

SUMMARY
Homeostatic signaling systems are thought to interface with other forms of plasticity to ensure flexible yet stable levels of neurotransmission. The role of neurotransmitter receptors in this process, beyond mediating neurotransmission itself, is not known. Through a forward genetic screen, we have identified the Drosophila kainate-type ionotropic glutamate receptor subunit DKaiR1D to be required for the retrograde, homeostatic potentiation of synaptic strength. DKaiR1D is necessary in presynaptic motor neurons, localized near active zones, and confers robustness to the calcium sensitivity of baseline synaptic transmission. Acute pharmacological blockade of DKaiR1D disrupts homeostatic plasticity, indicating that this receptor is required for the expression of this process, distinct from developmental roles. Finally, we demonstrate that calcium permeability through DKaiR1D is necessary for baseline synaptic transmission, but not for homeostatic signaling. We propose that DKaiR1D is a glutamate autoreceptor that promotes robustness to synaptic strength and plasticity with active zone specificity.
Graphical Abstract
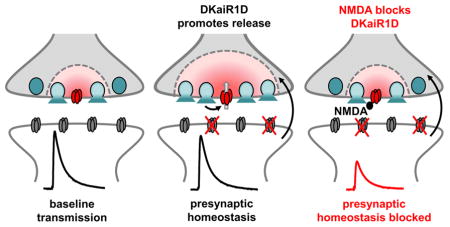
INTRODUCTION
The nervous system is endowed with potent and adaptive homeostatic signaling systems that maintain stable functionality despite the myriad changes that occur during neural development and maturation (Davis and Muller, 2015; Pozo and Goda, 2010). The importance of homeostatic regulation in the nervous system is underscored by associations with a variety of neurological diseases (Wondolowski and Dickman, 2013), yet the genes and mechanisms involved remain enigmatic. A powerful model of presynaptic homeostatic plasticity has been established at the Drosophila neuromuscular junction (NMJ), a model glutamatergic synapse with molecular machinery that parallels central synapses in mammals. Here, genetic and pharmacological manipulations that reduce postsynaptic (muscle) glutamate receptor function trigger a trans-synaptic, retrograde feedback signal to the neuron that increases presynaptic release to precisely compensate for this perturbation (Frank, 2013). This process is referred to as presynaptic homeostatic potentiation (PHP), because the expression mechanism requires a presynaptic increase in neurotransmitter release.
In recent years, forward and candidate genetic approaches have revealed several new and unanticipated genes necessary for PHP expression (Frank, 2013). While perturbations to the glutamate receptors in muscle are crucial events in the induction of PHP (Frank et al., 2006; Petersen et al., 1997), whether other ionotropic glutamate receptors (iGluRs) function in PHP or are even expressed at the Drosophila NMJ is unknown. Finally, although evidence has emerged that homeostatic modulation is synapse-specific (Davis and Goodman, 1998; Newman et al., 2017), no roles for neurotransmitter receptors or other factors have been found to enable the presynaptic tuning of release efficacy at individual synapses.
We have identified the kainate-type iGluR subunit DKaiR1D to be necessary for PHP expression at the Drosophila NMJ. We find that DKaiR1D is necessary for the calcium sensitivity of baseline synaptic transmission, as well as for the acute and chronic expression of homeostatic potentiation. Recently, the functional reconstitution of DKaiR1D was achieved in heterologous cells, revealing that these receptors form homomeric calcium permeable channels with atypical pharmacological properties compared to their vertebrate homologs (Li et al., 2016). We find that DKaiR1D is expressed in the nervous system and not the muscle, is present near presynaptic active zones, and is required specifically in motor neurons to enable the robustness of baseline neurotransmission and homeostatic plasticity. We propose that glutamate activates DKaiR1D at presynaptic release sites to translate autocrine activity into the robust stabilization of synaptic strength with active zone specificity.
RESULTS
DKaiR1D encodes a neural kainate-type glutamate receptor subunit
In the course of an electrophysiology-based, forward genetic screen to isolate genes necessary for PHP expression (Dickman and Davis, 2009), we identified a mutant that failed to homeostatically increase presynaptic release following acute application of Philanthotoxin-433 (PhTx), a drug that specifically blocks postsynaptic glutamate receptors at the Drosophila NMJ (Frank et al., 2006). Within 10 mins following application of this antagonist, mEPSP amplitudes are reduced but EPSP amplitudes are maintained at baseline values because of a homeostatic increase in presynaptic release (Frank et al., 2006). This mutation contained a transposon insertion into an intronic region of a gene, DKaiR1D, predicted to encode an ionotropic, kainate-type glutamate receptor subunit (Fig. 1A). We named this allele DKaiR1D1. We identified a second, independent transposon inserted into a coding exon of DKaiR1D (now named DKaiR1D2), as well as a deficiency which removes the entire open reading frame.
Figure 1. DKaiR1D, a neural kainate-type glutamate receptor subunit, is required in motor neurons for the acute and long term expression of presynaptic homeostatic potentiation.

(A) Diagram of two transposon insertions in the DKaiR1D locus (DKaiR1D1 and DKaiR1D2), and a deficiency that removes this entire locus (DKaiR1DDf; Df(3R)BSC819). (B) in situ hybridization of Drosophila embryos reveals that DKaiR1D is expressed in the central nervous system, with no apparent expression in other tissues. Sense strand DNA probe is used as the control. (C) Immunoblot analysis confirms nervous system expression of DKaiR1D protein and demonstrates that both mutant alleles are protein nulls. (D) Representative EPSP and mEPSP traces from electrophysiological recordings of wild type (w1118) and DKaiR1D mutant synapses (DKaiR1D2: w1118;Mi{ET1}CG3822MB01010) before and after PhTx application at 0.4 mM extracellular Ca2+. EPSP amplitude fails to return to baseline levels in DKaiR1D mutants following PhTx application because there is no homeostatic increase in presynaptic release (quantal content). (E) Quantification of mEPSP amplitude and quantal content values after PhTx treatment, normalized to baseline values of the same genotype. (F) Representative EPSP and mEPSP traces of motor neuron rescue (MN rescue: w;OK6-Gal4/UAS-DKaiR1D;DKaiR1D2) and muscle rescue (muscle rescue: w;G14-Gal4/UAS-DKaiR1D;DKaiR1D2) by tissue-specific expression of DKaiR1D in the DKaiR1D2 mutant background following PhTx application at 0.4 mM extracellular Ca2+. (G) Quantification of mEPSP and quantal content values normalized to baseline values of the same genotype. (H) The chronic expression of PHP, induced by loss of the postsynaptic GluRIIA receptor subunit, requires DKaiR1D, and can be restored by presynaptic expression of DKaiR1D. Quantification of mEPSP amplitude and quantal content values at 0.4 mM extracellular Ca2+ in the indicated mutant genotypes (w;GluRIIASP16 and w;GluRIIASP16;DKaiR1D2) and MN rescue (w;OK6-Gal4,GluRIIASP16/UAS-DKaiR1D, GluRIIASP16;DKaiR1D2). (I) Representative EPSP and mEPSP traces of wild type and DKaiR1D mutant synapses incubated with NSTX-3, another neurotoxin that targets Drosophila postsynaptic glutamate receptors. NSTX-3 was either applied alone or together with PhTx at 0.4 mM extracellular Ca2+. Note that while NSTX-3 induces PHP at wild-type NMJs, PHP fails to be expressed in DKaiR1D mutants. (J) Quantification of mEPSP amplitude and quantal content after NSTX-3 application (as well as with and without PhTx), normalized to baseline values of the same genotype. No significant differences were observed between NSTX-3 and PhTx treatments in wild type or DKaiR1D mutant synapses. Error bars indicate ±SEM. Asterisks indicate statistical significance using one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparison test: (*) p<0.05; (**) p<0.01; (***) p<0.001; (ns) not significant. Detailed statistical information for represented data (mean values, SEM, n, p) is shown in Table S1.
Phylogenetic analysis revealed that DKaiR1D is distinct from the five iGluR subunits expressed in the Drosophila muscle that drive the postsynaptic response to presynaptic glutamate release (Li et al., 2016). There is evidence that DKaiR1D functions in the adult fly visual system (Karuppudurai et al., 2014), but DKaiR1D has not been investigated at the NMJ and its expression pattern is unknown. We therefore performed in situ hybridization in embryos to determine DKaiR1D mRNA expression, which demonstrated that DKaiR1D was exclusively expressed in the nervous system (Fig. 1B). Finally, we generated an antibody against DKaiR1D which revealed a single 95 KDa band by immunoblot analysis, corresponding to the predicted size of DKaiR1D (Fig. 1C). This band was observed in larval brain lysates, while no detectable signal was found in lysates made from larval muscle (Fig. 1C). Further, both DKaiR1D1 and DKaiR1D2 alleles are protein nulls, as no expression was detected in brain lysates from these mutants. Thus, we have identified two independent null alleles of DKaiR1D, a kainate receptor expressed in the nervous system and putatively required for presynaptic homeostatic potentiation.
DKaiR1D is required in motor neurons for the acute and chronic expression of presynaptic homeostatic potentiation
iGluRs have been shown to function in baseline synaptic transmission, contributing to both postsynaptic currents and presynaptic facilitation (Lerma and Marques, 2013). We therefore characterized baseline synaptic transmission in addition to homeostatic plasticity in DKaiR1D mutants. We observed no significant change in mEPSP amplitude in DKaiR1D mutants, consistent with DKaiR1D being expressed presynaptically and not present in the muscle to mediate the postsynaptic responsiveness to glutamate release (Fig. 1D). Baseline EPSP amplitudes were slightly reduced in DKaiR1D mutants. However, following acute inhibition of postsynaptic glutamate receptors by application of PhTx, mEPSP values were reduced, and EPSP amplitudes were also reduced, indicating no adaptive increase in presynaptic release (quantal content) in either mutant allele alone, or in mutant alleles in trans to a deficiency that removes the entire DKaiR1D locus (Fig. 1D,E). Next, we asked if DKaiR1D expression is necessary cell autonomously in motor neurons for PHP expression. Expression of DKaiR1D specifically in motor neurons restored PHP expression in DKaiR1D mutants, while PHP remained disrupted when DKaiR1D was expressed in muscle (Fig. 1F,G). Thus, DKaiR1D expression is necessary in motor neurons for the acute expression of PHP.
PhTx was shown to inhibit reconstituted DKaiR1D homomers in vitro (Li et al., 2016), so we performed several experiments to test whether PhTx influenced DKaiR1D receptors in vivo. First, mutations in the postsynaptic GluRIIA receptor subunit is a genetic means of inducing PHP expression, independently of PhTx application. Loss of GluRIIA leads to a chronic reduction in mEPSP amplitude throughout larval development and a robust compensatory increase in presynaptic release (Fig. 1H). In GluRIIA, DKaiR1D double mutants, we observed reduced mEPSPs but no increase in presynaptic release compared to DKaiR1D mutants alone, confirming that DKaiR1D is required for PHP expression over chronic time scales and, importantly, independently of PhTx application (Fig. 1H). Further, PHP expression is restored in GluRIIA, DKaiR1D mutants when DKaiR1D expression is driven in motor neurons (Fig. 1H), as expected. Next, we confirmed that application of PhTx to GluRIIA mutants does not impact mEPSP amplitudes, while PHP is robustly expressed (Fig. S1D–H), as shown previously (Frank et al., 2006). This indicates that the only physiological target of PhTx in the conditions we are using are GluRIIA-containing postsynaptic glutamate receptors. Finally, we used a second drug to block postsynaptic GluRIIA-containing receptors, NSTX-3 (Frank et al., 2006), which has no reported specificity for DKaiR1D receptors. Application of NSTX-3 reduced mEPSP values to the same level in the absence or presence of PhTx in both wild type and DKaiR1D mutant synapses (Fig. 1I,J and Fig. S1A–C). Importantly, while PHP was robustly expressed at wild-type NMJs following NSTX-3 application, PHP was blocked in DKaiR1D mutants, and PhTx application had no additional impact (Fig. 1I,J and Fig. S1A–C). Taken together, there is no evidence that DKaiR1D receptors are targets of PhTx in vivo, and PHP expression requires DKaiR1D independently of PhTx application.
Finally, we also examined synaptic growth and structure in DKaiR1D mutants by immunostaining synaptic structures at the NMJ. We found no significant difference in the number or area of synaptic boutons or active zones in DKaiR1D mutants compared with controls (Fig. S2). This indicates there are no obvious changes to synaptic growth or structure in DKaiR1D mutants that may contribute to the inability to express PHP. Thus, DKaiR1D is a glutamate receptor required in motor neurons for both the acute and chronic expression of presynaptic homeostatic potentiation.
Altered calcium cooperativity and short term plasticity in DKaiR1D mutants
Presynaptic iGluRs modulate neurotransmission in rodent systems, particularly during high levels of activity (Kamiya, 2002; Pinheiro and Mulle, 2008). We therefore examined baseline synaptic transmission in DKaiR1D mutants in more detail. Recording in reduced extracellular calcium (0.2 mM) revealed a decrease in EPSP amplitude in DKaiR1D mutants compared to wild type (Fig. 2A) and an apparent increase in the calcium cooperativity of synaptic transmission (Fig. 2B). We went on to probe short term plasticity in lowered extracellular calcium, where wild-type synapses show a moderate facilitation in synaptic transmission when stimulated at 20 Hz (Fig. 2C). Consistent with reduced initial release probability in DKaiR1D mutants, facilitation was markedly increased, as has been observed in other homeostatic mutants (Dickman and Davis, 2009; Younger et al., 2013). Together, these experiments reveal increased calcium cooperativity and facilitation at DKaiR1D mutant synapses.
Figure 2. Altered calcium cooperativity and short term plasticity in DKaiR1D mutant synapses.

(A) Representative electrophysiological recordings at 0.2 mM extracellular Ca2+ reveals reduced baseline transmission in DKaiR1D2 mutants. (B) Quantal content in wild type, DKaiR1D1, and DKaiR1D2 mutants plotted as a function of extracellular Ca2+concentration on logarithmic scales. Note that DKaiR1D mutants have increased apparent slopes (wild type= 1.002; DKaiR1D1=2.248 (***), and DKaiR1D2=2.126 (***)). (C) Increased short term facilitation is observed in DKaiR1D2 mutants. EPSP values at each stimulus are normalized to the starting EPSP value during a train of 50 stimuli at 20 Hz. (D) Representative traces of homeostatically depressed synapses induced by presynaptic overexpression of the vesicular glutamate transporter alone (vGlut-OE: w;OK371-Gal4/UAS-vGlut) or in combination with the DKaiR1D mutation (DKaiR1D+vGlut-OE: w;OK371-Gal4/UAS-vGlut;DKaiR1D2). Note the increased mEPSP amplitude but normal EPSP amplitude, indicating a homeostatic decrease in quantal content. (E) Quantification of mEPSP amplitude and quantal content values normalized as a percentage of wild-type values. (F) Representative mEPSP and EPSP traces of the indicated genotypes at 0.2 mM extracellular Ca2+ concentrations. Note the large reduction in EPSP amplitude in DKaiR1D+vGlut-OE. Error bars indicate ±SEM. Asterisks indicate statistical significance using one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparison test: (*) p<0.05; (**) p<0.01; (***) p<0.001; (ns) not significant. Detailed statistical information for represented data (mean values, SEM, n, p) is shown in Table S1.
An inverse process to PHP, referred to as presynaptic homeostatic depression (PHD), has been demonstrated at the Drosophila NMJ. Here, excess glutamate release is observed through overexpression of the vesicular glutamate transporter (vGlut; (Daniels et al., 2004)). This leads to increased synaptic vesicle and quantal size but normal EPSP amplitude, due to a homeostatic decrease in presynaptic release (Daniels et al., 2004; Gavino et al., 2015). Some have speculated that PHD utilizes a presynaptic glutamate receptor as part of a homeostatic feedback sensor and signaling mechanism (Daniels et al., 2004; Frank, 2013), and we tested whether DKaiR1D receptors may subserve this role. Overexpression of vGlut (vGlut-OE) in motorneurons led to the expected increase in mEPSP amplitude and homeostatic reduction in quantal content (Fig. 2D,E). Similarly, in DKaiR1D mutants overexpressing vGlut (DKaiR1D+vGlut-OE), mEPSP amplitudes were increased and quantal content was similarly reduced. Finally, in lowered extracellular calcium, quantal content and EPSP amplitudes were further reduced in DKaiR1D+vGlut-OE mutants compared to vGlut-OE alone (Fig. 2F). Both DKaiR1D mutants and vGlut-OE lead to independent reductions in release probability, so a further reduction at lowered extracellular calcium when these manipulations are combined may be expected, given that vGlut-OE involves a reduction in presynaptic calcium influx (Gavino et al., 2015). Thus DKaiR1D, while necessary for PHP expression, is dispensable for the expression of PHD.
Elevated extracellular calcium restores PHP expression in DKaiR1D mutants
Since DKaiR1D mutants exhibited a pronounced reduction in release at lowered extracellular calcium, we next probed baseline synaptic transmission and PHP at elevated extracellular calcium using two-electrode voltage clamp (TEVC). As expected, baseline transmission was unperturbed in DKaiR1D mutants in this condition. However, PHP expression was now restored in DKaiR1D mutants, with GluRIIA, DKaiR1D double mutants showing similar EPSC amplitudes compared with GluRIIA mutants alone (Fig. 3A,B). This suggests that the requirement of DKaiR1D in both PHP and baseline transmission is sensitive to extracellular calcium, where lowered calcium highlights the importance of DKaiR1D in facilitating baseline neurotransmission and PHP, while elevated calcium circumvents the need for DKaiR1D in these processes.
Figure 3. Elevated extracellular Ca2+ restores PHP expression in DKaiR1D mutants.

(A) Representative EPSC traces of the indicated genotypes. Although PHP is blocked in DKaiR1D mutants at lower extracellular calcium (0.4 mM), PHP expression is restored in high calcium (3 mM). (B) Quantification of EPSC amplitudes in the indicated genotypes. (C) Representative EPSC traces of 30 stimuli at 60 Hz. (D) Estimated RRP sizes for the indicated genotypes under baseline and in GluRIIA mutant backgrounds. (E) Recordings from wild type and DKaiR1D mutants using high sucrose (420 nM for 3s, red bar) to evoke vesicle release. (F) Quantification of mEPSP events per second as a measure of the hypertonic sucrose-sensitive vesicle pool. Error bars indicate ±SEM. Asterisks indicate statistical significance using one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparison test: (*) p<0.05; (**) p<0.01; (***) p<0.001; (ns) not significant. Detailed statistical information for represented data (mean values, SEM, n, p) is shown in Table S1.
One key expression mechanism underlying PHP is a homeostatic increase in the readily releasable synaptic vesicle pool (RRP). This pool is defined as the number of vesicles available for immediate release, and has been shown to be homeostatically modulated following PhTx application and necessary for the expression of PHP (Muller et al., 2012; Weyhersmuller et al., 2011). The state of the RRP has not been determined in GluRIIA mutants. We therefore investigated the RRP in wild type, GluRIIA, and DKaiR1D mutants, as well as in GluRIIA; DKaiR1D double mutants. Using TEVC in 3 mM extracellular calcium, we estimated the RRP size by back extrapolating the cumulative EPSC amplitude following 30 stimuli at 60 Hz (Fig. 3C,D; see methods). Both GluRIIA and GluRIIA; DKaiR1D double mutants exhibited a robust increase in the RRP relative to baseline (Fig. 3D), consistent with no defect in PHP expression in DKaiR1D mutants at high extracellular calcium following homeostatic challenge. Finally, we controlled for the possibility that the vesicle pool size may change in DKaiR1D mutants at high and low calcium conditions. We measured the sucrose-sensitive synaptic vesicle pool using hypertonic sucrose in zero extracellular calcium, and observed no significant difference compared with controls (Fig. 3E,F). Together, this indicates that while PHP expression is completely blocked in lowered extracellular calcium, elevated calcium restores the expression of PHP along with the expected RRP modulation in DKaiR1D mutants.
DKaiR1D localizes to synaptic neuropil and presynaptic terminals
The most obvious sources of glutamate that would activate DKaiR1D in motor neurons are at dendrites, where glutamatergic inputs may signal to postsynaptic DKaiR1D receptors, or at presynaptic terminals, where DKaiR1D receptors at or near release sites may respond to synaptically released glutamate in an autocrine mechanism. Indeed, DKaiR1D has been suggested to function postsynaptically in dendrites in the Drosophila visual system (Karuppudurai et al., 2014), while rodent kainate and NMDA receptors function presynaptically near active zones to modulate synaptic transmission (Bouvier et al., 2015; Chittajallu et al., 1996; Pinheiro et al., 2007). To determine the subcellular expression of DKaiR1D, we used antibody staining in third-instar larvae. The polyclonal antibody we generated against DKaiR1D revealed specific expression in a broad, synapse-rich region of the central nervous system in the larval ventral nerve cord (Fig. 4A). This signal highly overlapped with the active zone marker BRP, and was absent in DKaiR1D mutants (Fig. 4A,B), indicating that in the central nervous system, DKaiR1D traffics to synaptic neuropil. While the majority of synaptic inputs onto motor neurons are cholinergic (Baines and Bate, 1998; Baines et al., 1999; Daniels et al., 2008), and the glutamate that is released onto motor neurons is thought to be inhibitory through activation of glutamate-gated chloride channels (Rohrbough and Broadie, 2002), we cannot rule out the possibility that DKaiR1D may be present in the dendrites of motor neurons.
Figure 4. Endogenous DKaiR1D receptors localize to synaptic neuropil and to presynaptic terminals when overexpressed.

Representative images of the ventral nerve cord (VNC) in wild type (A) and DKaiR1D mutants (B) immunostained with anti-DKaiR1D and anti-BRP. DKaiR1D is enriched in synapse-rich areas of the neuropil (highlighted by BRP signal), and is absent in DKaiR1D mutants. (C) DKaiR1D puncta are observed near BRP positive active zones at presynaptic NMJ terminals when overexpressed in motor neurons (DKaiR1D-OE: w;OK6-Gal4/UAS-DKaiR1D) and immunostained with anti-DKaiR1D and anti-BRP. Quantification of BRP and DKaiR1D puncta density in DKaiR1D-OE (D), percent BRP puncta co-localized with DKaiR1D puncta in wild-type synapses and DKaiR1D-OE (E), and fluorescence intensities of BRP and DKaiR1D puncta normalized to wild type backgrounds (F). Error bars indicate ±SEM. Asterisks indicate statistical significance using t-test: (*) p<0.05; (**) p<0.01; (***) p<0.001; (ns) not significant. Detailed statistical information for represented data (mean values, SEM, n, p) is shown in Table S1.
We then stained NMJs to determine whether DKaiR1D was present at presynaptic terminals of motor neurons. We were unable to detect a consistent endogenous DKaiR1D signal in wild-type motor neuron terminals (data not shown). However, following overexpression of DKaiR1D in motor neurons, we observed a punctate signal at presynaptic NMJ terminals, which localized at or near active zones labeled by BRP (Fig. 4C–F). Interestingly, only a subset of BRP positive active zones colocalized with DKaiR1D puncta (Fig. 4E), suggesting a heterogeneity of DKaiR1D presence and position relative to individual active zones. We observed no significant changes in the density or intensity of BRP signals at active zones following DKaiR1D overexpression (Fig. 4D–F, Table S1). Given that overexpressed DKaiR1D can traffic near active zones, we examined active zone ultrastructure in DKaiR1D mutants, but did not detect any significant changes in NMJ ultrastructure in DKaiR1D mutants (Fig. S3). Together, this demonstrates that DKaiR1D traffics to synapses in central neuropil and to presynaptic NMJ terminals where it could, in principle, respond to synaptically released glutamate.
Glutamate uncaging triggers calcium influx at active zones through DKaiR1D
DKaiR1D receptors were demonstrated to be calcium permeable in vitro (Li et al., 2016), and we next sought to utilize calcium imaging to test for the functional presence of endogenous presynaptic DKaiR1D activity at active zones. Conventional presynaptic calcium imaging uses action-potential evoked stimulation to elicit presynaptic calcium influx through voltage gated calcium channels at active zones (Muller and Davis, 2012; Yao et al., 2017). This standard approach would be unlikely to specifically detect the calcium signal through DKaiR1D receptors for two reasons. First, much less calcium is passed through DKaiR1D receptors compared to the voltage gated calcium channels that trigger synaptic vesicle release (illustrated in Fig. S4). Second, conventional calcium imaging necessarily measures the spatially averaged calcium signal across the entire synaptic bouton, and not the specific signal at individual active zones. Further, the contribution of calcium through DKaiR1D receptors is likely non-uniform, given the heterogeneity of DKaiR1D expression and localization relative to active zones (Fig. 4). Thus, we sought an alternative means to image presynaptic calcium influx through DKaiR1D at active zones independently of action potential-evoked activity.
We developed a genetically encoded ratiometric calcium indicator targeted specifically to active zones. We first engineered a transgene in which we fused GCaMP6s (Chen et al., 2013) to the red-shifted, calcium insensitive fluorophore mCherry (Fig. 5A). This enables the ratiometric analysis of the calcium-dependent GCaMP6s signal to that of a constant mCherry signal. This indicator would also permit the visualization of active zones at rest if targeted to release sites. To target this indicator specifically to active zones, we inserted a short fragment of the active zone scaffold BRP to the GCaMP6s::mCherry fusion. Fluorophores fused to BRP-short (BRPs) localize specifically to active zones at the Drosophila NMJ (Schmid et al., 2008). Expression of this transgene in motor neurons revealed co-localization of BRP, mCherry, and GCaMP6s signals, as expected (Fig. 5B). Finally, to uncouple presynaptic calcium influx through voltage-gated calcium channels and DKaiR1D receptors, we used photo-uncaging of glutamate instead of electrical stimulation to trigger extracellular glutamate release.
Figure 5. Glutamate uncaging induces calcium influx through DKaiR1D receptors at active zones.

(A) Schematic illustrating the design of the BRPs::mCherry::GCaMP6s active zone calcium indicator. (B) mCherry and GCaMP6s (immunostained with anti-GFP) localize to BRP-positive active zones (immunostained with anti-BRP) when this indicator is expressed in motor neurons (w;OK6-Gal4/UAS-BRPs::mCherry::GCaMP6s). (C) Glutamate uncaging at motor neuron terminals expressing BRPs::mCherry::GCaMP6s reveals a GCaMP signal at individual active zones (visualized with mCherry fluorescence) in 2 mM extracellular calcium. No change in the GCaMP signal is observed when glutamate is uncaged in the presence of the DKaiR1D antagonist NMDA (1 mM). (D) Quantification of GCaMP and mCherry fluorescence intensities (ΔF/F) at individual active zones following glutamate photo-uncaging in 2 mM extracellular Ca2+, 0 mM extracellular Ca2+, 2 mM Ca2+ plus NMDA (1 mM extracellular NMDA), and 2 mM Ca2+ in DKaiR1D2 mutants. (E) Quantification of GCaMP6s/mCherry fluorescence ratios at 2 mM and 0 mM extracellular Ca2+. (F) Quantification of the percentage of synapses responding to glutamate photo uncaging (+405 nm) imaged in 2 mM extracellular Ca2+ with and without 1 mM NMDA and in DKaiR1D2 mutants. Error bars indicate ±SEM. Asterisks indicate statistical significance using one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparison test: (*) p<0.05; (**) p<0.01; (***) p<0.001; (ns) not significant. Detailed statistical information for represented data (mean values, SEM, n, p) is shown in Table S1.
Glutamate uncaging at motor neuron terminals expressing BRPs::mCherry::GCaMP6s revealed a GCaMP6s signal at individual mCherry-positive active zones in 2 mM calcium (Fig. 5C,D,E). These calcium transients did not occur spontaneously in the absence of photo-stimulation, and consistently failed to be evoked in 0 mM extracellular calcium (Fig. 5D,E). Indeed, we found that only a subset of mCherry-positive active zones were responsive to photo-stimulation (Fig. 5C,F), consistent with DKaiR1D receptors being heterogeneously localized at BRP-positive active zones. Lastly, to test whether this signal was due to glutamate activating DKaiR1D receptors rather than an alternative glutamate-responsive target at presynaptic terminals, we took advantage of both DKaiR1D pharmacology and genetics. NMDA was reported to be an antagonist of DKaiR1D in heterologous systems (Li et al., 2016), and we confirmed this in vivo (Fig. 6). We therefore repeated glutamate uncaging in the presence of NMDA in the extracellular saline to block DKaiR1D receptors. NMDA application reduced the frequency of calcium signal responses after photo-uncaging from over 75% to below 20% (Fig. 5F). We also genetically validated that these calcium transients consistently fail to be evoked in DKaiR1D mutant synapses (Fig. 5D,F). Together, these experiments demonstrate that endogenous DKaiR1D receptors are capable of passing calcium in response to extracellular glutamate near presynaptic active zones at the Drosophila NMJ.
Figure 6. Acute pharmacological blockade of DKaiR1D disrupts the expression but not induction of PHP.

(A) NMDA application to wild-type NMJs reduces baseline transmission in 0.2 mM extracellular calcium, but has no effect on DKaiR1D mutants in this condition. Representative mEPSP and EPSP traces in wild type and DKaiR1D2 mutants recorded in baseline or 1 mM NMDA added to 0.2 mM Ca2+ saline. Quantification of EPSP (B) and mEPSP (C) amplitudes in the conditions indicated. (D) NMDA application to wild-type NMJs blocks PHP expression. Representative mEPSP and EPSP traces recorded in 1 mM NMDA and 0.4 mM Ca2+ in wild type and DKaiR1D mutants with or without PhTx application. (E) Quantification of mEPSP and quantal content values following PhTx application in the indicated genotypes and conditions. (F) DKaiR1D is not required for the induction of PHP. Representative traces and quantification of wild-type NMJs incubated in NMDA before rapidly washing out NMDA and recording. (G) Reversible blockade of PHP expression by NMDA. Representative traces and quantification of wild-type NMJs following PhTx application and NMDA washout. Note that the motor axon is severed and the central nervous system is removed before all recordings. Error bars indicate ±SEM. Asterisks indicate statistical significance using one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparison test: (*) p<0.05; (**) p<0.01; (***) p<0.001; (ns) not significant. Detailed statistical information for represented data (mean values, SEM, n, p) is shown in Table S1.
Acute pharmacological blockade of DKaiR1D disrupts the expression, but not induction, of PHP
Heterologous expression of DKaiR1D was recently achieved, where DKaiR1D was determined to form homomeric channels, to be activated by glutamate, and to be calcium permeable (Li et al., 2016). Interestingly, NMDA, an agonist for NMDA-type vertebrate glutamate receptors, was found to be a reversible antagonist of DKaiR1D channels (Li et al., 2016). We therefore tested whether NMDA could serve as an acute pharmacological antagonist of DKaiR1D activity in vivo using the semi-intact Drosophila NMJ preparation. Importantly, we sever the motor nerve from the cell body prior to application of NMDA in this preparation. Thus, acute blockade of DKaiR1D by NMDA would provide conclusive insight into whether this receptor was functioning in the dendrites or presynaptic terminals of motor neurons during baseline synaptic transmission and homeostatic plasticity, and resolve whether DKaiR1D had developmental roles or was necessary for the acute induction and/or expression of PHP.
We previously observed a substantial reduction in baseline EPSP amplitude in 0.2 mM extracellular calcium in DKaiR1D mutant synapses (Fig. 2). If NMDA is indeed an antagonist of DKaiR1D in vivo, we reasoned that acute application of NMDA to a wild-type preparation in similar conditions should reduce EPSP amplitudes to DKaiR1D mutant levels. While acute application of NMDA to wild-type synapses did not affect mEPSP amplitude (Fig. 6C and Fig. S5), EPSP amplitudes were reduced to similar levels as observed in DKaiR1D mutants at low calcium (0.2 mM) (Fig. 6A,B). We went on to test whether the acute blockade of DKaiR1D by NMDA, after severance of the motor nerve, disrupts the expression of PHP. At moderate extracellular calcium (0.4 mM), application of NMDA had no effect on baseline transmission in wild-type synapses, as expected (Fig. 6D). However, following incubation with PhTx, NMDA completely disrupted the expression of PHP in wild-type synapses (Fig. 6D,E). NMDA application had no impact on DKaiR1D mutants in this condition (PHP was blocked with no additional changes, Fig. 6E). Finally, we specifically tested whether DKaiR1D activity was needed for the induction and/or expression of PHP. First, we applied NMDA to semi-intact preparations before, during, and following PhTx application, then washed out NMDA and recorded mEPSP and EPSP values. PHP was restored to control levels within 2 mins of NMDA washout (Fig. 6F), demonstrating that DKaiR1D activity is not necessary for the acute induction of PHP. To test the reversibility and necessity of DKaiR1D for the expression of PHP, we applied NMDA to wild-type synapses after PhTx application, recorded reduced EPSP values, then washed out NMDA and found EPSP values returned to baseline levels, restoring PHP expression (Fig. 6G). Together, this demonstrates that DKaiR1D activity at presynaptic terminals is necessary for the acute and rapid expression, but not induction, of presynaptic homeostatic potentiation.
We went on to perform several controls for the specificity of DKaiR1D pharmacology. First, application of NMDA at moderately elevated extracellular calcium (1 mM) had no effect on baseline transmission (Fig. S5C), as expected, while PHP expression was greatly diminished (Fig. S5E). In addition, DKaiR1D heterozygous mutants exhibited an increased sensitivity to NMDA over a range of concentrations compared with wild type (Fig. S5D), consistent with DKaiR1D being a specific target of NMDA in vivo at the larval NMJ. Notably, AP5 also functions as an antagonist of DKaiR1D (Li et al., 2016), and we found that acute application of AP5 indeed disrupts PHP expression, with DKaiR1D heterozygous mutants exhibiting an increased sensitivity to AP5 (Fig. S5F–H). The blockade of DKaiR1D by both NMDA and AP5 excludes any possibility of NMDA receptors influencing synaptic physiology at the Drosophila NMJ in our assay, since one is an NMDA receptor agonist and the other is a competitive antagonist. We did observe that AP5 was less effective at disrupting homeostatic plasticity than NMDA, consistent with NMDA being a more potent antagonist in vitro (Li et al., 2016). However, we find that both NMDA and AP5 are effective at lower concentrations in vivo than might be expected from their actions in vitro, suggesting there are pharmacological differences for DKaiR1D receptors in vivo compared to in vitro. Thus, NMDA is a specific antagonist of DKaiR1D receptors in vivo.
Calcium permeability through DKaiR1D is necessary for baseline transmission but not PHP expression
The unedited versions of vertebrate kainate and AMPA receptors are calcium permeable (Lerma and Marques, 2013; Pinheiro and Mulle, 2008) and DKaiR1D forms calcium permeable channels when expressed in heterologous cells (Li et al., 2016). We therefore tested the importance of calcium influx through DKaiR1D in driving baseline presynaptic release as well as presynaptic homeostatic potentiation. To accomplish this, we engineered a Q604R mutation in DKaiR1D that renders this channel calcium impermeable in heterologous cells (Li et al., 2016). This calcium impermeable DKaiR1D transgene (referred to as DKaiR1DR) was tested for the ability to rescue baseline and homeostatic synaptic function compared with the native, calcium permeable DKaiR1DQ transgene when expressed in motor neurons of DKaiR1D mutants.
We first tested baseline synaptic release at 0.2 mM extracellular calcium. First, we confirmed that overexpression of DKaiR1DQ and DKaiR1DR transgenes in motor neurons display similar expression and localization (data not shown). Expression of the calcium permeable DKaiR1DQ transgene in motor neurons of the DKaiR1D mutant restored wild-type EPSP amplitudes, while expression of DKaiR1DR had no effect on reduced EPSP amplitudes (Fig. 7A,B). This demonstrates that calcium permeability through DKaiR1D is necessary for proper baseline presynaptic release. We then performed the same experiment in elevated extracellular calcium (0.4 mM) following PhTx application to test whether calcium permeability through DKaiR1D was similarly necessary for the homeostatic potentiation of presynaptic release. Following PhTx application, PHP expression was rescued when DKaiR1DQ was expressed in DKaiR1D mutants. Surprisingly, PHP was also fully restored following DKaiR1DR expression in motor neurons (Fig. 7C,D). Importantly, this demonstrates that expression of DKaiR1DR is functional, in that it can fully restore PHP expression to similar levels as observed with DKaiR1DQ expression. These results suggest that calcium influx through DKaiR1D serves to potentiate presynaptic release in low extracellular calcium conditions, but that calcium influx through DKaiR1D is not necessary to enable the expression of PHP.
Figure 7. Calcium permeability through DKaiR1D receptors is necessary for baseline transmission but not for PHP expression.

(A) Calcium permeability through DKaiR1D is necessary to potentiate baseline release at low extracellular calcium. Representative mEPSP and EPSP traces in indicated genotypes. Motor neuron rescue with DKaiR1DQ (w;OK6-Gal4/UAS-DKaiR1DQ; DKaiR1D2) or DKaiR1DR (w;OK6-Gal4/UAS-DKaiR1DR;DKaiR1D2) transgenes expressed in DKaiR1D2 mutant backgrounds. (B) Quantification of EPSP amplitudes in indicated genotypes. (C) Calcium permeability through DKaiR1D is not required for PHP expression. Representative traces of indicated genotypes. (D) Quantification of indicated genotypes following PhTx application. Error bars indicate ±SEM. Asterisks indicate statistical significance using one-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparison test: (*) p<0.05; (**) p<0.01; (***) p<0.001; (ns) not significant. Detailed statistical information for represented data (mean values, SEM, n, p) is shown in Table S1.
DISCUSSION
We have revealed a role for presynaptic glutamatergic signaling modulating baseline and homeostatic neurotransmitter release at the Drosophila NMJ. This unexpected role for iGluRs in sensing glutamate at presynaptic terminals indicates an autocrine mechanism that responds to glutamate release to adaptively modulate presynaptic activity at individual active zones.
Autocrine mechanisms for iGluRs in modulating presynaptic neurotransmitter release
Glutamate receptors have diverse functions in modulating presynaptic excitability and short term plasticity in addition to their established roles in postsynaptic excitation (Lerma and Marques, 2013). Similar to what we observe with DKaiR1D at the Drosophila NMJ, rodent iGluRs also localize to presynaptic active zone, are activated by high concentrations of glutamate, and can modulate release during single action potentials (McGuinness et al., 2010; Pinheiro et al., 2007; Schmitz et al., 2000). This suggests conserved autocrine modulatory mechanisms shared between these systems.
Rodent autoreceptors are known to modulate presynaptic activity on rapid time scales (Kamiya, 2002; Schmitz et al., 2000), including potentiating release during single action potentials (McGuinness et al., 2010; Scott et al., 2008). Activation of presynaptic iGluRs can modulate presynaptic voltage and calcium influx in less than 3.5 milliseconds (McGuinness et al., 2010; Scott et al., 2008). In these cases, most of the impact on release is likely to derive from a calcium store-dependent mechanism, or from modulation of the action potential during the repolarization phase, when most of the calcium influx that drives vesicle release occurs (Schneggenburger and Rosenmund, 2015). In a similar fashion, activation of presynaptic DKaiR1D during a single action potential could lead to a rapid additional source of presynaptic calcium influx from DKaiR1D itself and/or through modulation of presynaptic membrane potential to drive increased vesicle release (Fig. S4). Voltage imaging at the Drosophila NMJ has found the half width of the action potential waveform to be ~5 milliseconds (Ford and Davis, 2014), sufficient time to be modulated through such a mechanism. Therefore, dynamic changes in voltage or calcium influx at or near active zones could, in principle, drive additional vesicle release during a single action potential. This modulation may be restricted to nearby active zones and compartments relative to the site of glutamate release. Indeed, presynaptic kainate autoreceptors have the capacity to confer short-range, synapse-specific modulation to synaptic transmission (Scott et al., 2008), while presynaptic ligand-gated ion channels in C. elegans can also rapidly modulate synaptic transmission (Takayanagi-Kiya et al., 2016). Local activation of DKaiR1D could, therefore, subserve a powerful and flexible means of tuning presynaptic efficacy at or near individual release sites.
Rapid, synapse-specific modulation during presynaptic homeostatic plasticity
How does DKaiR1D promote the expression of presynaptic homeostatic plasticity? In contrast to the role of DKaiR1D in baseline release discussed above, our data indicates that the DKaiR1D-dependent mechanism that drives presynaptic homeostatic potentiation is calcium independent. This implies two changes to DKaiR1D functionality that are unique to homeostatic adaptation compared to baseline transmission. First, because presynaptic release is acutely potentiated following application of PhTx, the activity, levels, and/or localization of DKaiR1D receptors must change to acquire a novel influence on neurotransmitter release following PHP induction. The activity of synaptic glutamate receptors can change through associations with additional subunits and auxiliary factors such as Neto (Kim et al., 2012; Straub et al., 2011). Furthermore, various forms of plasticity are expressed through dynamic changes in the levels and localization of glutamate receptors trafficking between active zones and endosome pools or extra-synaptic membrane (Anggono and Huganir, 2012; Kneussel and Hausrat, 2016; Yan et al., 2013). Indeed, when DKaiR1D is overexpressed in motor neurons, it rescues baseline transmission and PHP expression while localizing to heterogenous puncta of varying distances relative to active zones. Notably, there is evidence that DKaiR1D interacts with other glutamate receptor subunits in vivo (Karuppudurai et al., 2014), which may contribute to the pharmacological differences observed in this study compared with the in vitro characterization (Li et al., 2016), and may also be targets of modulation during PHP.
Second, calcium signaling through DKaiR1D differentially drives baseline release and homeostatic plasticity. Therefore, mechanisms distinct from calcium permeability of the channel must contribute to PHP expression. One possibility is that DKaiR1D signals through an undefined metabotropic mechanism during PHP (Petrovic et al., 2017; Rozas et al., 2003; Rutkowska-Wlodarczyk et al., 2015), which might contribute to the ability of the calcium impermeable DKaiR1DR transgene, with reduced conductance (Li et al., 2016), to rescue PHP expression. Alternatively, an attractive possibility is that following PHP induction, glutamate released from nearby active zones may dynamically modulate the presynaptic membrane potential and/or action potential waveform to promote additional synaptic vesicle release. Indeed, small, sub-threshold depolarizations of the presynaptic resting potential, as small as 5 mV, are sufficient to induce a two-fold increase in presynaptic release (Awatramani et al., 2005). The timescale of this activity could occur within a few milliseconds as discussed above, and studies at the Drosophila NMJ have revealed that glutamate is released from single synaptic vesicles over time scales of milliseconds (Pawlu et al., 2004). Interestingly, ENaC channels have been proposed to enable PHP expression through changes in the presynaptic membrane potential (Younger et al., 2013), and such a mechanism could be shared by DKaiR1D, but gated by glutamate release at individual active zones. Thus, DKaiR1D may serve to homeostatically modulate presynaptic release through modulation of presynaptic voltage and, intriguingly, with active zone specificity.
Glutamate signaling and homeostatic synaptic plasticity
Our characterization of DKaiR1D has revealed the first role for presynaptic glutamate signaling in homeostatic plasticity. In the mammalian central nervous system, glutamate signaling drives the adaptive regulation of postsynaptic AMPA receptor insertion and removal, known as homeostatic scaling (Turrigiano, 2008). Further, kainate receptors were recently demonstrated to regulate postsynaptic homeostatic scaling (Yan et al., 2013). Together with our present study, these results demonstrate that glutamatergic signaling through kainate receptors orchestrate the potent and adaptive homeostatic control of synaptic strength on both sides of the synapse. Future studies will reveal the integration between synaptic glutamate signaling and other forces that modulate synaptic strength to enable robust, flexible, and stable neurotransmission.
EXPERIMENTAL PROCEDURES
Fly Stocks
Drosophila stocks were raised at 25°C on standard molasses food. The w1118 strain is used as the wild-type control unless otherwise noted, as this is the genetic background of the transgenic lines and other genotypes used in this study. The DKaiR1D mutant stocks DKaiR1D1 (PBac{WH}CG3822f03502) and DKaiR1D2 (Mi{ET1}CG3822MB01010) as well as the DKaiR1D deficiency (Df(3R)BSC819) were obtained from the Bloomington Drosophila Stock Center.
Molecular Biology
We obtained an EST (RE06730) encoding the entire DKaiR1D open reading frame from the Berkeley Drosophila Genome Project (www.fruitfly.org). We inserted the DKaiR1D cDNA into the pACU2 vector (Han et al., 2011) using standard cloning methods. pACU2-Brps::mCherry::GCaMP6s was generated by PCR amplifying the region coding for the Brp-short sequence (Brps), mCherry sequence, and the GCaMP6s sequence, and cloned using Gibson Assembly.
Immunochemistry
Third-instar larvae were dissected in ice cold 0 Ca2+ HL-3 and immunostained as described (Chen et al., 2017). The following antibodies were used: mouse anti-Synapsin, 3C11 (1:10; Developmental Studies Hybridoma Bank; DSHB); mouse anti-Bruchpilot (BRP), (nc82; 1:100; DSHB); affinity-purified rabbit anti-GluRIII (1:2000; (Marrus et al., 2004)); rabbit anti-GFP (1:1000; A-11122; Invitrogen) and rat anti-DKaiR1D (1:1000). To generate polyclonal antibodies against DKaiR1D, we engineered a recombinant protein consisting of amino acids 31–219 and an N-terminal 10X-His tag. Recombinant protein was injected into 3 rats by PrimmBiotech, Inc. (Boston, MA), and polyclonal antibodies were affinity purified.
Western Blotting
Third-instar larval brain and muscle tissue extracts (50 animals of each genotype) were prepared and used for immunoblotting as previously described (Chen et al., 2017). DKaiR1D (1:1000) and α-Tubulin (1:2000; JLA20, DSHB) primary antibodies were used with a 1:5000 dilution of horseradish peroxidase-conjugated anti-rat or anti-mouse secondary antibodies (Jackson ImmunoResearch).
Confocal imaging and analysis
Samples were imaged using a Nikon A1R Resonant Scanning Confocal microscope equipped with NIS Elements software and a 100x APO 1.4NA oil immersion objective using separate channels with laser lines 488 nm, 561 nm, and 637 nm. The general analysis toolkit in the NIS Elements software was used to quantify BRP and DKaiR1D puncta, density, and intensity by applying the same intensity thresholds and filters to binary layers on each of the three channels for each genotype compared. BRP and DKaiR1D puncta were counted within a synapse area labeled by HRP. BRP and DKaiR1D intensities were normalized to the HRP signal intensity, then normalized to wild-type values. Density and intensity measurements based on confocal images were taken from at least twelve synapses acquired from at least six different animals.
For Ca2+ imaging experiments, third-instar larvae were dissected and incubated in ice-cold HL3 containing 2 mM Ca2+ and 10 mM MNI-caged glutamate (#1490, Tocris, resuspended in H2O) in the absence or presence of 1 mM NMDA (Abcam). Control experiments were performed in 0 mM Ca2+ saline. GCaMP and mCherry signals were measured at mCherry-positive active zones of type-1b and 1s boutons synapsing onto muscle 6/7 of abdominal segments A2/A3. Live imaging was performed using a Nikon A1R Resonant Scanning Confocal microscope equipped with NIS Elements software. Band scanning at a resonance frequency of 113 fps (512 × 86 pixels) was performed across the field of view. One synapse (4–12 boutons) was imaged each session. Measurements based on calcium imaging were taken from at least 12 synapses (approximately 100 boutons) from at least 6 animals.
Electrophysiology
All dissections and recordings were performed as previously described (Chen et al., 2017). Larvae were incubated with or without philanthotoxin-433 (Sigma; 20 μM) and resuspended in HL-3 for 10 mins, as described (Dickman and Davis, 2009; Frank et al., 2006). Larvae were also incubated with or without NSTX-3 (Enzo Life Sciences; 20 μM in 0.1% acetic acid) and resuspended in HL-3 for 15 mins, as described (Frank et al., 2006). For the acute blockade of DKaiR1D by NMDA or AP5, larvae were dissected and following 10 min incubation with PhTx, the central nervous system was removed and the larvae were incubated with 1 mM NMDA (Abcam, ab120052, resuspended in dH20) or 5 mM AP5 (ab120003, resuspended in dH20) for 5 mins, with recordings performed in the continued presence of NMDA or AP5. The readily releasable pool (RRP) size was estimated by examining cumulative EPSC amplitudes while recording using a two-electrode voltage clamp (TEVC) configuration (Muller et al., 2012; Schneggenburger et al., 1999; Weyhersmuller et al., 2011).
Statistical Analysis
All data are presented as mean +/−SEM. Data were compared using either a one-way ANOVA, followed by Tukey’s multiple-comparison test, or using a Student’s t-test (where specified), analyzed using Graphpad Prism or Microsoft Excel software, and with varying levels of significance assessed as p<0.05 (*), p<0.01 (**), p<0.001 (***), ns=not significant. See Table S1 for additional statistical details and values.
Supplementary Material
Acknowledgments
The authors declare no competing financial interests. We acknowledge Chi-Hon Lee and Mark Mayer (NIH, Bethesda, MD) for communicating unpublished results about DKaiR1D calcium permeability and pharmacology. We thank Manfred Heckmann (University of Wurzburg, Germany) for insightful discussions on kainate receptor biophysics. We also thank members of the Dickman lab for comments and discussions, and Surbhi Trivedi for technical support. We acknowledge the Developmental Studies Hybridoma Bank (Iowa, USA) for antibodies and the Bloomington Drosophila Stock Center for fly stocks. This work was supported by a grant from the National Institutes of Health (NS019546) and research fellowships from the Alfred P. Sloan, Ellison Medical, Whitehall, Mallinckrodt, and Klingenstein-Simons Foundations to DKD.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and one table and can be found with this article online.
AUTHOR CONTRIBUTIONS
B.K. and D.K.D. designed the experiments and wrote the manuscript. B.K. performed all experiments, with J.W. contributing early electrophysiology experiments. Y.L. obtained and communicated findings on DKaiR1D pharmacology before publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22:461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awatramani GB, Price GD, Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron. 2005;48:109–121. doi: 10.1016/j.neuron.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Baines RA, Bate M. Electrophysiological development of central neurons in the Drosophila embryo. J Neurosci. 1998;18:4673–4683. doi: 10.1523/JNEUROSCI.18-12-04673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Robinson SG, Fujioka M, Jaynes JB, Bate M. Postsynaptic expression of tetanus toxin light chain blocks synaptogenesis in Drosophila. Curr Biol. 1999;9:1267–1270. doi: 10.1016/s0960-9822(99)80510-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier G, Bidoret C, Casado M, Paoletti P. Presynaptic NMDA receptors: Roles and rules. Neuroscience. 2015;311:322–340. doi: 10.1016/j.neuroscience.2015.10.033. [DOI] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ma W, Zhang S, Paluch J, Guo W, Dickman DK. The BLOC-1 Subunit Pallidin Facilitates Activity-Dependent Synaptic Vesicle Recycling. eNeuro. 2017:4. doi: 10.1523/ENEURO.0335-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, Vignes M, Dev KK, Barnes JM, Collingridge GL, Henley JM. Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature. 1996;379:78–81. doi: 10.1038/379078a0. [DOI] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Gelfand MV, Collins CA, DiAntonio A. Visualizing glutamatergic cell bodies and synapses in Drosophila larval and adult CNS. J Comp Neurol. 2008;508:131–152. doi: 10.1002/cne.21670. [DOI] [PubMed] [Google Scholar]
- Davis GW, Goodman CS. Synapse-specific control of synaptic efficacy at the terminals of a single neuron. Nature. 1998;392:82–86. doi: 10.1038/32176. [DOI] [PubMed] [Google Scholar]
- Davis GW, Muller M. Homeostatic control of presynaptic neurotransmitter release. Annual review of physiology. 2015;77:251–270. doi: 10.1146/annurev-physiol-021014-071740. [DOI] [PubMed] [Google Scholar]
- Dickman DK, Davis GW. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science. 2009;326:1127–1130. doi: 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford KJ, Davis GW. Archaerhodopsin voltage imaging: synaptic calcium and BK channels stabilize action potential repolarization at the Drosophila neuromuscular junction. J Neurosci. 2014;34:14517–14525. doi: 10.1523/JNEUROSCI.2203-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA. Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology. 2013 doi: 10.1016/j.neuropharm.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavino MA, Ford KJ, Archila S, Davis GW. Homeostatic synaptic depression is achieved through a regulated decrease in presynaptic calcium channel abundance. eLife. 2015:4. doi: 10.7554/eLife.05473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Jan LY, Jan YN. Enhancer-driven membrane markers for analysis of nonautonomous mechanisms reveal neuron-glia interactions in Drosophila. Proc Natl Acad Sci U S A. 2011;108:9673–9678. doi: 10.1073/pnas.1106386108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H. Kainate receptor-dependent presynaptic modulation and plasticity. Neurosci Res. 2002;42:1–6. doi: 10.1016/s0168-0102(01)00303-0. [DOI] [PubMed] [Google Scholar]
- Karuppudurai T, Lin TY, Ting CY, Pursley R, Melnattur KV, Diao F, White BH, Macpherson LJ, Gallio M, Pohida T, et al. A hard-wired glutamatergic circuit pools and relays UV signals to mediate spectral preference in Drosophila. Neuron. 2014;81:603–615. doi: 10.1016/j.neuron.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Bao H, Bonanno L, Zhang B, Serpe M. Drosophila Neto is essential for clustering glutamate receptors at the neuromuscular junction. Genes Dev. 2012;26:974–987. doi: 10.1101/gad.185165.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneussel M, Hausrat TJ. Postsynaptic Neurotransmitter Receptor Reserve Pools for Synaptic Potentiation. Trends Neurosci. 2016;39:170–182. doi: 10.1016/j.tins.2016.01.002. [DOI] [PubMed] [Google Scholar]
- Lerma J, Marques JM. Kainate receptors in health and disease. Neuron. 2013;80:292–311. doi: 10.1016/j.neuron.2013.09.045. [DOI] [PubMed] [Google Scholar]
- Li Y, Dharkar P, Han TH, Serpe M, Lee CH, Mayer ML. Novel Functional Properties of Drosophila CNS Glutamate Receptors. Neuron. 2016;92:1036–1048. doi: 10.1016/j.neuron.2016.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrus SB, Portman SL, Allen MJ, Moffat KG, DiAntonio A. Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J Neurosci. 2004;24:1406–1415. doi: 10.1523/JNEUROSCI.1575-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness L, Taylor C, Taylor RD, Yau C, Langenhan T, Hart ML, Christian H, Tynan PW, Donnelly P, Emptage NJ. Presynaptic NMDARs in the hippocampus facilitate transmitter release at theta frequency. Neuron. 2010;68:1109–1127. doi: 10.1016/j.neuron.2010.11.023. [DOI] [PubMed] [Google Scholar]
- Muller M, Davis GW. Transsynaptic control of presynaptic Ca(2)(+) influx achieves homeostatic potentiation of neurotransmitter release. Curr Biol. 2012;22:1102–1108. doi: 10.1016/j.cub.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Liu KS, Sigrist SJ, Davis GW. RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:16574–16585. doi: 10.1523/JNEUROSCI.0981-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman ZL, Hoagland A, Aghi K, Worden K, Levy SL, Son JH, Lee LP, Isacoff EY. Input-Specific Plasticity and Homeostasis at the Drosophila Larval Neuromuscular Junction. Neuron. 2017;93:1388–1404. e1310. doi: 10.1016/j.neuron.2017.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlu C, DiAntonio A, Heckmann M. Postfusional control of quantal current shape. Neuron. 2004;42:607–618. doi: 10.1016/s0896-6273(04)00269-7. [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Petrovic MM, Viana da Silva S, Clement JP, Vyklicky L, Mulle C, Gonzalez-Gonzalez IM, Henley JM. Metabotropic action of postsynaptic kainate receptors triggers hippocampal long-term potentiation. Nat Neurosci. 2017;20:529–539. doi: 10.1038/nn.4505. [DOI] [PubMed] [Google Scholar]
- Pinheiro PS, Mulle C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat Rev Neurosci. 2008;9:423–436. doi: 10.1038/nrn2379. [DOI] [PubMed] [Google Scholar]
- Pinheiro PS, Perrais D, Coussen F, Barhanin J, Bettler B, Mann JR, Malva JO, Heinemann SF, Mulle C. GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A. 2007;104:12181–12186. doi: 10.1073/pnas.0608891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbough J, Broadie K. Electrophysiological analysis of synaptic transmission in central neurons of Drosophila larvae. J Neurophysiol. 2002;88:847–860. doi: 10.1152/jn.2002.88.2.847. [DOI] [PubMed] [Google Scholar]
- Rozas JL, Paternain AV, Lerma J. Noncanonical signaling by ionotropic kainate receptors. Neuron. 2003;39:543–553. doi: 10.1016/s0896-6273(03)00436-7. [DOI] [PubMed] [Google Scholar]
- Rutkowska-Wlodarczyk I, Aller MI, Valbuena S, Bologna JC, Prezeau L, Lerma J. A proteomic analysis reveals the interaction of GluK1 ionotropic kainate receptor subunits with Go proteins. J Neurosci. 2015;35:5171–5179. doi: 10.1523/JNEUROSCI.5059-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A, Hallermann S, Kittel RJ, Khorramshahi O, Frolich AM, Quentin C, Rasse TM, Mertel S, Heckmann M, Sigrist SJ. Activity-dependent site-specific changes of glutamate receptor composition in vivo. Nat Neurosci. 2008;11:659–666. doi: 10.1038/nn.2122. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Frerking M, Nicoll RA. Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron. 2000;27:327–338. doi: 10.1016/s0896-6273(00)00040-4. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Rosenmund C. Molecular mechanisms governing Ca(2+) regulation of evoked and spontaneous release. Nat Neurosci. 2015;18:935–941. doi: 10.1038/nn.4044. [DOI] [PubMed] [Google Scholar]
- Scott R, Lalic T, Kullmann DM, Capogna M, Rusakov DA. Target-cell specificity of kainate autoreceptor and Ca2+-store-dependent short-term plasticity at hippocampal mossy fiber synapses. J Neurosci. 2008;28:13139–13149. doi: 10.1523/JNEUROSCI.2932-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub C, Hunt DL, Yamasaki M, Kim KS, Watanabe M, Castillo PE, Tomita S. Distinct functions of kainate receptors in the brain are determined by the auxiliary subunit Neto1. Nat Neurosci. 2011;14:866–873. doi: 10.1038/nn.2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi-Kiya S, Zhou K, Jin Y. Release-dependent feedback inhibition by a presynaptically localized ligand-gated anion channel. Elife. 2016:5. doi: 10.7554/eLife.21734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyhersmuller A, Hallermann S, Wagner N, Eilers J. Rapid active zone remodeling during synaptic plasticity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:6041–6052. doi: 10.1523/JNEUROSCI.6698-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wondolowski J, Dickman D. Emerging links between homeostatic synaptic plasticity and neurological disease. Front Cell Neurosci. 2013;7:223. doi: 10.3389/fncel.2013.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Yamasaki M, Straub C, Watanabe M, Tomita S. Homeostatic control of synaptic transmission by distinct glutamate receptors. Neuron. 2013;78:687–699. doi: 10.1016/j.neuron.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao CK, Liu YT, Lee IC, Wang YT, Wu PY. A Ca2+ channel differentially regulates Clathrin-mediated and activity-dependent bulk endocytosis. PLoS Biol. 2017;15:e2000931. doi: 10.1371/journal.pbio.2000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger MA, Muller M, Tong A, Pym EC, Davis GW. A presynaptic ENaC channel drives homeostatic plasticity. Neuron. 2013;79:1183–1196. doi: 10.1016/j.neuron.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.