

Phosphatidylcholine transport protein (PC-TP) is a highly specific phosphatidylcholine-binding protein that we previously showed to regulate hepatocellular nutrient metabolism through its interacting partner thioesterase superfamily member 2 (Them2). This study identifies a pathogenic role for PC-TP, independent of Them2, in the methionine- and choline-deficient diet model of experimental steatohepatitis. Our current observations suggest that PC-TP promotes liver injury by mediating the intermembrane transfer of phosphatidylcholines, thus stabilizing more pathogenic microvesicular lipid droplets.
Keywords: nonalcoholic steatohepatitis, lipid droplet, phospholipid, triglyceride, methionine- and choline-deficient diet
Abstract
Mice fed a methionine- and choline-deficient (MCD) diet develop steatohepatitis that recapitulates key features of nonalcoholic steatohepatitis (NASH) in humans. Phosphatidylcholine is the most abundant phospholipid in the surfactant monolayer that coats and stabilizes lipid droplets within cells, and choline is required for its major biosynthetic pathway. Phosphatidylcholine-transfer protein (PC-TP), which exchanges phosphatidylcholines among membranes, is enriched in hepatocytes. PC-TP also regulates fatty acid metabolism through interactions with thioesterase superfamily member 2. We investigated the contribution of PC-TP to steatohepatitis induced by the MCD diet. Pctp−/− and wild-type control mice were fed the MCD diet for 5 wk and were then euthanized for histopathologic and biochemical analyses, as well as determinations of mRNA and protein expression. Whereas all mice developed steatohepatitis, plasma alanine aminotransferase and aspartate aminotransferase activities were only elevated in wild-type mice, indicating that Pctp−/− mice were protected from MCD diet-induced hepatocellular injury. Reduced hepatotoxicity due to the MCD diet in the absence of PC-TP expression was further evidenced by decreased activation of c-Jun and reduced plasma concentrations of fibroblast growth factor 21. Despite similar total hepatic concentrations of phosphatidylcholines and other lipids, the relative abundance of microvesicular lipid droplets within hepatocytes was reduced in Pctp−/− mice. Considering that the formation of larger lipid droplets may serve to protect against lipotoxicity in NASH, our findings suggest a pathogenic role for PC-TP that could be targeted in the management of this condition.
NEW & NOTEWORTHY Phosphatidylcholine-transfer protein (PC-TP) is a highly specific phosphatidylcholine-binding protein that we previously showed to regulate hepatocellular nutrient metabolism through its interacting partner thioesterase superfamily member 2 (Them2). This study identifies a pathogenic role for PC-TP, independent of Them2, in the methionine- and choline-deficient diet model of experimental steatohepatitis. Our current observations suggest that PC-TP promotes liver injury by mediating the intermembrane transfer of phosphatidylcholines, thus stabilizing more pathogenic microvesicular lipid droplets.
nonalcoholic fatty liver disease (NAFLD) is a common condition that affects 20–30% of the United States population and is strongly associated with obesity and type 2 diabetes (52). A subset of these patients develop nonalcoholic steatohepatitis (NASH), characterized by hepatic steatosis in association with inflammation and cell death plus fibrosis, which may lead to important clinical consequences including cirrhosis and hepatocellular carcinoma (52).
Mouse models are commonly utilized for studying the pathogenesis of NASH and for the preclinical testing of potential therapies (28). Although none faithfully recapitulates the human disease, each has relative strengths and can be leveraged to study specific aspects of NASH pathogenesis (37). When fed a methionine- and choline-deficient (MCD) diet, mice develop histopathologic lesions that appear quite similar to NASH in humans (44). An important drawback of this model is that the mice do not concomitantly develop obesity and peripheral insulin resistance (48). These features can instead be captured by caloric-rich diets or a variety of genetic modifications (28); however, the typical shortcoming of such models is the failure to develop significant steatohepatitis, cell death, and fibrosis.
Phosphatidylcholine is the predominant phospholipid in most mammalian membranes as well as in the surfactant monolayer that stabilizes lipid droplets within cells (16). Phosphatidylcholine is also important in the packaging and export of triglycerides from the liver in the form of very low density lipoproteins (VLDL; 67). The major biosynthetic route for phosphatidylcholine synthesis within cells is referred to as the Kennedy pathway, which is characterized by the incorporation of choline into diacylglycerol molecules. Within the liver, phosphatidylcholine can also be formed via three sequential methylations of phosphatidylethanolamine. This reaction is catalyzed by the enzyme phosphatidylethanolamine-N-methyl transferase (PEMT; 16). In humans and mice, either dietary choline deficiency or genetic deficiencies in PEMT activity can limit hepatic phosphatidylcholine biosynthesis and result in hepatosteatosis (69).
Phosphatidylcholine transfer protein (PC-TP; synonym StarD2) is a highly specific phosphatidylcholine-binding protein that is enriched in the liver and oxidative tissues (23). PC-TP complexes with phosphatidylcholine molecules in a 1:1 stoichiometry and catalyzes the intermembrane transfer and exchange of phosphatidylcholines in vitro (65). PC-TP does not influence the biosynthetic rate of phosphatidylcholines in cells (1), and whether it functions as a phospholipid exchange/transfer protein in vivo is unclear (66). Upon complexing with phosphatidylcholine, PC-TP binds and enhances the enzymatic activity of the thioesterase superfamily member 2 (Them2), a mitochondria-associated long-chain acyl-CoA thioesterase (synonym Acot13). The PC-TP/Them2 complex regulates nutrient and energy homeostasis apparently by controlling rates of fatty acid oxidation and incorporation into glycerolipids (22, 26, 50). In this context, PC-TP appears to function as a sensor of the fatty acyl composition of phosphatidylcholines in surrounding membranes, which, in turn, regulates cellular fatty acid metabolism and insulin signaling by modulating Them2 activity (11, 26). Consistent with this hypothesis, the genetic ablation of either PC-TP or Them2 mitigates hepatic insulin resistance (11, 53).
Because choline deficiency reduces phosphatidylcholine biosynthesis within the liver, the current study was designed to examine a potential role for PC-TP in the development of MCD diet-induced steatohepatitis. We observed reduced markers of hepatic injury in Pctp−/−, but not Them2−/− mice. Although both strains developed steatohepatitis, the histologic pattern of steatosis in Pctp−/− mice was characterized by a reduction in microvesicular lipid droplets. This occurred largely in the absence of changes in hepatic lipid concentrations or compositions. When taken together, our results suggest that PC-TP regulates lipid droplet size by controlling the distribution of hepatic phosphatidylcholines.
MATERIALS AND METHODS
Mice.
Pctp−/− and Them2−/− mice and their respective wild-type (WT) controls on an FVB/NJ genetic background were described previously (21, 50). Age-matched adult (8–12 wk old) male mice were housed in a specific pathogen-free facility with a standard 12-h:12-h alternate light-dark cycle and were fed ad libitum either a standard rodent diet (PicoLab Rodent Diet 20-5053; LabDiet, St. Louis, MO) or a methionine- and choline-deficient diet (TD.90262; Harlan Laboratories, Madison, WI) with free access to drinking water. Groups were assigned randomly, with 3–5 mice housed per cage in individually ventilated cages (Tecniplast IVC rack systems) on pine chip bedding with cotton nesting squares. Body weight was measured twice per week. Liver and blood were harvested from fed mice at the initiation of the light cycle, following euthanasia using isoflurane. Tissues were snap frozen in liquid N2 and stored at −80°C. Protocols for animal use, treatment, and euthanasia were approved by the Institutional Animal Care and Use Committee of Harvard Medical School.
Biochemical assays.
Plasma activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured enzymatically using Stanbio Laboratory LiquiColor test kits no. 2930 and 2920, respectively (Stanbio, Boerne, TX). In preliminary studies, results were also validated using ALT and AST Activity Assay Kits MAK052 and MAK055, respectively (Sigma-Aldrich). Plasma fibroblast growth factor 21 (FGF21) concentrations were measured by immunoassay (Quantikine ELISA Rat/Mouse Immunoassay MF2100; R&D Systems, Minneapolis, MN). β-Hydroxybutyrate concentrations were measured enzymatically using a β-hydroxybutyrate LiquiColor kit (no. 2440-058; Stanbio). Hepatic malondialdehyde (MDA) concentrations were measured using a thiobarbituric acid-reactive substances (TBARS) assay kit, TCA method (no. 700870; Cayman Chemical, Ann Arbor, MI). Concentrations of triglycerides, nonesterified fatty acids, and cholesterol in the plasma and liver were determined enzymatically (Wako Diagnostics, Mountain View, CA; 62). Hepatic contents of hydroxyproline were determined using a Hydroxyproline Assay Kit (no. MAK008; Sigma-Aldrich), with the modification that samples were hydrolyzed overnight at 105°C and then filtered (0.2 µm).
Liver histology.
Freshly harvested liver samples were fixed in 4% paraformaldehyde, embedded in paraffin, cut into 5-µm sections, and stained with hematoxylin and eosin (36). Sirius red collagen staining was performed by first dewaxing, hydrating, and staining liver samples with Weigert’s hematoxylin for 10 min. This was followed by washing and staining with Direct Red (no. 365540; Sigma-Aldrich) for 1 h. After washing and dehydrating using 100% alcohol, samples were mounted using permount (SP15-500, Fisher Scientific). Histology slides were viewed and captured using a Zeiss Axio Imager M1 microscope and AxioVisionHR camera. NAFLD activity score (NAS) and fibrosis scores were assigned by a blinded observer (19). The relative abundance of microvesicular and macrovesicular lipid droplets was also scored in a blinded manner: three images per mouse were visually inspected and scored according to the distribution of macrovesicular and/or microvesicular steatosis; scores were averaged per mouse and displayed as a percentage.
Tolerance tests.
Tolerance tests to glucose and pyruvate were performed in mice fasted for 6 h. Twenty percent glucose [for glucose tolerance test (GTT)] or sodium pyruvate [for pyruvate tolerance test (PTT)] solutions were injected intraperitoneally at 1.5 g/kg body wt. Plasma glucose concentrations in tail blood were measured at baseline and every 15 min for 1 h using a glucometer (GE100 blood glucose monitoring system; GE Healthcare, Taichung, Taiwan). GTT and PTT were performed 3 and 4 wk, respectively, after initiation of the MCD diet. For the insulin tolerance test, 2 wk after initiation of MCD diet feeding, mice were fasted for 1.5 h followed by intraperitoneal injection with insulin at 0.25 U/kg body wt and measurement of blood glucose at baseline, 15 min, and 30 min. All tests were performed ~8 h following light cycle initiation.
Lipidomics.
Lipidomic analyses were performed using the Metabolomics Platform at the Broad Institute (Cambridge, MA; Dr. Clary Clish, Director). Briefly, liver samples (weight range, 24.6–107.3 mg) were homogenized in 4 µl of water per milligram of tissue weight using a bead mill (TissueLyser II; Qiagen, Valencia, CA), and then aqueous homogenates were aliquoted for lipid profiling. Lipids were extracted from homogenates (10 µl) using 190 µl of isopropanol containing 1-heptadecanoyl-2-hydroxy-sn-glycero-3-phosphocholine, 1,2-didodecanoyl-sn-glycero-3-phosphocholine, N-heptadecanoyl-d-erythro-sphingosine (Avanti Polar Lipids; Alabaster, AL), 1,2,3-triheptadecanoylglycerol, and 1,3-dipentadecanoylglycerol (Sigma-Aldrich) as internal standards. After centrifugation (10 min, 9,000 g, ambient temperature), supernatants (2 µl) were injected directly onto a 100 × 2.1-mm Acquity ethylene-bridged hybrid (BEH) C8 column (1.7 µm; Waters, Milford, MA). The column was eluted at a flow rate of 450 µl/min isocratically for 1 min at 80% mobile phase A (95:5:0.1 vol/vol/vol of 10 mM ammonium acetate:methanol:formic acid), followed by a linear gradient to 80% mobile phase B (99.9:0.1 vol/vol methanol:formic acid) over 2 min, a linear gradient to 100% mobile phase B over 7 min, and then 3 min at 100% mobile phase B. Mass spectrometry analyses were performed using electrospray ionization in the positive ion mode using full scan analysis over mass-to-charge ratio (m/z) 200–1,100 at 70,000 resolution and 3-Hz data acquisition rate. Additional mass spectrometry settings were as follows: ion spray voltage, 3.0 kV; capillary temperature, 320°C; probe heater temperature, 300°C; sheath gas, 50; auxiliary gas, 15; and S-lens radio frequency level, 60. Full scan raw data were processed using TraceFinder 3.1 software (Thermo Fisher Scientific; Waltham, MA).
Gene expression analysis.
Relative mRNA expression in liver homogenates was determined by quantitative PCR (qPCR) using SYBR Green Real-Time PCR Master Mix (Applied Biosystems/Thermo Fisher Scientific). RNA was extracted using TRIzol and cDNA synthesized using Superscript III First Strand Synthesis System (18080-051; Invitrogen). Genes were expressed relative to ribosomal protein L32 (RPL32). The nucleotide sequences of oligonucleotides used for qPCR can be found in Table 1.
Table 1.
Oligonucleotides used for qPCR
| Gene | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| Phosphatidylcholine transfer protein; Pctp | CCAGAGTATCTCGGCACCTC | ACGCTTTCACCATGTCCTC |
| Sterol regulatory element binding protein-1c; Srebp1c | TGGATTGCACATTTGAAGACAT | GGCCAGAGAAGCAGAAGAGA |
| Carbohydrate response element binding protein-alpha; Chrebpa | CGACACTCACCCACCTCTTC | TTGTTCAGCCGGATCTTGTC |
| Acetyl-CoA carboxylase; Acaca | GAGAGGGCTCAAGTCCTTCC | ACATCCACTTCCACACACGA |
| Fatty acid synthase; Fasn | TGGGTTCTAGCCAGCAGAGT | ACCACCAGAGACCGTTATGC |
| Diacylglycerol acyltransferase 3; Dgat1 | GGCCCAAGGTAGAAGAGGAC | GATCAGCATCACCACACACC |
| Diacylglycerol acyltransferase 4; Dgat2 | AGGATCTGCCCTGTCACG | GCCAGCCAGGTGAAGTAG |
| Glycerol-3-phosphate acyltransferase 3; Gpat3 | TACATGCCTCCCATGACTAG | ATCCGTTGCCCACGATCATC |
| Glycerol-3-phosphate acyltransferase 4; Gpat4 | TGTCTGGTTTGAGCGTTCTG | TTCTGGGAAGATGAGGATGG |
| Fibroblast growth factor 21; Fgf21 | CTGGGGGGTCTACCAAGCATA | CACCCAGGATTTGAATGACC |
| Monocyte chemoattractant protein-1; Mcp1 | GTTGGCTCAGCCAGATGCA | AGCCTACTCATTGGGATCATCTTG |
| Tumor necrosis factor-alpha; Tnfa | CACAAGATGCTGGGACAGTGA | TCCTTGATGGTGGTGCATGA |
| Matrix metalloproteinase-9; Mmp9 | TGTCTGGAGATTCGACTTGAAGTC | TGAGTTCCAGGGCACACCA |
| Transforming growth factor-beta; Tgfb | GTGTGGAGCAACATGTGGAACTCTA | TTGGTTCAGCCACTGCCGTA |
| Alpha-smooth muscle actin; Acta2 | TCCTGACGCTGAAGTATCCGATA | GGTGCCAGATCTTTTCCATGTC |
| c-jun | GCCAACCTCAGCTTCAA | CCTGAGACTCCATGTCGATAG |
| CCAAT/enhancer-binding protein; Chop | GGGCCATAGAACTCTGACTGGA | CATCCCAAAGCCCTCGCTCTCCA |
| Inositol-requiring enzyme 1-alpha; Ire1a | CCCAAATGTGATCCGCTACT | TTGAGAGAATGCAGGTGTGC |
| Spliced X-box-binding protein-1; Xbp1s | GAGTCCGCAGCAGGTG | GTGTCAGAGTCCATGGGA |
| Unspliced X-box-binding protein-1; Xbp1u | AAGAAACACGCTTGGGAATGG | ACTCCCCTTGGCCTCCAC |
| Activating transcription factor 6; Atf6 | GCCCAGACTGTTTTGCTCTC | CCCATACTTCTGGTGGCACT |
| Liver receptor homolog-1; Lrh1 | CCCTGCTGGACTACACGGTTT | GCTAATGGGAGATGTGACAAA |
| Polo-like kinase 3; Plk3 | ACCTACAGCACCGCCATATC | CGCAGGTAGTAGCGAACCTC |
| Sterol 12-α-hydroxylase; Cyp8b1 | GCCTTCAAGTATGATCGGTTCCT | GATCTTCTTGCCCGACTTGTAGA |
| Small heterodimer partner 1; Shp1 | AACCTGCCGTCCTTCTGCCA | AACCTGCCGTCCTTCTGCCA |
| Activating transcription factor 2; Atf2 | CTCCAGCTCACACAACTCCA | TGTTTCAGCTGTGCCACTTC |
| Thioesterase superfamily member 2; Them2 | TCGACCATGGCTCTAATGTG | TCCTGTGGGTCTTGTTGGTCA |
| Ribosomal protein L32; Rpl32 | CACCAGTCAGACCGATATG | TTCTCCGCACCCTGTTG |
Immunoblot analyses.
Immunoblot analyses were performed using standard techniques (11). Briefly, 20–40 µg of liver homogenate in radioimmunoprecipitation assay (RIPA) buffer were solubilized in Laemelli buffer containing Orange G and separated using 12% SDS-PAGE, along with a Chameleon Duo prestained protein ladder (LI-COR Biosciences). Proteins were transferred onto nitrocellulose membranes and blocked for 1 h using Odyssey blocking buffer (Odyssey no. 927-50000). Primary antibodies were specific to perilipin 2 (adipophilin) NH2 terminus diluted 1:1,000 (no. GP40; Progen Biotechnik, Heidelberg, Germany), PC-TP (24) diluted 1:3,000, phospho-stress-activated protein kinase/c-Jun NH2-terminal kinase (p-SAPK/JNK; Thr183/Tyr185) diluted 1:1,000 (no. 9255; Cell Signaling Technology), SAPK/JNK diluted 1:1,000 (no. 9252; Cell Signaling Technology), phospho-c-Jun (Ser63 and Ser 73) diluted 1:1,000 (no. 9261 and 3270, respectively; Cell Signaling Technology), c-Jun diluted 1:2,000 (no. 2315; Cell Signaling Technology), and GAPDH diluted 1:5,000 (IMG5143A; Imgenex). Secondary antibodies obtained from Agilent Technologies were IRDye 800CW goat (polyclonal) anti-rabbit IgG (no. 925-32211; LI-COR Biosciences) diluted 1:15,000, IRDye 800CW donkey (polyclonal) anti-guinea pig IgG (no. 925-32411; LI-COR Biosciences) diluted 1:15,000, and IRDye 680RD goat (polyclonal) anti-mouse IgG (no. 925-68070; LI-COR Biosciences) diluted 1:10,000. Membranes were imaged using Odyssey CLx Imaging System (LI-COR Biosciences). Proteins of interest were quantified relative to total protein using REVERT Total Protein Stain (no. 926-11013; LI-COR Biosciences). Phosphoproteins were quantified relative to their respective total protein bands. Samples were prepared at the same time and processed in parallel. Molecular weight (mw) markers are visible in the first lane of each image.
Statistical methods.
Two-tailed unpaired Student’s t-test or two-way ANOVA was used to determine statistical significance for 2 groups or >2 groups, respectively. P < 0.05 was considered to be statistically significant.
RESULTS
PC-TP contributes to hepatocellular injury and microvesicular steatosis in mice fed an MCD diet.
Weight loss, a characteristic feature of MCD diet feeding (56), was observed to similar extents in Pctp−/− and WT control mice (Fig. 1A). Pctp mRNA expression was elevated 2.5-fold in livers of MCD diet-fed WT mice compared with chow-fed controls (Fig. 1B), and protein abundance was similarly increased (Fig. 1C). Steatohepatitis was observed in both Pctp−/− and WT MCD diet-fed mice, including ballooning hepatocytes, inflammatory cell infiltrate, and marked microvesicular and macrovesicular steatosis (Fig. 1D). Mild characteristic perisinusoidal fibrosis was also noted in both genotypes by Sirius red staining (Fig. 1E). Application of NAS confirmed the presence of NASH (NAS ≥5), although no genotype-related differences were observed in either the total score (Fig. 1F) or its components (Fig. 1G). Moreover, we did not detect any differences in the degree or pattern of fibrosis between MCD diet-fed Pctp−/− and WT mice (Fig. 1H) or in the hepatic contents of hydroxyproline (Fig. 1I). We next assessed the extent of liver injury as reflected by the serum activities of ALT and AST. The activities of both ALT and AST were elevated in WT, but not Pctp−/− MCD diet-fed mice (Fig. 1, J and K). Pctp−/− mice also exhibited fewer hepatic microvesicular lipid droplets, with a trend toward increased abundance of macrovesicular lipid droplets compared with controls (Fig. 1L). Whereas the protein abundance of the lipid droplet (LD)-associated protein perilipin 2 was increased by the MCD diet, there were no differences attributable to genotype (Fig. 1M).
Fig. 1.

PC-TP contributes to hepatocellular injury and microvesicular steatosis in mice fed an MCD diet. WT and Pctp−/− mice were fed a chow or the MCD diet for 5 wk. A: body weight plotted as a function of time in mice fed the MCD diet. Influence of MCD diet on hepatic Pctp mRNA expression (B) and protein abundance (immunoblot and quantification; C) in WT mice. Representative hematoxylin and eosin- (D) and Sirius red (E)-stained liver images (×40 magnification; white bar = 50 µm). Arrows indicate areas of macrovesicular and microvesicular steatosis. NAFLD activity score (NAS; F) and components that comprise this score (G) in MCD diet-fed mice. NAFLD fibrosis score (H) and hepatic hydroxyproline concentrations (I) in MCD diet-fed mice. Plasma activities of alanine aminotransferase (ALT; J) and aspartate aminotransferase (AST; K). L: relative abundance of microvesicular and macrovesicular lipid droplets in the liver of MCD diet-fed mice. M: hepatic perilipin 2 (Plin2) protein abundance (immunoblot and relative quantification). Values represent means ± SE. A: n = 22–24 per group. B, C, and F–M: chow-fed mice, n = 4–5; MCD diet-fed mice, n = 13–15. *P < 0.05, Pctp−/− vs. WT; §P < 0.05, MCD vs. chow diet.
PC-TP does not alter hepatic lipid concentrations or compositions in MCD diet-fed mice.
On the basis of the differences observed in lipid droplet morphology, we assessed whether PC-TP altered hepatic lipid concentrations. Mass spectrometry was utilized to determine the relative abundance of each molecular species of phosphatidylcholine, diglyceride (DAG), triglyceride (TAG), and ceramide. Similar to previous reports (3, 41), hepatic phosphatidylcholine concentrations were reduced upon MCD diet feeding (Fig. 2A). Although not all molecular species were uniformly reduced by MCD diet, the changes observed in phosphatidylcholine molecular species were not influenced by PC-TP expression (Fig. 2B). Hepatic DAG levels were elevated by MCD diet in both genotypes (Fig. 2C), and although the total DAG levels did not differ between Pctp−/− and WT mice, hepatic concentrations of two of the most abundant DAGs, C36:3 and C36:4, were higher in Pctp−/− mice (Fig. 2D). Total hepatic TAG and ceramide levels were also elevated in mice fed a MCD diet compared with chow-fed mice. However, there were no differences attributable to PC-TP expression in either the hepatic concentrations or compositions (Fig. 2, E–H). In keeping with these findings, hepatic mRNA expression of the lipogenic transcription factors sterol regulatory element binding protein-1c (Srebp1c) and carbohydrate response element binding protein (Chrebpa), as well as the selected targets acetyl-CoA carboxylase (Acaca) and fatty acid synthase (Fasn), were not altered by PC-TP expression (Fig. 2I). Neither were there differences between Pctp−/− and WT mice in the mRNA expression of diacylglycerol acyltransferase (Dgat) 1 or 2, nor in glycerol-3-phosphate acyltransferase (Gpat) 3 or 4, each of which contribute to glycerolipid biogenesis (64; Fig. 2J).
Fig. 2.

PC-TP does not alter hepatic lipid concentrations or compositions in MCD diet-fed mice. WT and Pctp−/− mice were fed a chow or MCD diet for 5 wk. Hepatic concentrations of total (A) and molecular species of (B) phosphatidylcholine, total (C) and molecular species of (D) diglyceride, total (E) and molecular species of (F) triglyceride, and total (G) and molecular species of (H) ceramide were quantified by mass spectrometry and are displayed as arbitrary units (AU). For each lipid molecular species, the number to the left of the colon indicates the total number of carbons, and the number to the right of the colon denotes the total number of unsaturations. Hepatic gene expression of transcription factors and enzymes that regulate lipid metabolism (I) and lipid biogenesis (J). Concentrations of plasma triglycerides (K) and cholesterol (L) as well as hepatic cholesterol (M) were assessed enzymatically. Values represent means ± SE. A–H: n = 4–5. I–M: n = 13–15. *P < 0.05, Pctp−/− vs. WT; §P < 0.05, MCD vs. chow diet.
Hepatic phosphatidylcholines are critical for the production and secretion of triglyceride-rich VLDL particles (68). Their reduced secretion in response to the MCD diet contributes to the hepatic steatosis in this model (47). Although we did not measure VLDL secretion rates directly, the absence of genotype-dependent differences in the steady-state concentrations of plasma triglycerides and cholesterol (Fig. 2, K and L), as well as hepatic triglycerides (Fig. 2E) and cholesterol (Fig. 2M), argues against changes in VLDL metabolism as a basis for differences in hepatic lipid droplet morphology in Pctp−/− mice.
PC-TP alters hepatocellular stress pathways during MCD diet.
In human NASH (10, 35) and in MCD diet-induced steatohepatitis in mice (12, 57), fibroblast growth factor 21 (FGF21), which promotes the β-oxidation of fatty acids, is upregulated as a protective response against hepatocellular injury (12). In Pctp−/− mice fed the MCD diet, hepatic mRNA expression and plasma FGF21 concentrations were reduced compared with WT mice (Fig. 3, A and B), which is consistent with the finding of a lower degree of liver injury. Presumably because both FGF21 and PC-TP promote the β-oxidation of fatty acids within the liver (12, 26), MCD diet-fed Pctp−/− mice also exhibited reduced plasma concentrations of β-hydroxybutyrate, which indicates reduced fatty acid oxidation (Fig. 3C). This occurred in the absence of changes in the steady-state concentrations of hepatic free fatty acids (Fig. 3D). Considering the role for oxidative stress in the development of steatohepatitis (40), we measured hepatic malondialdehyde (MDA) levels, but no genotype-dependent differences were observed (Fig. 3E).
Fig. 3.

PC-TP alters hepatocellular stress pathways during MCD diet. WT and Pctp−/− mice were fed a chow or MCD diet for 5 wk before measurements of fibroblast growth factor 21 (FGF21) mRNA expression in liver (A) and FGF21 protein concentration in plasma (B). Plasma concentration of β-hydroxybutyrate (C), hepatic concentration of free fatty acids (D), hepatic malondialdehyde (MDA) concentrations (E), and hepatic expression of genes central to cellular stress, inflammation, and fibrogenesis (F). Immunoblot of liver homogenate (G) and relative quantification of phospho-JNK over total JNK (H) and phospho-c-Jun Ser63 (I) and Ser73 (J) over total c-Jun. K: hepatic expression of genes that reflect endoplasmic reticulum (ER) stress. L: hepatic mRNA expression of liver receptor homolog-1 (LRH-1), along with downstream transcriptional targets. M: tolerance tests to glucose (GTT), insulin (ITT), and pyruvate (PTT). Values represent means ± SE. Chow-fed mice, n = 4–5; MCD diet-fed mice, n = 13–15. *P < 0.05, Pctp−/− vs. WT; §P < 0.05, MCD vs. chow diet.
Expression of alpha-smooth muscle actin (α-SMA) is a surrogate marker of hepatic stellate cell activation and directly linked to early fibrogenesis (2). We found that α-SMA mRNA (Fig. 3F), but not protein (data not shown), was lower in livers of MCD diet-fed Pctp−/− compared with WT mice. The expression of transforming growth factor-β (Tgfb) mRNA was also unchanged. Although Pctp−/− mice exhibited reduced hepatic mRNA expression of the inflammatory cytokine monocyte chemoattractant protein-1 (Mcp1), Kupffer cell activation appeared to be unaffected since the mRNA expression of neither tumor necrosis factor-α (Tnfa) nor matrix metalloproteinase-9 (Mmp9; 40) was influenced by PC-TP expression. mRNA expression levels of c-jun were reduced in livers of Pctp−/− mice fed a MCD diet.
A mitogen-activated protein kinase, c-Jun NH2-terminal kinase (JNK) mediates many cellular stress responses through the activator protein-1 (AP1) component c-Jun (8). As evidenced by its phosphorylation, both p54 and p46 splice variants of JNK were modestly activated in the absence of PC-TP in the livers of both chow- and MCD diet-fed mice (Fig. 3, G and H, and data not shown; 51). We also observed a marked increase in the total abundance of c-Jun in Pctp−/− mice, irrespective of diet (Fig. 3G). However, this was not accompanied by increases in phosphorylation of c-Jun at the JNK target sites Ser63 and Ser73, so that activation of c-Jun was markedly reduced in the absence of PC-TP expression (Fig. 3, G, I, and J).
Chronic endoplasmic reticulum (ER) stress results in activation of the unfolded protein response, which can, in turn, activate the JNK/AP1 pathway (71). However, in MCD diet-fed mice, CCAAT/enhancer-binding protein (C/EBP)-homologous protein (Chop) mRNA expression was similarly elevated in WT and Pctp−/− mice, and there were no genotype-specific alterations in other ER stress markers (Fig. 3K).
A stress resolution pathway initiated by liver receptor homolog-1 (LRH-1) that is independent of the unfolded protein response has recently been demonstrated (38). Additionally, LRH-1-deficient mice are resistant to MCD diet-induced inflammation and liver damage through regulation of methyl pools (63). To determine whether PC-TP may act to regulate LRH-1 transcriptional pathways, we assessed the mRNA expression of LRH-1 and its downstream targets polo-like kinase 3 (Plk3), sterol 12-alpha-hydroxylase (Cyp8B1), and small heterodimer partner (Shp). Although Plk3 mRNA expression was normalized in MCD diet-fed Pctp−/− mice relative to WT, the other LRH-1 transcription targets were unchanged (Fig. 3L). Furthermore, hepatic concentrations of dilauroyl-sn-glycero-3-phosphocholine (C30:0 PC), an activating ligand of LRH-1 (33), were not affected by PC-TP expression (Fig. 2B).
Because inflammation and cell stress can promote hepatic insulin resistance (32), we tested whether PC-TP expression altered glucose homeostasis. However, we did not observe any difference between MCD diet-fed WT and Pctp−/− mice in their responses to a glucose, insulin, or pyruvate challenge (Fig. 3M).
PC-TP-interacting protein Them2 does not promote hepatic injury in response to MCD diet.
In contrast to PC-TP, MCD diet feeding suppressed Them2 mRNA expression, and this suppression was partly reversed in Pctp−/− mice (Fig. 4A). MCD diet-fed Them2−/− mice developed steatohepatitis (Fig. 4, B–D) but did not exhibit reduced activities of plasma ALT and AST (Fig. 4, E and F) or alterations in lipid droplet morphology (Fig. 4G) compared with WT controls. Plasma β-hydroxybutyrate levels and hepatic MDA were also unchanged (Fig. 4, H and I). These experiments demonstrate a role for PC-TP, independent of Them2, in enabling steatohepatitis induced by MCD diet feeding.
Fig. 4.

PC-TP-interacting protein Them2 does not promote hepatic injury in response to the MCD diet. A: hepatic Them2 mRNA expression in WT and Pctp−/− mice fed a MCD diet for 5 wk. WT and Them2−/− mice were fed a chow or the MCD diet for 5 wk. B: representative images of hematoxylin and eosin-stained liver sections (×40 magnification; white bar = 50 µm). NAFLD activity score (NAS; C) and NAS components (D). Plasma activities of alanine aminotransferase (ALT; E) and aspartate aminotransferase (AST; F). G: relative abundance of macrovesicular and microvesicular lipid droplets in livers of MCD diet-fed mice. Plasma concentration of β-hydroxybutyrate (H) and hepatic malondialdehyde (MDA; I). Values represent means ± SE. A: n = 7–15. C–I: n = 3–7. *P < 0.05, Pctp−/− vs. WT; §P < 0.05, MCD vs. chow diet.
DISCUSSION
This study focused on the role of PC-TP in the development of murine experimental steatohepatitis induced by an MCD diet. Although steatohepatitis occurred in the absence of PC-TP, Pctp−/− mice were relatively protected from liver injury. This was potentially explained by a reduction in microvesicular lipid droplets.
The hydrophobic cores of cytoplasmic lipid droplets are stabilized by both specialized proteins and phospholipids (29). These phospholipids are amphipathic lipid molecules comprising hydrophobic fatty acyl chains that associate with the core lipids and polar head groups that interact with the aqueous cytosol, forming a membrane monolayer. Phosphatidylcholine is the most abundant phospholipid on the lipid droplet surface, and its cylindrical shape tends to prevent the fusion of lipid droplets (64). Most lipid droplet surface phosphatidylcholine molecules are synthesized by the Kennedy biosynthetic pathway (30, 60). The rate-limiting enzyme CTP:phosphocholine cytidylyltransferase (CCT) is targeted to expanding lipid droplets, whereas other enzymes in the pathway, including CDP-cholinephosphotransferase (CPT), which catalyzes the final step in phosphatidylcholine synthesis, are located in the ER (30). When existing lipid droplets expand, there is an increased requirement for phospholipids to coat the surface of the enlarging hydrophobic core. However, the mechanism by which phosphatidylcholine is supplied to expanding cytosolic lipid droplets remains unknown (64). Because spontaneous transfer rates of phospholipids between membrane monolayers are negligible (54), their redistribution in response to changes in lipid droplet size requires either membrane trafficking or the activity of lipid transfer proteins (25). The first scenario requires that newly formed phosphatidylcholine molecules be transferred via lateral diffusion within the ER membrane onto lipid droplets via specific contact sites between the two organelles (17). Another possibility is that a phosphatidylcholine lipid transfer protein (e.g., PC-TP) could transfer phosphatidylcholine from the ER membranes to lipid droplet surfaces. Because of its cylindrical shape, the relative abundance of phosphatidylcholines on lipid droplet surfaces largely determines the surface curvature and the likelihood that lipid droplets will fuse (59). The coalescence of lipid droplets reduces the overall surface area as well as the requirement for phospholipids (64). Similar to the process that occurs in the plasma when plasma phospholipid transfer protein removes excess phospholipids from the surface of triglyceride-depleted remnant particles and transfers them to HDL particles (4), PC-TP might function within the hepatocyte to redistribute phospholipids from the ER or other lipid droplet surfaces and thereby regulate lipid droplet morphology (see Fig. 5). The observation of reduced microvesicular lipid droplets in livers of Pctp−/− mice, despite the same degree of hepatic triglyceride accumulation, suggests a role for PC-TP in the redistribution of phosphatidylcholine between cellular membranes to stabilize smaller lipid droplets. In the absence of PC-TP, lipid droplets may have lacked sufficient phosphatidylcholine molecules on their surfaces to maintain stability and therefore coalesced to reduce the surface-to-volume ratios.
Fig. 5.
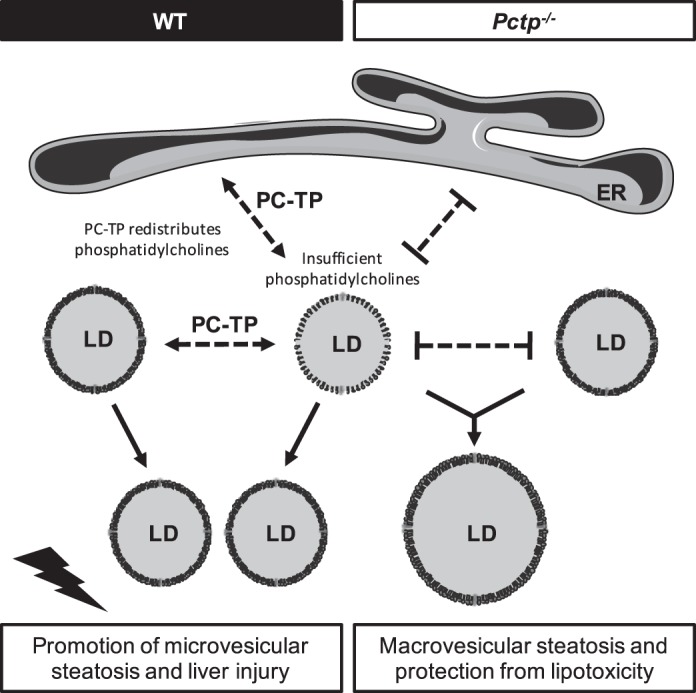
Schematic diagram: MCD diet reduces hepatic phosphatidylcholine concentrations. Under these limiting conditions, PC-TP is upregulated and functions to redistribute phosphatidylcholine molecules between the endoplasmic reticulum (ER) and lipid droplets (LDs) with sufficient or excess phosphatidylcholine, to preserve the stability of small LDs that promote liver injury. In the absence of PC-TP, smaller lipid droplets with insufficient phosphatidylcholine on their surfaces coalesce into larger lipid droplets that are less metabolically active and hepatotoxic.
The complement of hepatic lipid droplet-associated proteins, distinct from other mammalian tissues, are modified by diet and metabolic status, and they influence lipid droplet size (5, 29). In this connection, PC-TP associates with lipid droplets in the livers of high-fat diet-fed mice (27), further implicating its possible role in the distribution of phosphatidylcholine molecules at the surface of expanding lipid droplets. Our current observations suggest that PC-TP stabilizes microvesicular lipid droplets by mediating the intermembrane transfer of phosphatidylcholines from the ER or other organelles.
Triglyceride accumulation in hepatic lipid droplets appears to defend against the cellular toxicity of free fatty acids (46). Indeed, macrovesicular steatosis is the most common manifestation of benign hepatosteatosis. By contrast, microvesicular steatosis is more commonly associated with NASH, including ballooning hepatocyte death and more advanced fibrosis (13, 58). Experimental animal models have further demonstrated that microvesicular steatosis reduces regenerative capacity, which may underscore its prevalence in advanced NASH (42). The proteomes of microvesicular and macrovesicular lipid droplets are also distinct. Whereas the lipid droplet protein perilipin 1 is expressed predominantly on larger lipid droplets, perilipin 2 is more characteristically associated with microvesicular lipid droplets and is observed in ballooning hepatocytes in the setting of NASH (15). Both perilipins 1 and 2 influence lipid droplet size; however, hepatic perilipin 2 contributes to MCD diet-induced steatohepatitis and fibrosis (41). Whereas hepatic levels of perilipin 2 were higher in the mice fed the MCD diet, there were no differences attributable to PC-TP expression. This suggests a mechanism by which PC-TP acts independently of lipid droplet-associated proteins to distribute phosphatidylcholines among microvesicular lipid droplets. When taken together, we propose that PC-TP impedes the transition of smaller to larger lipid droplets and thereby negates a protective mechanism that defends against lipotoxicity. The concept that a lack of PC-TP forces lipid droplets to coalesce because more limited phosphatidylcholines are not readily redistributed could lead to an asymmetric distribution of phospholipid on small vs. larger lipid droplet. However, because triglycerides are forced to redistribute when phosphatidylcholines cannot, a detailed knowledge of lipid compositions, even if it were technically feasible to purify intact lipid droplets of different sizes, would be insufficient to support or refute this hypothesis. Further studies are required to elucidate the role of PC-TP in the regulation of lipid droplet morphology and how alterations in lipid droplet size influence the natural history of liver disease.
Studies in humans have correlated the activation of JNK and c-Jun with hepatosteatosis and steatohepatitis (9). However, studies in mouse models have demonstrated that JNK may also protect against liver injury depending on the context and timing of its activation (7, 14, 18). In Pctp−/− and WT mice, we observed modest elevations in phosphorylation of JNK; however, in the absence of PC-TP, c-Jun phosphorylation was not increased. Reduced activation of c-Jun by JNK could explain, at least in part, the reduced liver injury we observed in the absence of PC-TP expression. Although factors that typically activate JNK and c-Jun (i.e., free fatty acids, oxidative stress, Tnf-α, and ER stress; 55) were not altered in Pctp−/− mice, the dissociation of c-Jun protein and mRNA levels in both chow- and MCD diet-fed mice suggests that PC-TP may increase the protein stability of c-Jun and potentially alter its responsiveness to activated JNK. Future studies exploring the influence of PC-TP on the activation of JNK and c-Jun may shed new light on the relationship between lipid accumulation and stress signaling pathways within the liver.
Our prior studies have revealed that PC-TP binds and activates Them2 at the mitochondrial membrane (25). This leads to increased fatty acid oxidation during fasting, which, in turn, supports hepatic gluconeogenesis (26). The PC-TP/Them2 complex increases fatty acid channeling into the glycerolipid synthetic pathway (39). This, in turn, suppresses insulin action (11) by increasing the synthesis of DAG molecules, which activates protein kinase C epsilon to reduce insulin receptor activity (43). Whereas total hepatic concentrations of the major lipid classes were unchanged in MCD diet-fed Pctp−/− mice, we did observe genotype-specific differences in DAG molecular species in response to the MCD diet: C36:3 and C36:4 were elevated in livers of Pctp−/− mice. Glucose, insulin, and pyruvate tolerance tests showed that PC-TP expression did not influence glucose metabolism. However, the development of obesity and peripheral insulin resistance is not a feature of MCD diet-induced steatohepatitis (49). As a result, the potential metabolic consequences of the altered acyl DAGs in this study remain unclear.
We previously examined the influence of genetic ablation, as well as chemical inhibition, of PC-TP on the development of high-fat diet-induced fatty liver in mice (53). Although the absence of PC-TP expression or its chemical inhibition markedly reduced hepatic glucose production, it had no effect on the development of hepatic steatosis, liver histology, or plasma ALT activities. Similarly, the genetic ablation of PC-TP in leptin-deficient ob/ob mice did not influence hepatic lipid concentrations, but did decrease hepatic glucose production (31). By contrast, Them2-deficient mice are protected from high-fat diet-induced hepatic steatosis (20). Although the PC-TP/Them2 complex regulates hepatic glucose and lipid metabolism, the absence of Them2 expression did not protect against steatohepatitis or changes in lipid droplet morphology. The findings support the likelihood that PC-TP acts independently of Them2 to promote MCD diet-induced steatohepatitis.
In the MCD mouse model of steatohepatitis, PC-TP mRNA and protein expression were upregulated in the setting of reduced hepatic phosphatidylcholine concentrations. Hepatic phosphatidylcholine concentrations are also reduced in NASH in humans (45), and a systematic analysis including 11 individual microarray experiments indicated that PC-TP mRNA was upregulated in livers of NASH patients compared with healthy controls (70). Additional studies will be required to ascertain hepatic phosphatidylcholine concentrations relative to PC-TP in human NAFL and NASH.
A limitation of the present study is that experiments were conducted using male mice exclusively. Whereas MCD diet-induced steatohepatitis is more robust in male than female mice (34), female mice do develop features of steatohepatitis following 5 wk of feeding (6). Although gender-specific phenotypes attributable to hepatic PC-TP have not been demonstrated (61), it is not known whether similar protection from experimental steatohepatitis occurs in female mice.
In summary, PC-TP ablation did not protect against hepatosteatosis or improve histopathological features of experimental steatohepatitis; however, biochemical analysis revealed protection against MCD diet-induced hepatocellular injury. Most notably, this manifested as reduced elevation of ALT and AST activities in the plasma from Pctp−/− mice, albeit the increase in aminotransferase activities was modest compared with other MCD diet-feeding studies (48, 63). Protection from liver injury was further supported by demonstrating blunted elevations in plasma and hepatic expression of FGF21 and marked reductions in c-Jun activation. The influence of PC-TP was independent of its interactions with Them2 and appeared to be related to the redistribution of phosphatidylcholines within hepatocytes. Taken together with prior studies that showed a role for PC-TP in excess hepatic glucose metabolism and insulin resistance in the setting of overnutrition, these data suggest an additional mechanism by which PC-TP may contribute to the pathogenesis of NAFLD.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants 48873 and 56626. H. T. Nicholls was supported by the American Association for the Study of Liver Diseases Foundation Pinnacle Research Award in Liver Disease.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.T.N. and D.E.C. conceived and designed research; H.T.N. performed experiments; H.T.N., J.L.H., and D.E.C. analyzed data; H.T.N., J.L.H., and D.E.C. interpreted results of experiments; H.T.N. and D.E.C. prepared figures; H.T.N. and D.E.C. drafted manuscript; H.T.N. and D.E.C. edited and revised manuscript; H.T.N., J.L.H., and D.E.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Clary Clish, Amy Deik, and Kevin Bullock for mass spectrometry analysis and Jessica Wu and Jiunn Song for technical assistance.
REFERENCES
- 1.Baez JM, Barbour SE, Cohen DE. Phosphatidylcholine transfer protein promotes apolipoprotein A-I-mediated lipid efflux in Chinese hamster ovary cells. J Biol Chem 277: 6198–6206, 2002. doi: 10.1074/jbc.M106799200. [DOI] [PubMed] [Google Scholar]
- 2.Carpino G, Morini S, Ginanni Corradini S, Franchitto A, Merli M, Siciliano M, Gentili F, Onetti Muda A, Berloco P, Rossi M, Attili AF, Gaudio E. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig Liver Dis 37: 349–356, 2005. doi: 10.1016/j.dld.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Chiappini F, Desterke C, Bertrand-Michel J, Guettier C, Le Naour F. Hepatic and serum lipid signatures specific to nonalcoholic steatohepatitis in murine models. Sci Rep 6: 31587, 2016. doi: 10.1038/srep31587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen DE, Fisher EA. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin Liver Dis 33: 380–388, 2013. doi: 10.1055/s-0033-1358519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crunk AE, Monks J, Murakami A, Jackman M, Maclean PS, Ladinsky M, Bales ES, Cain S, Orlicky DJ, McManaman JL. Dynamic regulation of hepatic lipid droplet properties by diet. PLoS One 8: e67631, 2013. doi: 10.1371/journal.pone.0067631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Csak T, Bala S, Lippai D, Kodys K, Catalano D, Iracheta-Vellve A, Szabo G. MicroRNA-155 deficiency attenuates liver steatosis and fibrosis without reducing inflammation in a mouse model of steatohepatitis. PLoS One 10: e0129251, 2015. doi: 10.1371/journal.pone.0129251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cubero FJ, Zoubek ME, Hu W, Peng J, Zhao G, Nevzorova YA, Al Masaoudi M, Bechmann LP, Boekschoten MV, Muller M, Preisinger C, Gassler N, Canbay AE, Luedde T, Davis RJ, Liedtke C, Trautwein C. Combined activities of JNK1 and JNK2 in hepatocytes protect against toxic liver injury. Gastroenterology 150: 968–981, 2016. doi: 10.1053/j.gastro.2015.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Czaja MJ. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends Endocrinol Metab 21: 707–713, 2010. doi: 10.1016/j.tem.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorn C, Engelmann JC, Saugspier M, Koch A, Hartmann A, Müller M, Spang R, Bosserhoff A, Hellerbrand C. Increased expression of c-Jun in nonalcoholic fatty liver disease. Lab Invest 94: 394–408, 2014. doi: 10.1038/labinvest.2014.3. [DOI] [PubMed] [Google Scholar]
- 10.Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, Badman MK, Martinez-Chantar ML, Maratos-Flier E. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 139: 456–463, 2010. doi: 10.1053/j.gastro.2010.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ersoy BA, Tarun A, D’Aquino K, Hancer NJ, Ukomadu C, White MF, Michel T, Manning BD, Cohen DE. Phosphatidylcholine transfer protein interacts with thioesterase superfamily member 2 to attenuate insulin signaling. Sci Signal 6: ra64, 2013. doi: 10.1126/scisignal.2004111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisher FM, Chui PC, Nasser IA, Popov Y, Cunniff JC, Lundasen T, Kharitonenkov A, Schuppan D, Flier JS, Maratos-Flier E. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology 147: 1073–1083, 2014. doi: 10.1053/j.gastro.2014.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther 67: 101–154, 1995. doi: 10.1016/0163-7258(95)00012-6. [DOI] [PubMed] [Google Scholar]
- 14.Fuest M, Willim K, MacNelly S, Fellner N, Resch GP, Blum HE, Hasselblatt P. The transcription factor c-Jun protects against sustained hepatic endoplasmic reticulum stress thereby promoting hepatocyte survival. Hepatology 55: 408–418, 2012. doi: 10.1002/hep.24699. [DOI] [PubMed] [Google Scholar]
- 15.Fujii H, Ikura Y, Arimoto J, Sugioka K, Iezzoni JC, Park SH, Naruko T, Itabe H, Kawada N, Caldwell SH, Ueda M. Expression of perilipin and adipophilin in nonalcoholic fatty liver disease; relevance to oxidative injury and hepatocyte ballooning. J Atheroscler Thromb 16: 893–901, 2009. doi: 10.5551/jat.2055. [DOI] [PubMed] [Google Scholar]
- 16.Gibellini F, Smith TK. The Kennedy pathway: de novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 62: 414–428, 2010. doi: 10.1002/iub.354. [DOI] [PubMed] [Google Scholar]
- 17.Grippa A, Buxó L, Mora G, Funaya C, Idrissi FZ, Mancuso F, Gomez R, Muntanyà J, Sabidó E, Carvalho P. The seipin complex Fld1/Ldb16 stabilizes ER-lipid droplet contact sites. J Cell Biol 211: 829–844, 2015. doi: 10.1083/jcb.201502070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasselblatt P, Rath M, Komnenovic V, Zatloukal K, Wagner EF. Hepatocyte survival in acute hepatitis is due to c-Jun/AP-1-dependent expression of inducible nitric oxide synthase. Proc Natl Acad Sci USA 104: 17105–17110, 2007. doi: 10.1073/pnas.0706272104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itagaki H, Shimizu K, Morikawa S, Ogawa K, Ezaki T. Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice. Int J Clin Exp Pathol 6: 2683–2696, 2013. [PMC free article] [PubMed] [Google Scholar]
- 20.Kang HW, Niepel MW, Han S, Kawano Y, Cohen DE. Thioesterase superfamily member 2/acyl-CoA thioesterase 13 (Them2/Acot13) regulates hepatic lipid and glucose metabolism. FASEB J 26: 2209–2221, 2012. doi: 10.1096/fj.11-202853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang HW, Ozdemir C, Kawano Y, LeClair KB, Vernochet C, Kahn CR, Hagen SJ, Cohen DE. Thioesterase superfamily member 2/Acyl-CoA thioesterase 13 (Them2/Acot13) regulates adaptive thermogenesis in mice. J Biol Chem 288: 33376–33386, 2013. doi: 10.1074/jbc.M113.481408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang HW, Ribich S, Kim BW, Hagen SJ, Bianco AC, Cohen DE. Mice lacking Pctp/StarD2 exhibit increased adaptive thermogenesis and enlarged mitochondria in brown adipose tissue. J Lipid Res 50: 2212–2221, 2009. doi: 10.1194/jlr.M900013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang HW, Wei J, Cohen DE. PC-TP/StARD2: of membranes and metabolism. Trends Endocrinol Metab 21: 449–456, 2010. doi: 10.1016/j.tem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanno K, Wu MK, Agate DS, Fanelli BJ, Wagle N, Scapa EF, Ukomadu C, Cohen DE. Interacting proteins dictate function of the minimal START domain phosphatidylcholine transfer protein/StarD2. J Biol Chem 282: 30728–30736, 2007. doi: 10.1074/jbc.M703745200. [DOI] [PubMed] [Google Scholar]
- 25.Kanno K, Wu MK, Scapa EF, Roderick SL, Cohen DE. Structure and function of phosphatidylcholine transfer protein (PC-TP)/StarD2. Biochim Biophys Acta 1771: 654–662, 2007. doi: 10.1016/j.bbalip.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawano Y, Ersoy BA, Li Y, Nishiumi S, Yoshida M, Cohen DE. Thioesterase superfamily member 2 (Them2) and phosphatidylcholine transfer protein (PC-TP) interact to promote fatty acid oxidation and control glucose utilization. Mol Cell Biol 34: 2396–2408, 2014. doi: 10.1128/MCB.01601-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan SA, Wollaston-Hayden EE, Markowski TW, Higgins L, Mashek DG. Quantitative analysis of the murine lipid droplet-associated proteome during diet-induced hepatic steatosis. J Lipid Res 56: 2260–2272, 2015. doi: 10.1194/jlr.M056812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohli R, Feldstein AE. NASH animal models: are we there yet? J Hepatol 55: 941–943, 2011. doi: 10.1016/j.jhep.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 29.Krahmer N, Farese RV Jr, Walther TC. Balancing the fat: lipid droplets and human disease. EMBO Mol Med 5: 973–983, 2013. doi: 10.1002/emmm.201100671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krahmer N, Guo Y, Wilfling F, Hilger M, Lingrell S, Heger K, Newman HW, Schmidt-Supprian M, Vance DE, Mann M, Farese RV Jr, Walther TC. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab 14: 504–515, 2011. doi: 10.1016/j.cmet.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krisko TI, LeClair KB, Cohen DE. Genetic ablation of phosphatidylcholine transfer protein/StarD2 in ob/ob mice improves glucose tolerance without increasing energy expenditure. Metabolism 68: 145–149, 2017. doi: 10.1016/j.metabol.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leclercq IA, Lebrun VA, Stärkel P, Horsmans YJ. Intrahepatic insulin resistance in a murine model of steatohepatitis: effect of PPARgamma agonist pioglitazone. Lab Invest 87: 56–65, 2007. doi: 10.1038/labinvest.3700489. [DOI] [PubMed] [Google Scholar]
- 33.Lee JM, Lee YK, Mamrosh JL, Busby SA, Griffin PR, Pathak MC, Ortlund EA, Moore DD. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature 474: 506–510, 2011. doi: 10.1038/nature10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee YH, Kim SH, Kim SN, Kwon HJ, Kim JD, Oh JY, Jung YS. Sex-specific metabolic interactions between liver and adipose tissue in MCD diet-induced non-alcoholic fatty liver disease. Oncotarget 7: 46959–46971, 2016. doi: 10.18632/oncotarget.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Fang Q, Gao F, Fan J, Zhou J, Wang X, Zhang H, Pan X, Bao Y, Xiang K, Xu A, Jia W. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J Hepatol 53: 934–940, 2010. doi: 10.1016/j.jhep.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 36.Long JZ, Svensson KJ, Tsai L, Zeng X, Roh HC, Kong X, Rao RR, Lou J, Lokurkar I, Baur W, Castellot JJ Jr, Rosen ED, Spiegelman BM. A smooth muscle-like origin for beige adipocytes. Cell Metab 19: 810–820, 2014. doi: 10.1016/j.cmet.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Machado MV, Michelotti GA, Xie G, Almeida Pereira T, Boursier J, Bohnic B, Guy CD, Diehl AM. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One 10: e0127991, 2015. doi: 10.1371/journal.pone.0127991. [Corrigendum. PLoS One. 10: June 2015, e0132315. doi:. ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mamrosh JL, Lee JM, Wagner M, Stambrook PJ, Whitby RJ, Sifers RN, Wu SP, Tsai MJ, Demayo FJ, Moore DD. Nuclear receptor LRH-1/NR5A2 is required and targetable for liver endoplasmic reticulum stress resolution. eLife 3: e01694, 2014. doi: 10.7554/eLife.01694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minami A, Cooper DE, Coleman RA, Cohen DE. Thioesterase superfamily member 2 channels fatty acids to glycerol-3-phosphate acyltransferase 1: role in hepatic insulin resistance. In: Experimental Biology Late-Breaking Abstracts, Boston 2015. Bethesda, MD: American Association of Anatomists, 2015, p. 8 (LB173). [Google Scholar]
- 40.Mu YP, Ogawa T, Kawada N. Reversibility of fibrosis, inflammation, and endoplasmic reticulum stress in the liver of rats fed a methionine-choline-deficient diet. Lab Invest 90: 245–256, 2010. doi: 10.1038/labinvest.2009.123. [DOI] [PubMed] [Google Scholar]
- 41.Najt CP, Senthivinayagam S, Aljazi MB, Fader KA, Olenic SD, Brock JR, Lydic TA, Jones AD, Atshaves BP. Liver-specific loss of Perilipin 2 alleviates diet-induced hepatic steatosis, inflammation, and fibrosis. Am J Physiol Gastrointest Liver Physiol 310: G726–G738, 2016. doi: 10.1152/ajpgi.00436.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oleszczuk A, Spannbauer M, Tannapfel A, Blüher M, Hengstler J, Pietsch UC, Schuhmacher A, Wittekind C, Hauss JP, Schön MR. Regenerative capacity differs between micro- and macrovesicular hepatic steatosis. Exp Toxicol Pathol 59: 205–213, 2007. doi: 10.1016/j.etp.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 43.Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510: 84–91, 2014. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pickens MK, Yan JS, Ng RK, Ogata H, Grenert JP, Beysen C, Turner SM, Maher JJ. Dietary sucrose is essential to the development of liver injury in the methionine-choline-deficient model of steatohepatitis. J Lipid Res 50: 2072–2082, 2009. doi: 10.1194/jlr.M900022-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46: 1081–1090, 2007. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 46.Ramadori P, Kroy D, Streetz KL. Immunoregulation by lipids during the development of non-alcoholic steatohepatitis. Hepatobiliary Surg Nutr 4: 11–23, 2015. doi: 10.3978/j.issn.2304-3881.2015.01.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res 49: 1068–1076, 2008. doi: 10.1194/jlr.M800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol 40: 47–51, 2004. doi: 10.1016/j.jhep.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 49.Rizki G, Arnaboldi L, Gabrielli B, Yan J, Lee GS, Ng RK, Turner SM, Badger TM, Pitas RE, Maher JJ. Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J Lipid Res 47: 2280–2290, 2006. doi: 10.1194/jlr.M600198-JLR200. [DOI] [PubMed] [Google Scholar]
- 50.Scapa EF, Pocai A, Wu MK, Gutierrez-Juarez R, Glenz L, Kanno K, Li H, Biddinger S, Jelicks LA, Rossetti L, Cohen DE. Regulation of energy substrate utilization and hepatic insulin sensitivity by phosphatidylcholine transfer protein/StarD2. FASEB J 22: 2579–2590, 2008. doi: 10.1096/fj.07-105395. [DOI] [PubMed] [Google Scholar]
- 51.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology 43: 163–172, 2006. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 52.Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. J Gastroenterol Hepatol 28, Suppl 1: 68–76, 2013. doi: 10.1111/jgh.12212. [DOI] [PubMed] [Google Scholar]
- 53.Shishova EY, Stoll JM, Ersoy BA, Shrestha S, Scapa EF, Li Y, Niepel MW, Su Y, Jelicks LA, Stahl GL, Glicksman MA, Gutierrez-Juarez R, Cuny GD, Cohen DE. Genetic ablation or chemical inhibition of phosphatidylcholine transfer protein attenuates diet-induced hepatic glucose production. Hepatology 54: 664–674, 2011. doi: 10.1002/hep.24393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith R, Tanford C. The critical micelle concentration of l-α-dipalmitoylphosphatidylcholine in water and water-methanol solutions. J Mol Biol 67: 75–83, 1972. doi: 10.1016/0022-2836(72)90387-7. [DOI] [PubMed] [Google Scholar]
- 55.Solinas G, Becattini B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol Metab 6: 174–184, 2016. doi: 10.1016/j.molmet.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stanković MN, Mladenović DR, Duričić I, Šobajić SS, Timić J, Jorgačević B, Aleksić V, Vučević DB, Ješić-Vukićević R, Radosavljević TS. Time-dependent changes and association between liver free fatty acids, serum lipid profile and histological features in mice model of nonalcoholic fatty liver disease. Arch Med Res 45: 116–124, 2014. doi: 10.1016/j.arcmed.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka N, Takahashi S, Zhang Y, Krausz KW, Smith PB, Patterson AD, Gonzalez FJ. Role of fibroblast growth factor 21 in the early stage of NASH induced by methionine- and choline-deficient diet. Biochim Biophys Acta 1852: 1242–1252, 2015. doi: 10.1016/j.bbadis.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tandra S, Yeh MM, Brunt EM, Vuppalanchi R, Cummings OW, Unalp-Arida A, Wilson LA, Chalasani N; NASH Clinical Research Network (NASH CRN) . Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J Hepatol 55: 654–659, 2011. doi: 10.1016/j.jhep.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thiam AR, Farese RV Jr, Walther TC. The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol 14: 775–786, 2013. doi: 10.1038/nrm3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vance DE. Phospholipid methylation in mammals: from biochemistry to physiological function. Biochim Biophys Acta 1838: 1477–1487, 2014. doi: 10.1016/j.bbamem.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 61.van Helvoort A, de Brouwer A, Ottenhoff R, Brouwers JF, Wijnholds J, Beijnen JH, Rijneveld A, van der Poll T, van der Valk MA, Majoor D, Voorhout W, Wirtz KW, Elferink RP, Borst P. Mice without phosphatidylcholine transfer protein have no defects in the secretion of phosphatidylcholine into bile or into lung airspaces. Proc Natl Acad Sci USA 96: 11501–11506, 1999. doi: 10.1073/pnas.96.20.11501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.VanPatten S, Ranginani N, Shefer S, Nguyen LB, Rossetti L, Cohen DE. Impaired biliary lipid secretion in obese Zucker rats: leptin promotes hepatic cholesterol clearance. Am J Physiol Gastrointest Liver Physiol 281: G393–G404, 2001. [DOI] [PubMed] [Google Scholar]
- 63.Wagner M, Choi S, Panzitt K, Mamrosh JL, Lee JM, Zaufel A, Xiao R, Wooton-Kee R, Ståhlman M, Newgard CB, Borén J, Moore DD. Liver receptor homolog-1 is a critical determinant of methyl-pool metabolism. Hepatology 63: 95–106, 2016. doi: 10.1002/hep.28124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilfling F, Haas JT, Walther TC, Farese RV Jr. Lipid droplet biogenesis. Curr Opin Cell Biol 29: 39–45, 2014. doi: 10.1016/j.ceb.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wirtz KW. Phospholipid transfer proteins. Annu Rev Biochem 60: 73–99, 1991. doi: 10.1146/annurev.bi.60.070191.000445. [DOI] [PubMed] [Google Scholar]
- 66.Wu MK, Hyogo H, Yadav SK, Novikoff PM, Cohen DE. Impaired response of biliary lipid secretion to a lithogenic diet in phosphatidylcholine transfer protein-deficient mice. J Lipid Res 46: 422–431, 2005. doi: 10.1194/jlr.M400387-JLR200. [DOI] [PubMed] [Google Scholar]
- 67.Yao ZM, Vance DE. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem 263: 2998–3004, 1988. [PubMed] [Google Scholar]
- 68.Yao ZM, Vance DE. Reduction in VLDL, but not HDL, in plasma of rats deficient in choline. Biochem Cell Biol 68: 552–558, 1990. doi: 10.1139/o90-079. [DOI] [PubMed] [Google Scholar]
- 69.Zeisel SH. Choline: critical role during fetal development and dietary requirements in adults. Annu Rev Nutr 26: 229–250, 2006. doi: 10.1146/annurev.nutr.26.061505.111156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y, Baker SS, Baker RD, Zhu R, Zhu L. Systematic analysis of the gene expression in the livers of nonalcoholic steatohepatitis: implications on potential biomarkers and molecular pathological mechanism. PLoS One 7: e51131, 2012. doi: 10.1371/journal.pone.0051131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng GF, Cai Z, Meng XK, Zhang Y, Zhu W, Pang XY, Dou L. Unfolded protein response mediated JNK/AP-1 signal transduction, a target for ovarian cancer treatment. Int J Clin Exp Pathol 8: 6505–6511, 2015. [PMC free article] [PubMed] [Google Scholar]