

Abstract
We assessed effects of acute volume reductions induced by administration of diuretics in rats. Direct block of Na+ transport produced changes in urinary electrolyte excretion. Adaptations to these effects appeared as alterations in the expression of protein for the distal nephron Na+ transporters NCC and ENaC. Two hours after a single injection of furosemide (6 mg/kg) or hydrochlorothiazide (HCTZ; 30 mg/kg) Na+ and K+ excretion increased but no changes in the content of activated forms of NCC (phosphorylated on residue T53) or ENaC (cleaved γ-subunit) were detected. In contrast, amiloride (0.6 mg/kg) evoked a similar natriuresis that coincided with decreased pT53NCC and increased cleaved γENaC. Alterations in posttranslational membrane protein processing correlated with an increase in plasma K+ of 0.6–0.8 mM. Decreased pT53NCC occurred within 1 h after amiloride injection, whereas changes in γENaC were slower and were blocked by the mineralocorticoid receptor antagonist spironolactone. Increased γENaC cleavage correlated with elevation of the surface expression of the subunit as assessed by in situ biotinylation. Na depletion induced by 2 h of furosemide or HCTZ treatment increases total NCC expression without affecting ENaC protein. However, restriction of Na intake for 10 h (during the day) or 18 h (overnight) increased the abundance of both total NCC and of cleaved α- and γENaC. We conclude that the kidneys respond acutely to hyperkalemic challenges by decreasing the activity of NCC while increasing that of ENaC. They respond to hypovolemia more slowly, increasing Na+ reabsorptive capacities of both of these transporters.
Keywords: NCC, ENaC, furosemide, hydrochlorothiazide, amiloride, hyperkalemia
the kidneys control extracellular levels of both Na+ and K+ by regulating transport of the electrolytes between urine and blood. Independent management of their levels in plasma is accomplished at least in part through coordinated control of coupled NaCl reabsorption through the cotransporter NCC in the distal convoluted tubule (DCT) and Na+ reabsorption coupled to K+ secretion involving the epithelial Na channel (ENaC) in more distal nephron segments (25, 32, 33, 50). For control of Na+ the two transport systems are regulated in parallel, whereas to manage K+ they are modulated in opposite directions. For example, to specifically increase distal K+ secretion the kidneys may transport less Na+ through NCC and more through ENaC.
The mineralocorticoid aldosterone is a major regulator of ENaC (47). The adrenal cortex releases the hormone in response to either volume depletion or hyperkalemia (17). Aldosterone stimulates channel activity at least in part through increased surface expression of the protein subunits (9, 22). A hallmark of this regulatory pathway is cleavage of the α and γ subunits of ENaC (7, 10, 23).
Aldosterone administration may also increase the abundance of NCC protein (3, 20, 44) although in some studies this effect was small compared with that on ENaC processing (14). Furthermore a recent study showed that deletion of the mineralocorticoid receptor in DCT cells did not affect NCC abundance (5). Angiotensin II (ANG II) also appears to upregulate NCC through aldosterone-independent mechanisms (37, 43), although other studies suggest that the effects of chronic ANG II infusion are mediated at least in part by a fall in plasma K+ (45). Thus either of these hormones could stimulate the cotransporter during volume depletion. ANG II may work at least in part through the WNK/SPAK kinase system which regulates the phosphorylation of the cotransporter (1, 18). The phosphorylation state of NCC also decreases rapidly in response to elevated plasma K+ (35, 39), apparently through a direct effect of K+ on the NCC-expressing cells (34, 42).
Our goal in this study was to evaluate the responses of both the NCC and ENaC systems to acute volume depletion. Specifically we asked whether the two transporters are modulated under these conditions and if so whether they are up- or downregulated in concert. Using a mathematical model of the DCT cell (48, 49) to estimate changes in cell Cl− we also evaluated the idea that changes in cell Cl− in DCT cells evoked by alterations in Cl− entry across the apical membrane might alter NCC phosphorylation through a feedback-inhibition process.
METHODS
Animals.
All procedures using animals were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Sprague-Dawley rats (Charles River Laboratories, Kingston, NY) weighing from 150 – 200 g were raised free of viral infections and fed standard rat chow. Most studies were carried out with female rats except for those in Fig. 10 which used both females and males. In a few cases animals were fed overnight with a Na-deficient diet containing 0.004% Na by weight or a matched diet supplemented with 1% NaCl (MP Biomedicals, Solon, OH). Animals were treated with furosemide (6 mg/kg), hydrochlorothiazide (HCTZ) (30 mg/kg), amiloride (0.6 mg/kg), or combinations of amiloride with furosemide or HCTZ. Furosemide and HCTZ were dissolved in DMSO, and 0.2 ml of this solution was mixed with 1.8 ml saline and injected into the peritoneum. Controls were treated with vehicle (DMSO) added to the saline, which was used to prevent volume depletion in control animals. Amiloride was dissolved in H2O and added to 1.8 ml saline for injection. For measurements of urinary excretion of Na and K, animals were kept in metabolic cages with free access to drinking water but without food. Urine was collected for 1, 2, or 5 h after drug administration. Animals were euthanized, blood was drawn from the abdominal aorta. The Na and K concentrations in the collected urine and plasma were measured using a flame photometer (Instrumentation Laboratory IL Model 943).
Fig. 10.

Effects of overnight (o/n) Na depletion on ENaC and pNCC expression. Right: Western blots showing expression of ENaC subunits and NCC and pNCC for animals fed either a control or low-Na diet overnight. In this blot the two left-hand lanes in each group were from female rats, whereas the two right-hand lanes used males. Females had a higher expression of NCC protein but changes were similar in the two sexes. Left: densitometric analysis. Data represent means ± SE for 8 animals. *Statistical significance (P < 0.05).
Some animals were pretreated with the mineralocorticoid antagonist spironolactone before the administration of amiloride. For these experiments we first tested the effects of 30 mg/kg spironolactone on the biochemical changes in ENaC induced by 30 μg/kg aldosterone. Aldosterone or aldosterone plus spironolactone dissolved in 0.2 ml polyethylene glycol (PEG-300) were injected subcutaneously. Kidneys were harvested 5 h after injection and prepared for immunoblotting. To test the dependence of amiloride effects on the mineralocorticoid receptor, 30 mg/kg spironolactone or vehicle was injected subcutaneously 2 h before the administration of amiloride intraperitoneally. Kidneys were harvested 2 h later.
In situ biotinylation.
Biotinylation of membrane proteins followed a modification of a protocol described previously (8). Rats were anesthetized with ketamine 90 mg/kg plus xylazine 4 mg/kg ip. The abdominal cavity was opened and the aorta cannulated below the renal arteries. The kidneys were perfused by gravity with ice-cold solutions at a rate of ~10 ml/min. The vena cava was punctured to allow fluid outflow. The aorta was then ligated above the renal arteries, as were the superior mesenteric and hepatic arteries. The animal was euthanized by opening the chest and cutting the cardiac apex. The rat was placed on ice and the abdominal cavity was kept at <7°C with a steady stream of ice-cold 150 mM NaCl solution over the left kidney. Flow rates through the urethra were 0.2–0.5 ml/min.
The kidneys were perfused first for 5–10 min with PBS, containing (in mM) 140 NaCl, 4.5 KCl, 10 Na2HPO4, at pH 8.0 and containing 50 USP units heparin/dl, then for 20–25 min with PBS + 0.5 mg/ml sulfosuccinimidyl-2-[biotinamido]ethyl-1,3-dithiopropionate (sulfo-NHS-biotin, Campbell Science, Rockville IL), and finally for 30 min with TBS, containing (in mM) 120 NaCl, 4.5 KCl, 25 Tris at pH 8.0 to quench the reaction of the reagent with the tissue and remove excess biotin.
The left kidney was removed, minced with a razor blade, and homogenized with a tight-fitting Dounce in 8 ml of lysis buffer, containing (in mM) 250 sucrose, 10 triethanolamine HCl, 1.6 ethanolamine and 0.5 EDTA, phosphatase inhibitors (10 NaF, 3 Na orthovanadate, 10 Na4 pyrophosphate), and 60 μl protease inhibitors cocktail (Sigma-Aldrich) at pH 7.4. The time between excision of the kidney and the beginning of homogenization was 1–2 min. The homogenate was sieved with a 100-μm nylon mesh to separate intact pieces of tissue. The filtrate was centrifuged at 100,000 g for 2 h to sediment a total membrane fraction. This pellet was resuspended in 2 ml of lysis buffer, aliquoted, and frozen at −80°C. Protein concentrations were measured with a BCA kit (Pierce). For isolation of biotinylated proteins, 3 mg of protein was solubilized in 1.5 ml of solubilization buffer, containing (in mM) 100 mM NaCl, 50 mM Tris, 5 mM EDTA, 3% Triton X-100, phosphatase inhibitors, 1 μg/ml leupeptin (Sigma-Aldrich), 0.1 mg/ml PMSF (Sigma-Aldrich) at pH 7.4 along with 0.4 ml of a 50% suspension of UltraLink Neutravidin beads (Pierce Chemical) and gently rocked overnight at 4°C. The beads were washed twice with solubilization buffer with 1% Triton X-100, twice with high-salt solubilization buffer containing (in mM) 500 NaCl, 50 Tris, 5 EDTA, 0.1% Triton X-100, and twice with no-salt buffer containing (in mM) 10 Tris, at pH 7.4. After a 2-min centrifugation the fluid over the beads was aspirated and proteins were eluted from the beads with 60 μl of 500 mM DTT at 70°C for 15 min. The eluate was collected after centrifugation at 3,000 rpm for 5 min, and mixed with 20 μl of 4X sample buffer. Forty microliters of this mixture was loaded into one lane of a polyacrylamide gel for electrophoresis.
Previous studies established that intracellular membrane proteins (golgin, calnexin) are not labeled by this procedure (9).
Whole kidney samples were prepared for electrophoresis with 0.2–0.5 mg of protein in 65 μl lysis buffer, 25 μl LDS Sample Buffer, and 10 μl Sample Reducing Agent and heated at 70°C for 10 min. Each lane of the gel was loaded with 30 μg (β and γENaC, NCC and pT53NCC) or 40 μg (αENaC) total protein.
Immunoblotting.
The whole kidney and cell-surface samples were electrophoresed on 4–12% Bis-Tris gels (Invitrogen) and the proteins were transferred electrophoretically to PVDF membranes. After blocking, membranes were incubated overnight at 4°C with primary antibodies against α, β, or γ ENaC at 1:1,000 dilution as described in Ref. 7. Anti-rabbit IgG conjugated with alkaline phosphatase was used as a secondary antibody. Bound antibody was visualized on autoradiography film (HyBlot CL, Denville Scientific) using a chemiluminescence substrate (Western Breeze, Invitrogen). Semiquantitative densitometry of protein bands was performed with background subtraction using AdobePhotoshop CS5. After films were developed blots were stained with Coomassie blue to assess protein loading in individual lanes, which varied by <10%.
Antibodies.
Polyclonal antibodies against the β and γ subunits of rat ENaC were based on short peptide sequences in the carboxy termini as described previously (7, 24). Antisera were purified using peptide-linked agarose bead affinity columns (Sulfolink Kit, Pierce Biotechnology). The antibody against the NH2 terminus of mouse αENaC (39) was a gift of Prof. Johannes Loffing (Univ. of Zürich). The antibody against NCC (30) was a gift of Prof. Alicia McDonough (Univ. of Southern California). The antibody against the phospho-T53 form of NCC was described previously (3).
Statistics.
Statistical significance between two groups was assessed by unpaired Student’s t-tests. P < 0.05 was considered significant.
RESULTS
Effects of single doses of diuretics on urinary Na+ and K+ excretion, as well as the expression of the putative active forms of ENaC and NCC, are shown in Figs. 1–4, and summarized in Table 1. Administration of the loop diuretic furosemide (Fig. 1) or the thiazide diuretic HCTZ (Fig. 2) produced large increases in Na+ excretion and smaller changes in K+ excretion during 2 h following the injection. Despite the kaliuresis plasma K+ concentrations remained constant. To see if the extracellular volume depletion activated ENaC we assessed the degree of cleavage γENaC protein using Western blot analysis. We detected no significant changes in the abundance of either the 80-kDa full-length or the 65-kDa cleaved form of the subunit.
Fig. 1.

Effects of a single injection of furosemide (6 mg/kg) on renal electrolyte excretion and the expression of ENaC and NCC. Furosemide markedly increased Na+ excretion and K+ excretion over a 2-h period although plasma K+ was not affected. Western blot analysis was performed on microsomes from kidneys homogenized on ice with protease and phosphatase inhibitors within 2 min of excision. Blots showed no measureable changes in ENaC but significant increases in full-length but not pT53NCC. Data represent means ± SE for 4 animals. *Statistical significance (P < 0.05).
Fig. 4.

Effects of a single injection of furosemide (6 mg/kg) + amiloride (0.6 mg/kg) or HCTZ (30 mg/kg) + amiloride (0.6 mg/kg) on renal electrolyte excretion and the expression of γENaC and pNCC. In both conditions Na+ excretion increased while K+ excretion decreased over a 2-h period; plasma K+ increased in both cases. With both treatments levels of cleaved γENaC increased, while those of pT53NCC decreased. Data represent means ± SE for 4 animals. *Statistical significance (P < 0.05).
Table 1.
Effect of acute Na depletion protocols on ENaC and NCC
| Protocol | ΔNa, μmol | ΔK, μmol | Δ[K]o, mM | γENaC (cleaved) | totNCC | pT53MCC |
|---|---|---|---|---|---|---|
| Furosemide (2 h) | −430 | −130 | ↔ | ↔ | ↑ | ↔ |
| HCTZ (2 h) | −310 | −180 | ↔ | ↔ | ↑ | ↔ |
| Amiloride (1 h) | −150 | +120 | +0.62 | ↔ | ↔ | ↓ |
| Amiloride (2 h) | −560 | +140 | +0.80 | ↑ | ↔ | ↓ |
| Amiloride + furosemide | −760 | +108 | +0.91 | ↑ | ↔ | ↓ |
| Amiloride + HCTZ | −950 | +140 | +0.84 | ↑ | ↔ | ↓ |
| Na depletion (8 h) | −400 | −960 | ↔ | ↑ | ↑ | ↑ |
| Na depletion (overnight) | −320 | +1,540 | +0.47 | ↑ | ↑ | ↔ |
ΔNa and ΔK represent the net gain (+) or loss (−) of electrolytes in experimental animals relative to controls over the indicated time period. Δ[K]o is the change in plasma [K+] between experimental and control animals at the end of the time period. The other columns indicate significant changes [increase (↑) or decrease (↓)] relative to controls in the content of protein for the cleaved form (65 kDa) of γENaC protein, the total content of NCC protein, and the content of pT53NCC protein. ↔, No significant change relative to control.
Fig. 2.

Effects of a single injection of HCTZ (30 mg/kg) on renal electrolyte excretion and the expression of γENaC and pT53NCC. HCTZ markedly increased Na+ excretion and K+ excretion over a 2-h period although plasma K+ was not affected. Western blot analysis showed no significant effects on ENaC but significant increases in full-length but not pT53NCC. Data represent means ± SE for 4 animals. *Statistical significance (P < 0.05).
Effects on NCC were more difficult to predict. Volume depletion might upregulate NCC through either ANG II (2, 29, 30, 37, 43) or aldosterone (20, 44). However, recent evidence suggests that elevated cell Cl− can inhibit NCC phosphorylation (42). This idea predicts that the cotransporter could respond differently to administration of HCTZ, which will directly decrease Cl− entry into the DCT cells, and furosemide, which should, if anything, increase Cl− entry by enhancing the delivery of NaCl to the distal nephron (see discussion). Total NCC increased in response to both diuretics. However, we found no consistent changes in the abundance of pNCC, assessed as pT53NCC (Figs. 1 and 2).
The kidneys responded very differently to amiloride administration. This diuretic produced comparable increases in Na+ excretion (Fig. 3) but, as expected, decreased rather than increased K+ excretion. As a result plasma K+ increased significantly from 3.6 to 4.4 mM. Significant effects on the transport proteins also occurred. The abundance of the cleaved forms of αENaC and γENaC increased markedly, while pT53NCC levels decreased. The latter effect reflects the degree of phosphorylation of the transporter since the overall NCC protein expression did not change.
Fig. 3.

Effects of a single injection of amiloride (0.6 mg/kg) on renal electrolyte excretion and the expression of ENaC and NCC. Amiloride increased Na+ excretion and decreased K+ excretion over a 2-h period; plasma K+ increased significantly. Western blot analysis revealed significant increases in the cleaved form of α- and both total and cleaved γENaC, and decreased pT53NCC levels. Data represent means ± SE for 4 animals. *Statistical significance (P < 0.05).
We also assessed the effects of combined treatment with amiloride and the more proximally acting diuretics (Fig. 4). Furosemide + amiloride or HCTZ + amiloride evoked a large natriuresis with inhibition of K+ excretion, implying that inhibition of K+ secretion by amiloride also blocked the kaliuresis associated with furosemide or HCTZ. In both cases plasma K+ increased by ~0.8 mM. Changes in transport proteins were similar to those induced by amiloride alone; cleaved γENaC increased while pT53NCC decreased. The major difference was the absence of an increase in total NCC.
Increases in cleaved γENaC correlate with enhanced surface expression of the 3 ENaC subunits under a variety of circumstances (9, 10, 12). To see if the relatively rapid changes observed in Figs. 2 and 3 were also associated with ENaC trafficking we assessed surface expression after a similar 2-h treatment with amiloride using in situ biotinylation of the kidneys (Fig. 5). As reported previously (9) most of the γENaC in the surface membrane fraction is in the cleaved form. Expression of this form increased threefold in response to amiloride, an increase comparable in magnitude to that evoked by chronic salt depletion or aldosterone administration (9, 11). Thus significant changes in trafficking can occur over a 2-h period. The decreases in pNCC observed in Figs. 3 and 4 were not detected in either the total membrane or surface fractions from these kidneys (not shown). One interpretation is that changes in NCC phosphorylation may have reversed during the perfusion of the kidneys with solution containing a standard (4.5 mM) extracellular [K+].
Fig. 5.
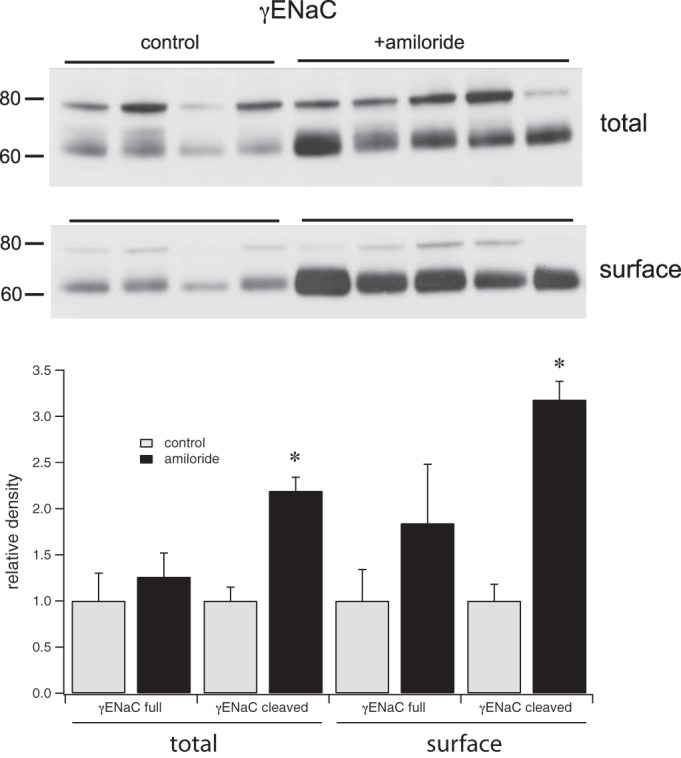
Effects of a single injection of amiloride (0.6 mg/kg) on surface expression of γENaC. Western blots of total membrane and surface fractions from animals treated with amiloride or vehicle for 2 h. Amiloride treatment significantly increased the overall and surface expression of the cleaved form of γENaC. Data represent means ± SE for 4 animals. *Statistical significance (P < 0.05).
The observed decreases in pNCC may reflect direct effects of increased plasma K+ on the DCT cells expressing the transporter (34, 42). Hyperkalemia could in principle also directly affect ENaC. Alternatively, activation of the channels could be secondary to increases in circulating aldosterone levels resulting from K+-dependent steroidogenesis in the adrenal cortex. We tested these possibilities using a shorter exposure to amiloride, reasoning that effects of aldosterone that require mRNA and protein synthesis would be minimal at this time. The 1-h treatment with amiloride elicited an increase in plasma K+ of 0.6 mM and a concomitant fall in pNCC (Fig. 6). In contrast the amount of cleaved γENaC did not increase, indicating that this effect results from activation of a slower pathway. We tested whether this might involve aldosterone signaling in animals pretreated with the mineralocorticoid receptor antagonist spironolactone. The drug blocked the increases in cleaved αENaC and γENaC, as well as the antinatriuresis in response to an acute administration of aldosterone (Fig. 7). It also prevented cleavage resulting from a 2-h challenge with amiloride (Fig. 8). We expected that decreased pNCC would persist under these conditions. However a large variability in pNCC expression in this set of experiments precludes a meaningful analysis.
Fig. 6.

Rapid effects of a single injection of amiloride (0.6 mg/kg) on ENaC and NCC. Right: Western blots showing expression of ENaC and NCC in microsomes from kidneys collected 1 h after administration of amiloride or vehicle. Amiloride treatment significantly decreased the expression of pT53NCC but did not alter that of total NCC or ENaC. Data represent means ± SE for 4 animals. *Statistical significance (P < 0.05).
Fig. 7.

Effects of spironolactone on urinary electrolyte excretion and ENaC and NCC expression. Rats were given a single injection of either aldosterone alone or aldosterone + spironolactone. Spironolactone increased Na+ excretion but had no significant effect on K+ excretion. Spironolactone decreased the level of both total and cleaved αENaC and of cleaved γENaC observed 5 h after administration of aldosterone. Data represent means ± SE for 4 animals. *Statistical significance (P < 0.05).
Fig. 8.

Effects of spironolactone on the response to amiloride. Rats were pretreated with spironolactone for 2 h before administration of amiloride (0.6 mg/kg) or vehicle. Spironolactone prevented the increase in cleaved γENaC as well as the decrease in pT53NCC in response to amiloride administration.
To see if more prolonged volume depletion could affect ENaC or NCC processing we first tested the effects of a 5-h administration of HCTZ. This also failed to change either cleaved γENaC or pT53NCC (not shown). We then further extended the period of Na+ depletion to 9 h. Kidneys were obtained from paired animals in the morning and in the early evening. Because the animals ate minimally during the daytime this protocol was nearly equivalent to a 9-h fast and mimicked the normal diurnal pattern of food (and Na+) consumption. However, to eliminate variations in Na+ intake we fed the animals with a low-Na+ diet during this period. By the evening both ENaC and NCC levels had increased. This was observed for all the ENaC subunits, both full-length and cleaved, and for both the total and the phosphorylated form of NCC (Fig. 9). This shows that day-to-day modulation of these transporters involves changes in protein expression and processing, and that NCC and ENaC increase in parallel under these conditions, minimizing Na+ excretion.
Fig. 9.

Effects of diurnal Na depletion on ENaC and pNCC expression. Right: Western blots showing expression of ENaC subunits and NCC and pNCC before and after dietary Na restriction for 9 h. Left: densitometric analysis. Data represent means ± SE for 8 animals. *Statistical significance (P < 0.05).
Some of these changes could be driven by diurnal rhythms rather than by changes in extracellular volume per se (31). To dissociate these factors we studied animals fed either a low-Na+ or a matched control diet overnight (Fig. 10). In this case protein expression was assessed at the same time of day (morning) in both groups. This protocol also elicited increases in cleaved α- and γENaC and in total NCC, suggesting that these effects are attributable, at least in part, to a change in overall Na (and extracellular volume) balance. However, the increases in total βENaC and full-length α and γENaC observed with the daytime protocol did not occur with overnight Na+ restriction. This indicates that these proteins could be under diurnal control, or that they were affected by the reduced intake of other nutrients such as K+ that occurs during the diurnal fast. With overnight Na+ depletion the levels of pNCC did not increase in parallel with total NCC, indicating that the fraction of phosphorylated cotransporter declined. A small but significant increase in plasma K+ (Table 1) might be responsible for the decreased phosphorylation state. The elevated K+ could result from reduced Na+ delivery to the K+-secreting parts of the nephron, although we cannot rule out other contributing factors.
DISCUSSION
Protection against acute increases in K involves antiparallel changes in NCC and ENaC.
In response to amiloride treatment, the kidneys responded to a modest acute increase in plasma K+ by downregulating NCC and upregulating ENaC. Inhibition of NCC entails dephosphorylation of the cotransporter protein and confirms previous results obtained with an acute oral or intravenous K load (35, 39). Changes observed in our study were elicited by relatively small (<1 mM) increases in plasma K+. The decreases in pNCC abundance occurred within 1 h and did not involve changes in the overall expression of the protein, implying a change in the phosphorylation state of the transporter.
Furosemide and HCTZ increased both Na+ and K+ excretion. This effect of HCTZ has been well documented in the rat (4, 21) and presumably results from increased delivery of NaCl and fluid to downstream K+-secreting segments (46). In a recent report Hunter and colleagues did not observe kaliuresis in response to HCTZ in the mouse (19). One possible reason for the difference is that in mice a higher fraction of K+ secretion is through an amiloride-insensitive mechanism (51) than in rats (13). Despite the increased excretion rates, these diuretics had no effect on plasma [K+] over this time course, consistent with the unchanged levels of pT53NCC.
Activation of ENaC was documented as an increased abundance of the cleaved form of the γENaC (and αENaC) subunits. Consistent with previous results (9, 10, 12) these effects correlated with an increase in surface expression of the channel. This response was somewhat slower than that of NCC dephosphorylation, requiring 2 h of hyperkalemic challenge. Nevertheless it documents a rapid change in ENaC processing and trafficking.
Mechanism of NCC regulation can be dissociated from changes in cell [Cl−].
The rapid response of pNCC to amiloride-induced hyperkalemia is compatible with the hypothesis that changes in plasma K+ within the physiological range regulate renal NCC directly without the requirement of other circulating factors. Dephosphorylation of NCC was also observed in isolated kidneys (34), renal slices (34), and cultured cells heterologously expressing the cotransporter (42).
To account for these effects, Ellison and colleagues proposed a mechanism in which changes in extracellular K+ produce parallel changes in intracellular Cl− concentration ([Cl−]in) through alterations in the basolateral membrane voltage and in the driving force for outward KCl cotransport (42). High [Cl−]in modulates WNK kinase activity, ultimately reducing SPAK-dependent NCC phosphorylation. The findings that phosphorylation state of NCC is not affected by either furosemide or thiazide may be hard to reconcile with the idea that pNCC levels are entirely dependent on antiparallel changes in [Cl−]in. A mathematical model of the DCT cell (48) predicts that administration of amiloride, which increases plasma K+ from 3.6 to 4.4 mM, would increase [Cl−]in by ~16% (Fig. 11A), in agreement with previous work (42). However, inhibition of NCC blocks Cl− (and Na+) entry, decreasing [Cl−]in for all values of lumen Na+ and peritubular K+ (Fig. 11B). In the presence of hydrochlorothiazide, the hyperkalemia induced by amiloride again raised [Cl−]in by 15%, but that concentration was still lower than under control conditions (in the absence of either diuretic.) We observed no effect of a near-maximal dose of HCTZ on NCC phosphorylation. Furthermore the model predicts that the net effect of combining HCTZ and amiloride is to decrease [Cl−]in, whereas we observed decreased pNCC levels. Finally, the model indicates that furosemide, through increases in luminal [Na+] and [Cl−], will increase [Cl−]in modestly, due to the low apparent Km values for transport (7 mM for Na+ and 7–9 mM for Cl−) under in vivo conditions (27).
Fig. 11.

Correlation of pT53NCC with plasma [K+] and estimated cell [Cl−] in DCT. Simulations were made with an epithelial model of the DCT (48, 49). In A and B, lumen NaCl is varied from 35 to 95 mM, and peritubular KCl from 3 to 7 mM. For each input pair, [Cl−]in is indicated as a heat map, from orange to green (high to low [Cl−]in). Contour lines for constant [Cl−]in are indicated; where nearly horizontal, they document greater sensitivity of [Cl−]in to peritubular K+ (compared with luminal Na+). A: calculations under control conditions. Markers denote values obtained for control conditions and for treatment with furosemide and/or amiloride. B: calculations for 90% reduction of NCC activity. Markers denote values obtained for treatment with HCTZ ± amiloride. C: expression of pT53NCC in animals treated with furosemide, HCTZ and/or amiloride is plotted vs. plasma [K+] measured at the end of the treatment. The line is from regression analysis of chronic experiments [redrawn from Terker et al. (41) with permission from Elsevier. Copyright 2016 Elsevier]. D: the same data plotted as a function of cell [Cl−]. The line represents the activity of WNK1 as a function of cell [Cl−] [redrawn from Terker et al. (41) with permission from Elsevier. Copyright 2016 Elsevier].
Figure 11C plots the relative change in pT53NCC as a function of measured values of plasma [K+]. The correlation exactly matches that previously reported for more chronic alterations in plasma [K+] (41). Figure 11D plots the same data as a function of DCT [Cl−]in based on the model. No correlation is evident. For reference the effect of [Cl]in on WNK4 activity (41) is also shown as a dashed line.
A caveat to these conclusions is that changes in [Cl−]in were predicted and not measured directly, and the model employed may not be complete. Furthermore the narrow range of predicted [Cl−]in relative to the effects of the anion on WNK kinase activity (see Fig. 11) may explain the lack of correlation with measured pNCC levels. In any case this suggests that factors other than [Cl−]in are likely to also be involved. This conclusion is in agreement with results on kidney tissue slices in which dephosphorylation of NCC occurred in response to increased extracellular K+ even when increases in cell [Cl−] were suppressed by low extracellular [Cl−] (34). Increased extracellular [K+] could alter pNCC levels in a Cl−-independent fashion through activation of the Ca2+-dependent phosphatase calcineurin. Inhibitors of this enzyme blocked the NCC dephosphorylation elicited by an acute oral potassium load (38).
Aldosterone may mediate acute ENaC regulation by extracellular K+.
Increases in γENaC cleavage and surface expression are similar to those observed in response to an acute administration of aldosterone (11). Since hyperkalemia can stimulate adrenal aldosterone production (17) this hormone is a likely candidate for mediating the observed effects of amiloride. The requirement for changes in mRNA and protein synthesis in the response to the mineralocorticoid could account for the finding that changes in ENaC were slower than those involving NCC phosphorylation. Increased ENaC cleavage in response to an oral K load was also delayed relative to decreased NCC phosphorylation (39). Inhibition of amiloride-induced γENaC cleavage by the mineralocorticoid-receptor antagonist spironolactone is also compatible with an effect mediated by aldosterone.
More prolonged volume depletion involves parallel increases in NCC and ENaC.
A 2-h treatment with furosemide and HCTZ induced a small but significant increase in the total NCC protein (Figs. 1 and 2). This was observed in the absence of changes in plasma [K+] and may be a response to acute volume depletion. The diuretics induced a net loss of Na+ of 430 and 310 μmol, respectively (Table 1), corresponding to decreases of extracellular volume of 2–3 ml. This upregulation occurred without a significant change in ENaC protein.
A comparable loss of Na+ induced by reduced Na+ intake during the day or overnight did increase γENaC cleavage. The diurnal fasting also produced an increase in the abundance of the full-length form γENaC as well as that of βENaC; neither acute amiloride nor overnight Na+ restriction led to these changes, despite similar increases in the cleaved forms of the α- and γENaC subunits. It is possible that the additional changes observed in this protocol arise from alterations in gene expression arising from intrinsic diurnal rhythms rather than from losses of extracellular volume (26, 28). Diurnal patterns of urinary salt and water excretion are altered by disruption of genes governing circadian rhythms (31), as is the transcription of a number of genes including NCC and αENaC (16, 36). In mice, levels of pNCC but not overall NCC protein show diurnal fluctuations (40). Although it was not measured in our study, previous work has documented a diurnal pattern of plasma aldosterone concentrations that coincides with the increased levels of distal Na+ transport proteins observed here (6, 15).
During the diurnal fasting period we also documented increases in overall as well as pNCC. In contrast, with overnight Na+ depletion we observed an elevation of total NCC protein without a change in pNCC. This difference may also reflect diurnal rhythms. However, it might also arise from alterations in K+ balance. During the diurnal period K+ balance was negative (Table 1), whereas during overnight Na+ restriction it was positive, due to increased food intake, with a slightly increased plasma K+ (Table 1). In any case it is likely that dietary Na+ restriction over these time periods induces compensatory increases in both NCC and ENaC activities that increase Na+ reabsorption in response to decreased extracellular volume.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant RO1-DK-099284.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.F. and L.G.P. conceived and designed research; G.F., L.Y., A.M.W., and L.G.P. performed experiments; G.F., A.M.W., and L.G.P. analyzed data; G.F., S.U., A.M.W., and L.G.P. interpreted results of experiments; G.F., L.Y., S.U., A.M.W., and L.G.P. edited and revised manuscript; G.F., L.Y., S.U., A.M.W., and L.G.P. approved final version of manuscript; A.M.W. and L.G.P. prepared figures; L.G.P. drafted manuscript.
REFERENCES
- 1.Alessi DR, Zhang J, Khanna A, Hochdörfer T, Shang Y, Kahle KT. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 7: re3, 2014. doi: 10.1126/scisignal.2005365. [DOI] [PubMed] [Google Scholar]
- 2.Ashek A, Menzies RI, Mullins LJ, Bellamy CO, Harmar AJ, Kenyon CJ, Flatman PW, Mullins JJ, Bailey MA. Activation of thiazide-sensitive co-transport by angiotensin II in the cyp1a1-Ren2 hypertensive rat. PLoS One 7: e36311, 2012. doi: 10.1371/journal.pone.0036311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S. Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int 74: 1403–1409, 2008. doi: 10.1038/ki.2008.451. [DOI] [PubMed] [Google Scholar]
- 4.Costanzo LS. Comparison of calcium and sodium transport in early and late rat distal tubules: effect of amiloride. Am J Physiol Renal Fluid Electrolyte Physiol 246: F937–F945, 1984. [DOI] [PubMed] [Google Scholar]
- 5.Czogalla J, Vohra T, Penton D, Kirschmann M, Craigie E, Loffing J. The mineralocorticoid receptor (MR) regulates ENaC but not NCC in mice with random MR deletion. Pflugers Arch 468: 849–858, 2016. doi: 10.1007/s00424-016-1798-5. [DOI] [PubMed] [Google Scholar]
- 6.Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, Emoto N, Okuno Y, Tsujimoto G, Kanematsu A, Ogawa O, Todo T, Tsutsui K, van der Horst GT, Okamura H. Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med 16: 67–74, 2010. doi: 10.1038/nm.2061. [DOI] [PubMed] [Google Scholar]
- 7.Ergonul Z, Frindt G, Palmer LG. Regulation of maturation and processing of ENaC subunits in the rat kidney. Am J Physiol Renal Physiol 291: F683–F693, 2006. doi: 10.1152/ajprenal.00422.2005. [DOI] [PubMed] [Google Scholar]
- 8.Frindt G, Ergonul Z, Palmer LG. Surface expression of epithelial Na channel protein in rat kidney. J Gen Physiol 131: 617–627, 2008. doi: 10.1085/jgp.200809989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frindt G, Ergonul Z, Palmer LG. Surface expression of epithelial Na channel protein in rat kidney. J Gen Physiol 131: 617–627, 2008. doi: 10.1085/jgp.200809989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frindt G, Gravotta D, Palmer LG. Regulation of ENaC trafficking in rat kidney. J Gen Physiol 147: 217–227, 2016. doi: 10.1085/jgp.201511533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frindt G, Palmer LG. Acute effects of aldosterone on the epithelial Na channel in rat kidney. Am J Physiol Renal Physiol 308: F572–F578, 2015. doi: 10.1152/ajprenal.00585.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol 299: F890–F897, 2010. doi: 10.1152/ajprenal.00323.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frindt G, Palmer LG. K+ secretion in the rat kidney: Na+ channel-dependent and -independent mechanisms. Am J Physiol Renal Physiol 297: F389–F396, 2009. doi: 10.1152/ajprenal.90528.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frindt G, Palmer LG. Surface expression of sodium channels and transporters in rat kidney: effects of dietary sodium. Am J Physiol Renal Physiol 297: F1249–F1255, 2009. doi: 10.1152/ajprenal.00401.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez-Sanchez C, Holland OB, Higgins JR, Kem DC, Kaplan NM. Circadian rhythms of serum renin activity and serum corticosterone, prolactin, and aldosterone concentrations in the male rat on normal and low-sodium diets. Endocrinology 99: 567–572, 1976. doi: 10.1210/endo-99-2-567. [DOI] [PubMed] [Google Scholar]
- 16.Gumz ML, Stow LR, Lynch IJ, Greenlee MM, Rudin A, Cain BD, Weaver DR, Wingo CS. The circadian clock protein Period 1 regulates expression of the renal epithelial sodium channel in mice. J Clin Invest 119: 2423–2434, 2009. doi: 10.1172/JCI36908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall JE, Brands MW. The renin-angiotensin-aldosterone systems. In: The Kidney: Physiology and Pathophysiology, edited by Seldin DW, Giebisch G. New York: Raven, 1992, p. 1455–1504. [Google Scholar]
- 18.Hoorn EJ, Nelson JH, McCormick JA, Ellison DH. The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol 22: 605–614, 2011. doi: 10.1681/ASN.2010080827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunter RW, Craigie E, Homer NZ, Mullins JJ, Bailey MA. Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am J Physiol Renal Physiol 306: F457–F467, 2014. doi: 10.1152/ajprenal.00339.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB, Knepper MA. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci USA 95: 14552–14557, 1998. doi: 10.1073/pnas.95.24.14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunau RT Jr, Weller DR, Webb HL. Clarification of the site of action of chlorothiazide in the rat nephron. J Clin Invest 56: 401–407, 1975. doi: 10.1172/JCI108105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loffing J, Pietri L, Aregger F, Bloch-Faure M, Ziegler U, Meneton P, Rossier BC, Kaissling B. Differential subcellular localization of ENaC subunits in mouse kidney in response to high- and low-Na diets. Am J Physiol Renal Physiol 279: F252–F258, 2000. [DOI] [PubMed] [Google Scholar]
- 23.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCormick JA, Yang CL, Ellison DH. WNK kinases and renal sodium transport in health and disease: an integrated view. Hypertension 51: 588–596, 2008. doi: 10.1161/HYPERTENSIONAHA.107.103788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mills JN, Stanbury SW. Persistent 24-hour renal excretory rhythm on a 12-hour cycle of activity. J Physiol 117: 22–37, 1952. [PMC free article] [PubMed] [Google Scholar]
- 27.Monroy A, Plata C, Hebert SC, Gamba G. Characterization of the thiazide-sensitive Na(+)-Cl(-) cotransporter: a new model for ions and diuretics interaction. Am J Physiol Renal Physiol 279: F161–F169, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Moore-Ede MC, Herd JA. Renal electrolyte circadian rhythms: independence from feeding and activity patterns. Am J Physiol Renal Fluid Electrolyte Physiol 232: F128–F135, 1977. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen MT, Han J, Ralph DL, Veiras LC, McDonough AA. Short-term nonpressor angiotensin II infusion stimulates sodium transporters in proximal tubule and distal nephron. Physiol Rep 3: e12496, 2015. doi: 10.14814/phy2.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen MT, Lee DH, Delpire E, McDonough AA. Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol 305: F510–F519, 2013. doi: 10.1152/ajprenal.00183.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nikolaeva S, Pradervand S, Centeno G, Zavadova V, Tokonami N, Maillard M, Bonny O, Firsov D. The circadian clock modulates renal sodium handling. J Am Soc Nephrol 23: 1019–1026, 2012. doi: 10.1681/ASN.2011080842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmer LG, Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol 10: 676–687, 2015. doi: 10.2215/CJN.12391213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Penton D, Czogalla J, Loffing J. Dietary potassium and the renal control of salt balance and blood pressure. Pflugers Arch 467: 513–530, 2015. doi: 10.1007/s00424-014-1673-1. [DOI] [PubMed] [Google Scholar]
- 34.Penton D, Czogalla J, Wengi A, Himmerkus N, Loffing-Cueni D, Carrel M, Rajaram RD, Staub O, Bleich M, Schweda F, Loffing J. Extracellular K(+) rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl(-) -dependent and independent mechanisms. J Physiol 594: 6319–6331, 2016. doi: 10.1113/JP272504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rengarajan S, Lee DH, Oh YT, Delpire E, Youn JH, McDonough AA. Increasing plasma [K+] by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am J Physiol Renal Physiol 306: F1059–F1068, 2014. doi: 10.1152/ajprenal.00015.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richards J, Ko B, All S, Cheng KY, Hoover RS, Gumz ML. A role for the circadian clock protein Per1 in the regulation of the NaCl co-transporter (NCC) and the with-no-lysine kinase (WNK) cascade in mouse distal convoluted tubule cells. J Biol Chem 289: 11791–11806, 2014. doi: 10.1074/jbc.M113.531095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, Gamba G. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci USA 106: 4384–4389, 2009. doi: 10.1073/pnas.0813238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shoda W, Nomura N, Ando F, Mori Y, Mori T, Sohara E, Rai T, Uchida S. Calcineurin inhibitors block sodium-chloride cotransporter dephosphorylation in response to high potassium intake. Kidney Int 91: 402–411, 2017. doi: 10.1016/j.kint.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- 40.Susa K, Sohara E, Isobe K, Chiga M, Rai T, Sasaki S, Uchida S. WNK-OSR1/SPAK-NCC signal cascade has circadian rhythm dependent on aldosterone. Biochem Biophys Res Commun 427: 743–747, 2012. doi: 10.1016/j.bbrc.2012.09.130. [DOI] [PubMed] [Google Scholar]
- 41.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 89: 127–134, 2016. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Lubbe N, Lim CH, Fenton RA, Meima ME, Jan Danser AH, Zietse R, Hoorn EJ. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int 79: 66–76, 2011. doi: 10.1038/ki.2010.290. [DOI] [PubMed] [Google Scholar]
- 44.van der Lubbe N, Lim CH, Meima ME, van Veghel R, Rosenbaek LL, Mutig K, Danser AH, Fenton RA, Zietse R, Hoorn EJ. Aldosterone does not require angiotensin II to activate NCC through a WNK4-SPAK-dependent pathway. Pflugers Arch 463: 853–863, 2012. doi: 10.1007/s00424-012-1104-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Veiras LC, Han J, Ralph DL, McDonough AA. Potassium supplementation prevents sodium chloride cotransporter stimulation during angiotensin II hypertension. Hypertension 68: 904–912, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Velázquez H, Wright FS. Control by drugs of renal potassium handling. Annu Rev Pharmacol Toxicol 26: 293–309, 1986. doi: 10.1146/annurev.pa.26.040186.001453. [DOI] [PubMed] [Google Scholar]
- 47.Verrey F, Hummler E, Schild L, Rossier BC. Mineralocorticoid action in the aldosterone-sensitive distal nephron (4th ed.). In: The Kidney: Physiology and Pathophysiology, edited by Alpern RJ, Hebert SC. Burlington, MA: Academic, 2008, p. 889–924. doi: 10.1016/B978-012088488-9.50035-8. [DOI] [Google Scholar]
- 48.Weinstein AM. A mathematical model of rat distal convoluted tubule. I. Cotransporter function in early DCT. Am J Physiol Renal Physiol 289: F699–F720, 2005. doi: 10.1152/ajprenal.00043.2005. [DOI] [PubMed] [Google Scholar]
- 49.Weinstein AM. A mathematical model of rat distal convoluted tubule. II. Potassium secretion along the connecting segment. Am J Physiol Renal Physiol 289: F721–F741, 2005. doi: 10.1152/ajprenal.00044.2005. [DOI] [PubMed] [Google Scholar]
- 50.Weinstein AM. Potassium excretion during antinatriuresis: perspective from a distal nephron model. Am J Physiol Renal Physiol 302: F658–F673, 2012. doi: 10.1152/ajprenal.00528.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L, Frindt G, Lang F, Kuhl D, Vallon V, Palmer LG. SGK1-dependent ENaC processing and trafficking in mice with high dietary K intake and elevated aldosterone. Am J Physiol Renal Physiol 312: F65–F76, 2017. doi: 10.1152/ajprenal.00257.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]