reduces the interaction between endogenous KEAP1 and CUL3 in 293T and MCF7 cells. 293T (left) or MCF7 (right) cells were treated with 50 μM 5SGlcNAc (24 h), and WCLs were analyzed by IP/Western blot. Arrowhead indicates CUL3, arrow indicates KEAP1.
- 5SGlcNAc reduces the interaction between expressed CUL3 and KEAP1. 293T cells were transfected as indicated for 24 h and then treated with 50 μM 5SGlcNAc for additional 48 h. WCLs were analyzed by IP/Western blot. n = 3, error bars represent standard deviation, and P‐values were calculated by Student's t‐test, **P < 0.01. Arrow indicates HA‐tagged KEAP1.
Source data are available online for this figure.
Figure EV5. The KEAP1 S104 glycosylation site is broadly conserved across the KLHL family.
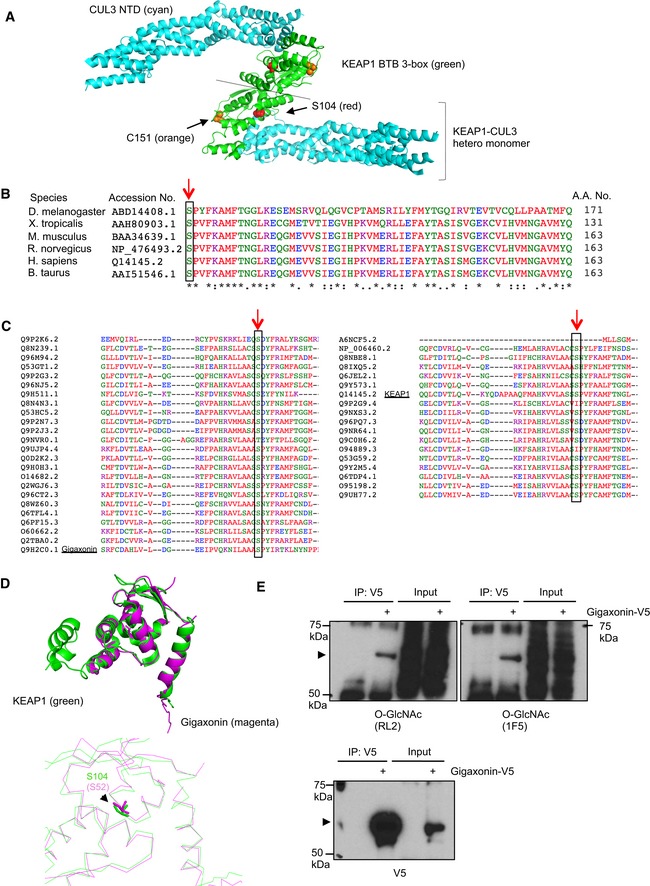
- Modeling of the KEAP1 BTB 3‐box (green) and CUL3 (cyan) NTD hetero‐tetramer complex, based on the recently reported KEAP1–CUL3 heterodimer complex (PDB: 5NLB). S104 of KEAP1 is shown in red. C151 of KEAP1 is shown in orange.
- Amino acid sequence alignment of KEAP1 orthologs in different species reveals that S104 (red arrow) is highly conserved across evolution.
- S104 of KEAP1 is conserved in most human KLHL family proteins. Amino acid sequence alignment of 42 human KLHL proteins. The cognate residues of KEAP1 S104 are indicated by red arrows and black rectangles.
- Structural alignments of KEAP1 (green, PDB: 5NLB) and gigaxonin (magenta, PDB: 2PPI) BTB domains. KEAP1 S104 and gigaxonin S52 are nearly superimposable (bottom panel).
- Gigaxonin‐V5 from stably transduced 293T cells was analyzed by IP/Western blot. Arrowheads indicate V5‐tagged gigaxonin.
Source data are available online for this figure.
The recently reported structure of the KEAP1 BTB–CUL3 complex (PDB: 5NLB) suggests that S104 does not directly contact CUL3 (Fig EV5A). However, the addition of a bulky O‐GlcNAc moiety at S104 may induce a local conformational change in the BTB domain that reduces CUL3 binding and/or NRF2 ubiquitination. Indeed, a potentially analogous form of regulation has been reported at the KEAP1 C151 residue in the BTB domain: C151 is also distant from the KEAP1/CUL3 and KEAP1/KEAP1 interaction interfaces (PDB: 5NLB; Fig EV5A), but modification of C151 by electrophilic small molecules disables KEAP1‐mediated NRF2 ubiquitination (Eggler et al, 2009; Hur et al, 2010). Therefore, modification of residues in the BTB domain, by either glycosylation or electrophilic attack, may induce changes in KEAP1 that impair CUL3‐dependent NRF2 ubiquitination. Based on our data (Figs 5 and EV5A, and Appendix Fig S3J and K) and the available structural information (PDB: 5NLB), we concluded that a major biochemical effect of KEAP1 S104 glycosylation is to promote its productive interaction with CUL3, leading to efficient downstream ubiquitination of NRF2.
Correlation between NRF2, O‐GlcNAc, and glucose levels
To understand the physiological importance of NRF2 regulation by O‐GlcNAcylation, we tested whether NRF2 protein levels are regulated by OGT in response to glucose level fluctuations, since glucose flux through the HBP gives rise to UDP‐GlcNAc (Hart et al, 2011; Bond & Hanover, 2015). First, we confirmed that global O‐GlcNAc levels correlate with glucose concentrations in MCF7 and MDA‐MB‐231 cells (Fig 6A and B), in keeping with the well‐established role for OGT in nutrient sensing (Hart et al, 2011; Bond & Hanover, 2015). Consistent with an earlier report (Cullinan & Diehl, 2004), NRF2 protein accumulated during glucose deprivation (Fig 6A and B). Interestingly, the effects of glucose deprivation on both O‐GlcNAc and NRF2 levels were reversed by the addition of N‐acetylglucosamine (GlcNAc; Fig 6B) or glucosamine (GlcN; Fig 6C), which increase UDP‐GlcNAc levels by bypassing the rate‐limiting step of the HBP (Hart et al, 2011; Bond & Hanover, 2015). Importantly, inhibiting OGA also prevented hypoglycemia‐induced NRF2 upregulation (Fig 6D). Moreover, KEAP1 O‐GlcNAcylation, as recognized by two different monoclonal O‐GlcNAc antibodies, was reduced by glucose deprivation (Fig 6E). Finally, we also observed decreased KEAP1–CUL3 interaction during glucose deprivation (Fig 6F). These data suggest that glucose deprivation reduces KEAP1 glycosylation and decreases the productive KEAP1–CUL3 interaction, leading to NRF2 pathway activation.
Figure 6. KEAP1 O‐GlcNAcylation and NRF2 stability are regulated by glucose levels.
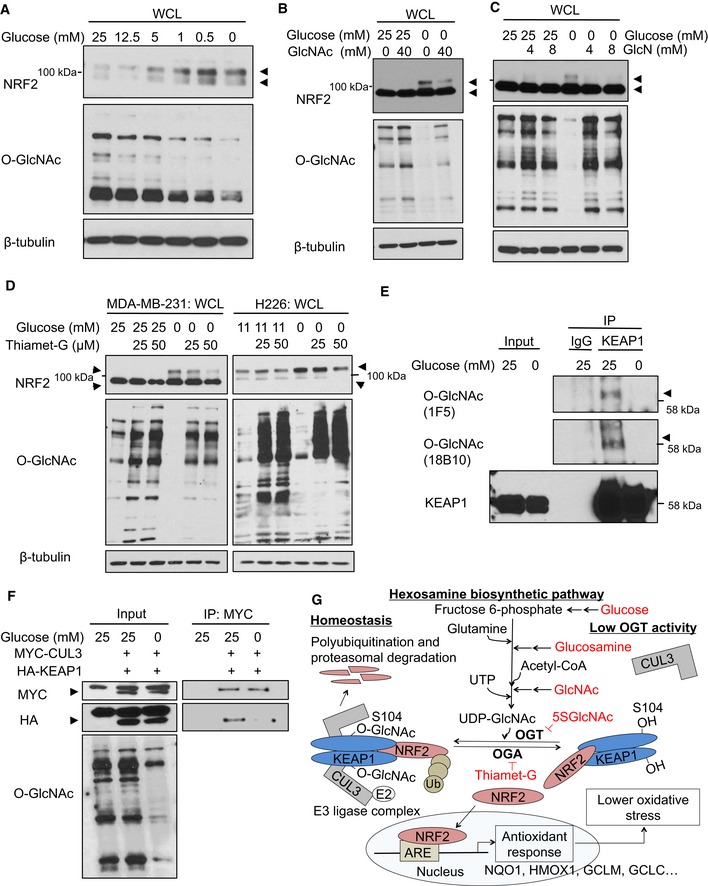
-
ANRF2 protein levels anti‐correlate with both glucose levels and global O‐GlcNAcylation. MCF7 cells were incubated with the indicated glucose concentrations for 12 h, and WCLs were analyzed by Western blot. Arrows indicate NRF2.
-
B, CGlcNAc (B) or GlcN (C) supplementation suppresses NRF2 induction by glucose deprivation. MDA‐MB‐231 cells were incubated with the indicated levels of glucose and GlcNAc (B) or GlcN (C) for 16 h, and WCLs were analyzed by Western blot. Arrows indicate NRF2.
-
DBlocking OGA activity abolishes NRF2 induction by glucose deprivation. MDA‐MB‐231 or H226 cells were treated with Thiamet‐G for 8 or 24 h, respectively, and then subjected to glucose deprivation for additional 16 or 24 h, respectively. WCLs were analyzed by Western blot. Arrows indicate NRF2.
-
EGlucose deprivation reduces KEAP1 O‐GlcNAcylation. MDA‐MB‐231 cells were incubated with 25 or 0 mM glucose for 16 h and lysates were analyzed by IP/Western blot. Arrow indicates O‐GlcNAcylated KEAP1.
-
FGlucose deprivation reduces the KEAP1–CUL3 interaction. MDA‐MB‐231 cells were transfected with HA‐KEAP1 and MYC‐CUL3 for 24 h and then subjected to the indicated glucose concentrations for additional 12 h. Lysates were analyzed by IP/Western blot. Arrow indicates overexpressed MYC‐CUL3 or HA‐KEAP1, respectively.
-
GProposed model, in which nutrient‐sensitive KEAP1 S104 O‐GlcNAcylation promotes its productive interaction with CUL3 to mediate NRF2 ubiquitination and destruction. Please see text for details.
Source data are available online for this figure.
Based on our results, we propose that OGT is required to restrain NRF2 through the O‐GlcNAcylation of KEAP1 on S104, promoting its optimal interaction with CUL3 for efficient NRF2 ubiquitination (Fig 6G). Upon a reduction in global O‐GlcNAcylation—through OGT inhibition, glucose deprivation, or other mechanisms—KEAP1 is deglycosylated, especially at S104, which impairs its interaction with CUL3 and reduces NRF2 ubiquitination and degradation. Accumulated NRF2 protein then translocates into the nucleus to initiate a characteristic antioxidant response (Fig 6G). Because NRF2 can be regulated by glucose‐dependent changes in O‐GlcNAcylation (Fig 6A–E), we propose that OGT is a signaling conduit that links nutrient status to downstream stress responses.
Discussion
We have combined genetic, biochemical, and chemical biology approaches to discover a novel link between OGT and the NRF2 antioxidant pathway via the nutrient‐sensitive O‐GlcNAcylation of KEAP1 S104. This regulation is conserved among disparate human cell lines, suggesting that O‐GlcNAcylation is broadly required to restrain the NRF2 pathway in the absence of oxidative stress. Furthermore, our results indicate that glycosylation of KEAP1 at S104 promotes its productive interaction with CUL3 for optimal polyubiquitination and degradation of NRF2, providing a molecular mechanism for our observations. Among the 11 potential O‐GlcNAcylated sites we identified on KEAP1, S104 is the most functionally relevant in this context, since the unglycosylatable S104A mutant KEAP1 is less able to interact with CUL3 and repress NRF2 activity (Fig 5). However, other KEAP1 glycosylation sites may regulate its activity toward NRF2 or other substrates in response to stimuli or conditions that have not yet been tested. Our work has revealed a new functional connection between O‐GlcNAcylation and NRF2 signaling, pinpointed KEAP1 as an important glycoprotein substrate in this regulation, and elucidated the biochemical effects of O‐GlcNAc in promoting the productive KEAP1–CUL3 interaction and NRF2 ubiquitination and degradation.
KEAP1 O‐GlcNAcylation and ubiquitination of substrates
KEAP1 associates with CUL3 through its BTB domain and part of its intervening region (IVR) domain (Kobayashi et al, 2004; Furukawa & Xiong, 2005), whereas the KEAP1 Kelch motifs interact with substrates (e.g., NRF2; Itoh et al, 1999; Komatsu et al, 2010). We found that the interaction between KEAP1 and CUL3 (but not KEAP1 and NRF2) is affected by O‐GlcNAcylation of KEAP1 at S104 (Fig 5E and F). Therefore, we propose a model in which O‐GlcNAcylation of KEAP S104 is necessary for its productive interaction with CUL3 and the efficient ubiquitination of its substrates, through either enhanced KEAP1–CUL3 binding, optimized KEAP1 conformation, or both (Fig 6G). While NRF2 is the best‐known substrate of the KEAP1–CUL3 complex, several studies have suggested that IKKβ is also a target (Lee et al, 2009; Kim et al, 2010). Thus, it will be interesting to determine in future studies whether IKKβ and/or other KEAP1 substrates also accumulate in response to OGT inhibition. Additionally, while the candidate O‐GlcNAc sites on the KEAP1 Kelch domains did not have significant effects on NRF2 regulation in our experiments (Appendix Fig S3D and F), they may contribute to the binding of other KEAP1/CUL3 substrates (Mulvaney et al, 2016), or may regulate KEAP1 in response to other signals. Future work will focus on the potential functional consequences and downstream effects of individual KEAP1 O‐GlcNAc sites beyond S104.
O‐GlcNAcylation and NRF2 activity
Previous reports demonstrated that oxidative stress modulates cysteine residues of KEAP1, leading to conformational changes and reduced ubiquitination of bound NRF2, as described in the hinge and latch model (Zhang & Hannink, 2003; Wakabayashi et al, 2004; Zhang et al, 2004; Kobayashi et al, 2006). Here, we found that even without oxidative stress, O‐GlcNAcylation of KEAP1 S104 is a novel mechanism of restraining NRF2 signaling (Figs 4 and 5, and Appendix Fig S3). O‐GlcNAcylation is regulated by the availability of glucose and glutamine, which are precursors for the biosynthesis of UDP‐GlcNAc by the HBP (Hart et al, 2011; Bond & Hanover, 2015), and our data demonstrate that NRF2 is induced when global O‐GlcNAcylation is reduced either by OGT inhibition (Figs 1, 2, 3) or by glucose starvation (Fig 6). The fact that we did not observe a reciprocal decrease in NRF2 signaling in the transcriptional response to OGA inhibition (Fig 1B) could be due to constitutively low levels of NRF2 and high levels of KEAP1 O‐GlcNAcylation under homeostasis conditions. This hypothesis is supported by our observation of a robust interaction between KEAP1 and OGT in unstressed cells (Fig 4D).
Since intracellular glucose levels influence global O‐GlcNAc (Fig 6; Hart et al, 2011; Bond & Hanover, 2015), glucose availability may regulate the NRF2‐mediated antioxidant response through the O‐GlcNAcylation of KEAP1 and perhaps other substrates. For instance, although we did not detect evidence of NRF2 or CUL3 O‐GlcNAcylation in our experimental system (Fig 4A and B), it is possible that protein is either glycosylated at an undetectable stoichiometry or is robustly glycosylated under different experimental conditions. In future studies, it will be interesting to determine whether other components of the NRF2 pathway are also OGT substrates. Importantly, however, we showed that the KEAP1 S104A mutation abolishes the NRF2 response to OGT inhibition in our systems (Fig 5B and C), indicating that other OGT substrates are not necessary to explain these effects in this specific context.
Interestingly, recent studies have identified cross‐talk between O‐GlcNAcylation and the NRF2 pathway in different species and experimental models. For example, SKN‐1, the NRF ortholog in Caenorhabditis elegans, is O‐GlcNAcylated in response to oxidative stress (Li et al, 2017a). Although we did not detect evidence of NRF2 O‐GlcNAcylation under homeostasis conditions (Fig 4A), human NRF proteins may be glycosylated in response to oxidative stress or other triggers. In addition, in murine macrophages, NRF2 activation was found to positively regulate OGT at transcriptional level (Li et al, 2017b), though we failed to detect an analogous effect in human breast cancer cells treated with known NRF2 activators (data not shown). Recently, Kang et al (2016) observed that TET1‐mediated recruitment of OGT to the NRF2 promoter regulates NRF2 levels in 5‐fluorouracil‐resistent colon cancer cells. However, in our experiments, we observed no effect of OGT or OGA inhibition on NRF2 mRNA (Fig 2E and Appendix Fig S1C). More studies are required to fully elucidate the transcriptional cross‐talk between the OGT and NRF2 pathways.
Our findings suggest that altered O‐GlcNAcylation regulates cellular redox balance through NRF2‐mediated antioxidant signaling in a KEAP1‐dependent manner under homeostasis. Consistent with our model, a recent report demonstrated that deletion of OGT in murine neurons causes the upregulation of several oxidative stress genes, although the specific O‐GlcNAcylated protein(s) responsible was not identified (Wang et al, 2016). Understanding the impact of O‐GlcNAc dynamics on antioxidant regulators (Lee et al, 2016) under physiologically relevant hypoglycemic, hyperglycemic, or oxidative conditions will be an interesting focus of future work.
Tumor microenvironment and therapeutic potential
Hypoxia, lactic acidosis, and hypoglycemia are common features of solid tumors due to poor perfusion (Chen et al, 2008; Gatza et al, 2011; Tang et al, 2012). These tumor microenvironmental stresses activate several transcription factors, including HIF1 (Harris, 2002), MondoA‐Mlx (Chen et al, 2010), and ATF3 (Tang et al, 2012), which in turn trigger a wide range of stress responses, drive oncogenesis, and confer resistance to various treatments (Castells et al, 2012). In particular, glucose deprivation confers resistance to various chemotherapeutic agents (Ledoux et al, 2003; Komurov et al, 2012). Interestingly, recent work demonstrated a functional connection between O‐GlcNAcylation and the HIF1 pathway in tumor cell signaling (Ferrer et al, 2014). Our results suggest that stresses that reduce OGT activity, such as glucose deprivation, may induce NRF2 activation (Figs 1B and 6) and confer chemoresistance via increased antioxidant signaling (Syu et al, 2016). Indeed, we found that the gene expression signatures of low OGT activity and high NRF2 pathway activation are strongly correlated in several human tumor datasets (Fig 1C), suggesting a potential in vivo connection between these pathways in cancer that will be tested in future studies. Although NRF2 plays a significant role in tumorigenesis and treatment resistance, as a transcription factor, it has not traditionally been considered a readily druggable target. Our results describe a novel mechanistic connection between O‐GlcNAcylation and NRF2 and may offer new opportunities to improve the treatment responses of solid tumors under microenvironmental stresses by targeting the OGT/NRF2 axis.
KLHL family and O‐GlcNAcylation
KEAP1 belongs to the Kelch‐like (KLHL) family, which is an evolutionarily conserved group with 42 human members (Dhanoa et al, 2013). Most KLHL proteins have a BTB/POZ domain, a BACK domain, and several Kelch motifs. While the function and regulation of most KLHL proteins are not well understood, their importance is demonstrated by various human diseases associated with mutations in KLHL genes (Dhanoa et al, 2013). For example, the mutations of KEAP1 (KLHL19) or gigaxonin (KLHL16) lead to various cancers (Singh et al, 2006) and giant axonal neuropathy (GAN; Bomont et al, 2000), respectively. Interestingly, S104 of human KEAP1 is conserved in the KEAP1 orthologs of diverse animal species (Fig EV5B), and in 37 of 42 human KLHL proteins (Fig EV5C; Sievers et al, 2011). In KLHL11, the corresponding position is a threonine residue, which would also permit glycosylation by OGT. In addition, mutations in the cognate residue of KEAP1 S104 in gigaxonin, serine 52 (Fig EV5C), cause a subset of GAN (Bomont et al, 2000; Mahammad et al, 2013; Boizot et al, 2014), and the structures of the KEAP1 and gigaxonin BTB domains, including the positions of Ser104/Ser52, are nearly identical (Fig EV5D). These facts suggest that gigaxonin and perhaps other KLHL proteins could be regulated by O‐GlcNAcylation at the KEAP1 S104‐cognate residue, or other sites. In a first step toward testing this possibility, we found that gigaxonin is O‐GlcNAcylated (Fig EV5E). Therefore, O‐GlcNAcylation may be a conserved mode of regulating the KLHL family of proteins. This hypothesis will be an important focus of future studies. Given the conservation of KEAP1 S104 among most KLHL family members, the glycosylation of KLHL proteins may provide a general mechanism for coupling nutrient sensing to proteostasis in a variety of physiological contexts.
Materials and Methods
Cell culture
MDA‐MB‐231, MCF7, ZR‐75‐1, BT474, A549, H838, 293T, H226, and RCC4 cells were obtained from the Duke Cell Culture Facility and maintained at 37°C supplied with 5% CO2. H1299 cells containing a yellow fluorescent protein (YFP) gene retrovirally inserted into intron 2 of the NQO1 gene (130207PL1G9‐NQO1) were a gift from Dr. Uri Alon and the Kahn Protein Dynamics group (Sigal et al, 2006; Cohen et al, 2009). Cell lines were cultured in either regular DMEM or RPMI medium with 10% fetal bovine serum (FBS), HEPES, and penicillin/streptomycin according to the instructions from the American Type Culture Collection. For glucose deprivation medium, cells at 90% confluency were first washed with phosphate‐buffered saline (PBS) twice and incubated with DMEM (ThermoFisher, DMEM, no glucose 11966) supplemented with 1× sodium pyruvate, HEPES, FBS, and penicillin/streptomycin, as well as indicated glucose concentrations (Sigma).
OGT/OGA inhibitors and azidosugar
Ac45SGlcNAc (abbreviated 5SGlcNAc or 5SG) was synthesized and purified as described (Gloster et al, 2011). Thiamet‐G was synthesized and purified by the Duke Small Molecule Synthesis Facility as described (Yuzwa et al, 2008). Ac4GalNAz (abbreviated GalNAz) was synthesized essentially as described (Boyce et al, 2011) by Tandem Sciences.
Co‐immunoprecipitation (co‐IP)
For co‐IP of endogenous proteins, 293T and MCF7 cells were treated with 5SGlcNAc (50 μM, 24 h) and lysed in Pierce IP buffer (1% Triton X‐100, 150 mM NaCl, 1 mM EDTA, and 25 mM Tris–HCl pH 7.5) supplemented with protease and phosphatase inhibitors. Cell lysates were incubated with anti‐CUL3 antibody overnight at 4°C. Then, protein G beads (Dynabeads, ThermoFisher) were added for an additional 8 h with gentle rotation at 4°C. For HA‐KEAP1 and MYC‐CUL3 interaction experiments, 293T cells transfected (Mirus LT1) with HA‐KEAP1 and MYC‐CUL3 were treated with 5SGlcNAc (50 μM, 48 h) and harvested in Pierce IP buffer (with the addition of 5 μM PUGNAc to inhibit hexosaminidase activity). For MYC‐CUL3 and HA‐tagged wild‐type KEAP1 or S104A KEAP1 interaction experiments, H838 or 293T cells transfected with the indicated constructs (32–48 h post‐transfection) were lysed in Pierce IP buffer. Lysates were incubated with MYC or HA antibody for 8 h with rotation at 4°C, and then, protein G beads were added for additional overnight rotation at 4°C. For KEAP1–CUL3 interaction experiments under glucose deprivation, 80% confluent MDA‐MB‐231 were first transfected with the indicated constructs (Invitrogen, Lipofectamine 2000) for 24 h. The cells were then washed twice in PBS and incubated with low‐ or high‐glucose medium for an additional 12 h before harvesting in Pierce IP buffer. After IPs, protein G beads were washed three times (1% Triton X‐100, 300 mM NaCl, 1 mM EDTA, and 25 mM Tris–HCl pH 7.5) and eluted in 2× SDS–PAGE sample buffer for Western blot analysis. For OGT and KEAP1 interaction experiments, 293T cells were transfected with MYC‐OGT and HA‐KEAP1 for 48 h, harvested in Pierce lysis buffer, and used for co‐IP as described. The proteins of interest were then evaluated by Western blot using either chemiluminescence (Thermo Scientific SuperSignal Pico) or LI‐COR Odyssey imager (IRDye).
YFP‐NQO1 induction assay using IncuCyte ZOOM
The reporter cell line H1299 stably expressing YFP‐NQO1, cultured in RPMI medium supplemented with 10% FBS, was plated onto 48‐well tissue‐culture microplates (8,000 cells/well) and allowed to adhere overnight. DMSO, MLN4924, or 5SGlcNAc was added at the indicated concentrations, and the plates were immediately transferred into the IncuCyte ZOOM platform (Essen BioScience), housed inside a cell incubator at 37°C/5% CO2, until the end of the assay. Two fields per well from five technical replicates were imaged with phase and green (400 ms acquisition) channels. Images were taken using a 10× objective lens every 1 h for 45 h and then analyzed using the IncuCyte Basic Software. Automated image processing on the fluorescence and phase channels was accomplished by applying an appropriate processing definition. In phase contrast, cell segmentation was achieved by applying a mask to exclude cells from background. An area filter was applied to exclude objects below 75 μm2. Green channel background noise was subtracted with the Top‐Hat method of background non‐uniformity correction with a radius of 100 μm and a threshold of 0.6 green corrected units (GCU). Fluorescence signal was quantified applying a mask. Total Green Object Integrated Intensity (GCU × μm2/Image) was normalized by confluence and further normalized by mean values obtained for untreated cells. Data presented are from one experiment with five technical replicates and are reported as mean ± standard deviation (SD). Data were reproduced in a biological replicate with five technical replicates.
In vivo ubiquitination
293T cells transfected with MYC‐NRF2 and HA‐Ub were treated with DMSO vehicle or 5SGlcNAc (50 μM, 48 h). The cells were then harvested in RIPA buffer (Sigma, 150 mM NaCl, 1.0% IGEPAL CA‐630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0). Lysates were diluted (final 0.1% SDS) for incubation with anti‐MYC antibody and then protein G beads at 4°C with gentle rotation. For endogenous NRF2‐Ub assays, MCF7 cells were first treated with DMSO vehicle or 5SGlcNAc (50 μM) for 42 h, and MG132 (10 μM) was added for an additional 6 h of incubation. Cells were lysed in 1% SDS RIPA buffer and sonicated before IP. To IP endogenous NRF2, NRF2 antibody (Abcam 62352) was incubated with diluted cell lysates (final 0.1% SDS) overnight at 4°C, and the protein G beads were added for an additional 8 h of incubation at 4°C. After incubation, the beads were washed in IP buffer three times and boiled in 2× Western sample buffer. Sample buffer‐eluted proteins were separated by SDS–PAGE and analyzed by Western blot with the indicated antibodies.
GalNAz labeling and GlcNAc competition assays
MCF7 and MDA‐MB‐231 cells were incubated with DMSO or 100 μM GalNAz for 24 h. In brief, cells were lysed and fractionated into nuclear and cytosolic fractions for click reactions (1 h at room temperature with gentle rotation; 5 mM sodium ascorbate, 25 μM alkyne‐biotin, 100 μM TBTA, and 1 mM CuSO4). After click reactions, lysates were precipitated and washed in cold methanol to remove unreacted alkyne‐biotin, resuspended in 4 M guanidine/PBS, and incubated with NeutrAvidin beads overnight at 4°C to capture biotin‐conjugated proteins. The beads were then washed three times each with the following buffers: 4 M guanidine/PBS; 5 M NaCl/H2O; 6 M urea/PBS; and 1% SDS/PBS. Bound proteins were eluted by boiling in 2× SDS–PAGE sample buffer and analyzed by Western blot (Boyce et al, 2011). For GlcNAc competition assays, 293T cells transfected with HA‐KEAP1 were lysed and IP‐ed with HA antibody as above. Equal amounts of precipitated protein samples were loaded, separated by SDS–PAGE, transferred onto the same PVDF membrane, and blocked in 5% BSA/1× Tris‐buffered saline with Tween (TBST; 0.1% Tween‐20). For competition controls, O‐GlcNAc antibodies were pre‐incubated with 1 M GlcNAc (Sigma) in 5% BSA/TBST at room temperature for 2–3 h before incubation with membrane overnight at 4°C (Sakabe et al, 2010). Blots performed with or without GlcNAc competition were then evaluated by enhanced chemiluminescence using identical exposure times.
Quantitative real‐time PCR and microarray analysis
RNAs collected by RNeasy kit (Qiagen) were used for reverse transcription by the SuperScript II kit (Invitrogen). The primers used for real‐time PCR are listed in the Appendix. Analyses of microarray data and in vivo correlation were performed according to previous reports (Gatza et al, 2010; Keenan et al, 2015). Detailed information is provided in Appendix Supplementary Materials and Methods.
Author contributions
P‐HC, TJS, JW, PFS, FK, and FT performed the experiments. MH and BMS synthesized Ac45SGlcNAc. ES performed MS‐based O‐GlcNAc site mapping of KEAP1. BJB created the structural modeling figures. P‐HC, TJS, MB, and J‐TC designed and interpreted the experiments and wrote and revised the manuscript. All authors reviewed and approved the finished manuscript. MB and JTC supervised all aspects of the work. JRM and MBM provided reagents and manuscript discussion.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We are grateful for critical discussions and technical support from members of the Chi and Boyce labs. We thank C. Toleman (Boyce lab) for His‐OGT protein. We acknowledge the financial support from the Pilot Project programs of Duke Cancer Institute, the NIH (CA125618 to JTC and GM118847 to MB), the Department of Defense (W81XWH‐12‐1‐0148, W81XWH‐14‐1‐0309, W81XWH‐15‐1‐0486 to JTC), and Central Michigan University and Cottrell College Scholar Award #22525 from the Research Corporation for Science Advancement (to BMS). MB is a Scholar of the Rita Allen Foundation. PHC is a Hung‐Taiwan Duke fellow. Research reported in this publication was supported by Duke O'Brien Center for Kidney Research (DOCK) (to MB and JTC) supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number P30DK096493.
The EMBO Journal (2017) 36: 2233–2250
Contributor Information
Michael Boyce, Email: michael.boyce@duke.edu.
Jen‐Tsan Chi, Email: jentsan.chi@duke.edu.
References
- Adam J, Hatipoglu E, O'Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, Stevens M, Fischer R, Carmeliet P, Maxwell PH, Pugh CW, Frizzell N, Soga T, Kessler BM, El‐Bahrawy M, Ratcliffe PJ et al (2011) Renal cyst formation in Fh1‐deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20: 524–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam J, Wicks C, Stewart D, Gong P, Touchard C, Otterbein S, Choi AM, Burow ME, Tou J (2000) Mechanism of heme oxygenase‐1 gene activation by cadmium in MCF‐7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J Biol Chem 275: 27694–27702 [DOI] [PubMed] [Google Scholar]
- Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N (1996) Rapamycin blocks the phosphorylation of 4E‐BP1 and inhibits cap‐dependent initiation of translation. EMBO J 15: 658–664 [PMC free article] [PubMed] [Google Scholar]
- Boizot A, Talmat‐Amar Y, Morrogh D, Kuntz NL, Halbert C, Chabrol B, Houlden H, Stojkovic T, Schulman BA, Rautenstrauss B, Bomont P (2014) The instability of the BTB‐KELCH protein Gigaxonin causes Giant Axonal Neuropathy and constitutes a new penetrant and specific diagnostic test. Acta Neuropathol Commun 2: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tuysuz B, Landrieu P, Hentati F, Koenig M (2000) The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet 26: 370–374 [DOI] [PubMed] [Google Scholar]
- Bond MR, Hanover JA (2013) O‐GlcNAc cycling: a link between metabolism and chronic disease. Annu Rev Nutr 33: 205–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, Hanover JA (2015) A little sugar goes a long way: the cell biology of O‐GlcNAc. J Cell Biol 208: 869–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, Carrico IS, Ganguli AS, Yu SH, Hangauer MJ, Hubbard SC, Kohler JJ, Bertozzi CR (2011) Metabolic cross‐talk allows labeling of O‐linked beta‐N‐acetylglucosamine‐modified proteins via the N‐acetylgalactosamine salvage pathway. Proc Natl Acad Sci USA 108: 3141–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning P, Sorrell FJ, Bullock AN (2015) Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med 88: 101–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castells M, Thibault B, Delord JP, Couderc B (2012) Implication of tumor microenvironment in chemoresistance: tumor‐associated stromal cells protect tumor cells from cell death. Int J Mol Sci 13: 9545–9571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JY, Kwong M (2000) Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic‐leucine zipper protein. Biochem Biophys Acta 1517: 19–26 [DOI] [PubMed] [Google Scholar]
- Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, Dewhirst M, West M, Chi JT (2008) The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet 4: e1000293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Merl D, Peterson CW, Wu J, Liu PY, Yin H, Muoio DM, Ayer DE, West M, Chi JT (2010) Lactic acidosis triggers starvation response with paradoxical induction of TXNIP through MondoA. PLoS Genet 6: e1001093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, Chen F, Feiler H, Tokuyasu T, Kingsley C, Dairkee S, Meng Z, Chew K, Pinkel D, Jain A, Ljung BM et al (2006) Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 10: 529–541 [DOI] [PubMed] [Google Scholar]
- Chou TY, Hart GW, Dang CV (1995) c‐Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J Biol Chem 270: 18961–18965 [DOI] [PubMed] [Google Scholar]
- Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA, Callahan JF, Carr R, Concha N, Kerns JK, Qi H, Sweitzer T, Ward P, Davies TG (2014) Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS One 9: e98896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AA, Kalisky T, Mayo A, Geva‐Zatorsky N, Danon T, Issaeva I, Kopito RB, Perzov N, Milo R, Sigal A, Alon U (2009) Protein dynamics in individual human cells: experiment and theory. PLoS One 4: e4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, Diehl JA (2004) PERK‐dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem 279: 20108–20117 [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA (2004) The Keap1‐BTB protein is an adaptor that bridges Nrf2 to a Cul3‐based E3 ligase: oxidative stress sensing by a Cul3‐Keap1 ligase. Mol Cell Biol 24: 8477–8486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanoa BS, Cogliati T, Satish AG, Bruford EA, Friedman JS (2013) Update on the Kelch‐like (KLHL) gene family. Hum Genomics 7: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B III, Stockwell BR (2012) Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell 149: 1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doss JF, Jonassaint JC, Garrett ME, Ashley‐Koch AE, Telen MJ, Chi JT (2016) Phase 1 study of a sulforaphane‐containing broccoli sprout homogenate for sickle cell disease. PLoS One 11: e0152895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggler AL, Small E, Hannink M, Mesecar AD (2009) Cul3‐mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem J 422: 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, Vocadlo DJ, Seagroves TN, Reginato MJ (2014) O‐GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF‐1 pathway. Mol Cell 54: 820–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer CM, Reginato MJ (2014) Cancer metabolism: cross talk between signaling and O‐GlcNAcylation In Cancer genomics and proteomics: methods and protocols, Wajapeyee N. (ed.), pp 73–88. New York, NY: Springer New York; [DOI] [PubMed] [Google Scholar]
- Ferrer CM, Sodi VL, Reginato MJ (2016) O‐GlcNAcylation in cancer biology: linking metabolism and signaling. J Mol Biol 428: 3282–3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa M, Xiong Y (2005) BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3‐Roc1 ligase. Mol Cell Biol 25: 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, Morrow JD, Freeman ML (2007) Novel n‐3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem 282: 2529–2537 [DOI] [PubMed] [Google Scholar]
- Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, Datto MB, Kelley M, Mathey‐Prevot B, Potti A, Nevins JR (2010) A pathway‐based classification of human breast cancer. Proc Natl Acad Sci USA 107: 6994–6999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatza ML, Kung HN, Blackwell KL, Dewhirst MW, Marks JR, Chi JT (2011) Analysis of tumor environmental response and oncogenic pathway activation identifies distinct basal and luminal features in HER2‐related breast tumor subtypes. Breast Cancer Res 13: R62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ (2011) Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol 7: 174–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AL (2002) Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer 2: 38–47 [DOI] [PubMed] [Google Scholar]
- Hart GW, Slawson C, Ramirez‐Correa G, Lagerlof O (2011) Cross talk between O‐GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 80: 825–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CJ, Thimmulappa RK, Singh A, Blake DJ, Ling G, Wakabayashi N, Fujii J, Myers A, Biswal S (2009) Nrf2‐regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic Biol Med 46: 443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hast BE, Cloer EW, Goldfarb D, Li H, Siesser PF, Yan F, Walter V, Zheng N, Hayes DN, Major MB (2014) Cancer‐derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res 74: 808–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB (2002) Phosphorylation of Nrf2 at Ser‐40 by protein kinase C regulates antioxidant response element‐mediated transcription. J Biol Chem 277: 42769–42774 [DOI] [PubMed] [Google Scholar]
- Huerta C, Jiang X, Trevino I, Bender CF, Ferguson DA, Probst B, Swinger KK, Stoll VS, Thomas PJ, Dulubova I, Visnick M, Wigley WC (2016) Characterization of novel small‐molecule NRF2 activators: structural and biochemical validation of stereospecific KEAP1 binding. Biochem Biophys Acta 1860: 2537–2552 [DOI] [PubMed] [Google Scholar]
- Hur W, Sun Z, Jiang T, Mason DE, Peters EC, Zhang DD, Luesch H, Schultz PG, Gray NS (2010) A small‐molecule inducer of the antioxidant response element. Chem Biol 17: 537–547 [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino‐terminal Neh2 domain. Genes Dev 13: 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang ES, Han D, Park J, Kwak TK, Oh MA, Lee SA, Choi S, Park ZY, Kim Y, Lee JW (2008) O‐GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic beta cells. Exp Cell Res 314: 2238–2248 [DOI] [PubMed] [Google Scholar]
- Kang KA, Piao MJ, Ryu YS, Kang HK, Chang WY, Keum YS, Hyun JW (2016) Interaction of DNA demethylase and histone methyltransferase upregulates Nrf2 in 5‐fluorouracil‐resistant colon cancer cells. Oncotarget 7: 40594–40620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL (2013) The Keap1‐Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol 1: 45–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keembiyehetty C, Love DC, Harwood KR, Gavrilova O, Comly ME, Hanover JA (2015) Conditional knock‐out reveals a requirement for O‐linked N‐Acetylglucosaminase (O‐GlcNAcase) in metabolic homeostasis. J Biol Chem 290: 7097–7113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan MM, Liu B, Tang X, Wu J, Cyr D, Stevens RD, Ilkayeva O, Huang Z, Tollini LA, Murphy SK, Lucas J, Muoio DM, Kim SY, Chi JT (2015) ACLY and ACC1 regulate hypoxia‐induced apoptosis by modulating ETV4 via alpha‐ketoglutarate. PLoS Genet 11: e1005599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S (2007) Cell survival responses to environmental stresses via the Keap1‐Nrf2‐ARE pathway. Annu Rev Pharmacol Toxicol 47: 89–116 [DOI] [PubMed] [Google Scholar]
- Kim J‐E, You D‐J, Lee C, Ahn C, Seong JY, Hwang J‐I (2010) Suppression of NF‐κB signaling by KEAP1 regulation of IKKβ activity through autophagic degradation and inhibition of phosphorylation. Cell Signal 22: 1645–1654 [DOI] [PubMed] [Google Scholar]
- Kinch L, Grishin NV, Brugarolas J (2011) Succination of Keap1 and activation of Nrf2‐dependent antioxidant pathways in FH‐deficient papillary renal cell carcinoma type 2. Cancer Cell 20: 418–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Itoh K, Suzuki T, Osanai H, Nishikawa K, Katoh Y, Takagi Y, Yamamoto M (2002) Identification of the interactive interface and phylogenic conservation of the Nrf2‐Keap1 system. Genes Cells 7: 807–820 [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3‐based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24: 7130–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M (2006) Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol 26: 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12: 213–223 [DOI] [PubMed] [Google Scholar]
- Komurov K, Tseng JT, Muller M, Seviour EG, Moss TJ, Yang L, Nagrath D, Ram PT (2012) The glucose‐deprivation network counteracts lapatinib‐induced toxicity in resistant ErbB2‐positive breast cancer cells. Mol Syst Biol 8: 596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Tian W, Whitman SA, Zhang DD (2013) The predicted molecular weight of Nrf2: it is what it is not. Antioxid Redox Signal 18: 91–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux S, Yang R, Friedlander G, Laouari D (2003) Glucose depletion enhances P‐glycoprotein expression in hepatoma cells: role of endoplasmic reticulum stress response. Cancer Res 63: 7284–7290 [PubMed] [Google Scholar]
- Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA (2003) Identification of the NF‐E2‐related factor‐2‐dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem 278: 12029–12038 [DOI] [PubMed] [Google Scholar]
- Lee DF, Kuo HP, Liu M, Chou CK, Xia W, Du Y, Shen J, Chen CT, Huo L, Hsu MC, Li CW, Ding Q, Liao TL, Lai CC, Lin AC, Chang YH, Tsai SF, Li LY, Hung MC (2009) KEAP1 E3 ligase‐mediated downregulation of NF‐kappaB signaling by targeting IKKbeta. Mol Cell 36: 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Miller D, Henry R, Paruchuri VD, O'Meally RN, Boronina T, Cole RN, Zachara NE (2016) Combined antibody/lectin‐enrichment identifies extensive changes in the O‐GlcNAc sub‐proteome upon oxidative stress. J Proteome Res 15: 4318–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leturcq M, Lefebvre T, Vercoutter‐Edouart AS (2017) O‐GlcNAcylation and chromatin remodeling in mammals: an up‐to‐date overview. Biochem Soc Trans 45: 323–338 [DOI] [PubMed] [Google Scholar]
- Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley‐Usmar VM (2004) Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J 378: 373–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BA, Hanover JA (2014) O‐GlcNAc and the epigenetic regulation of gene expression. J Biol Chem 289: 34440–34448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Johnson D, Calkins M, Wright L, Svendsen C, Johnson J (2005) Stabilization of Nrf2 by tBHQ confers protection against oxidative stress‐induced cell death in human neural stem cells. Toxicol Sci 83: 313–328 [DOI] [PubMed] [Google Scholar]
- Li H, Liu X, Wang D, Su L, Zhao T, Li Z, Lin C, Zhang Y, Huang B, Lu J, Li X (2017a) O‐GlcNAcylation of SKN‐1 modulates the lifespan and oxidative stress resistance in Caenorhabditis elegans . Sci Rep 7: 43601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang Z, Li L, Gong W, Lazenby AJ, Swanson BJ, Herring LE, Asara JM, Singer JD, Wen H (2017b) Myeloid‐derived cullin 3 promotes STAT3 phosphorylation by inhibiting OGT expression and protects against intestinal inflammation. J Exp Med 214: 1093–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Parker GJ, Cooksey RC, Soesanto Y, Evans M, Jones D, McClain DA (2007) Chronic hexosamine flux stimulates fatty acid oxidation by activating AMP‐activated protein kinase in adipocytes. J Biol Chem 282: 7172–7180 [DOI] [PubMed] [Google Scholar]
- Ma Z, Vosseller K (2013) O‐GlcNAc in cancer biology. Amino Acids 45: 719–733 [DOI] [PubMed] [Google Scholar]
- Ma Z, Vosseller K (2014) Cancer metabolism and elevated O‐GlcNAc in oncogenic signaling. J Biol Chem 289: 34457–34465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahammad S, Murthy SN, Didonna A, Grin B, Israeli E, Perrot R, Bomont P, Julien JP, Kuczmarski E, Opal P, Goldman RD (2013) Giant axonal neuropathy‐associated gigaxonin mutations impair intermediate filament protein degradation. J Clin Investig 123: 1964–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD (2006) Dimerization of substrate adaptors can facilitate cullin‐mediated ubiquitylation of proteins by a “Tethering” mechanism: a two‐site interaction model for the Nrf2‐Keap1 complex. J Biol Chem 281: 24756–24768 [DOI] [PubMed] [Google Scholar]
- Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, Bergh J (2005) An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA 102: 13550–13555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J (2005) Genes that mediate breast cancer metastasis to lung. Nature 436: 518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehlenkamp JD, Johnson JA (1999) Activation of antioxidant/electrophile‐responsive elements in IMR‐32 human neuroblastoma cells. Arch Biochem Biophys 363: 98–106 [DOI] [PubMed] [Google Scholar]
- Mulvaney KM, Matson JP, Siesser PF, Tamir TY, Goldfarb D, Jacobs TM, Cloer EW, Harrison JS, Vaziri C, Cook JG, Major MB (2016) Identification and characterization of MCM3 as a Kelch‐like ECH‐associated protein 1 (KEAP1) substrate. J Biol Chem 291: 23719–23733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan S, Andrali SS, Cantrell JE (2010) Modulation of transcription factor function by O‐GlcNAc modification. Biochem Biophys Acta 1799: 353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M (2006) Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell 21: 689–700 [DOI] [PubMed] [Google Scholar]
- Palaniappan KK, Hangauer MJ, Smith TJ, Smart BP, Pitcher AA, Cheng EH, Bertozzi CR, Boyce M (2013) A chemical glycoproteomics platform reveals O‐GlcNAcylation of mitochondrial voltage‐dependent anion channel 2. Cell Rep 5: 546–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, Liu ET, Miller L, Nordgren H, Ploner A, Sandelin K, Shaw PM, Smeds J, Skoog L, Wedren S, Bergh J (2005) Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population‐based cohorts. Breast Cancer Res 7: R953–R964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML (2008) Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol 21: 705–710 [DOI] [PubMed] [Google Scholar]
- Sakabe K, Wang Z, Hart GW (2010) β‐N‐acetylglucosamine (O‐GlcNAc) is part of the histone code. Proc Natl Acad Sci USA 107: 19915–19920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A (2006) Glycogen synthase kinase‐3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem 281: 14841–14851 [DOI] [PubMed] [Google Scholar]
- Sangokoya C, Telen MJ, Chi JT (2010) microRNA miR‐144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 116: 4338–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider‐Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO (2010) Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6: 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafi R, Iyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD (2000) The O‐GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci USA 97: 5735–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal A, Milo R, Cohen A, Geva‐Zatorsky N, Klein Y, Alaluf I, Swerdlin N, Perzov N, Danon T, Liron Y, Raveh T, Carpenter AE, Lahav G, Alon U (2006) Dynamic proteomics in individual human cells uncovers widespread cell‐cycle dependence of nuclear proteins. Nat Methods 3: 525–531 [DOI] [PubMed] [Google Scholar]
- Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV, Biswal S (2006) Dysfunctional KEAP1–NRF2 interaction in non‐small‐cell lung cancer. PLoS Med 3: e420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawson C, Hart GW (2011) O‐GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer 11: 678–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET (2003) Breast cancer classification and prognosis based on gene expression profiles from a population‐based study. Proc Natl Acad Sci USA 100: 10393–10398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart D, Killeen E, Naquin R, Alam S, Alam J (2003) Degradation of transcription factor Nrf2 via the ubiquitin‐proteasome pathway and stabilization by cadmium. J Biol Chem 278: 2396–2402 [DOI] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D (2016) Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63: 173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykiotis GP, Bohmann D (2010) Stress‐activated cap'n'collar transcription factors in aging and human disease. Sci Signal 3: re3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syu JP, Chi JT, Kung HN (2016) Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget 7: 14659–14672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Lucas JE, Chen JL, LaMonte G, Wu J, Wang MC, Koumenis C, Chi JT (2012) Functional interaction between responses to lactic acidosis and hypoxia regulates genomic transcriptional outputs. Cancer Res 72: 491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Keenan MM, Wu J, Lin CA, Dubois L, Thompson JW, Freedland SJ, Murphy SK, Chi JT (2015) Comprehensive profiling of amino acid response uncovers unique methionine‐deprived response dependent on intact creatine biosynthesis. PLoS Genet 11: e1005158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Ding CK, Wu J, Sjol J, Wardell S, Spasojevic I, George D, McDonnell DP, Hsu DS, Chang JT, Chi JT (2016a) Cystine addiction of triple‐negative breast cancer associated with EMT augmented death signaling. Oncogene https://doi.org/10.1038/onc.2016.394 [DOI] [PubMed] [Google Scholar]
- Tang X, Wu J, Ding CK, Lu M, Keenan MM, Lin CC, Lin CA, Wang CC, George D, Hsu DS, Chi JT (2016b) Cystine deprivation triggers programmed necrosis in VHL‐deficient renal cell carcinomas. Cancer Res 76: 1892–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt C (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res 45: D158–D169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A, Hu M, Chan DA, Ethier SP, van‘t Veer LJ, Polyak K, McCormick F, Gray JW (2013) Glutamine sensitivity analysis identifies the xCT antiporter as a common triple‐negative breast tumor therapeutic target. Cancer Cell 24: 450–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M (2006a) Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two‐site molecular recognition model. Mol Cell Biol 26: 2887–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Kobayashi A, Katsuoka F, Yamamoto M (2006b) Two‐site substrate recognition model for the Keap1‐Nrf2 system: a hinge and latch mechanism. Biol Chem 387: 1311–1320 [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova‐Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P (2004) Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA 101: 2040–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer‐van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA (2005) Gene‐expression profiles to predict distant metastasis of lymph‐node‐negative primary breast cancer. Lancet 365: 671–679 [DOI] [PubMed] [Google Scholar]
- Wang AC, Jensen EH, Rexach JE, Vinters HV, Hsieh‐Wilson LC (2016) Loss of O‐GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc Natl Acad Sci USA 113: 15120–15125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR (2007) RAS‐RAF‐MEK‐dependent oxidative cell death involving voltage‐dependent anion channels. Nature 447: 864–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, Cho JW (2006) Modification of p53 with O‐linked N‐acetylglucosamine regulates p53 activity and stability. Nat Cell Biol 8: 1074–1083 [DOI] [PubMed] [Google Scholar]
- Yang YR, Song M, Lee H, Jeon Y, Choi E‐J, Jang H‐J, Moon HY, Byun H‐Y, Kim E‐K, Kim DH, Lee MN, Koh A, Ghim J, Choi JH, Lee‐Kwon W, Kim KT, Ryu SH, Suh P‐G (2012) O‐GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell 11: 439–448 [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, Vocadlo DJ (2008) A potent mechanism‐inspired O‐GlcNAcase inhibitor that blocks phosphorylation of tau in vivo . Nat Chem Biol 4: 483–490 [DOI] [PubMed] [Google Scholar]
- Zhang DD, Hannink M (2003) Distinct cysteine residues in Keap1 are required for Keap1‐dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 23: 8137–8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M (2004) Keap1 is a redox‐regulated substrate adaptor protein for a Cul3‐dependent ubiquitin ligase complex. Mol Cell Biol 24: 10941–10953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Tan EP, VandenHull N, Peterson KR, Slawson C (2014) O‐GlcNAcase expression is sensitive to changes in O‐GlcNAc homeostasis. Front Endocrinol 5: 206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipper LM, Mulcahy RT (2002) The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem 277: 36544–36552 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6