

Many RNA sequences form more than one stable secondary or tertiary structure under physiological conditions. As these conformations frequently exchange with one another in a period of minutes or seconds, solution-based methods are often inadequate to resolve differences in their structure and activity. This unit describes methods for separating RNA conformers by native polyacrylamide gel electrophoresis. The activity of each electrophoretic species is assayed while the RNA is still immobilized in the gel matrix. The protocols given here were initially developed to discriminate between active and inactive conformations of the self-splicing RNA from Tetrahymena thermophila (Emerick and Woodson, 1994), but can be applied to a wide variety of catalytic RNAs, riboswitches, and RNA-protein complexes.
Native polyacrylamide gel electrophoresis is used to separate 32P- or dye-labeled RNA on the basis of shape or hydrodynamic radius (see Basic Protocol 1). Once optimal electrophoretic separation has been achieved, the catalytic activity or ligand binding of each conformer is determined in situ by soaking substrates into the gel matrix (see Basic Protocol 2). Alternatively, the RNA can be eluted from the gel and analyzed directly (see Alternate Protocol). The secondary structure of the RNA is probed by addition of base-modification reagents to the RNA in the gel (see Basic Protocol 3).
CAUTION: Procedures using radioactive materials require personnel training and licensing to comply with safety and regulatory standards.
NOTE: Care must be taken to avoid introducing ribonucleases into the samples. All solutions should be prepared with deionized water (18 MΩ) that is free of pyrogens and organic contaminants. This can be achieved by in-line purification system (ideally tissue culture grade) or by purchasing molecular biology or HPLC grade water. Solutions should be sterilized by filtration (0.2 μm) or autoclaved. Gloves must be worn while handling samples and preparing solutions, and pipet tips and sample tubes must be free of nucleases. This can usually be achieved by purchasing good quality machine-packaged disposable plasticware, avoiding contact with bare skin, and storing disposables in clean, dust-free containers.
BASIC PROTOCOL 1. NATIVE GEL ELECTROPHORESIS OF 32P-LABELED RNA
This protocol was developed to separate different structural forms of a 657-nt self-splicing RNA (Emerick and Woodson, 1994). The protocol can be adapted to shorter transcripts by increasing the percentage of the polyacrylamide gel. Once the best conditions for separating the conformers of interest have been determined, this protocol is used to prepare the RNA for further analysis as described in Basic Protocols 2 and 3.
Materials
70% ethanol
40% (w/v) 29:1 acrylamide/bisacrylamide, 4°C
10× THEM buffer (see recipe and notes)
10% (w/v) ammonium persulfate, 4°C
TEMED, 4°C
32P-labeled or dye-labeled RNA, desalted (see Support Protocol 1)
30 mM MgCl2
5× glycerol loading buffer (see recipe)
20 × 20–cm glass plates with 0.5-mm spacers and comb (1 or 2 sets)
Vacuum source and 50-mL side-arm flask (for degassing)
20 × 20–cm vertical gel electrophoresis apparatus with recirculating cooling reservoir (e.g., ThermoFisher Owl Scientific Penguin 10DS or equivalent)
Refrigerated recirculating bath to connect to gel apparatus
Spatula or razor blade
3MM filter paper (Whatman)
Vacuum gel drying apparatus
Phosphorstorage or other gel imager, or X-ray film for autoradiography
CAUTION: Acrylamide and N,N′-methylenebisacrylamide are neurotoxins and suspected carcinogens. Preparation of solutions with these compounds should be performed in a well-ventilated fume hood, and extreme precautions taken to minimize contact with solids or solutions.
Cast native polyacrylamide gel
-
1
Thoroughly clean two glass plates and remove any streaks with 70% ethanol.
-
2
Assemble glass plates, placing 0.5-mm spacers between glass on each side. Clamp the sides of the sandwich with aluminum binder clamps and seal bottom with tape.
The bottom of plates may be left open. The gel solution will be retained by capillary action. -
3
Mix the following in a graduated cylinder (25 mL total volume per gel):
3.75 mL 40% 29:1 acrylamide/bisacrylamide
2.5 mL 10×· THEM buffer
250 μL 10% ammonium persulfate
-
Deionized water up to 25 mL.
The final composition is 6% polyacrylamide. For a 400 nt RNA, use 8% polyacrylamide; for a 200 nt. RNA, use 10% polyacrylamide.
-
4
Transfer to 50-mL side-arm flask and degas mixture by swirling 1 to 3 min under a gentle vacuum (water aspirator).
-
5
Add 16 μL TEMED and swirl to initiate polymerization. Immediately pour solution into the top of the casting frame while holding glass plates at a very slight incline with respect to the bench.
It is important to pour the solution evenly to avoid introducing bubbles. -
6
Insert comb into top of frame to form desired number of sample wells; allow gel to polymerize in a horizontal position (i.e., ~20 min).
Pre-run gel
-
7
Remove comb, flushing wells with water or running buffer. Remove any tape from bottom of gel. Place gel on electrophoresis apparatus as instructed by manufacturer.
-
8
Prepare 1×· THEM buffer by diluting 10×· stock with deionized water. Add sufficient 1×· THEM buffer to upper and lower chambers to cover top and bottom of gel.
The MgCl2 concentration in the running buffer should be adjusted to the minimum needed to stabilize the folded RNA. -
9
Pre-run at 15 W per gel for 15 to 30 min. Adjust temperature of circulating bath to maintain surface of plates at 10°C.
Prechilling the bath decreases the time required for gel to reach desired temperature. Bath temperatures of −5° to 0°C may be required to cool gel sufficiently; add 50% ethylene glycol to bath to prevent freezing.
CAUTION: Personnel should be shielded from beta radiation during steps 10 to 14.
Prepare and load RNA samples
-
10
Add the following to a 0.5-mL microcentrifuge tube (8 μL total volume):
2 μL 5×· folding buffer
1 μL 32P-labeled RNA (100,000 cpm) or Cyanine5 (Cy5)-labeled RNA (200 fmol).
5 μL deionized H2O.
-
11
Place 2 μL of 30 mM MgCl2 on lid of microcentrifuge tube and close firmly.
The drop of buffer will remain on the lid by surface tension. -
12
To renature RNA, place closed tube in a heating block at 95°C for 1 min. Transfer to microcentrifuge and immediately spin for 1 min. Place on ice.
For unrenatured RNA samples, omit the incubation at 95°C.The drop of MgCl2 will mix with the warm RNA solution at the bottom of the tube during centrifugation. This procedure minimizes metal ion–catalyzed hydrolysis of the RNA during the high-temperature incubation. Place the heating block next to the microcentrifuge to minimize the time required to transfer the samples.The Tetrahymena and Azoarcus ribozymes are renatured by incubating at 50 °C for 20 min in the desired folding buffer with MgCl2. -
13
Add 2.5 μL of 5×· glycerol loading buffer and mix well. Place samples on ice.
-
14
Load 2 μL of each sample into wells of native gel and run at 15 W per gel for 5 to 6 hr at 10°C.
For the best resolution, the sample should be no more than 2 to 3 mm deep after loading. The electrophoresis time will vary with the size of the RNA and the percentage of polyacrylamide. Gels are typically run until xylene cyanol FF is at the bottom of the gel or beyond.
Detect 32P or dye-labeled bands of RNA
-
15
Unclamp gel from apparatus and remove spacers. Gently pry the glass plates apart with a thin spatula or razor blade and remove one glass plate.
Do this step as soon as the run is complete. -
16
Transfer gel from the second glass plate to dry Whatman 3MM filter paper cut a little larger than the gel. Cover completely with plastic wrap, and dry 15 to 30 min under vacuum using a heated gel-drying apparatus.
Do not do this step if planning to do two-dimensional gel electrophoresis (Basic Protocol 2). -
17
Expose dried gel to X-ray film or a phosphor-storage screen overnight to obtain an image of the radioactive bands. Gels containing fluorescently-labeled RNA should be scanned with an appropriate imager (e.g. Typhoon or equivalent).
ALTERNATE PROTOCOL. NATIVE GEL ELECTROPHORESIS OF DYE-LABELED RNA
As an alternative to radiolabeled RNA, the RNA can be labeled by hybridization with a dye-labeled oligonucleotide complementary to a 3′ extension of the RNA sequence, and detected in the gel using fluorescent scanners. This protocol was developed for detecting ribosomal RNA-protein complexes. Fluorophores can be directly attached to RNAs and RNA-binding proteins, however, using commercially available activated dyes such as NHS-esters or maleimides. An advantage of fluorophore labeling is that different subunits of a complex can be labeled with different dyes, allowing for multi-color detection of the components. A disadvantage of this approach is its slightly lower sensitivity (40–50 fmol), compared to 32P-labeled RNA (2–10 fmol).
Additional Materials (also see Basic Protocol 1)
In vitro transcribed RNA, with unstructured region suitable for binding oligonucleotides
Synthetic RNA oligonucleotide, labeled with Cy5, Cy3, Alexa 488, FAM or other appropriate dye (~20 nt), free of unconjugated dye and desalted
HK buffer (see recipe)
Typhoon or other fluorescence imager
Heat block or water bath
Hybridize RNA with dye-labeled oligonucleotide and refold
-
1
Incubate 10–100 nM RNA with complementary dye-labeled oligonucleotide (5 nM, 10 μL) in HK buffer at 70 °C for 5 min.
-
2
Cool to 25 °C for 5 min.
For direct detection of RNA, proceed to step 5. -
3
Refold RNA-oligomer complex at 37 °C in 0–20 mM MgCl2.
-
4
Add Cy3-labeled RNA binding protein (15 nM or as appropriate) if desired, and incubate another 15 min at 37 °C.
-
5
Mix 8 μL sample with 2 μL glycerol loading buffer, and load 5 μL on a pre-chilled native polyacrylamide gel (see Basic Protocol 1).
If the RNA is labeled with Cy5, xylene cyanol tracking dye should be omitted to avoid interference with Cy5 detection. -
6
Run the native gel (see Basic Protocol 1).
-
7
Remove top gel from plate and cover in clean plastic wrap. Scan the wrapped gel with a Typhoon or similar imager, using the appropriate excitation laser.
BASIC PROTOCOL 2. ASSAY OF CATALYTIC ACTIVITY BY TWO-DIMENSIONAL GEL ELECTROPHORESIS
Following separation of RNA conformers by native gel electrophoresis (see Basic Protocol 1), the catalytic activity of each species is determined by addition of substrate while the RNA is immobilized in the gel matrix. The products are analyzed by two-dimensional electrophoresis under denaturing conditions (Branch et al., 1989). The spliced products are detected as shorter RNAs on the denaturing gel.
Materials
0.1 mM GTP in folding buffer (see recipe)
2×· urea loading buffer (see recipe)
40% (w/v) 29:1 acrylamide/bisacrylamide, 4°C
10×· TBE buffer (APPENDIX 2A)
Urea
10% (w/v) ammonium persulfate
TEMED
Bed of ice (size of gel) X-ray film
Razor blades
Glass plates (e.g., 20 × 20 cm or larger)
Additional reagents and equipment for native gel electrophoresis (see Basic Protocol 1)
Excise samples from native polyacrylamide gel (first dimension)
-
8
Separate RNA conformers by native gel electrophoresis (see Basic Protocol 1, steps 1 to 15). In step 10, use ≥500,000 cpm per sample (1 pmol Cy5-labeled RNA) and reduce the volumes by half (i.e., total 5 μL). Load 3 to 5 μL.
Skipping wells will make it easier to excise the bands from gel without cross-contamination in step 4. -
9
After removing top glass plate, spot 1–2 μL 32P-ATP diluted with loading buffer in 2 or 3 corners of the gel to mark them. Cover gel with plastic wrap. Place gel (glass side down) on a bed of ice.
The gel should be maintained on ice when possible during the following steps to retard diffusion of the RNA in the gel. Steps 3 to 5 should be performed as quickly as possible. -
10
Expose the gel to a phosphorstorage screen for 20–30 min in a dark room or light-tight box. Scan the screen and print out an image of the gel on white paper or transparency film.
-
11
Align gel (glass side down) over the printed image of the gel, using the corner dots as a guide. Using the image to locate the RNA, excise each lane with a new razor blade, being certain to include the entire region of the lane that contains the bands of interest.
Invert an acrylic radiation shield over gel to block 32P radiation and protect samples from airborne ribonucleases.The gel pieces should be ~0.6 × 4 cm. -
12
Transfer gel pieces to a glass plate (e.g., 20 × 20 cm or larger), laying them horizontally along the bottom edge as shown in right panel of Figure 11.4.1.
Forceps should be RNase-free or flamed with ethanol just before use.
Figure 11.4.1.
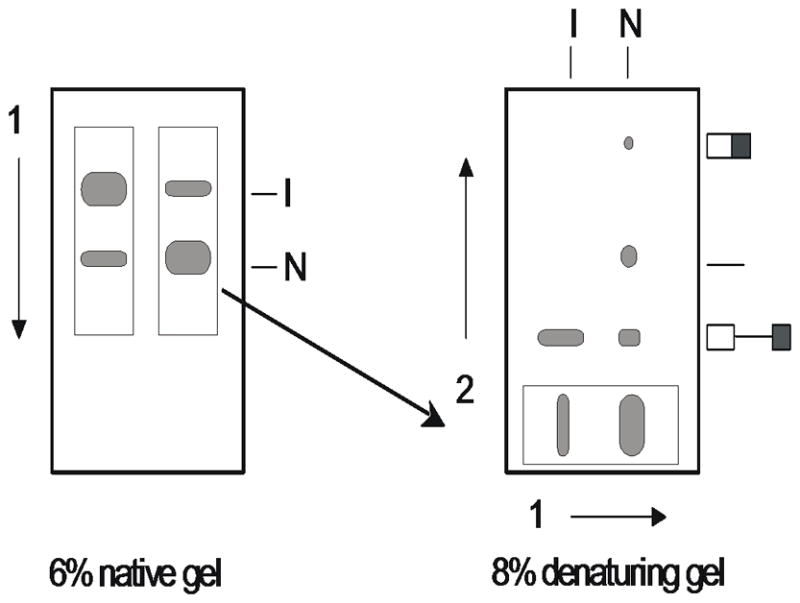
Two-dimensional gel electrophoresis of self-splicing RNA. After native gel electrophoresis (first dimension), the lane is excised and placed on the bottom of the second casting frame. The active form of the RNA (N) is spliced when GTP substrate is added to the gel slice. The products are resolved in a denaturing polyacrylamide gel (second dimension).
Perform self-splicing in situ
-
13
Pipet 5 μL 0.1 mM GTP in folding buffer as evenly as possible over the surface of each gel piece. Allow to stand 2 min at room temperature.
Use the appropriate substrate or cofactor for the RNA of interest in lieu of GTP. -
14
Add 10 μL of 2× urea loading buffer.
Urea quenches the self-splicing reaction.
Assemble and run the denaturing polyacrylamide gel (second dimension)
-
15
Complete assembly of gel frame for the second-dimension electrophoresis by placing spacers on each side and a second glass plate on top of samples from step 7. Clamp sides of frame and seal bottom with tape.
It may be necessary to use spacers that are slightly thicker than those used for the first-dimension gel to avoid squashing the gel pieces. -
16
Prepare solution for an 8% denaturing sequencing gel (25 mL total volume for 0.5 mm × 20 ×· 20 cm).
5 mL 40% 29:1 acrylamide/bisacrylamide
2.5 mL 10×· TBE buffer
12 g urea
Deionized water up to 25 mL.
Dissolve urea at 50 to 65°C. Transfer to a sidearm flask and degas.
-
17
Add 0.5 mL of 10% ammonium persulfate and 20 μL TEMED to begin polymerization.
-
18
Immediately pour solution into gel frame from top, allowing solution to run slowly down the side and then across the bottom, covering gel slices without introducing air bubbles.
It is not necessary to insert a comb in the top of gel. -
19
After gel has polymerized, clamp it on gel box with first-dimension samples at the bottom. Add 1·× TBE buffer to upper and lower reservoirs.
-
20
Connect the positive electrode to top of gel and the negative electrode to the bottom.
The gel is run in reverse so that the RNA migrates from bottom to top. -
21
Run at 25 to 30 W (40° to 50°C) until the xylene cyanol has run off the top of gel.
-
22
Disassemble, dry, and expose gel to a scanner or to X-ray film (see Basic Protocol 1, steps 15 to 17).
ALTERNATE PROTOCOL. ELUTION OF SPLICED PRODUCTS FROM NATIVE POLYACRYLAMIDE GELS
As an alternative to two-dimensional electrophoresis, the molecular weight of the RNAs in a native gel band may be determined by eluting the RNA from the gel matrix. The activity of the conformer can be determined by adding substrate to the gel slice before eluting the RNA. After elution, the RNA is concentrated by ethanol precipitation and analyzed on a denaturing polyacrylamide gel.
Additional Materials (also see Basic Protocols 1 and 2)
TEN buffer (see recipe)
10 mg/mL carrier tRNA (Sigma)
70% and 100% ethanol, −20°C
TE buffer, pH 7.5 (APPENDIX 2A)
Rocker or orbital mixer
4% to 6% denaturing polyacrylamide gel (see Basic Protocol 2)
Recover RNA from native polyacrylamide gel
-
1
Separate RNA conformers by native gel electrophoresis (see Basic Protocol 1, steps 1 to 14), using 200,000 cpm [32P]-RNA or 50–100 fmol Cy5-labeled RNA per sample.
-
2
Excise bands of RNA from native gel (see Basic Protocol 2, steps 1 to 4). Instead of excising the entire lane, individually remove each band to be analyzed and place in separate 1.5- or 2-mL microcentrifuge tubes.
-
3
Place 1 to 2 μL of 0.1 mM GTP or desired substrate in folding buffer on gel slice and incubate for 2 min, room temperature. Add 2 to 4 μL of 2× urea loading buffer.
It is useful to prepare control samples, omitting this step, to determine whether any reaction occurred before native gel electrophoresis. -
4
Freeze excised gel slices on dry ice 1 to 5 min. Thaw at room temperature or in a 25°C water bath.
A freeze/thaw cycle improves recovery of RNA. -
5
Add a sufficient volume of TEN buffer to cover gel piece (e.g., 0.3 to 0.6 mL). Place tightly sealed tubes on a rocker or orbital shaker and soak overnight at 4°C.
In some cases, 30 to 60 min at 65°C improves recovery of RNA. -
6
Microcentrifuge 1 min at 10,000 ×·; g, room temperature. Carefully transfer supernatant to a clean tube.
Avoid transferring pieces of polyacrylamide.Solution may be filtered using a small spin-filtration device (0.45μm) that is nuclease free. -
7
Add a small amount of TEN buffer (e.g., 50 to 100 μL) to gel slices, mix thoroughly, and remove as in step 6. Pool supernatants.
Elution of radiolabeled RNA from gel is easily checked using a standard survey meter.
Concentrate RNA by ethanol precipitation
-
8
Precipitate RNA by adding 1 μL of 10 mg/mL carrier tRNA and three volumes ice-cold 100% ethanol to supernatants. Chill at −20°C 1 to 2 hr or overnight, or at −70°C for 10 min.
-
9
Recover RNA by microcentrifuging 20 to 30 min at 10,000 × g, 4°C. Discard supernatant with a pipet, being careful to avoid pellet.
-
10
Wash pellet with 70% ethanol and dry under vacuum.
Analyze RNA by denaturing polyacrylamide gel electrophoresis
-
11
Dissolve dried pellets in 2 μL water and add 2 μL of 2× urea loading buffer.
Adjust volumes as required so that the concentration of 32P in each sample is approximately equal. -
12
Prepare one or more reference samples by diluting the original RNA sample (or molecular weight markers) in TE buffer, pH 7.5, so that the concentration of radioactivity matches that of the samples in step 11. Add an equal concentration of 2× urea loading buffer.
-
13
Load 4 μL of each sample into the wells of a 20 × 20–cm 4% to 6% denaturing polyacrylamide gel (see Basic Protocol 2, steps 9 to 10) and run at 30 W, 50°C, until xylene cyanol FF tracking dye reaches the bottom.
Samples may be heated 1 min at 80°C before loading if desired.
BASIC PROTOCOL 3. CHEMICAL MODIFICATION OF RNA IN A NATIVE POLYACRYLAMIDE GEL
After native gel electrophoresis, RNA is modified in situ by dimethyl sulfate (DMS), kethoxal, or N-methylisatoic anhydride (NMIA). DMS reacts with unpaired adenosines (N1) and cytosines (N3). Kethoxal reacts with the N1 and N2 of unpaired guanosines. NMIA acylates the 2′OH of single-stranded riboses (SHAPE). The position of the modified bases is determined by primer extension with reverse transcriptase.
Additional Materials (also see Basic Protocols 1 and 2 and Alternate Protocol)
Unlabeled RNA (see Support Protocol 2; see also UNIT 6.1)
Dimethyl sulfate (DMS, Sigma Aldrich; obtain fresh and store at −20°C)
50% and 100% ethanol
Stop buffer A (see recipe)
70 mg/mL kethoxal (ICN Biochemicals or Sigma Aldrich) in 20% ethanol (store at −20°C)
Stop buffer B (see recipe)
100 mM (17.7 mg/mL) N-methylisatoic anhydride (NMIA, Molecular Probes) in anhydrous DMSO (store at −20 °C with desiccant).
Buffered phenol (APPENDIX 2A)
24:1 (v/v) chloroform/isoamyl alcohol
Additional reagents and equipment for primer extension (see Support Protocol 3) and for sequencing (UNIT 6.1 and APPENDIX 3B)
CAUTION: Dimethyl sulfate (DMS) is a carcinogen and should be handled in a fume hood with gloves that are impermeable to nonpolar solvents.
Separate RNA conformers by native gel electrophoresis
-
1
Prepare a native gel (see Basic Protocol 1, steps 1 to 9).
-
2
Prepare 2 samples of 32P-labeled RNA (100,000 cpm) or dye-labeled RNA in 10 μL folding buffer (see Basic Protocol 1, steps 10 to 12).
This will serve as a marker for the unlabeled RNA. -
3
Prepare 4 samples containing 2 pmol each unlabeled RNA in the same manner.
-
4
Load samples into adjacent wells of a native polyacrylamide gel (see Basic Protocol 1, steps 13 to 15) with the labeled RNA on the outside lanes. When electrophoresis is complete, disassemble gel and obtain an image (see Basic Protocol 2, steps 2 and 3).
-
5
Using the position of labeled RNA as a guide, excise the regions of adjacent lanes containing unlabeled RNA that contain the bands of interest (see Basic Protocol 2, step 2).
This step and all subsequent steps are carried out on ice. -
6
Transfer each gel slice (containing a single band) to a separate 1.5- or 2-mL microcentrifuge tube.
There should be three duplicate sets of gel slices containing unlabeled RNA.
Modify RNA with DMS
-
7
Dilute DMS 1:40 in 50% ethanol. Add 20 μL diluted DMS to each slice originating from one lane of native gel. Distribute as evenly as possible over the surface. Incubate 0.5 to 2 min on ice.
The concentration of DMS should be adjusted so that an even ladder of primer extension products is obtained in steps 16 to 19. -
8
Quench reaction with 300 μL stop buffer A.
Modify RNA with kethoxal
-
9
Apply 20 μL of 70 mg/mL kethoxal to each gel piece from a second lane of the native gel. Incubate 3 to 8 min on ice.
The incubation time should be adjusted as above. -
10
Quench reaction with 300 μL stop buffer B.
Modify RNA with NMIA
-
11
Dilute the NMIA stock solution with anhydrous DMSO to a working concentration that modifies ~100–200 nucleotides, determined from pilot assays.
-
12
Apply 20 μL NMIA working solution to each gel piece from a third lane of the native gel. Incubate 30–120 min on ice.
-
13
Add 300 μL stop buffer B.
Elute RNA from gel
-
14
Add 300 μL stop buffer B to each gel piece from the fourth lane of the native gel.
These will serve as unmodified controls. -
15
Soak gel pieces several hours or overnight at 4°C. Recover RNA (see Alternate Protocol, steps 5 to 6).
-
16
Extract supernatants once with an equal volume of buffered phenol. Microcentrifuge 1 min at 10,000 × g, 4°C, to separate layers and carefully transfer all of upper aqueous phase to a clean microcentrifuge tube.
-
17
Extract aqueous phases with 24:1 chloroform/isoamyl alcohol and transfer upper aqueous phase to a new set of microcentrifuge tubes.
-
18
Precipitate RNA with 3 volumes of 100% ethanol, wash pellets with 70% ethanol, and dry the pellets (see Alternate Protocol, steps 8 to 10).
RNA may be stored at −20°C as an ethanol suspension or as a dried pellet for up to a week.
Detect modified bases by primer extension
-
19
Dissolve RNA pellets in 2 to 10 μL water or 0.5× TE, so that their concentrations are roughly equal (neglecting any carrier RNA that may have been added).
The relative amount of RNA present in each band of native gel can be estimated by quantifying distribution of radioactive samples in step 4. -
20
Perform primer extension using Superscript III reverse transcriptase (see Support Protocol 3).
RNA that was not subjected to native gel electrophoresis can be used as a template for dideoxynucleotide sequencing reactions. If sufficient RNA is recovered from the native gel, each sample can be divided into aliquots and used for several primer extension reactions. -
21
Separate primer extension products on a 6% or 8% polyacrylamide sequencing gel (APPENDIX 3B) or by capillary electrophoresis.
-
22
Detect primer extension products by autoradiography or by using a phosphorstorage imaging screen. Compare primer extension pattern of modified RNAs to unmodified controls to determine which bands are specific for reactions with DMS or kethoxal. Determine relative intensity of specific primer extension bands for each RNA conformer by comparing products of RNA samples that were recovered from different regions of the native gel.
Differences in the extent of modification may be quantified using a phosphorimager or densitometer.
SUPPORT PROTOCOL 1. IN VITRO SYNTHESIS OF 32P-LABELED PRE-RNA WITH T7 RNA POLYMERASE
Uniformly radiolabeled pre-RNA is prepared by run-off transcription of plasmid DNA or oligonucleotide templates (Milligan and Uhlenbeck, 1980) with purified phage T7 RNA polymerase (Davanloo et al., 1984). The template DNA must be cleaved downstream of the desired RNA sequence with a restriction enzyme. Transcription is carried out for short times (i.e., 20 to 30 min) at 30°C to minimize self-splicing (Emerick and Woodson, 1993).
Materials
1 μg/μL plasmid DNA containing a T7 promoter, digested with appropriate restriction enzyme(s) (e.g. Feinbaum, 2000; Tabor, 2000), or 0.2 μg/μL PCR product containing a T7 promoter.
10× T7 RNAP buffer (see recipe)
20 mM spermidine–HCl (store at −20°C) 10× NTPs “low A” (see recipe)
10 to 20 μCi [α-32P] ATP (store at −20°C)
500 to 1000 U/μL T7 RNA polymerase (store at −20°C) TE buffer, pH 7.5 (APPENDIX 2A) or another buffer
30°C water bath
Size exclusion spin column
Liquid scintillation counter
Scintillation fluid and vials
NOTE: Buffers and reagents should be kept on ice after thawing. Steps 1 to 4 are carried out at 4°C except where noted.
Transcribe RNA
-
1
Add the following to a 1.5-mL microcentrifuge tube (40 μL total volume):
1 μL 1 μg/μL digested plasmid DNA or 1–2 μL 0.2 μg/μL PCR product
4 μL 10× T7 RNAP buffer
4 μL 20 mM spermidine–HCl
4 μL 10× NTPs “low A”
1 to 2 μL 10 μCi/μL [α-32P] ATP
-
25 to 24 μL H2O.
For RNAs that are not self-splicing, the additional spermidine should be omitted.
-
2
Mix gently by tapping with finger. Spin briefly in a microcentrifuge to bring contents to the bottom of tube, if necessary.
-
3
Add 1 μL of 500 to 1000 U/μL T7 RNA polymerase. Mix well.
-
4
Incubate at 30°C for 20 to 30 min.
Short reaction times reduce accumulation of spliced products. For RNAs that do not self-cleave, better yields will be obtained by incubating 1 hr at 37°C.
Remove unincorporated nucleotide triphosphates
-
5
While the transcription reaction is proceeding, prepare a spin column at room temperature for each sample. Pre-equilibrate column with TE buffer, pH 7.5 (or another buffer). After the column has drained, spin 3 min at 3000 × g, room temperature, to remove all excess buffer. Avoid trapping air bubbles in column matrix.
Drain column in clean microcentrifuge tube and avoid touching tip. -
6
When transcription reaction is complete, immediately load entire sample onto top of spin column.
-
7
Spin 5 min at 3000 × g, room temperature, using a clean RNase-free tube to collect sample.
-
8
Discard column as solid radioactive waste.
-
9
Determine the yield of RNA by counting 1 or 2 μL eluate in a liquid scintillation counter. Place RNA on ice, or store at −20°C.
One should obtain approximately 105 cpm/μL.The RNA may also be purified by denaturing gel electrophoresis.
SUPPORT PROTOCOL 2. LARGER SCALE IN VITRO SYNTHESIS OF UNLABELED PRE-RNA WITH T7 RNA POLYMERASE
Larger quantities (i.e., 0.5 to 2 nmol) of unlabeled RNA may prepared by increasing the scale of in vitro transcription reactions. Abortive initiation products and unincorporated nucleotide triphosphates are removed by spin microconcentrators (ultrafiltration). For some applications, unwanted products must be removed by preparative polyacrylamide gel electrophoresis.
Materials
1 μg/μL digested plasmid DNA (10 μg total) or 5 μg total PCR product DNA
10× T7 RNAP buffer (see recipe)
1 M spermidine–HCl
50 or 100 mM each of ATP, CTP, GTP, and UTP
~1000 U/μL T7 RNA polymerase
0.5 M EDTA (APPENDIX 2A)
3 M sodium acetate, pH 5.0 (APPENDIX 2A)
100% ethanol
Microconcentrators with appropriate MWCO (eg Amicon Ultra 2 mL centrifugal filters)
15-mL sterile polypropylene culture tubes
Transcribe RNA
-
1
Add the following to a 15-mL sterile culture tube (2 mL total volume):
10 μL 1 μg/μg digested plasmid DNA or 0.5 μg/μL PCR DNA
200 μL 10 × T7 RNAP buffer
1 μL 1 M spermidine–HCl
20 μL each of 100 mM ATP, CTP, GTP, and UTP (1 mM each NTP final)
-
1.70 mL H2O.
For RNAs that are not self-splicing, spermidine should be omitted.
-
2
Mix thoroughly by vortexing. Centrifuge briefly in a benchtop centrifuge to bring contents to bottom of tube.
-
3
Add 10 μL concentrated T7 RNA polymerase. Mix well (do not vortex).
-
4
Incubate 1 hr at 30°C or 2 hr at 37 °C for non-self-cleaving RNAs.
-
5
Add 5 μL 0.5 M EDTA and mix well.
Remove unincorporated nucleotide triphosphates
-
6
Recover RNA by placing the sample in a spin concentrator. Follow the manufacturer’s instructions and suggested MWCO for ultrafitration of DNA.
Avoid concentrating RNA to above 5 mg/mL unless it is known to resist aggregation. -
7
Dilute sample with 1–2 mL TEN buffer, and concentrate again.
-
8
Repeat step 7 if needed to remove last traces of nucleotides and abortive initiation products.
In step 8 and following, the RNA can be desalted by diluting several times with low salt buffer. -
9
Determine final yield from the absorbance at 260 nm.
SUPPORT PROTOCOL 3. DETECTION OF MODIFIED BASES BY PRIMER EXTENSION
The positions of modified nucleotides are determined by extension of a 32P-labeled primer with reverse transcriptase using the modified RNA from Basic Protocol 3 as a template. Positions of the modified bases are determined by running the primer extension reactions on a sequencing gel alongside sequencing lanes prepared using ddNTPs for chain termination. Differences in the secondary and tertiary structure of the RNA conformers probed in Basic Protocol 3 can be determined by comparing the intensities of the termination products. Refer to UNIT 6.1 for an alternate protocol and more comprehensive discussion of primer extension methods. The modification pattern of the RNA can also be determined by extension of a dye-labeled primer and capillary electrophoresis (Wilkinson et al., 2006; Vasa et al., 2008) or by high-throughput sequencing (Lucks et al., 2011).
Additional Materials (also see Basic Protocols 1 and 3)
5–20 μL 1 pmol/μL 5′ 32P-labeled sequencing primer (UNIT 6.1)
Diluted RNA for primer extension (see Basic Protocol 3)
Unmodified RNA (250 nM) (see Support Protocol 2)
2.5 mM ddATP, ddCTP, ddGTP, and ddTTP solutions, in water
5× first strand buffer (Invitrogen; see recipe)
10 mM dNTP mix: 10 mM each of dATP, dCTP, dGTP, and dTTP in water
Superscript III reverse transcriptase (Invitrogen Life Technologies)
55° and 65°C water baths or heating blocks
Ethanol and salt for RNA precipitation
2× formamide loading buffer (see recipe)
Additional reagents and equipment for preparing and running 6% or 8% polyacrylamide sequencing gel (APPENDIX 3B)
NOTE: The following steps are carried out on ice except where indicated. Thawed reagents should be kept on ice throughout.
Anneal complementary primer to RNA
-
1
If desired, prepare the RNA template by denaturing at 95 °C for 1 min and immediately placing on ice.
This step is only necessary for highly structured RNA templates. -
2
For each sample, mix primer and template in a 0.5-mL microcentrifuge tube (total volume 12 μL):
10 μL diluted RNA (see Basic Protocol 3, step 16)
1 μL 1 pmol/μL 5′ 32P-labeled sequencing primer
-
1 μL deionized water.
It is helpful to carry out control reactions using 0.1 to 0.5 pmol RNA that has been modified with DMS, kethoxal or NMIA in solution (UNIT 6.1).
-
3
Incubate in a water bath or heating block at 65°C for 3 min and place immediately on ice. Microcentrifuge briefly at maximum speed before opening tubes. Keep samples on ice.
Some primer/template combinations require heating 2 min at 80°C.
Prepare sequencing reactions
-
4
Mix the following in a 0.5-mL tube (40.8 μL total):
8 μL unmodified RNA (2 pmol total)
5 μL 1 pmol/μL 5′ 32P-labeled primer
27.9 μL deionized water.
-
5
Anneal primer as in step 3.
-
6
Divide the annealing reaction from step 5 into five 8.16-μL aliquots.
-
7
To each aliquot add 3.84 μL either 2.5 mM ddATP, ddCTP, ddGTP, ddTTP, or water.
The four reactions containing dideoxynucleotide triphosphates serve as sequencing lanes; the reaction containing only buffer serves as a blank. The blank should have few or no extension products shorter than full-length, unless the RNA is partially degraded or has strong secondary structure that causes the reverse transcriptase to terminate.
Extend primer with reverse transcriptase
-
8
Prepare a RT cocktail by mixing the following (volumes are per sample):
4 μL 5× First strand synthesis buffer
1 μL 100 mM DTT
1 μL 10 mM dNTP mix
1.5 μL water
0.5 μL Superscript III.
Add the enzyme last. Resuspend enzyme thoroughly in the buffer, but do not vortex and avoid foaming.
-
9
Begin primer extension by adding 8 μL RT cocktail to each 12 μL reaction (steps 3 and 7). Immediately place each tube in a water bath or heat block at 55°C for 15 min or up to 1 hr for long RNAs.
-
10
Stop reactions by adding 20 μL 2× formamide loading buffer or by heating 85 °C for 5 min.
If desired, cDNA can be concentrated by ethanol precipitation after terminating the reaction with heat. Pellets should be dissolved in 5–10 μL 1× formamide loading buffer. Samples in loading buffer are now ready to be analyzed on a sequencing gel (see Basic Protocol 3, steps 18 and 19). Samples may be stored at −20°C for one week, if needed.
REAGENTS AND SOLUTIONS
Use deionized or distilled nuclease-free water in all recipes and protocol steps. To eliminate traces of RNase on glassware, rinse in RNase-free water and bake 2 hr or overnight at 150°C. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX.
First strand synthesis buffer, 5× (Invitrogen Superscript III)
100 μL 1 M Tris–HCl, pH 8.3 (APPENDIX 2A; 250 mM final)
150 μL 1 M KCl (375 mM final)
6 μL 1 M MgCl2 (15 mM final)
144 μL deionized water (400 μL total volume)
Store up to 6 months at −20°C
Folding buffer, 5×
250 L 1 M NaHEPES, pH 7.5 (final concentration 250 mM)
500 μL 1 M (NH4)2SO4 (final concentration 500 mM)
10 μL 0.5 M sodium EDTA, pH 8.3 (APPENDIX 2A; final concentration 5 mM)
240 μL distilled H2O (total volume 1 mL)
-
Store up to 1 year at −20°C
The composition of the folding buffer should be adjusted to maximize the function of the RNA of interest. For example, the RNA may be refolded in 25 mM Na-HEPES, pH 7.5, with or without 50 mM NaCl.
Formamide loading buffer, 2×
940 μL deionized formamide
40 μL 0.5 M EDTA, pH 8.3 (APPENDIX 2A; 20 mM final)
20 μL 2% (w/v) xylene cyanol
20 μL 2% (w/v) bromphenol blue
Store up to one year at −20°C
Glycerol loading buffer, 5×
500 μL glycerol
20 μL 2% (w/v) xylene cyanol
Bring up to 1 mL with H2O
Store up to 1 year at −20°C or 1 month at room temperature
GTP in folding buffer, 0.1 mM
Dilute a 100 mM stock solution of GTP to 0.1 mM with 1× folding buffer (see recipe) containing 6 mM MgCl2. Store in aliquots at −20° or −80°C.
Hepes-KCl (HK) buffer, 5×
400 L 1 M K-HEPES, pH 7.6 (final concentration 400 mM)
500 μL 3 M KCl (final concentration 1.5 M)
2.01 μL 2-mercaptoethanol (final concentration 30 mM)
98 μL distilled H2O (total volume 1 mL)
Store up to 6 months at −20°C
NTPs “low A,” 10×
Obtain concentrated stocks (e.g., 50 or 100 mM) of each nucleoside triphosphate (ATP, CTP, GTP, and UTP). These may be prepared by dissolving 100 mg solid in 2 mL water at 4°C. Adjust the pH to 7.0 using small aliquots (i.e., 10 to 50 μL) of concentrated NaOH. The pH may be monitored with a microelectrode or by spotting 5 μL onto calibrated pH indicator strips. Determine the concentration from the UV absorbance of a 1:1000 dilution, and adjust to the desired final concentration with additional water. Store in aliquots at −80°C.
-
To prepare working solution, mix 1.25 μL 100 mM ATP, 6.25 μL 100 mM CTP, 6.25 μL 100 mM GTP, 6.25 μL 100 mM UTP and bring final volume to 0.5 μL with water. Store at −20°C.
Final concentrations are 0.25 mM ATP and 1.25 mM each CTP, GTP, and UTP.
Stop buffer A
14 μL 2-mercaptoethanol (final concentration 0.2 M)
100 μL 3 M sodium acetate (APPENDIX 2A; final concentration 0.3 M)
40 μL 0.5 M sodium EDTA, pH 8.3 (APPENDIX 2A; final concentration 20 mM)
846 μL distilled H2O
Mix well and store up to 3 months at −20°C
Stop buffer B
100 μL 3 M sodium acetate (APPENDIX 2A; final concentration 0.3 M)
40 μL 0.5 M sodium EDTA, pH 8.3 (APPENDIX 2A; final concentration 20 mM)
860 μL distilled H2O.
Mix well and store up to 1 year at −20°C
T7 RNAP (RNA polymerase) buffer, 10×
400 μL 1 M Tris–HCl, pH 7.5 (APPENDIX 2A; final concentration 400 mM)
150 μL 1 M MgCl2 (APPENDIX 2A; final concentration 150 mM)
20 μL 1 M spermidine–HCl (final concentration 20 mM)
50 μL 1 M dithiothreitol (DTT, APPENDIX 2A; final concentration 50 mM)
380 μL distilled H2O (total volume 1 mL)
Mix well and store up to 3 months at −20°C
TEN (Tris/EDTA/NaCl) buffer
10 mM Tris–HCl, pH 7.5 (APPENDIX 2A)
1 mM disodium EDTA (APPENDIX 2A)
250 mM sodium chloride
Autoclave and store indefinitely at room temperature or up to 2 months after opening
THEM (Tris/HEPES/EDTA/MgCl2) buffer, 10×, pH 7.4
157.08 g HEPES (final concentration 660 mM)
41.14 g Tris base (final concentration 340 mM)
0.36 g disodium EDTA (final concentration 1 mM)
20.32 g MgCl2 (final concentration 100 mM) 1 L H2O (final volume)
-
Autoclave and store up to 1 year at room temperature
The MgCl2 content of the running buffer should be adjusted to the minimum needed to stabilize the folded RNA. For the Tetrahymena and Azoarcus ribozymes, 1× concentration is 3 mM MgCl2.
Urea loading buffer, 2×
1.1 g urea (final concentration 10 M)
4 μL 2% (w/v) xylene cyanol
980 μL distilled H2O
Dissolve urea at 50°C
Store in 0.5-mL aliquots up to 1 year at −20°C
COMMENTARY
Background Information
Native polyacrylamide gel electrophoresis is now well established as a method for characterizing stable nucleic acid–protein complexes (Fried and Crothers, 1981). Complexes that normally dissociate within several minutes were found to be stable during many hours of electrophoresis, apparently due to a “caging” effect of the polyacrylamide matrix (Fried and Crothers, 1981). More recently, this approach has been used to quantify the binding of small RNA substrates to the Tetrahymena ribozyme (Pyle et al., 1990). It has also been used to monitor conformational changes in large and small RNAs (e.g., LeCuyer and Crothers, 1993; Emerick and Woodson, 1994). The mobility of native RNA is correlated with its hydrodynamic radius (Orr et al., 1998), with compact structures migrating more rapidly than extended forms. Separation of different RNA conformers requires that they exchange more slowly than the RNA is transported through the polyacrylamide gel matrix. As a result, this method is most useful when analyzing RNA sequences that fold into defined tertiary structures. More recently, we have used this approach to analyze binding of ribosome proteins to fragments of ribosomal RNA (Abeysirigunawardena and Woodson, 2015).
Although these protocols are optimized for analysis of a 657-nt RNA, electrophoretic conditions can be readily varied depending on the RNA to be studied. The author has successfully used gels containing 4% to 12% polyacrylamide and 3 to 10 mM MgCl2. Nonionic substrates such as guanosine may be incorporated into the gel matrix. RNA folding intermediates or complexes that exchange on the 1-min time scale at room temperature can be resolved if the appropriate conditions are chosen. This method is not suitable for trapping short-lived intermediates, as ~15 sec is required for samples to enter the polyacrylamide matrix (Pan and Woodson, 1998).
Native gel electrophoresis is easily combined with a variety of methods for probing RNA structure, including chemical modification and photo-cross-linking (Branch et al., 1989). Chemical reagents are commonly used to map regions of the RNA that are accessible to the chemical probe (Ehresmann et al., 1987). NMIA and other SHAPE reagents selectively modify the ribose 2’OH of residues that are single-stranded and thus more flexible, or that are held in a highly reactive conformation by the RNA tertiary structure (Steen et al., 2012) or a protein (Peng et al., 2014). The modified positions are typically detected by extension of a complementary DNA primer with reverse transcriptase (Inoue and Cech, 1985; Moazed et al., 1986). More recently, high-throughput sequencing methods have been adapted to read out the structures of RNA in the entire transcriptome (reviewed in Kwok et al., 2015).
A number of organic and inorganic compounds and enzymes that modify nucleic acids have been used to footprint protein-DNA complexes within polyacrylamide gels (Law et al., 1987; Straney and Crothers, 1987), and these methods were extended to the analysis of RNA structure (Emerick and Woodson, 1994). The advantage of modifying RNA in the gel matrix is that the rate of conformational exchange remains low. The disadvantage is that reagents must diffuse through the gel, making it difficult to control reaction time. In addition, some reagents, such as diethylpyrocarbonate, are strongly inhibited by the gel components. An alternative approach that avoids some of these pitfalls is to modify the RNA before electrophoresis, as in modification interference (Pan and Woodson, 1998).
Critical Parameters
An important parameter that affects separation of different conformational forms of RNA by electrophoresis is whether or not the conformational change induces a significant change in the overall shape of the molecule (i.e., by altering tertiary interactions). Beyond this, it is important to ensure that the folded RNA is stable in the electrophoresis and sample buffers, and that the internal temperature of the gel remains below 12°C. For the Tetrahymena pre-RNA, the bands representing active and inactive conformers become blurred at 15°C, and are no longer separated at 20°C. When labeling long RNA by hybridizing with 32P- or dye-labeled oligonucleotides, the complex must be stable to electrophoresis, with Kd ≤ 5 nM. The oligomer binding site must be chosen so that it does not interfere with folding of the RNA or with ligand recognition. Two-dimensional electrophoresis and in situ chemical modification require extensive handling of RNA. Even very slight levels of ribonuclease contamination can result in undesirable background at the end of the experiment. To obtain interpretable results, the concentrations of chemical modifying reagents and times of exposure must be adjusted empirically for each RNA sequence, so that each molecule is modified only once on average.
Anticipated Results
Typical results from native gel electrophoresis of the Tetrahymena pre-RNA are illustrated in Figure 11.4.2. Addition of GTP and RNA substrates to the gel showed that only the more rapidly migrating species was competent to self-splice (Emerick and Woodson, 1994). Consistent with this result, nucleotides in the catalytic core of the intron were more susceptible to modification with DMS and kethoxal in the inactive conformer than in the active form. In general, a qualitative test of ribozyme activity is relatively straightforward, if a reasonable degree of separation in the native gel is obtained. In situ chemical modification of the RNA is more challenging, and one can expect that several trials will be required before the optimal conditions are found. An advantage of labeling the RNA with a fluorophore is that protein binding can be confirmed by co-localization of two fluorophores in the gel, if the protein is labeled with a dye that fluoresces in a different window of the spectrum. This illustrated for binding of protein S20 to a fragment of the 16S ribosomal RNA in Figure 11.4.3.
Figure 11.4.2.
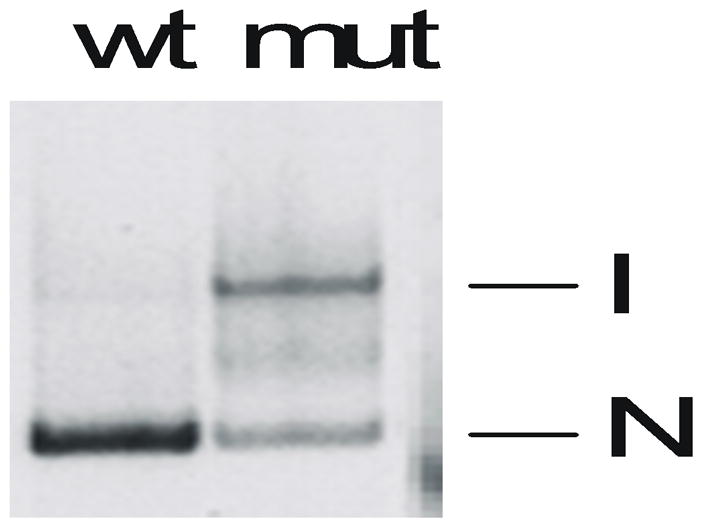
Native 6% polyacrylamide gel of 32P-labeled Tetrahymena pre-RNA (657-nt). The RNA was annealed at 95°C before loading. Abbreviations: I, inactive pre-RNA; N, native (active) RNA; wt, wild type; mut, mutation (170C) that destablizes the active RNA structure.
Figure 11.4.3.

Binding of 15 nM RNA•Cy3-oligomer + Cy5-S20 by native PAGE. Top, a 16S rRNA fragment is labeled by hydridization with Cy3-conjugated oligonucleotide. Protein S20 is labeled with Cy5. Bottom, Native PAGE with increasing S20-Cy5 shows colocalization of S20 with labeled RNA in the gel. Superposition of green, (532 nm) and red (650 nm) scans; Typhoon 9410.
Time Considerations
All of the protocols described here can be carried out in 1 to 3 days. Two-dimensional gel electrophoresis requires a continuous 10- to 12-hr block of time.
Acknowledgments
The author thanks the members of her research group who contributed to the following protocols over the years, especially S. Abeysirigunawardena, Y. Hao, S. Panja, and I. Sharma for help with recent updates.
This work was funded by NIH 1R01GM46686 and NIH 1R01GM60819.
Literature Cited
- Abeysirigunawardena SC, Woodson SA. Differential effects of ribosomal proteins and Mg2+ ions on a conformational switch during 30S ribosome 5′-domain assembly. RNA. 2015;21:1859–1865. doi: 10.1261/rna.051292.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branch AP, Benedfeld BJ, Paul CP, Robertson HP. Analysis of ultraviolet-induced RNA-RNA cross-links: A means for probing RNA structure-function relationships. Methods Enzymol. 1989;180:418–442. doi: 10.1016/0076-6879(89)80115-6. [DOI] [PubMed] [Google Scholar]
- Davanloo P, Rosenberg AH, Dunn JJ, Studier FW. Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1984;81:2035–2039. doi: 10.1073/pnas.81.7.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehresmann C, Baudin F, Mougel M, Romby P, Ebel JP, Ehresmann B. Probing the structure of RNAs in solution. Nucl Acids Res. 1987;15:9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerick VL, Woodson SA. Self-splicing of the Tetrahymena pre-rRNA is decreased by misfolding during transcription. Biochemistry. 1993;32:14062–14067. doi: 10.1021/bi00213a040. [DOI] [PubMed] [Google Scholar]
- Emerick VL, Woodson SA. Fingerprinting the folding of a group I precursor RNA. Proc Natl Acad Sci USA. 1994;91:9675–9679. doi: 10.1073/pnas.91.21.9675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinbaum R. Introduction to plasmid biology. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. John Wiley & Sons; New York: 2000. pp. 1.5.1–1.5.17. [DOI] [PubMed] [Google Scholar]
- Fried M, Crothers DM. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucl Acids Res. 1981;9:6505–6525. doi: 10.1093/nar/9.23.6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Cech TR. Secondary structure of the circular form of the Tetrahymena rRNA intervening sequence: A technique for RNA structure analysis using chemical probes and reverse transcriptase. Proc Natl Acad Sci USA. 1985;76:1670–1764. doi: 10.1073/pnas.82.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok CK, Tang Y, Assmann SM, Bevilacqua PC. The RNA structurome: transcriptome-wide structure probing with next-generation sequencing. Trends Biochem Sci. 2015;40:221–232. doi: 10.1016/j.tibs.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Law R, Kuwabara MD, Briskin M, Fasel N, Hermanson G, Sigman DS, Wall R. Protein-binding site at the immunoglobulin mu membrane polyadenylation signal: Possible role in transcription termination. Proc Natl Acad Sci USA. 1987;84:9160–9164. doi: 10.1073/pnas.84.24.9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCuyer KA, Crothers DM. The Leptomonas collosoma spliced leader RNA can switch between two alternate structural forms. Biochemistry. 1993;32:5301–5311. doi: 10.1021/bi00071a004. [DOI] [PubMed] [Google Scholar]
- Lucks JB, Mortimer SA, Trapnell C, Luo S, Aviran S, Schroth GP, Pachter L, Doudna JA, Arkin AP. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq) Proc Natl Acad Sci U S A. 2011;108:11063–1108. doi: 10.1073/pnas.1106501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1980;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- Moazed D, Stern S, Noller HF. Rapid chemical probing of conformation in 16 S ribosomal RNA and 30S ribosomal subunits using primer extension. J Mol Biol. 1986;187:399–416. doi: 10.1016/0022-2836(86)90441-9. [DOI] [PubMed] [Google Scholar]
- Orr JW, Hagerman PJ, Williamson JR. Protein and Mg2+-induced conformational changes in the S15 binding site of 16S ribosomal RNA. J Mol Biol. 1998;275:453–464. doi: 10.1006/jmbi.1997.1489. [DOI] [PubMed] [Google Scholar]
- Pan J, Woodson SA. Folding intermediates of a self-splicing RNA: Mispairing of the catalytic core. J Mol Biol. 1998;280:597–609. doi: 10.1006/jmbi.1998.1901. [DOI] [PubMed] [Google Scholar]
- Peng Y, Curtis JE, Fang X, Woodson SA. Structural model of an mRNA in complex with the bacterial chaperone Hfq. Proc Natl Acad Sci U S A. 2014;111:17134–17139. doi: 10.1073/pnas.1410114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle AM, McSwiggen JA, Cech TR. Direct measurement of oligonucleotide substrate binding to wild-type and mutant ribozymes from Tetrahymena. Proc Natl Acad Sci USA. 1990;87:8187–8191. doi: 10.1073/pnas.87.21.8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen KA, Rice GM, Weeks KM. Fingerprinting noncanonical and tertiary RNA structures by differential SHAPE reactivity. J Am Chem Soc. 2012;134:13160–13163. doi: 10.1021/ja304027m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straney DC, Crothers DM. Comparison of the open complexes formed by RNA polymerase at the Escherichia coli lac UV5 promoter. J Mol Biol. 1987;193:279–292. doi: 10.1016/0022-2836(87)90219-1. [DOI] [PubMed] [Google Scholar]
- Tabor S. DNA-dependent RNA polymerases. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. John Wiley & Sons; New York: 2000. pp. 3.8.1–3.8.4. [Google Scholar]
- Vasa SM, Guex N, Wilkinson KA, Weeks KM, Giddings MC. ShapeFinder: a software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA. 2008;14:1979–1990. doi: 10.1261/rna.1166808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc. 2006;1:1610–1616. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]