

Abstract
Photocatalytic formation of glycosidic bonds employing stable and readily accessible O-glycosyl derivatives of 2,2,6,6-tetramethylpiperidin-1-ol is presented that employs an iridium-based photocatalyst and blue LEDs. The reaction proceeds at room temperature and in the absence of additives other than 4 Å molecular sieves. Stereoselectivities are modest but nevertheless dependent on the anomeric configuration of the donor suggesting a substantial degree of concerted character.
Graphical Abstract
Photostimulated glycosylation revolves around the notion of single electron transfer from a suitable electron-rich glycosyl donor to a photostimulated one electron oxidant to give a radical cation-radical anion pair. In competition with back electron transfer, the radical cation expels a radical to give a glycosyl cation or oxocarbenium ion. The oxocarbenium ion is trapped directly either by the acceptor alcohol or by an anion present in the reaction mixture to form a reactive glycosylating species that undergoes reaction with the alcohol (Scheme 1), with the understanding that suitably protected systems may also engage in neighboring group participation. The expelled radical is typically considered to undergo degradation by radical-radical pathways such as dimerization. Alternatively, the expelled radical may suffer reduction by the radical anion resulting from the initial single electron transfer step, thereby rendering the process catalytic in the photostimulated oxidant, as envisaged by early workers in the field,1 and now commonly called photocatalysis. A related concept is the homolytic scission of the anomeric C-S bond in thioglycosides on irradiation with UV light through quartz, followed by oxidation of the formed anomeric radical to the oxocarbenium ion with a stoichiometric oxidant.2,3 Another approach is the use of photochemically stimulated strong acids with which to activate glycosyl donors through protonation and promote more conventional glycosylations.4–6
Scheme 1.

Photoglycosylation Concept
Noyori employed electron rich aryl glycosides (Scheme 1, RX = ArO) and dicyanobenzene as the photostimulated oxidant by irradiation with a 200 W Hg lamp,1 whereas subsequent work focused on the more readily oxidized thio- and selenoglycosides, with the use of charged photosensitizers to minimize back electron transfer,7 irradiation in the visible range of wavelengths,4,8 or both.9 More recently, a number of photocatalytic methods employing visible-light-stimulated transition metal complex-based photo-oxidants and thio- or selenoglycosides have been described.3,10–14 These latter processes employ readily available LEDs emitting visible light and offer broad functional group compatibility. Nevertheless, it is apparent that the simple photocatalytic mechanism envisaged by Noyori1 is not operative in most of these cases and that potent electrophilic species, either generated in situ or added in stoichiometric fashion, contribute to the activation of the glycosyl donor.3,10–14 Consequently, we were attracted by recent work on the photocatalytic generation of simple tertiary alkyl, allyl and benzyl cations from sterically hindered O-alkyl hydroxylamines that proceeds in the absence of stoichiometric additives in which the photocatalytic cycle is closed by reduction of the expelled nitroxyl radical (Scheme 2, R = t-Alkyl).15
Scheme 2.

Photocatalytic Cation Generation from Tempol Ethers and Tempol Glycosides
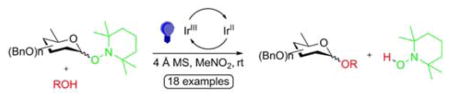
Encouraged by application of the process to O-tetrahydrofuranyl 2,2,6,6-tetramethylpiperidinoxide,15 we considered that this process might be adapted to photocatalytic glycosylation through the use of O-glycosyl 2,2,6,6-tetramethylpiperidinoxides16–17 as donors if the highly destabilizing effect of adjacent C-O bonds on cations18 and oxocarbenium ions19–23 could be overcome. Here we report the successful reduction of this concept of LED-energized photocatalytic glycosylation in the absence of added electrophiles to practice employing O-glycosyl 2,2,6,6-tetramethylpiperidinoxides (Scheme 2, R = glycosyl).
Three α-glycopyranosyl fluorides 1–3 were prepared according to literature methods,24–26 whereas 4 was accessed by hydrolysis of phenyl 2-azido-2-deoxy-3,4,6-tri-O-benzyl β-D-thioglucopyranoside 5 to the corresponding hemiacetal followed by treatment with HF.pyridine according to the method of Noyori et al. (Scheme 3).27
Scheme 3.

Synthesis of Glycosyl Fluoride 4
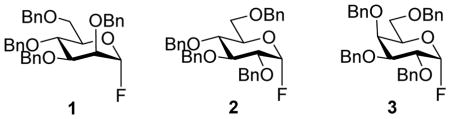
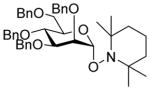
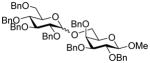
Glycosyl fluorides 1–4 were then coupled to the persistent radical Tempo with activation by BF3.OEt2 in acetonitrile at room temperature in the presence of tetramethylguanidine following the method of Sato.17 Manno-configured donor 1 gave a single α-glycoside 6, whereas gluco- and galacto configured glycosyl fluorides 2–4 afforded separable α,β mixtures of the corresponding glycosides 7–9 (Table 1) by a reaction whose mechanism remains to be clarified.
Table 1.
Synthesis of Tempol Glycosides
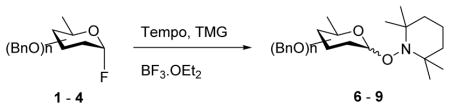
| ||
|---|---|---|
|
| ||
| entry | glycosyl fluorides | tempol glycosides (yield, %) |
| 1 | 1 | 6α (64) and 6β (0) |
| 2 | 2 | 7α (38) and 7β (37) |
| 3 | 3 | 8α (41) and 8β (30) |
| 4 | 4 | 9α (22) and 9β (63) |
Adapting the method of Knowles,15 we photolyzed 0.1 M nitromethane solutions of the various Tempol glycosides with blue LEDs through Pyrex at room temperature in the presence of powdered 4 Å MS, 5 mol % of iridium photocatalyst 10,15 and the model acceptor alcohols 11–13, monitoring with TLC or ESI MS.
Catalyst 10 was selected for this study as the optimum system in the Knowles work15 on tertiary cation generation. Similarly, nitromethane was chosen as solvent by extrapolation from that study;15 indeed, a brief investigation of acetonitrile as solvent revealed it to be far inferior. This preliminary investigation was limited to the use of Tempol derivatives following Knowles,15 but it is recognized that other hydroxylamine derivatives may eventually prove advantageous. Blank reactions conducted in the dark or by photolysis in the absence of 10 did not proceed and indicated the need for 10 and its photolytic activation. Experiments conducted in the absence of molecular sieves were marred by formation of significant amounts of the hydrolyzed donor (hemiacetals) but also proceeded significantly more slowly, suggesting involvement of the sieves in the reaction mechanism.
Table 2 reveals that good to excellent yields were obtained in all couplings conducted in this survey, with only modest differences in yield observed between the α- and β-anomers of a particular donor (compare Table 2, entries 4, 6, and 8 with 5, 7, and 9; and entries 10, 12, and 14 with 11, 13, and 15). Although the anomeric selectivity of the coupling reactions is modest, it is reproducibly a function of the anomeric configuration of the donor (compare Table 2, entries 4, 6, and 8 with 5, 7, and 9; and entries 10, 12, and 14 with 11, 13, and 15). Greater differences in selectivity between the two anomers of a given donor are observed with the more reactive cyclohexanol, which was additionally used in greater excess, and with the primary acceptor 12, than with the less reactive secondary acceptor 13. The implication is that glycosylation proceeds at least in part in a concerted manner in which displacement of the Tempo radical from the oxidatively activated donor is coupled with attack by the incoming acceptor. Such a concerted process can be interpreted as either a direct attack on the activated donor or an attack on the initially formed radical ion triplet before diffusive equilibration (Scheme 4). There is a steadily increasing body of kinetic evidence28–37 that a variety of stereoselective glycosylation reactions proceed by associative reaction pathways rather than by dissociatively free glycosyl oxocarbenium ions that have only been observed in super acidic media.22,23,38,39 In such classical glycosylation reactions, the associative pathways are more prevalent with more reactive acceptor alcohols,40–43 consistent with the observed pattern in Table 2. The hexafluorophosphate counterion is not considered to intervene directly in the reaction mechanism except for providing charge stabilization consistent with studies on the influence of a variety of counterions on the stereoselectivity of glycosylation reactions.38,44 The part of the reaction manifold that affords substitution with overall retention of configuration is best interpreted as taking place via solvent-separated ion triplets rather than via equilibration of the donor configuration followed by concerted displacement as no evidence was found for donor anomerization in these reactions. The root cause of the observed acceleration of reaction rate by the 4 Å MS is presumably associated with their basic character45 and the relative acidity46 of the hydroxylamine Tempol.
Table 2.
Photoglycosylation of Alcohols with Donors 6–9 and Catalytic 10
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | donor | acceptor (equiv) | photolysistime (h) | product | yield, %a α:β-ratiob |
| 1 |
 6α |
 11 (3) |
36 |
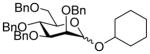 14 |
80, 1.3:1 |
| 2 | 6α |
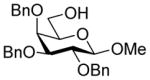 12 (1.5) |
48 |
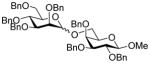 15 |
62, 1.5:1 |
| 3 | 6α |
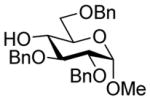 13 (1.5) |
60 |
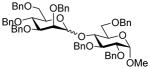 16 |
64, 1:1 |
| 4 |
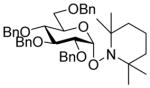 7α |
11 (3) | 36 |
 17 |
96, 1:2.5 |
| 5 |
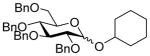
 7β |
11 (5) | 36 | 17 | 91, 1.2:1 |
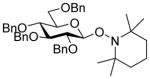
| 6 | 7α | 12 (1.5) | 48 |
 18 |
88, 1:1 |
| 7 | 7β | 12 (1.5) | 48 | 18 | 79, 1.3:1 |
| 8 | 7α | 13 (1.5) | 48 |
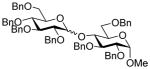 19 |
69, 1.3:1 |
| 9 | 7β | 13 (1.5) | 48 | 19 | 63, 1.6:1 |
| 10 |
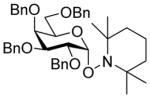 8α |
11 (3) | 36 |
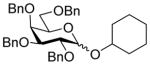 20 |
95, 1:1.6 |
| 11 |
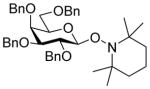 8β |
11 (3) | 36 | 20 | 87, 1.6:1 |
| 12 | 8α | 12 (1.5) | 48 |
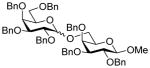 21 |
82, 1.2:1 |
| 13 | 8β | 12 (1.5) | 48 | 21 | 75, 1.4:1 |
| 14 | 8α | 13 (1.5) | 48 |
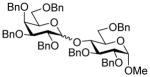 22 |
77, 1.6:1 |
| 15 | 8β | 13 (1.5) | 48 | 22 | 59, 1.6:1 |
| 16 |
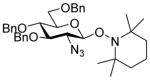 9β |
11 (3) | 36 |
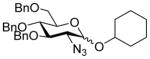 23 |
86, 1.8:1 |
| 17 | 9β | 12 (1.5) | 72 |
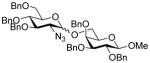 24 |
61, 1.4:1 |
| 18 | 9β | 13 (1.5) | 60 |
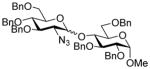 25 |
57, 2:1 |
Combined yield of the isolated anomers.
Anomeric ratios were determined by integration of the 1H NMR spectra of the crude photolysates before purification
Scheme 4.

Concerted and Radical Ion Triplet Mechanisms for Glycosylation
In summary, a new photocatalytic glycosylation method is presented that functions under irradiation through Pyrex by blue LEDs and in the absence of additives other than 10 and 4 Å MS. The method uses stable and readily accessible Tempol glycosides as donors and neither requires nor generates in situ any strongly electrophilic agents. Further work expanding the scope and selectivity of this novel glycosylation is underway in our laboratories.
Supplementary Material
Acknowledgments
We thank the NIH (GM62160) for support of this work, and acknowledge support from the NSF (MRI-084043) for the purchase of the 600 MHz NMR spectrometer in the Lumigen Instrument Center at Wayne State University.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Full experimental details and 1H and 13C NMR spectra for all new compounds (PDF)
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.7b00932.
References
- 1.Hashimoto S, Kurimoto I, Fujii Y, Noyori R. J Am Chem Soc. 1985;107:1427. [Google Scholar]
- 2.Mao RZ, Guo F, Xiong DC, Li Q, Duan J, Ye XS. Org Lett. 2015;17:5606. doi: 10.1021/acs.orglett.5b02823. [DOI] [PubMed] [Google Scholar]
- 3.Mao RZ, Xiong DC, Guo F, Li Q, Duan J, Ye XS. Org Chem Front. 2016;3:737. [Google Scholar]
- 4.Nakanishi M, Takahashi D, Toshima K. Org Biomol Chem. 2013;11:5079. doi: 10.1039/c3ob41143e. [DOI] [PubMed] [Google Scholar]
- 5.Iwata R, Uda K, Takahashi D, Toshima K. Chem Commun. 2014:10695. doi: 10.1039/c4cc04753b. [DOI] [PubMed] [Google Scholar]
- 6.Kimura T, Eto T, Takahashi D, Toshima K. Org Lett. 2016;18:3190. doi: 10.1021/acs.orglett.6b01404. [DOI] [PubMed] [Google Scholar]
- 7.Furuta T, Takeuchi K, Iwamura M. Chem Commun. 1996:157. [Google Scholar]
- 8.Griffin GW, Bandara NC, Clarke MA, Tsang WS, Garegg PJ, Oscarson S, Silwanis BA. Heterocycles. 1990;30:939. [Google Scholar]
- 9.Cumpstey I, Crich D. J Carbohydr Chem. 2011;30:469. [Google Scholar]
- 10.Wever WJ, Cinelli MA, Bowers AA. Org Lett. 2013;15:30. doi: 10.1021/ol302941q. [DOI] [PubMed] [Google Scholar]
- 11.Spell M, Wang X, Wahba AE, Conner E, Ragains J. Carbohydr Res. 2013;369:42. doi: 10.1016/j.carres.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Yuan X, Cheng S, Shi Y, Xue W. Synthesis. 2014;46:331. [Google Scholar]
- 13.Yu Y, Xiong DC, Mao RZ, Ye XS. J Org Chem. 2016;81:7134. doi: 10.1021/acs.joc.6b00999. [DOI] [PubMed] [Google Scholar]
- 14.Spell ML, Deveaux K, Bresnahan CG, Bernard BL, Sheffield W, Kumar R, Ragains JR. Angew Chem, Int Ed. 2016;55:6515. doi: 10.1002/anie.201601566. [DOI] [PubMed] [Google Scholar]
- 15.Zhu Q, Gentry EC, Knowles RR. Angew Chem, Int Ed. 2016;55:9969. doi: 10.1002/anie.201604619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kancharla PK, Kato T, Crich D. J Am Chem Soc. 2014;136:5472. doi: 10.1021/ja501276r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato S, Kumazawa T, Matsuba S, Onodera J-i, Aoyama M, Obara H, Kamada H. Carbohydr Res. 2001;334:215. doi: 10.1016/s0008-6215(01)00184-7. [DOI] [PubMed] [Google Scholar]
- 18.Lambert JB, Mark HW. J Am Chem Soc. 1978;100:2501. [Google Scholar]
- 19.Glaudemans CPJ, Fletcher HG. J Am Chem Soc. 1965;87:4636. [Google Scholar]
- 20.Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B. J Am Chem Soc. 1988;110:5583. [Google Scholar]
- 21.Douglas NL, Ley SV, Lucking U, Warriner SL. J Chem Soc, Perkin Trans. 1998;1:51. [Google Scholar]
- 22.Bohé L, Crich D. C R Chimie. 2011;14:3. [Google Scholar]
- 23.Martin A, Arda A, Désiré J, Martin-Mingot A, Probst N, Sinaÿ P, Jiménez-Barbero J, Thibaudeau S, Blériot Y. Nat Chem. 2015;8:186. doi: 10.1038/nchem.2399. [DOI] [PubMed] [Google Scholar]
- 24.Zeng J, Vedachalam S, Xiang S, Liu XW. Org Lett. 2011;13:42. doi: 10.1021/ol102473k. [DOI] [PubMed] [Google Scholar]
- 25.López JC, Uriel C, Guillamón-Martín A, Valverde S, Gómez AM. Org Lett. 2007;9:2759. doi: 10.1021/ol070753r. [DOI] [PubMed] [Google Scholar]
- 26.López JC, Bernal-Albert P, Uriel C, Valverde S, Gómez AM. J Org Chem. 2007;72:10268. doi: 10.1021/jo7018653. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi M, Hashimoto S, Noyori R. Chem Lett. 1984;13:1747. [Google Scholar]
- 28.Crich D. Acc Chem Res. 2010;43:1144. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 29.Huang M, Garrett GE, Birlirakis N, Bohé L, Pratt DA, Crich D. Nat Chem. 2012;4:663. doi: 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang M, Retailleau P, Bohé L, Crich D. J Am Chem Soc. 2012;134:14746. doi: 10.1021/ja307266n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adero PO, Furukawa T, Huang M, Mukherjee D, Retailleau P, Bohé L, Crich D. J Am Chem Soc. 2015;137:10336. doi: 10.1021/jacs.5b06126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Angelo KA, Taylor MS. J Am Chem Soc. 2016;138:11058. doi: 10.1021/jacs.6b06943. [DOI] [PubMed] [Google Scholar]
- 33.Khomutnyk YY, Argüelles AJ, Winschel GA, Sun Z, Zimmerman PM, Nagorny P. J Am Chem Soc. 2016;138:444. doi: 10.1021/jacs.5b12528. [DOI] [PubMed] [Google Scholar]
- 34.Wurst JM, Liu G, Tan DS. J Am Chem Soc. 2011;133:7916. doi: 10.1021/ja201249c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hosoya T, Takano T, Kosma P, Rosenau T. J Org Chem. 2014;79:7889. doi: 10.1021/jo501012s. [DOI] [PubMed] [Google Scholar]
- 36.Hosoya T, Kosma P, Rosenau T. Carbohydr Res. 2015;411:64. doi: 10.1016/j.carres.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 37.Kwan EE, Park Y, Besser HA, Anderson TL, Jacobsen EN. J Am Chem Soc. 2017;139:43. doi: 10.1021/jacs.6b10621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bohé L, Crich D. Carbohydr Res. 2015;403:489. doi: 10.1016/j.carres.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bohé L, Crich D. Nat Chem. 2016;8:99. doi: 10.1038/nchem.2436. [DOI] [PubMed] [Google Scholar]
- 40.Krumper JR, Salamant WA, Woerpel KA. J Org Chem. 2009;74:8039. doi: 10.1021/jo901639b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beaver MG, Woerpel KA. J Org Chem. 2010;75:1107. doi: 10.1021/jo902222a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Vorm S, Hansen T, Overkleeft HS, van der Marel GA, Codée JDC. Chem Sci. 2017;8:1867. doi: 10.1039/c6sc04638j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hagen B, Ali S, Overkleeft HS, van der Marel GA, Codée JDC. J Org Chem. 2017;82:848. doi: 10.1021/acs.joc.6b02593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hasty SJ, Ranade SC, Demchenko AV. Reports Org Chem. 2014;4:1. [Google Scholar]
- 45.Kartha KPR, Mukhopadhyay B, Field RA. Carbohydr Res. 2004;339:729. doi: 10.1016/j.carres.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 46.Bartmess JE. In: Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids. Rappoport Z, Liebman JF, editors. Vol. 2. Wiley; Chichester, U.K: 2011. pp. 115–143. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.