

Abstract
We report here the synthesis of the first Selenocysteine SPPS derivatives which bear TFA-labile sidechain protecting groups. New compounds Fmoc-Sec(Xan) and Fmoc-Sec(Trt) are presented as useful and practical alternatives to the traditional Fmoc-Sec derivatives currently available to the peptide chemist. From a bis Fmoc-protected selenocystine precursor, multiple avenues of diselenide reduction were attempted to determine the most effective method for subsequent attachment of the protecting group electrophiles. Our previously-reported one-pot reduction methodology was ultimately chosen as the optimal approach toward the synthesis of these novel building blocks, and both were easily obtained in high yield and purity. Fmoc-Sec(Xan) was discovered to be bench-stable for extended timeframes while the corresponding Fmoc-Sec(Trt) derivative appeared to detritylate slowly when not stored at -20°C. Both Sec derivatives were incorporated into single- and multiple-Sec-containing test peptides in order to ascertain the peptides’ deprotection behavior and final form upon TFA cleavage. Single-Sec-containing test peptides were always isolated as their corresponding diselenide dimers, while dual-Sec-containing peptide sequences were afforded exclusively as their intramolecular diselenides.
Keywords: Selenocysteine, Xanthenyl Protection, Sidechain Protection, Solid Phase Peptide synthesis
INTRODUCTION
Selenocysteine (Sec, U) is an uncommon proteinogenic amino acid whose significance in biological processes and biochemical structure is noteworthy. Considered colloquially to be the “21st Amino Acid”, Sec has utility as a structural component of active sites in various redox proteins [1], cellular signaling processes [2], and acts as a biological selenium donor when incorporated into Sec-bearing proteins [3, 4]. In biological systems, Selenocysteine is synthesized and ribosomally inserted into proteins through an expanded translational process involving the conversion of free Serine into Sec and its subsequent co-translational insertion into the linear sequence of the protein by way of a Selenocysteine Insertion Sequence (SECIS) element [5]. There are a growing number of known SeC-containing proteins such as selenoprotein P [4] and selenoprotein W [6] which utilize the unusual chemistry inherent to Sec in order to carry out diverse biochemical processes crucial to the smooth operation of biological systems.
As interest in the field of selenoproteins grows, the demand for analytical research samples of these biomolecules increases in kind. The growing importance of biologically-relevant Sec-containing peptides, particularly those which may contain non-proteinogenic amino acids or unnatural architecture, creates a niche in which these peptides can be more easily constructed through chemical synthesis rather than biological vectors. Solid phase peptide synthesis (SPPS) allows for the straightforward synthesis of Sec-containing peptides through the use of Boc or Fmoc SPPS building blocks in which the reactive selenol sidechains of the selenocysteine residues are protected throughout the linear construction of the peptide sequence. In recent years, Boc SPPS has given way to the more gentle and straightforward piperidine-driven Fmoc Nα-deprotection methodology [7] which typically utilizes orthogonal, acid-labile protecting groups on the reactive sidechains of the amino acid derivatives. At present, all commercially-available Fmoc derivatives of the standard amino acids bearing potentially-reactive sidechains are orthogonally protected with an acid-labile vector, mostly consisting of Trt/tBu architecture. Thus, treatment of the peptide resin with trifluoroacetic acid (TFA) at the completion of the linear sequence serves the dual purpose of liberating the completed peptide from the solid support as well as concurrent global deprotection of all sidechain protectants. Selenocysteine is the only SPPS derivative which does not possess TFA-labile sidechain deprotection.
Traditional Sec SPPS derivatives, whether of the Fmoc or Boc variety, have exclusively employed benzyl-type sidechain protection architecture [8–11] which (dependent upon the identity and degree of substitution pattern upon it) require a variety of harsh treatment conditions to effect their removal (Figure 1). More current Sec sidechain protection protocol has recently been reported which offers structural diversity and expanded orthognality when compared with these benzyl protectants in use for decades [12]. However, these blocking groups also require further treatment with other reagents in addition to standard TFA deprotection in order to fully remove them. Further, these additional steps can involve harsh or overly-toxic reagents, potentially altering the peptide product profile. Thus, it would be very advantageous for peptide chemists using selenocysteine to have access to an Fmoc Sec SPPS derivative in which the selenol sidechain could be completely and cleanly removed using a standard TFA/scavenger deprotection cocktail in analogous fashion to all other standard acid-labile sidechain protectants.
Figure 1.
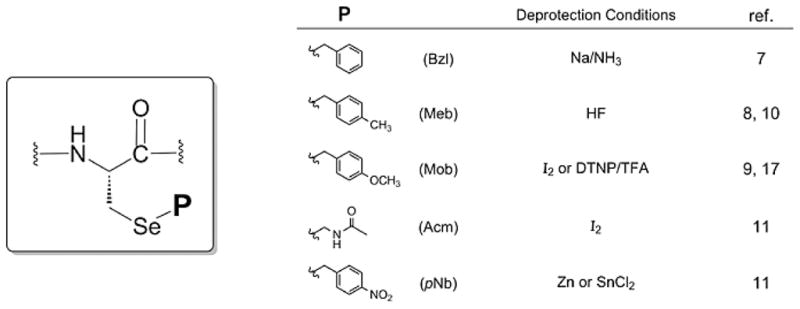
Traditional sidechain protection architecture for Sec SPPS derivatives.
Presented here are the results of a research effort which offers a practical solution to this problem, allowing a more streamlined approach toward the synthesis of Sec-containing peptides. The TFA-labile Xanthenyl (Xan) and Trityl (Trt) protecting groups were selected as candidates for this study (Figure 2) since they have both found use as sidechain protectants for their analogous cysteine SPPS residues [13, 14]. The building block derivatives Fmoc-Sec(Xan)-OH 2 and Fmoc-Sec(Trt)-OH 3 were easily synthesized from bis-Fmoc selenocystine intermediate 1 as reported previously [15, 16], and incorporated into a number of test peptide sequences to ascertain their potential and applicability toward Fmoc SPPS. Ease of deprotection using a standard TFA cocktail was studied in these test peptides, with careful observation toward product profile optimization during acidolytic cleavage.
Figure 2.
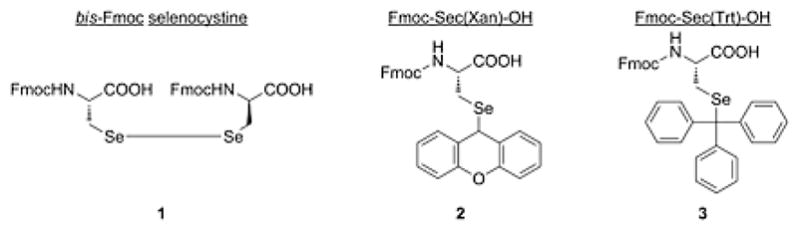
bis-Fmoc selenocystine synthetic intermediate 1 and Fmoc-Sec(Xan)-OH 2 and Fmoc-Sec(Trt)-OH 3 derivatives presented in this study.
MATERIALS AND METHODS
Materials
All standard Fmoc amino acid derivatives, O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), and 1-hydroxy-7-azabenzotriazole (HOAt) were purchased from RS Synthesis (Louisville, KY). Piperidine and N,N-disopropylcarbodiimide were purchased from Sigma-Aldrich (Milwaukee, WI). Novasyn TGR resin and Fmoc-OSu were purchased from Novabiochem (EMD Chemicals; Philadelphia, PA). L-Selenocystine, 9-hydroxyxanthene, trifluoroacetic acid, and all other solvents and reagents were purchased from Fisher Scientific (Pittsburgh, PA).
High Performance Liquid Chromatography (HPLC)
HPLC analysis was carried out on a Hitachi analytical HPLC system with L-7100 pumps, an L-7200 autosampler, an L-7400 UV detector, and an L-7000 interface using a Poroshell 120 column from Agilent (4.6 × 150 mm). Aqueous and organic phases were 99:1 water/acetonitrile (0.1% TFA) (Buffer A) and 90:10 acetonitrile/water (0.1% TFA) (Buffer B), respectively. Beginning with 100% Buffer A, a 1.0 mL/min gradient elution increase of 1% Buffer B/min for 55 min was used for all peptide chromatograms. Peptides were detected at 214 nm.
Mass Spectrometry
Samples were analyzed using an AB Sciex 4000 QTrap (AB Sciex, Framingham, MA) hybrid triple quadrupole/linear ion trap liquid chromatograph-mass spectrometer (LCMS). Positive electrospray ionization (ESI) was used as the ionization source. Source temperature was maintained at 400 °C. The mass spectrometer was operated in single quadrupole mode, scanning from 200 to 1000 da.
High Resolution Mass Spectrometry (HRMS)
HRMS spectra were obtained using a Thermo-Fisher Scientific LTQ Linear Quadrupole Ion Trap-Oribitrap Mass Spectrometer Plus Liquid Chromatography.
Nuclear Magnetic Resonance (NMR)
1H and 13C NMR spectra were obtained on a Bruker AXR 500 MHz high-field NMR spectrometer fitted with a direct detection liquid probe using standard pulse sequences. Spectra were obtained in d6 DMSO, and chemical shifts are given in ppm against the center DMSO multiplet peak at 2.51 ppm.
Synthesis of (Fmoc-Sec)2 (1)
Procedure and characterization as previously described [16].
Synthesis of Fmoc-Sec(Xan)-OH (2)
0.5 g (0.64 mmol) bis-Fmoc-L-selenocystine (1) and 820 mg (20 eq) finely powdered Zn were suspended in 10 mL diethyl ether followed by the addition of 4 mL 3M HCl using our own modification [16] from the procedure of Santi [17]. This mixture was allowed to stir vigorously for 45 min until the organic layer turned from light yellow to colorless. At the end of this time, the organic layer was drawn off and passed through a pad of granulated MgSO4 in order to remove residual water. The resultant supernatant was introduced to a 100 mL flask and the solvent was removed via evaporation under a stream of nitrogen with gentle warming of the flask. Following complete concentration, 5 ml dichloromethane and 95 μL (1.28 mmol) TFA was added with stirring according to the procedure of Barany [13]. 267 mg (1.34 mmol) 9-hydroxyxanthene was added to this stirring solution as a solid in small portions over 10 minutes and the mixture was allowed to stir for 1 hr. At the end of this time, much of the product had precipitated out of solution. 50 mL petroleum ether was added and the mixture was stirred vigorously for an additional 15 min at 0 °C. The resultant solid was then filtered, washing with 25 mL of ice-cold 3:1 petroleum ether/dichloromethane to yield pure Fmoc-Sec(Xan)-OH as an off-white solid with a bluish tinge (614 mg, 84%). mp 158–160 °C; [α]25D = −5.93° (c = 100 mm, acetone); 1H NMR (500 MHz, DMSO d6) δ 12.86 (br. s, 1H), 7.88 (d, J = 7.5 Hz, 2H), 7.77 (d, J = 8.0 Hz, 1H), 7.72 (dd, J = 3.0 Hz, J = 7.5 Hz, 2H), 7.46 (dd, J = 1.5 Hz, J = 7.5 Hz, 1H), 7.44-7.37 (m, 3H), 7.33-7.26 (m, 4H), 7.17-7.09 (m, 4H), 5.82 (s, 1H), 4.35-4.17 (m, 3H), 4.13-4.05 (m, 1H), 2.90 (dd, J = 5.0 Hz, J = 12.5 Hz, 1H), 2.80 (dd, J = 10.0 Hz, J = 12.5 Hz, 1H); 13C NMR (125 MHz, DMSO d6) δ 172.2, 155.8, 151.65, 151.62, 143.66, 143.65, 140.6, 128.7, 128.6, 128.4, 127.5, 127.0, 125.18, 125.16, 123.6, 123.5, 123.11, 123.09, 120.0, 116.2, 116.1, 65.7, 54.3, 46.5, 33.9, 25.5 ppm; IR (film) 1721 cm−1, 1260 cm−1, 760 cm−1; HRMS Calcd. for C31H25NO5SeNa: 594.0790. Found: 594.0783 (M+Na).
Synthesis of Fmoc-Sec(Trt)-OH (3)
Diselenide reduction of bis Fmoc Selenocystine 1 was carried out identically as for 2. To the reduced selenol residue was added 5 mL dichloromethane followed by 373 mg (1.34 mmol) trityl chloride in small solid portions over 10 minutes (with no TFA activator). After 1.5h, some solid was observed to have precipitated out of solution. At this time, 50 mL petroleum ether was added and the mixture was stirred vigorously for an additional 15 min at 0 °C. The resultant solid was then filtered, washing with 25 mL of ice-cold 3:1 petroleum ether/dichloromethane to yield pure Fmoc-Sec(Trt)-OH was isolated as a light yellow amorphous solid which required immediate storage at −20 °C (705 mg, 87%). mp 150–152 °C; [α]25D = +3.75° (c = 100 mm, acetone); 1H NMR (500 MHz, DMSO d6) δ 12.73 (br. s, 1H), 2.89 (d, J = 7.5 Hz, 2H), 7.80 (d, J = 8.5 Hz, 1H), 7.72 (d, J = 7.5 Hz, 2H), 7.43-7.38 (m, 2H), 7.36-7.20 (m, 17H), 4.35-4.20 (m, 3H), 4.02-3.95 (m, 1H), 2.78 (t, J = 10.5 Hz, 1H), 2.48 (dd, J = 5.0 Hz, J = 11.5 Hz, 1H) ; 13C NMR (125 MHz, DMSO d6) δ 172.0, 155.7, 147.7, 145.0, 143.7, 143.6, 140.6, 129.4, 128.0, 127.7, 127.6, 127.4, 127.0, 126.7, 126.5, 125.2, 120.0, 65.6, 64.1, 53.7, 46.5, 28.2 ppm; IR (film) 1722 cm−1, 1034 cm−1, 737 cm−1; HRMS Calcd. for C37H31NO4SeNa: 656.1311. Found: 656.1305 (M+Na).
Peptide Synthesis
All peptides were synthesized manually via Fmoc protocol on a 40 μmol scale using Novasyn™ TGR resin (0.29 mmol/g). All Sec amino acid derivatives were coupled using a five fold molar excess of Fmoc amino acid, DIC, and HOAt (with 5 min preactivation) in DMF for 2 hours. All other amino acid couplings were carried out using a five molar excess of Fmoc amino acid, HBTU, and HOAt (with 5 min preactivation) in 0.2 M DIEA/DMF for 1 hour. Fmoc deprotection was achieved using 20% piperidine/DMF (2 × 5 min). Cleavage of peptides from their resins was accomplished through treatment of the resin with 96:2:2 TFA/TIPS/H2O for 1.5 h. Following isolation, the TFA supernatant was evaporated to one tenth its original volume under a stream of nitrogen followed by precipitation of the crude peptide into cold anhydrous diethyl ether.
RESULTS AND DISCUSSION
A focus of this project was to optimize the synthetic pathway which would afford the desired Sec derivative in the most straightforward and facile manner. Literature preparations of Fmoc-Sec derivatives have traditionally followed two general reaction pathways, based upon whether the in situ - derived selenium nucleophile is delivered within the amino acid framework [10] or the protectant module [9, 18] (Figure 3). Based upon prior successful results from our laboratories [16, 18], we opted for the former approach, in which reduction of bis-Fmoc-protected selenocystine 1 [15, 16] provides a nucleophilic selenium species which we envisioned would condense with the appropriate protectant electrophile to afford the desired target molecules in a one-pot process. However, since the electrophiles in these cases are traditionally manifested as secondary and tertiary carbocations [13, 14] as opposed to the primary benzyl electrophiles commonly used for used for Sec sidechain protection [8–12], there was some uncertainty as to whether the in situ reduced Fmoc-Sec(SeH) intermediate would successfully attack them. As such, we elected to carry out a survey of different reduction conditions with concurrent electrophile attachment based upon analogous literature preparations.
Figure 3.
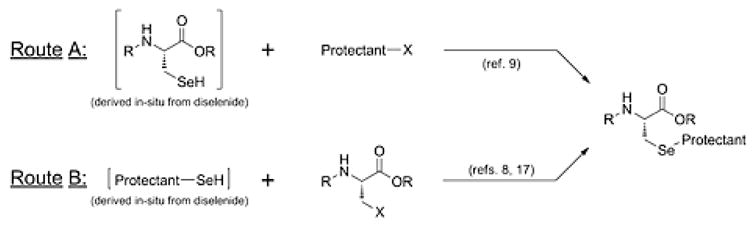
Traditional literature synthetic approaches toward Sec SPPS derivatives.
Ordinarily, matching reaction conditions to the analogous sulfur-containing systems (ie: cysteine) would appear to be a reasonable approach towards designing a comparable Sec synthetic methodology due to the very similar chemistry shared between the two chalcogen elements. In contrast to sulfur, however, the high oxidative potential of selenium and the propensity of a generated Sec selenol to quickly reestablish its diselenide form [19, 20] became the principal synthetic challenge to overcome. Care had to be taken to choose reaction conditions which allowed the reactive selenol to remain intact long enough to allow condensation with the appropriate protectant electrophiles. Subsequently, a survey of diselenide scission methodologies was undertaken in order to identify which would be most amenable to the current structural goal (Table 1).
Table 1.
|
| ||||
|---|---|---|---|---|
| Entry | Reducing Conditions | Sec Intermediate | Electrophile | Notes |
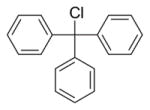
| 1 | LiBEt3H (ref. 17) |
|
|
Trace product formed. Selenol intermediate readily re-formed 1 under the basic reaction conditions. |
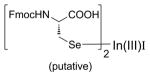
| 2 | In(I)I (refs. 21 & 22) |
|
|
No product formed. In(III)I diorganyl selenoate unreactive to the tertiary alkyl chloride electrophile. |
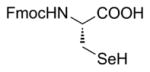
| 3a | Zn/HCl (refs. 15 & 16) |
|
|
84% Fmoc-Sec(Xan)-OH product isolated. |
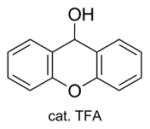
| 3b | Zn/HCl (refs. 15 & 16) |
|
|
87% Fmoc-Sec(Trt)-OH product isolated. |
One of the most common approaches of selenocystine reduction prior to Se derivatization is through aqueous borohydride reduction [10]. Our initial attempts using this methodology focused on our previously-reported anhydrous reduction conditions using Super Hydride (LiBEt3H) in THF [18] to generate the reactive selenium species (entry 1, Table 1). However, incubation of the generated intermediate with trityl chloride [14, 21] resulted in only a trace of Fmoc-Sec(Trt)-OH formation and nearly complete regeneration of the original bis Fmoc selenocystine 1 regardless of how stringent our efforts to exclude oxygen from the reaction vessel. The reasoning for this was undoubtedly the aforementioned propensity of free selenol to readily form its corresponding diselenide under alkaline conditions. This result led us to elect not to pursue other known diselenide reduction vectors which employ alkaline conditions such as phosphine [22] and hydrazine hydrate-alkali [23] and instead focus on methodologies which utilize non-basic conditions.
An interesting and promising approach toward diselenide reduction without the potential for impeding diselenide reformation was through the treatment of bis Fmoc selenocystine 1 with In(I) iodide to afford (following oxidative insertion) the corresponding indium(III)-dichalcogenate (Entry 2, Table 1). Ranu and coworkers showed that in-situ generated indium(III)-phenylseleno dichalcogenates condensed smoothly with a variety of alkyl halides to form a series of diorganyl selenides [24]. Further, Braga illustrated that formation and delivery of the corresponding alkylseleno derivative onto a corresponding electrophile was within the scope of the reaction [25]. In our hands, reaction of 1 with In(I)I appeared to circumvent the undesired reformation of starting material 1 (as evidenced by TLC analysis). However, treatment of the intermediate mixture with trityl chloride resulted in no formation of 3 whatsoever. Indeed, the TLC profile maintained constant even after prolonged reaction time, suggesting the putative In(III)I diorganylseleno intermediate was not sufficiently reactive to induce its condensation with a tertiary alkyl electrophile.
The Zn-mediated reductive methodology of Barany in his synthesis of the analogous Fmoc-Cys(Xan)-OH derivative [13] was an attractive approach to utilize for the synthesis of the corresponding Sec(Xan) analog 2, particularly because the acidic conditions required for the in-situ generation of the 9-Xanthenyl cation electrophile doubled as an environment in which diselenide reformation was sufficiently suppressed. Unfortunately, our initial efforts using Barany’s Zn/AcOH diselenide reduction conditions resulted in no product formation. However, aligning with the successes we have enjoyed previously [16], adapting the reduction environment to the biphasic Zn-mediated reductive system of Santi [17] was gratifyingly successful for Fmoc-Sec(Xan)-OH 2 (Entry 3a, Table 1). The analogous Fmoc-Sec(Trt)-OH derivative 3 was similarly obtained using the corresponding trityl chloride electrophile (Entry 3b, Table 1). Using this approach with our own modifications, both derivatives were isolated in yields exceeding 80%.
It was noticed that, upon languishing on the lab bench for more than 1 hour, Sec(Trt) derivative 3 began to take on an increasingly yellow tinge, signifying a slow rate of spontaneous detritylation. However, it was found that if derivative 3 were stored at −20 °C, it retained its original light yellow color indefinitely. Sec(Xan) derivative 2 on the other hand was found to be remarkably bench-stable for extended periods (> 1 month), retaining its diagnostic bluish tinge without evidence of spontaneous decomposition. Since shelf stability is an important consideration when weighing the merits and practical viability of new compounds such as these derivatives, it was decided that Fmoc-Sec(Xan)-OH 2 was the superior product, given that both derivatives produced comparable results when incorporated into peptide models (vide infra).
With Fmoc-Sec(Xan)-OH 2 and Fmoc-Sec(Trt)-OH 3 derivatives in hand, their incorporation into suitable peptide models became the next objective of this undertaking. We wanted to select peptide templates which would showcase the efficiency of TFA-mediated deprotection of Sec(Xan) and Sec(Trt) as well as to explore the fate of the liberated Sec selenol once the deprotected peptide was isolated. Importantly, we wished to design peptide sequences to help identify a niche for these TFA-labile protectants which would assist in solving enduring practical problems and challenges in the arena of Sec SPPS. For example, a major concern in the Fmoc synthesis of Sec-containing peptides is the alleged propensity of protected selenocysteine to deselenate during base-mediated peptide elongation and/or cleavage to form dehydroalanine (dHA) [26]. More generally, it was important to show that any peptides synthesized using these derivatives retained stereointegrity at the Sec residue.
The Cys-containing Stromelysin 1 matrix metalloproteinase inhibitor (MMP3-I) [27] of the sequence Ac-Arg-Cys-Gly-Val-Pro-Asp-NH2 was chosen as a general template in which to build the Sec-containing analog in order to demonstrate ease of construction of this template using Sec derivatives 2 and 3. While incorporation of 2 and 3 into their respective MMP3-1 sequences was carried out under neutral DIC/HOAt conditions, all remaining residues were coupled in a standard fashion under basic conditions (0.2 M DIEA/DMF) using HBTU/HOAt conditions. Additionally, no efforts were made to shorten Fmoc deprotection sequences from the standard length of time used for all other residues (2×5min). Liberation and deprotection of the peptides from the resin was carried out using a standard 96:2:2 v/v TFA/TIS/H2O cocktail for 1.5hr. The crude HPLC traces of both batches of the MMP3-I sequence showed a major peak comprising ~90% of the profile (Figure 4). Mass spectrometric (MS) analysis of this component showed it to be the MMP3-I diselenide dimer, presumably formed spontaneously upon cleavage of the peptide from the resin. MS analysis of the preceding neighboring peaks present in both chromatograms showed the existence of small amounts of enduring side products. Mass analysis of these peaks show evidence of two putative additional components, dHA adduct 5 and Se-OAc conjugate 6. Since these peaks are in the extreme minority in the overall HPLC profile, it can be surmised that any alternate reaction pathways encountered during synthesis and/or cleavage of this peptide were gratifyingly minimal. It should be further noted that the analogous Cys-containing MMP3-I sequence was similarly constructed, producing an HPLC profile which showed a similar high level of crude peptide purity as the Sec-containing version, but manifesting predictably as the free thiol rather than the disulfide dimer (data not shown).
Figure 4.
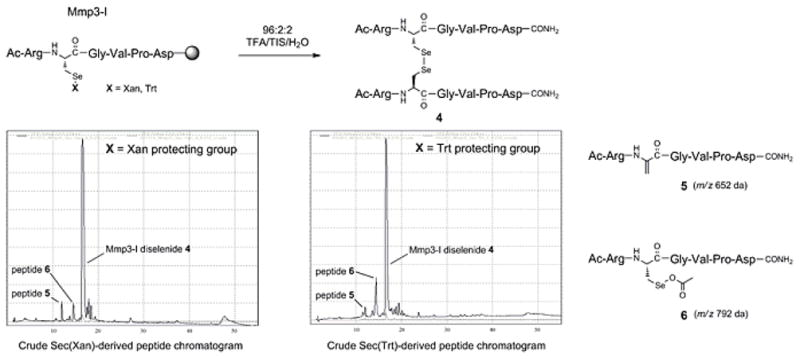
Chromatograms showing MS Peak analysis of crude Mmp3-I peptide isolates.
In order to offer the greatest demonstration of the abilities of derivatives 2 and 3 toward the successful synthesis of Sec-containing peptides without racemization or deselenation, we undertook the synthesis of Moroder’s Sec-containing glutaredoxin analog fragment Lys16 Grx 10–17 of the sequence Ac-Gly-Sec-Pro-Tyr-Sec-Val-Lys-Ala-NH2 [26], wherein both Sec residues were sidechain-protected with either Xan or Trt (Figure 5). This sequence was especially significant in that it was the model which was shown to be especially prone to deselenation under standard basic coupling and Fmoc deprotection conditions. The same traditional synthetic conditions as with the previously characterized MMP3-I analog were carried out in this synthesis, utilizing identical cleavage protocol. To our surprise, the resultant crude isolate was extremely homogenous, affording a single peak in the HPLC profile of the crude isolate (see chromatograms, Figure 5). MS analysis of this peak showed a mass consistent with the intramolecular diselenide, derived presumably from the spontaneous cyclization during cleavage. Again, it is noteworthy that the bis-Cys-containing analog of Lys16 Grx 10–17 was isolated as the reduced bis-thiol compound, and was able to be coaxed into its intramolecularly-cyclized disulfide form by 3 hr incubation in mild alkaline buffer (data not shown).
Figure 5.

HPLC chromatograms derived from crude Lys16 Grx 10 17 peptide isolates.
The successful results from the incorporation of derivatives 2 and 3 into the two test peptide sequences were in sharp contrast to previous literature-based outcomes concerning the use of Fmoc-based Sec derivatives in SPPS. Specifically, Moroder and coworkers reported a high degree of deselenation tendency in their synthesis of the Sec-containing Native Grx 10–17 analog [26] using Fmoc-Sec(Mob) as the building block in the sequence. The researchers surmised that the basic conditions of Fmoc amino acid coupling and 20% piperidine deprotection gave rise to significant amounts of dHA- and β-piperidyl-containing adducts derived from deselenated Sec, requiring them to optimize the synthetic conditions to use pentafluorophenyl ester amino acid building blocks and significantly shortened piperidine deprotection sequences. Concurrent with these augmented coupling conditions, Lys was substituted for Arg at the 16 position in order to avoid the challenging Arg(Pmc) deprotection in favor of the comparatively facile Lys(Boc) removal.
It is quite noteworthy that our synthesis of the identical peptide using Sec derivatives 2 and 3 under traditional (ie: standard basic) conditions produced crude product profiles which possessed few detectable side products. Indeed, the test peptide which did produce measurable side products (including the familiar dHA adduct) was the MMP3-I model. A closer look at the identities of the putative side products in the MMP3-I model suggest a specific mechanism for both of their genesis based upon partial Se oxidation during peptide elongation (Figure 6). A selenoxide formation event 9 during peptide synthesis followed by a Pummerer-style acetylation 10 [28] would provide the impetus to form dHA adduct 5 via base-induced elimination, driven by the ability to quench the positive charge on selenium. Alternatively, structure 10 might endure up until the cleavage/deprotection process, releasing xanthenyl cation 12 to afford Se-OAc conjugate 6 as the additional side product. It is postulated that Se oxidation and any potential dHA formation would have had to occur after the installation (and subsequent Fmoc deprotection) of the N-terminal Arg residue due to the absence of β-piperidyl adducts within the crude deprotection profile (Figure 4). Further, the presence of putative Se-OAc adduct 6 could be explained by the attack of intermediate 9 on Ac2O, the final step in the peptide elongation. While some of the putative mechanism just described may be speculative, it does offer a reasonable depiction of how side products 5 and 6 were formed during the synthesis of MMP3-I 4. Perhaps a more compelling question might be why the similar Lys16 Grx 10–17 peptide model 7 showed no evidence of dHA or Se-OAc adducts in its product profile, despite the fact that its synthesis was carried out under identical conditions, replete with an identical final acetylation step. Ongoing efforts in our laboratories are currently attempting to provide some answers to this question.
Figure 6.
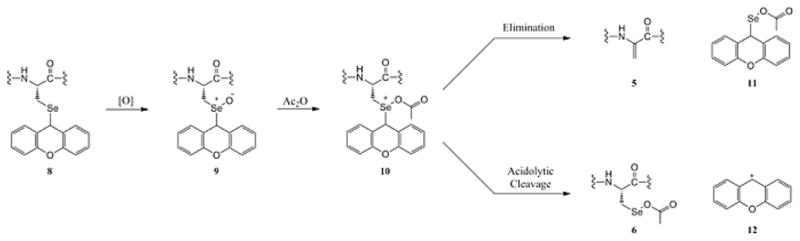
Possible mechanism to explain the existence of adducts 5 and 6 in MMP3-I test peptide product profile.
CONCLUSIONS
From the successful synthesis of the two preceding test peptides, it is evident that a TFA-labile vector of sidechain deprotection of Sec is a viable and effective methodology. Two Fmoc-Sec derivatives bearing TFA-labile sidechain protection, Fmoc-Sec(Xan) and Fmoc-Sec(Trt), have been synthesized and fully characterized. Of the two derivatives, Fmoc-Sec(Xan) was chosen as the more viable and practical compound due to its enhanced bench stability. Both derivatives were incorporated into test peptide systems which, following TFA deprotection, produced diselenide peptides in extremely high purity. Small amounts of dHA and Se-OAc peptide side products were observed in the crude deprotection profile of one of the peptides and a possible mechanism was put forth to account for their existence.
Supplementary Material
Acknowledgments
I would like to acknowledge and thank Dr. Robert J. Hondal (Department of Biochemistry; University of Vermont) for his invaluable ideas and guidance in the writing of this manuscript.
HRMS spectra were the result of support by the Vermont Genetics Network through grant number 8P20GM103449 from the INBRE program of the National Institute of General Medical Sciences (NIGMS), a component of the National Institutes of Health (NIH).
References
- 1.Mustacich D, Powis G. Thioredoxin reductase. Biochem J. 2000;346:1–8. [PMC free article] [PubMed] [Google Scholar]
- 2.Kohrle J. Local activation and inactivation of thyroid hormones: the deiodinase family. Mol Cell Endocrinol. 1999;151:103–19. doi: 10.1016/s0303-7207(99)00040-4. [DOI] [PubMed] [Google Scholar]
- 3.Lacourciere GM, Levine RL, Stadtman TC. Direct detection of potential selenium delivery proteins by using an Escherichia coli strain unable to incorporate selenium from selenite into proteins. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:9150–3. doi: 10.1073/pnas.142291199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burk RF, Hill KE. Selenoprotein-P - a Selenium-Rich Extracellular Glycoprotein. J Nutr. 1994;124:1891–7. doi: 10.1093/jn/124.10.1891. [DOI] [PubMed] [Google Scholar]
- 5.Walczak R, Westhof E, Carbon P, Krol A. A novel RNA structural motif in the selenocysteine insertion element of eukaryotic selenoprotein mRNAs. Rna. 1996;2:367–79. [PMC free article] [PubMed] [Google Scholar]
- 6.Kipp AP, Frombach J, Deubel S, Brigelius-Flohe R. Selenoprotein W as biomarker for the efficacy of selenium compounds to act as source for selenoprotein biosynthesis. Methods Enzymol. 2013;527:87–112. doi: 10.1016/B978-0-12-405882-8.00005-2. [DOI] [PubMed] [Google Scholar]
- 7.Carpino LA, Han GY. 9-Fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group. Journal of the American Chemical Society. 1970;92:5748–9. [Google Scholar]
- 8.Walter R, Du Vigneaud V. 6-Hemi-L-selenocystine-oxytocin and 1-deamino-6-hemi-L-selenocystine-oxytocin, highly potent isologs of oxytocin and 1-deamino-oxytocin. Journal of the American Chemical Society. 1965;87:4192–3. doi: 10.1021/ja01096a036. [DOI] [PubMed] [Google Scholar]
- 9.Metanis N, Keinan E, Dawson PE. Synthetic seleno-glutaredoxin 3 analogues are highly reducing oxidoreductases with enhanced catalytic efficiency. Journal of the American Chemical Society. 2006;128:16684–91. doi: 10.1021/ja0661414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koide T, Itoh H, Otaka A, Yasui H, Kuroda M, Esaki N, et al. Synthetic Study on Selenocystine-Containing Peptides. Chemical & pharmaceutical bulletin. 1993;41:502–6. doi: 10.1248/cpb.41.502. [DOI] [PubMed] [Google Scholar]
- 11.Gieselman MD, Xie LL, van der Donk WA. Synthesis of a selenocysteine-containing peptide by native chemical ligation. Organic Letters. 2001;3:1331–4. doi: 10.1021/ol015712o. [DOI] [PubMed] [Google Scholar]
- 12.Muttenthaler M, Ramos YG, Feytens D, de Araujo AD, Alewood PF. p-Nitrobenzyl protection for cysteine and selenocysteine: a more stable alternative to the acetamidomethyl group. Biopolymers. 2010;94:423–32. doi: 10.1002/bip.21502. [DOI] [PubMed] [Google Scholar]
- 13.Han YBG. Novel S-xanthenyl protecting groups for cysteine and their applications for the N-alpha-9 Fluorenylmethyloxycarbonyl (Fmoc) strategy of peptide synthesis. J Org Chem. 1997;62:3841–8. [Google Scholar]
- 14.Photaki I, Taylor-Papadimitriou J, Sakarellos C, Mazarakis P, Zervas L. On cysteine and cystine peptides. V. S-trityl and S-diphenylmethyl-cysteine and -cysteine peptides. Journal of the Chemical Society Perkin transactions 1. 1970;19:2683–7. doi: 10.1039/j39700002683. [DOI] [PubMed] [Google Scholar]
- 15.Agan M, Schroll A. Characterization of N,N′-bis[(9H-fluoren-9-ylmethoxy)carbonyl]-L-selenocystine. American Chemical Society; 2011. p. CHED-1064. [Google Scholar]
- 16.Flemer S. A Comprehensive One-Pot Synthesis of Protected Cysteine and Selenocysteine SPPS Derivatives. Protein Pept Lett. 2014 ePub ahead of print. [PubMed] [Google Scholar]
- 17.Claudio Santi SS, Testaferri Lorenzo, Tiecco Marcello. A simple zinc mediated preparation of selenols. Synlett. 2008:1471–4. [Google Scholar]
- 18.Schroll AL, Hondal RJ, Flemer S., Jr The use of 2,2′-dithiobis(5-nitropyridine) (DTNP) for deprotection and diselenide formation in protected selenocysteine-containing peptides. Journal of peptide science: an official publication of the European Peptide Society. 2012;18:155–62. doi: 10.1002/psc.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacob C, Giles GI, Giles NM, Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angewandte Chemie. 2003;42:4742–58. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 20.Moroder L. Isosteric replacement of sulfur with other chalcogens in peptides and proteins. Journal of peptide science: an official publication of the European Peptide Society. 2005;11:187–214. doi: 10.1002/psc.654. [DOI] [PubMed] [Google Scholar]
- 21.Marcucci E, Bayo-Puxan N, Tulla-Puche J, Spengler J, Albericio F. Cysteine-S-trityl a key derivative to prepare N-methyl cysteines. Journal of combinatorial chemistry. 2008;10:69–78. doi: 10.1021/cc7001588. [DOI] [PubMed] [Google Scholar]
- 22.Sakakibara M, Katsumata K, Watanabe Y, Toru T, Ueno Y. A convenient procedure for the preparation of organic selenides. Synthesis. 1992:377–89. [Google Scholar]
- 23.Grabel’nykh VA, Russavskaya NV, Korchevin NA, Deryagina EN. Reaction of Diethyl Diselenide with Dihaloalkanes in an Alkaline Reductive Medium. Russian Journal of General Chemistry. 2003;73:1243–5. [Google Scholar]
- 24.Ranu BC, Mandal T, Samanta S. Indium(I) Iodide-Mediated Cleavage of Diphenyl Diselenide. An Efficient One-Pot Procedure for the Synthesis of Unsymmetrical Diorganyl Selenides. Organic Letters. 2003;5:1439–41. doi: 10.1021/ol034178c. [DOI] [PubMed] [Google Scholar]
- 25.Braga AL, Schneider PH, Paixão MW, Deobald AM. Efficient synthesis of diorganyl selenides via cleavage of Se–Se bond of diselenides by indium(III) catalyst and zinc. Tetrahedron letters. 2006;47:7195–8. [Google Scholar]
- 26.Besse D, Moroder L. Synthesis of selenocysteine peptides and their oxidation to diselenide-bridged compounds. Journal of peptide science: an official publication of the European Peptide Society. 1997;3:442–53. doi: 10.1002/(SICI)1099-1387(199711)3:6%3C442::AID-PSC122%3E3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Hanglow AC, Lugo A, Walsky R, Finch-Arietta M, Lusch L, Visnick M, et al. Peptides based on the conserved predomain sequence of matrix metalloproteinases inhibit human stromelysin and collagenase. Agents and actions. 1993;39(Spec No):C148–50. doi: 10.1007/BF01972749. [DOI] [PubMed] [Google Scholar]
- 28.Hagiwara H, Kafuku K, Sakai H, Kirita M, Hoshi T, Suzuki T, et al. Domino Michael-seleno Pummerer type reaction (additive seleno Pummerer reaction) Journal of the Chemical Society, Perkin Transactions 1. 2000:2577–8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.