

Abstract
Dietary protein restriction has multiple benefits in kidney disease. Because protein intake is a major determinant of endogenous acid production, it is important that net acid excretion changes in parallel during changes in dietary protein intake. Dietary protein restriction decreases endogenous acid production and decreases urinary ammonia excretion, a major component of net acid excretion. Glutamine synthetase (GS) catalyzes the reaction of and glutamate, which regenerates the essential amino acid glutamine and decreases net ammonia generation. Because renal proximal tubule GS expression increases during dietary protein restriction, this could contribute to the decreased ammonia excretion. The purpose of the current study was to determine the role of proximal tubule GS in the renal response to protein restriction. We generated mice with proximal tubule-specific GS deletion (PT-GS-KO) using Cre-loxP techniques. Cre-negative (Control) and PT-GS-KO mice in metabolic cages were provided 20% protein diet for 2 days and were then changed to low-protein (6%) diet for the next 7 days. Additional PT-GS-KO mice were maintained on 20% protein diet. Dietary protein restriction caused a rapid decrease in urinary ammonia excretion in both genotypes, but PT-GS-KO blunted this adaptive response significantly. This occurred despite no significant genotype-dependent differences in urinary pH or in serum electrolytes. There were no significant differences between Control and PT-GS-KO mice in expression of multiple other proteins involved in renal ammonia handling. We conclude that proximal tubule GS expression is necessary for the appropriate decrease in ammonia excretion during dietary protein restriction.
Keywords: acid-base, ammonia, dietary protein, proximal tubule
renal ammonia1 metabolism has an important role in both acid-base homeostasis and nitrogen metabolism (33, 36, 39, 40). Renal ammonia excretion is the primary component of basal net acid excretion, and altered ammonia excretion is the primary component of the renal response to the majority of acid-base perturbations (38–40). In addition, because ammonia excretion necessarily involves nitrogen excretion, changes in ammonia excretion contribute to nitrogen balance (36).
Dietary protein restriction has multiple benefits in patients with chronic kidney disease (CKD) (4, 17, 19). One of the effects of dietary protein restriction is decreased endogenous acid production (5, 28). Because chronic metabolic acidosis is common in patients with CKD (13, 21, 39), appears to contribute to the progressive nephropathy observed (3, 6, 12), and predicts mortality in patients with CKD (11, 22, 27), one of the benefits of dietary protein restriction may relate to decreased endogenous acid production leading to decreased metabolic acidosis. However, it is also important to prevent the development of metabolic alkalosis that would result if decreased endogenous acid production were not balanced by decreased net acid excretion; metabolic alkalosis is associated with increased mortality in people with normal renal function and those with CKD (11, 22, 27). In addition, decreased dietary protein intake results in decreased dietary nitrogen loads. Thus, it is important to decrease urinary nitrogen loss to maintain nitrogen balance. Thus, understanding the mechanisms of renal response to decreased dietary protein intake is of fundamental importance.
Recent studies suggest that glutamine synthetase (GS) has an important role in renal acid-base and nitrogen metabolism. GS catalyzes the reaction of glutamate with , which decreases net ammoniagenesis (16, 32). Thus, GS can have an important role in ammonia metabolism and, by extension, in acid-base homeostasis. Because GS regenerates glutamine, an essential amino acid that is critical for nitrogen balance and for protein synthesis, GS may also have an important role in nitrogen homeostasis. GS is highly expressed in the renal proximal tubule, and its expression increases during dietary protein restriction (8, 15, 16, 32). This suggests ammonia recycling resulting from the increased GS expression during protein restriction could decrease net ammoniagenesis and thereby limit ammonia excretion.
The purpose of the current study was to determine whether proximal tubule GS has a role in the response to dietary protein restriction by examining the effects of its deletion on ammonia metabolism. Because GS is expressed in multiple cell types in the kidney (16, 32) and because expression in different cell types is regulated differently (32), we examined a model of proximal tubule-specific GS deletion (PT-GS-KO) that we reported recently (16). First, we determined whether PT-GS-KO alters urinary ammonia excretion in response to dietary protein restriction. Next, we determined whether the blunted response seen in PT-GS-KO mice was due to changes in urine pH and whether titratable acid excretion was also altered. Next, we determined whether the observed and specific effect on PT-GS-KO was related to effects of PT-GS-KO on expression of other proteins involved in renal ammonia metabolism. Finally, we confirmed that these changes in PT-GS-KO mice fed low-protein diets were specific to the low-protein diet and not a time-dependent effect. Our results show that intact proximal tubule GS expression, which increases during dietary protein restriction, is necessary for the normal urinary ammonia excretion response observed.
METHODS
Animals.
We generated male mice with PT-GS-KO as we described previously (16). Briefly, PT-GS-KO mice had loxP sites flanking exons 1 and 7 of the GS gene (GSfl/fl) and expressed Cre recombinase under control of a modified phosphoenolpyruvate carboxykinase (PEPCK)-Cre promoter (Cre) (26). This PEPCK promoter is a modified promoter that decreases hepatocyte PEPCK expression by ~60% and increases renal expression by ~10-fold (23, 26). Control mice were PEPCK-Cre negative littermates. In previous studies, we showed that this Cre-loxP approach results in proximal tubule-specific GS deletion and no alteration in hepatocellular GS expression (16). Because PEPCK-Cre is integrated on the X-chromosome, it is subject to X-chromosome inactivation in female mice. Thus, all mice used in these experiments were adult male mice averaging ~6 mo of age. We genotyped all mice using DNA extracted from tail-clip samples, as we described recently (16). The Institutional Animal Care and Use Committees of the University of Florida and the North Florida/South Georgia Veterans Health System approved all animal experiments. Trained personnel in the University of Florida College of Medicine Cancer and Genetics Transgenic Animal Core Facility oversaw all animal breeding. All mice were on the C57Bl/6 background strain.
Antibodies.
We used affinity-purified antibodies to Rhbg and Rhcg generated in our laboratory that we have characterized previously (1, 10, 14, 18, 31, 35). Norman Curthoys (Colorado State University) graciously provided antibodies to phosphate-dependent glutaminase (PDG) and H. Moo Kwon (Ulsan National Institute of Science and Technology, Ulsan, South Korea) graciously provided antibodies to Na+-K+-2Cl− transporter, isoform 2 (NKCC2). Antibodies to PEPCK were obtained from Cayman Chemical (Ann Arbor, MI), antibodies to GS were obtained from Abcam (Cambridge, MA), antibodies to Na+-H+ exchanger, isoform 3 (NHE3) were obtained from StressMarq Biosciences (Victoria, British Columbia), and antibodies to PDG were also obtained from ProteinTech Group (Rosemont, IL).
Protein diet.
Powdered semisynthetic diets with either normal (20%) or low (6%) protein content were obtained from Envigo (TD. 91352 and TD. 90016, respectively). Cre-negative (Control) and PT-GS-KO mice in metabolic cages (Tecniplast diuresis metabolic cage; Fisher Scientific) were fed 20% protein diet for 2 days and were then changed to low-protein (6%) diet for the next 7 days. Additional PT-GS-KO mice were maintained on 20% protein diet. Daily food intake was measured. At all times, animals were provided ad libitum access to water. Urine was collected under mineral oil, and body weights, urine volume, and pH were recorded daily. Urine samples were stored at −20°C until analyzed further.
Electrolyte measurements.
Urine ammonia was measured using a modification of a commercially available kit (A7553; Pointe Scientific, Canton, MI) as previously described (14). Urine pH was measured using a micro-pH electrode (ROSS semimicro pH, Orion 8115BN; Thermo Scientific). Blood was obtained by cannulation of the abdominal artery, drawn in a heparinized syringe, and immediately analyzed for Na+, K+, and concentration using a Siemens microanalytic blood gas analyzer (RAPIDLab 348 analyzer; Siemens). Urinary titratable acid was measured using methods we have previously described (14). Urea nitrogen was measured by a commercially available kit (B7552; Pointe Scientific) according to the manufacturer’s instructions, modified for use in 96-well plates. Urine samples were measured following dilution with deionized water.
Tissue preparation for immunolocalization.
Mice were anesthetized with inhalant isoflurane. The kidneys were preserved by in vivo cardiac perfusion with PBS (pH 7.4) followed by periodate-lysine-2% paraformaldehyde, cut transversely into several 2- to 3-mm-thick slices, and then immersed for 48 h at 4°C in the same fixative. Kidney samples from each animal were embedded in polyester wax made using polyethylene glycol 400 distearate (Polysciences, Warrington, PA) with 10% 1-hexadecanol, and 3-μm-thick sections were cut and mounted on gelatin-coated glass slides.
Immunohistochemistry.
Immunolocalization was accomplished using standard immunoperoxidase procedures. Briefly, sections were dewaxed in ethanol, rehydrated, heated in Trilogy (Cell Marque, Rocklin, CA) to 88°C for 30 min and then to 96°C for 30 min, cooled for 30 min, and rinsed in PBS. Endogenous peroxidase activity was blocked by incubating the sections in 3% H2O2 in distilled water for 45 min. Sections were blocked for 15 min with Serum-Free Protein Block (DakoCytomation) and then incubated at 4°C overnight with primary antibody. Sections were washed in PBS and incubated for 30 min with polymer-linked peroxidase-conjugated goat anti-rabbit IgG (MACH2; Biocare Medical, Concord, CA), washed again with PBS, and then exposed to diaminobenzidine for 5 min. Sections were washed in distilled water, dehydrated with xylene, mounted, and observed by light microscopy. Comparisons of labeling were made only between sections of the same thickness from the same immunohistochemistry experiment. Sections were examined on a Leica DM2000 microscope and photographed using a Leica DFC425 digital camera and Leica DFC Twain Software and LAS application suite (Leica Microsystems, Buffalo Grove, IL). Color correction was performed using Adobe Photoshop software (Adobe Systems, San Jose, CA).
Protein preparation.
Mice were anesthetized with inhalant isoflurane, and tissues were rinsed by in vivo cardiac perfusion with PBS (pH 7.4). The right renal vasculature was clamped, the right kidney was rapidly removed, and the cortex, outer stripe of the outer medulla, and inner stripe of the outer medulla were isolated rapidly under a dissection microscope. All samples were snap-frozen in liquid nitrogen and stored frozen at −70°C until used. Tissues were homogenized in T-PER Tissue Protein Extraction Reagent (Pierce Biotechnology) using microtube pestles (USA Scientific, Ocala, FL), and protein was extracted according to the manufacturer’s recommended procedures. An aliquot was obtained for protein determination using a BCA assay, and the remainder was stored frozen at −70°C until used.
Immunoblot analysis.
Five to ten micrograms of renal protein were electrophoresed on 10% PAGE ReadyGel (Bio-Rad, Hercules, CA). Gels were then transferred electrophoretically to nitrocellulose membranes, blocked with 5 g/dl nonfat dry milk in Blotto buffer (50 mM Tris, 150 mM NaCl, 5 mM Na2EDTA, and 0.05% Tween 20, pH 7.6), and incubated at 4°C overnight with primary antibody diluted in nonfat dry milk. Loading and transfer equivalence were assessed with Ponceau S staining. After being washed, membranes were exposed to secondary antibody, goat anti-rabbit IgG (Cell Signaling Technology, Beverly, MA), conjugated to horseradish peroxidase at a dilution of 1:5,000. Sites of antibody-antigen reaction were visualized by using enhanced chemiluminescence (SuperSignal West Pico Substrate; Pierce) and a Kodak Image Station 440CF digital imaging system. Band density was normalized such that mean density in the same region (cortex or outer stripe of the outer medulla) in control tissues was 100.0. The absence of saturation was confirmed by examining pixel intensity distribution in all immunoblots.
Statistics.
Results are presented as means ± SE. When we performed repeated measurements over time, statistical significance for the primary independent variable was determined using general linear model with repeated-measures analysis (IBM SPSS Statistics, version 24). If statistical significance for the independent variable was present, statistical analysis at individual time points was determined using Student’s t-test. Immunoblot analysis data were tested for normality using the Shapiro-Wilk test (IBM SPSS Statistics, version 24). When data were not normally distributed, expression was compared using nonparametric analysis of independent samples (IBM SPSS, version 24). When the alternative hypothesis that the data were not normally distributed could be rejected, data analysis was performed using Student’s t-test. P < 0.05 was taken as statistically significant; n refers to the number of animals studied.
RESULTS
Effect of PT-GS-KO on urinary ammonia response to dietary protein restriction.
The first goal of these studies was to determine whether GS expression in the proximal tubule was necessary for the normal response to dietary protein restriction. To address this issue we compared the response of mice with PT-GS-KO with that of Control mice with intact proximal tubule GS expression. Table 1 shows the mean value of daily body weight, food intake, and plasma electrolyte values in Control and PT-GS-KO mice. There was no significant difference in any of these parameters between Control and PT-GS-KO mice.
Table 1.
Physiological parameters during dietary protein restriction
| Parameter | Control | PT-GS-KO | P Value |
|---|---|---|---|
| Mean daily body wt, g/day | 32.2 ± 0.6 (12) | 32.7 ± 0.9 (12) | NS |
| Mean daily food intake, g/day | 5.9 ± 0.1 (12) | 5.7 ± 0.2 (12) | NS |
| Serum Na+, mmol/l | 150.5 ± 1.0 (12) | 151.4 ± 1.1 (9) | NS |
| Serum K+, mmol/l | 3.88 ± 0.08 (12) | 3.84 ± 0.14 (9) | NS |
| Serum , mmol/l | 21.9 ± 0.5 (12) | 21.9 ± 0.7 (12) | NS |
| Blood urea nitrogen, mg/dl | 20.3 ± 3.0 (6) | 19.0 ± 0.9 (4) | NS |
Values ae means ± SE; nos. in parentheses are no. of animals in each group. PT-GS-KO, proximal tubule-specific glutamine synthetase deletion; NS, not significant.
In response to dietary protein restriction there was a rapid decrease in urinary ammonia excretion in both Control and PT-GS-KO mice (Fig. 1). However, in PT-GS-KO mice the decrease in urinary ammonia excretion was blunted significantly (P < 0.005, n = 12 mice/genotype). During days 2–7 of dietary protein restriction, urinary ammonia excretion was decreased from baseline by 56 ± 5% in PT-GS-KO mice vs. 74 ± 3% observed in Control mice (P < 0.01, n = 12 in each group). This indicates that GS expression in the proximal tubule, which normally increases with dietary protein restriction (15), is necessary for the appropriate change in urinary ammonia excretion.
Fig. 1.
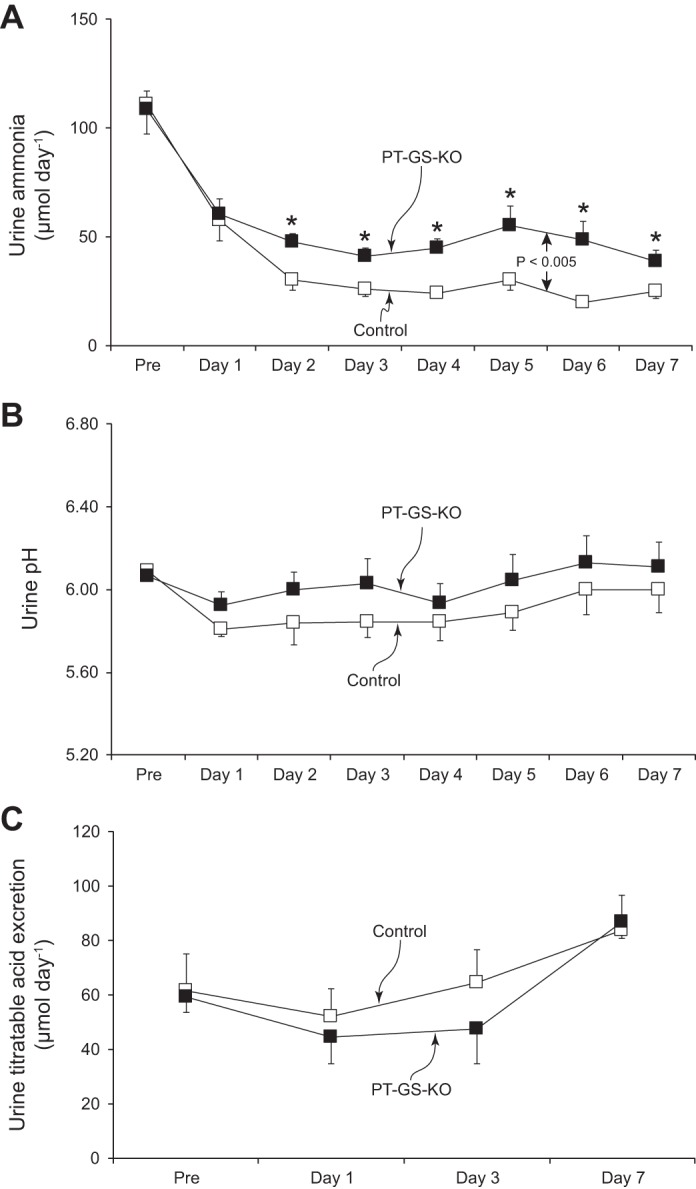
Effect of proximal tubule-specific glutamine synthetase deletion (PT-GS-KO) on urinary ammonia, pH, and titratable acid excretion response to dietary protein restriction. A: changes in urinary ammonia excretion. Initiation of a 6% protein diet caused a rapid decrease in urinary ammonia excretion in both Control and PT-GS-KO genotypes, but the decrease was blunted significantly in PT-GS-KO mice compared with Control mice (P < 0.005). *P < 0.05 vs. Control mice on an individual day; n = 12 mice/genotype. B: changes in urine pH. Urine pH was not different between genotypes and did not change significantly in either genotype during dietary protein restriction; n = 12 mice/genotype. C: titratable acid excretion. There were no time-dependent changes in titratable acid excretion in either genotype during dietary protein restriction and no difference between groups; n = 6 mice/genotype.
Because dietary protein restriction might alter urine pH, which could alter ammonia excretion, we determined whether PT-GS-KO altered urinary pH during dietary protein restriction. Figure 1B shows these results; proximal tubule GS deletion did not alter the urine pH response to a low-protein diet [P = not significant (NS), n = 12 in each genotype]. Thus, proximal tubule GS expression is necessary for the normal decrease in urinary ammonia excretion in response to dietary protein restriction, and the difference induced by its deletion is not mediated by changes in urine pH.
Effect of PT-GS-KO on titratable acid excretion during dietary protein restriction.
We next determined whether the role of proximal tubule GS in the ammonia response to dietary protein restriction was specific to ammonia or reflected a generalized effect on renal net acid excretion. Figure 1C shows these findings. Dietary protein restriction, which decreases endogenous acid production, does not alter titratable acid excretion (P = NS). Proximal tubule GS deletion, despite significant effects on ammonia excretion, did not affect the titratable acid excretion response to dietary protein restriction.
Effect of dietary protein restriction on proteins involved in ammoniagenesis in PT-GS-KO mice.
Dietary protein restriction alters expression of many proteins involved in renal ammonia metabolism (15). One possible explanation of the above findings is that proximal tubule GS expression is necessary for normal expression of other proteins involved in renal ammonia metabolism and that deleting proximal tubule GS induced abnormal expression of one or more of these proteins, thereby causing the observed changes in ammonia excretion. To explore this possibility, we examined expression of candidate proteins. There was no significant difference in the expression of major proteins involved in renal ammonia metabolism, including PDG, PEPCK, Rhbg, NHE3, and NKCC2 (Fig. 2, n = 6 mice/genotype). Thus, differences in expression in any of these key proteins involved in renal ammonia metabolism do not explain the inhibition of the ammonia excretion in response to dietary protein restriction that proximal tubule-specific GS deletion causes.
Fig. 2.

Effect of PT-GS-KO on phosphate-dependent glutaminase (PDG), phosphoenolpyruvate carboxykinase (PEPCK), Rhbg, Na+-H+ exchanger, isoform 3 (NHE3), and Na+-K+- 2Cl− transporter, isoform 2 (NKCC2) in dietary protein restriction. Immunoblot analyses of renal cortical and medullary protein homogenates from Control and PT-GS-KO mice fed 6% protein diet for 7 days are shown. There were no significant differences between Control and PT-GS-KO mice in expression of multiple proteins involved in renal ammonia handling, including PDG, PEPCK, Rhbg, NHE3, and NKCC2. Immunoblot data were not normally distributed for the following proteins: PDG in the outer stripe of the outer medulla (OSOM), PEPCK in cortex and OSOM, Rhbg in cortex and OSOM, and NKCC2. Nonparametric analysis was used to compare median expression levels. NS, not significant. Student’s t-test was used for the remaining analyses. Values are means ± SE; n = 6 mice/genotype.
Effect of PT-GS-KO on urinary nitrogen excretion during dietary protein restriction.
Nitrogen conservation is critical to maintenance of total body muscle and protein mass during dietary protein restriction. Urinary nitrogen is almost exclusively in the form of urea and ammonia (36). Thus, the finding that proximal tubule GS deletion alters ammonia nitrogen excretion during dietary protein restriction raises the question as to whether this effect is sufficient to alter total urinary nitrogen excretion. In response to dietary protein restriction, urinary urea nitrogen excretion did not differ significantly between Control and PT-GS-KO mice (Control, 19.9 ± 2.8 mg/day; PT-GS-KO, 19.8 ± 4.7 mg/day; P = NS, n = 4 and 5, respectively). Total urinary nitrogen excretion in the form of urea or ammonia also did not differ significantly between Control and PT-GS-KO mice (Control, 1,452 ± 205 µmol N/day; PT-GS-KO, 1,452 ± 347 µmol N/day, P = NS, n = 4 and 5, respectively). Thus, proximal tubule GS expression is critical for regulation of urinary ammonia excretion but not for changes in total urinary nitrogen excretion during dietary protein restriction.
Physiological parameters under dietary protein restriction.
An additional possible explanation of our findings is that PT-GS-KO has nonspecific time-dependent changes in urinary ammonia excretion in mice in metabolic cage studies. Our next set of studies examined this possibility. Mice with PT-GS-KO were placed in metabolic cages, fed a 20% protein diet for 2 days, and then were randomized to either 6% protein diet or 20% protein diet for 7 days. Table 2 shows physiological parameters after completion of this dietary protocol. There were no significant differences in body weight, mean daily food intake, serum sodium, serum potassium, or serum . Blood urea nitrogen (BUN) was significantly lower in mice receiving the low-protein diet, consistent with decreased protein intake.
Table 2.
Physiological parameters of PT-GS-KO mice on 20 vs. 6% protein diet
| Parameter | 20% Protein Diet | 6% Protein Diet | P Value |
|---|---|---|---|
| Mean daily body wt, g/day | 35.3 ± 1.1 (5) | 36.1 ± 0.8 (6) | NS |
| Mean daily food intake, g/day | 5.6 ± 0.2 (5) | 6.0 ± 0.3 (6) | NS |
| Serum Na+, mmol/l | 146.9 ± 1.6 (5) | 143.7 ± 1.6 (6) | NS |
| Serum K+, mmol/l | 4.56 ± 0.24 (5) | 4.56 ± 0.34 (6) | NS |
| Serum , mmol/l | 19.3 ± 0.5 (4) | 20.0 ± 0.5 (6) | NS |
| Blood urea nitrogen, mg/dl | 31.4 ± 5.7 (5) | 18.5 ± 2.2 (6) | <0.05 |
Values are means ± SE; nos. in parentheses are no. of animals in each group. NS, not significant.
Dietary protein restriction decreases urinary ammonia excretion in wild-type mice (15, 38). This was also observed in mice with PT-GS-KO (P < 0.001 for comparison of mice fed 20% vs. 6% protein diets, n = 5 fed 20% protein and n = 6 fed 6% protein diet, Fig. 3A). The decrease in urinary ammonia excretion was maximal on day 3 and continued through the next 4 days of dietary protein restriction without further change. The mean reduction in urinary ammonia excretion in mice with proximal tubule GS deletion in response to 6% protein diet was a 53 ± 16% reduction over the last 4 days compared with baseline ammonia excretion. In mice continued on 20% protein diet, there was no significant change in urinary ammonia excretion. These results indicate that mice with PT-GS-KO are able to decrease urinary ammonia excretion in response to dietary protein restriction and that the observed changes are not a nonspecific time-dependent response. Importantly, the observed decrease, ~53%, is substantially different from the ~85% reduction we observed previously in wild-type mice (15) and the ~74% reduction observed in Control mice (see above).
Fig. 3.
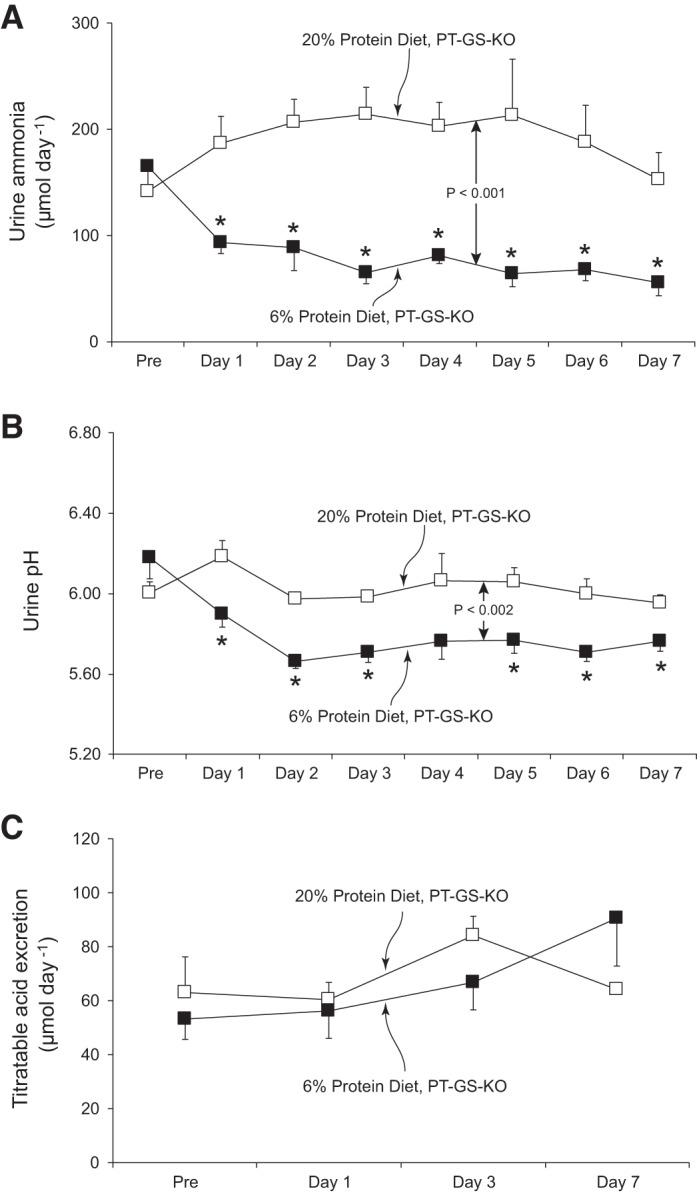
Urinary ammonia and acid excretion response to dietary protein restriction in PT-GS-KO mice. A: changes in urinary ammonia excretion. In the animals fed 20% protein diet, there were no significant time-dependent changes in urinary ammonia excretion. In mice fed 6% protein diet, urine ammonia excretion decreased and was significantly different from in mice fed 20% protein diet (P < 0.005). Thus, the changes in ammonia excretion in PT-GS-KO mice in response to dietary protein restriction are specific to the change in protein intake and are not a nonspecific time-dependent change. *P < 0.05 vs. 20% protein diet on individual day; n = 5 and 6 for 20% and 6% protein diets, respectively. B: changes in urine pH. There were no significant changes in urine pH in time-control studies of PT-GS-KO mice fed 20% protein diet. In mice fed 6% protein diet, urine pH was significantly different from in mice fed 20% protein diet (P < 0.005). *P < 0.05 vs. 20% protein diet in PT-GS-KO mice on an individual day; n = 5 and 6 for 20% and 6% protein diets, respectively. C: titratable acid excretion. There were no time-dependent changes in titratable acid excretion in PT-GS-KO mice fed 20% protein diet and 6% protein diet; n = 5, 20% protein diet and n = 6, 6% protein diet.
Because an important determinant of urinary ammonia excretion is urine pH, we determined whether the decreased ammonia excretion resulted from changes in urine pH. Figure 3B shows the results of this analysis. Urine pH was significantly more acidic in PT-GS-KO mice fed 6% protein than those fed 20% protein diet (P < 0.002, n = 5 fed 20% and n = 6 fed 6% protein diet). Because a more acidic, i.e., lower pH, urine would promote increased urinary ammonia excretion, the decreased ammonia excretion in response to 6% protein diet cannot be ascribed to the observed changes in urinary pH. Thus, mice with PT-GS-KO are able to decrease urinary ammonia excretion through a mechanism independent of urine pH, but the observed decrease is less than that observed previously in wild-type mice.
Effect of dietary protein restriction on net acid excretion.
Because ammonia excretion is an important component of renal net acid excretion, we determined the effect of dietary protein restriction on a second major component of net acid excretion, titratable acid excretion. Figure 3C shows effects of dietary protein restriction on titratable acid excretion in mice with PT-GS-KO. Titratable acid excretion did not change significantly after induction of dietary protein restriction (P = NS, n = 5 fed 20% and n = 6 fed 6% protein diet). These results therefore indicate that mice with PT-GS-KO are able to decrease ammonia excretion in response to the low-protein diet through a mechanism that cannot be explained by changes in urine pH and that is distinguishable from the lack of effect of low-protein diet on titratable acid excretion in this model.
Effect of dietary protein restriction on urea nitrogen metabolism.
Maintenance of nitrogen balance during conditions of decreased protein intake requires a parallel decrease in urinary nitrogen excretion. A major component of nitrogen excretion is in the form of urea. Figure 4A shows that dietary protein restriction significantly altered urinary urea nitrogen in mice with PT-GS-KO (P < 0.02, n = 5 fed 20% and n = 6 fed 6% protein diet). There was no significant change over time in urinary urea nitrogen in mice provided a 20% protein diet (P = NS by paired t-test, n = 5), whereas a 6% protein diet resulted in significantly less urea nitrogen excretion (P < 0.02 on day 1 and P < 0.001 on day 7 vs. “Pre”, n = 6). The decreased urinary urea nitrogen excretion was partially due to decreased urea clearance (20% protein diet, 198.8 µl/min; 6% protein diet, 88.5 µl/min; P < 0.01, n = 3 and 5, respectively), and partially due to lower blood urea nitrogen concentrations (20% protein diet, 31.4 mg/dl; 6% protein diet, 18.5 mg/dl, P < 0.05, n = 5 and 6, respectively).
Fig. 4.
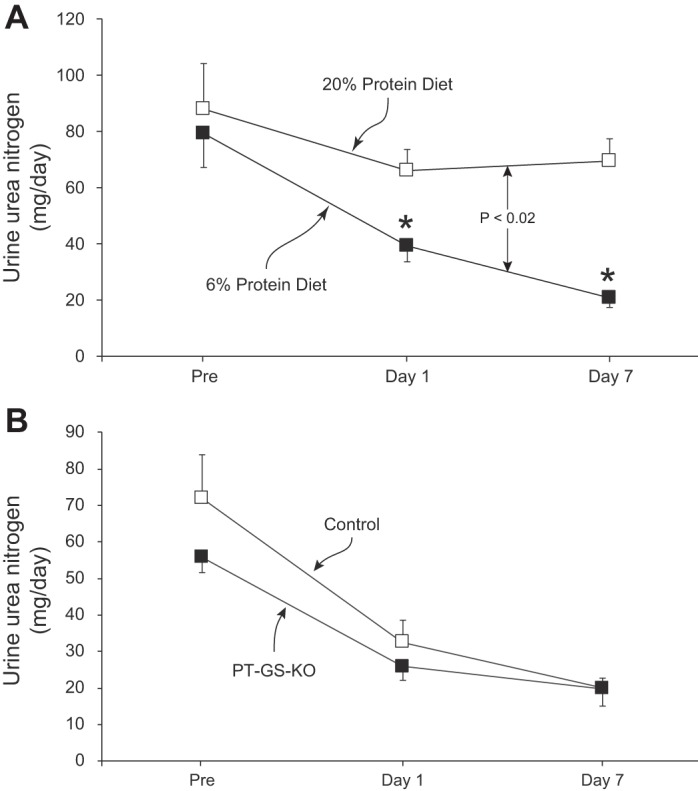
Urinary urea nitrogen excretion in response to dietary protein restriction. A: in PT-GS-KO mice, there were no significant time-dependent changes during 20% protein diet. In response to dietary protein restriction, urinary urea nitrogen excretion decreased rapidly and was significantly less in PT-GS-KO mice fed 6% protein diet than those fed 20% protein (P < 0.02). *P < 0.05 vs. 20% protein diet on an individual day; n = 6 in each group. B: comparison of the ability of Control and PT-GS-KO mice to alter urinary urea nitrogen excretion in response to dietary protein restriction. Urea nitrogen excretion did not differ significantly in the two genotypes, either under basal conditions or in response to a 6% protein diet; n = 6 in each genotype.
We then examined whether the ability to decrease urinary urea nitrogen excretion required proximal tubule GS expression. As shown in Fig. 4B, urinary urea nitrogen excretion did not differ significantly between Control and PT-GS-KO mice, either under basal conditions or in response to dietary protein restriction (P = NS, n = 6 in each genotype). Thus, during dietary protein restriction, where dietary nitrogen intake decreases, there is decreased urinary nitrogen excretion in the form of both ammonia and urea. However, in contrast to the findings examining ammonia excretion, the change in urea nitrogen excretion does not require proximal tubule GS expression.
Effect of dietary protein restriction on other proteins involved in ammonia metabolism.
The finding that dietary protein restriction decreases urinary ammonia excretion even in mice with PT-GS-KO suggests that mechanisms in addition to GS contribute to this response. In fact, previous studies showed that the renal response in normal mice to a low-protein diet involves a coordinated response of multiple proteins involved in ammonia metabolism (15). A similar process appears to be involved in mice with PT-GS-KO. Both PDG expression and PEPCK expression decreased significantly, which would decrease renal ammoniagenesis. These results are summarized in Fig. 5. The adaptive response in expression of ammoniagenic enzymes appears limited to the cortex. Expression of neither of these proteins changed significantly in the outer stripe of the outer medulla.
Fig. 5.

Effect of dietary protein restriction on proximal tubule proteins involved in ammonia generation in PT-GS-KO mice. Immunoblot analyses of renal homogenates. A: in the cortex, dietary protein restriction significantly decreased PEPCK expression. B: cortical PDG expression decreased significantly in response to dietary protein restriction. Immunoblot data were not normally distributed for PEPCK in cortex, and nonparametric analysis was used to compare median expression levels. Student’s t-test was used for remaining analyses. Values are means ± SE; n = 5, 20% protein diet and n = 6, 6% protein diet.
The model of PT-GS-KO used in these studies does not result in complete proximal tubule GS deletion (16). Immunoblot analysis showed that dietary protein restriction caused a small, but statistically significant, increase in GS expression in the renal cortex in this model (Fig. 6). Immunohistochemistry showed that this response involved increased immunolabel in proximal tubule cells that did not exhibit cell-specific GS gene deletion. This increase is similar to that which occurs in mice with intact proximal tubule GS expression (15). The residual proximal tubule GS expression, which dietary protein restriction increases, indicates the current studies likely underestimate the quantitative role of proximal tubule GS in the renal response to dietary protein restriction in normal animals.
Fig. 6.

Effect of dietary protein restriction on residual glutamine synthetase expression in PT-GS-KO mice. Previous studies showed residual glutamine synthetase expression in mice with PT-GS-KO, both in the proximal tubule and in distal epithelial cells (16). Left, immunoblot analyses show that dietary protein restriction significantly increases residual glutamine synthetase expression in the renal cortex. Data were normally distributed in both cortex and OSOM, and Student’s t-test was used for statistical analysis. Right, immunohistochemistry findings in PT-GS-KO fed 20% or 6% protein diet for 7 days. Glutamine synthetase expression in proximal tubule cells that was not deleted by this gene-targeting approach is evident (*). GS immunolabel intensity in these proximal tubule cells was increased in response to 6% protein diet. Results are representative of findings in 6 mice in each group. CD, collecting duct segments.
Effect of PT-GS-KO on Rhcg and Rhbg expression on dietary protein restriction.
Renal ammonia excretion involves coordinated transport of NH3 and by specific membrane proteins in specific renal epithelial cells (37–39, 41). The collecting duct secretes 60–80% of urinary ammonia, and the Rhesus glycoproteins Rhbg and Rhcg are necessary for normal ammonia excretion (7, 34). The current studies show dietary protein restriction in PT-GS-KO mice decreased Rhcg expression in the inner stripe of the outer medulla (Fig. 7). There were no observable changes in Rhbg expression, either by immunohistochemistry or by immunoblot analysis (data not shown). These findings show decreased ammonia excretion is partly due to parallel changes in Rhcg expression in medullary collecting duct intercalated cells.
Fig. 7.

Effect of PT-GS-KO on Rhcg and Rhbg expression on dietary protein restriction. Top, Rhcg immunolabel in PT-GS-KO mice fed 20% or 6% protein diet. In PT-GS-KO mice, dietary protein restriction decreased Rhcg expression in inner stripe of the outer medulla intercalated cells (arrows) and principal cells (unlabeled). Bottom, Rhbg immunolabel in PT-GS-KO mice fed 20% or 6% protein diet. There was no observable difference in Rhbg expression in the inner stripe of the outer medulla intercalated cells in response to dietary protein restriction in PT-GS-KO mice.
DISCUSSION
The current studies examine the role of GS expression in the proximal tubule GS in the renal response to decreases in dietary protein content. Genetic deletion of GS from proximal tubule epithelial cells significantly blunts the expected change in urinary ammonia excretion. This effect is independent of changes in urine pH and is specific to urinary ammonia excretion, since titratable acid excretion was not significantly altered. Moreover, proximal tubule GS deletion did not alter expression of other proteins that are involved in the integrated renal response to dietary protein restriction. Finally, although GS expression is necessary for the decrease in ammonia nitrogen excretion during dietary protein restriction, it does not alter urea nitrogen excretion. Thus, GS expression in the proximal tubule is important for the decrease in urinary ammonia excretion in response to dietary protein restriction. These findings significantly advance our understanding of the molecular mechanisms of renal ammonia metabolism and excretion.
Dietary protein restriction exerts specific stresses on acid-base and nitrogen homeostasis that require renal responses. Protein intake, particularly proteins with high content of sulfur-containing amino acids such as cysteine, generates endogenous acid loads (30). Changes in dietary protein thus alter endogenous acid production (5), requiring changes in renal net acid excretion to maintain acid-base homeostasis. Failure of this to occur causes either metabolic acidosis or metabolic alkalosis, conditions that are associated with increased mortality in humans (27). Changes in nitrogen intake also necessitate changes in urinary nitrogen excretion to maintain nitrogen balance, which is required for normal health (4, 36). Because renal ammonia excretion is a critical component of net acid excretion and nitrogen excretion, understanding the molecular mechanisms regulating ammonia excretion in response to dietary protein restriction is important.
Renal ammonia metabolism has been considered classically as a unidirectional process that culminates in renal urinary ammonia excretion. In this paradigm, there is regulated amino acid uptake, primarily of glutamine, amino acid metabolism, resulting in generation of equimolar amounts of and , and regulated transport of ammonia, in the form of both NH3 and , resulting in urinary ammonia excretion. The current study, in combination with previous studies, shows that this paradigm should be expanded to include a role of ammonia recycling via the enzyme GS.
GS has important roles in ammonia metabolism under a variety of conditions. Genetic deletion of proximal tubule GS expression alters basal ammonia metabolism, indicating a role under basal conditions (16). Metabolic acidosis decreases renal GS expression (2, 14, 42), and this change in the proximal tubule is necessary for the normal increase in ammonia excretion (16). Hypokalemia decreases proximal tubule GS expression in parallel with increases in urinary ammonia excretion (32), indicating that, in another model, increased ammonia excretion involves a parallel decrease in expression of the ammonia-recycling protein, GS. Finally, dietary protein restriction increases GS expression in the proximal tubule (15), and this expression is necessary for the expected decrease in ammonia excretion (current study). Thus, GS-mediated ammonia recycling in the proximal tubule is necessary for basal ammonia metabolism and for adaptive changes that increase and decrease ammonia excretion.
The quantitative role of GS in the response to dietary protein restriction is difficult to determine accurately. In mice with PT-GS-KO, the dietary protein restriction-induced decrease in urinary ammonia excretion is blunted by ~20% compared with Control mice (current study). However, the PT-GS-KO model used in the current studies results in subtotal proximal tubule GS deletion (16). Under basal conditions, these mice exhibit adaptive increases in proximal tubule GS expression in proximal tubule epithelial cells in which gene deletion did not occur (16) and a further increase in this residual proximal tubule cell GS expression in response to dietary protein restriction (current study). Thus, proximal tubule GS expression is necessary for at least 20% of the change in urinary ammonia excretion in response to dietary protein restriction, but in normal mice the GS contribution is probably greater.
Alterations in renal ammonia metabolism in response to dietary protein restriction involve a number of adaptive responses. We showed previously that dietary protein restriction decreased ammonia excretion by ~85% (15), similar to the decrease of ~75% in Control mice in the current study. This change was substantially greater than the observed change in any of several other proteins involved in renal ammonia metabolism, including PDG, PEPCK, NHE3, NKCC2, Rhbg, and Rhcg (15). The current study shows that GS expression, which increases in response to dietary protein restriction (15), is a critical component of this response, and, in addition, provides further support for the conclusion that the dramatic decrease in ammonia excretion is the result of coordinated changes in multiple proteins, rather than a single protein serving as the primary regulatory mechanism.
Urinary nitrogen excretion decreases in response to dietary protein restriction. This involves decreases in both urea and ammonia excretion. Urea excretion decreases in response to both a decreased urea clearance and because of decreased blood urea levels. Urea clearance primarily reflects glomerular filtration rate (GFR), and changes in protein intake acutely regulate GFR. This parallel decrease in both urea and ammonia excretion serves to facilitate nitrogen balance during dietary protein restriction. Quantitatively, nitrogen in the form of urea accounts for 97–98% of total urinary nitrogen excretion during dietary protein restriction. Thus, although GS regenerates the essential amino acid, glutamine, and GS expression increases during dietary protein restriction, the quantitative change in urinary nitrogen excretion in the form of ammonia caused by PT-GS-KO does not substantially alter net nitrogen balance. This is due to finding that GS is not the only mechanism regulating ammonia excretion under these conditions and because nitrogen excretion in the form of urea is substantially greater than in the form of ammonia.
GS is expressed in the liver, where it is important in hepatocellular ammonia metabolism. In the liver, GS is expressed specifically in the perivenous hepatocyte (20, 25, 29, 35), and studies using human hepatocytes suggest hepatocellular GS-mediated ammonia metabolism is a high-affinity low-capacity process (9). Hepatocellular-specific GS deletion causes development of significant hyperammonemia (24). Hepatocellular GS is unlikely, however, to explain the observations in the current study. First, plasma ammonia and filtered ammonia do not contribute significantly to urinary ammonia excretion (7, 39, 40). Second, although PEPCK is expressed in hepatocytes, we showed previously that the model of PEPCK-Cre-driven GS deletion used in this study does not alter perivenous hepatocyte GS expression (16). Thus, the observations in the current study in regard to the role of proximal tubule GS expression on the ammonia excretion response to dietary protein restriction should not be ascribed to hepatocellular GS expression.
GS is found also in intercalated cells in the renal collecting duct (32). In response to hypokalemia, where there is development of mild metabolic alkalosis and increased urinary ammonia excretion, intercalated cell GS expression increases (32). In response to proximal tubule GS deletion, GS expression in intercalated cells in the outer medullary collecting duct increases (16), which may function to minimize changes in urinary ammonia excretion that would otherwise occur as a result of decreased proximal tubule ammonia “recycling.” Thus, intercalated cell GS expression may have an important role in regulating urinary ammonia excretion. However, it is important also to note that proximal tubule and intercalated cell GS expression appears to be regulated differently, at least in response to hypokalemia (32). Understanding the specific mechanisms underlying this differential regulation and, by extension, the different physiological roles of GS in different renal epithelial cells is an important issue for future studies.
In a previous study, we found that proximal tubule GS deletion was associated with adaptive changes in several other proteins involved in renal ammonia metabolism and with a small increase in urinary ammonia excretion (16). In contrast, basal ammonia excretion did not differ between Control and PT-GS-KO mice in the current study. Our previous study quantified basal ammonia excretion while mice were receiving routine mouse chow (16), whereas in the current study basal ammonia excretion was determined while mice were receiving semisynthetic chow with an equivalent, 20%, protein content. Thus, the minor differences in the two diets mice received during basal measurements may explain the differences in effects of PT-GS-KO on basal ammonia excretion.
In summary, proximal tubule GS expression has a critical role in decreasing renal ammonia excretion and hence the renal acid-base response to dietary protein restriction. GS affects a specific component of net acid excretion, that is, ammonia excretion, since titratable acid excretion does not change with proximal tubule GS deletion, and the effect is not dependent on urine pH. Thus, proximal tubule GS has an important role in renal ammonia metabolism under basal conditions, decreased expression is necessary for the response to metabolic acidosis, and increased expression is necessary for the response to dietary protein restriction. These findings demonstrate that proximal tubule GS has a critical role in renal ammonia metabolism.
GRANTS
Funding from the National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK-045788 and R01-DK-107798), the Department of Veterans Affairs (1I01BX000818), and the Gatorade Trust distributed by the University of Florida College of Medicine supported these studies.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.-W.L. and I.D.W. conceived and designed research; H.-W.L., G.O., and M.E.H. performed experiments; H.-W.L., G.O., M.E.H., J.W.V., and I.D.W. analyzed data; H.-W.L., J.W.V., and I.D.W. interpreted results of experiments; H.-W.L. and I.D.W. prepared figures; H.-W.L. drafted manuscript; H.-W.L., G.O., M.E.H., J.W.V., and I.D.W. edited and revised manuscript; H.-W.L., G.O., M.E.H., J.W.V., and I.D.W. approved final version of manuscript.
Footnotes
Ammonia exists in two molecular forms, NH3 and , which are in equilibrium with each other. In this report, we use the term ammonia to refer to the combination of both molecular forms. When referring to a specific molecular species, we specifically state either NH3 or .
REFERENCES
- 1.Bishop JM, Verlander JW, Lee HW, Nelson RD, Weiner AJ, Handlogten ME, Weiner ID. Role of the Rhesus glycoprotein, Rh B glycoprotein, in renal ammonia excretion. Am J Physiol Renal Physiol 299: F1065–F1077, 2010. doi: 10.1152/ajprenal.00277.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conjard A, Komaty O, Delage H, Boghossian M, Martin M, Ferrier B, Baverel G. Inhibition of glutamine synthetase in the mouse kidney: a novel mechanism of adaptation to metabolic acidosis. J Biol Chem 278: 38159–38166, 2003. doi: 10.1074/jbc.M302885200. [DOI] [PubMed] [Google Scholar]
- 3.de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 20: 2075–2084, 2009. doi: 10.1681/ASN.2008111205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franch HA, Mitch WE. Navigating between the Scylla and Charybdis of prescribing dietary protein for chronic kidney diseases. Annu Rev Nutr 29: 341–364, 2009. doi: 10.1146/annurev-nutr-080508-141051. [DOI] [PubMed] [Google Scholar]
- 5.Frassetto LA, Todd KM, Morris RC Jr, Sebastian A. Estimation of net endogenous noncarbonic acid production in humans from diet potassium and protein contents. Am J Clin Nutr 68: 576–583, 1998. [DOI] [PubMed] [Google Scholar]
- 6.Goraya N, Wesson DE. Does correction of metabolic acidosis slow chronic kidney disease progression? Curr Opin Nephrol Hypertens 22: 193–197, 2013. doi: 10.1097/MNH.0b013e32835dcbbe. [DOI] [PubMed] [Google Scholar]
- 7.Hamm LL, Simon EE. Roles and mechanisms of urinary buffer excretion. Am J Physiol 253: F595–F605, 1987. [DOI] [PubMed] [Google Scholar]
- 8.Handlogten ME, Osis G, Lee HW, Romero MF, Verlander JW, Weiner ID. NBCe1 expression is required for normal renal ammonia metabolism. Am J Physiol Renal Physiol 309: F658–F666, 2015. doi: 10.1152/ajprenal.00219.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Häussinger D, Lamers WH, Moorman AF. Hepatocyte heterogeneity in the metabolism of amino acids and ammonia. Enzyme 46: 72–93, 1992. [DOI] [PubMed] [Google Scholar]
- 10.Kim HY, Verlander JW, Bishop JM, Cain BD, Han KH, Igarashi P, Lee HW, Handlogten ME, Weiner ID. Basolateral expression of the ammonia transporter family member Rh C glycoprotein in the mouse kidney. Am J Physiol Renal Physiol 296: F543–F555, 2009. doi: 10.1152/ajprenal.90637.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovesdy CP, Anderson JE, Kalantar-Zadeh K. Association of serum bicarbonate levels with mortality in patients with non-dialysis-dependent CKD. Nephrol Dial Transplant 24: 1232–1237, 2009. doi: 10.1093/ndt/gfn633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kraut JA. Effect of metabolic acidosis on progression of chronic kidney disease. Am J Physiol Renal Physiol 300: F828–F829, 2011. doi: 10.1152/ajprenal.00074.2011. [DOI] [PubMed] [Google Scholar]
- 13.Kraut JA, Madias NE. Metabolic acidosis of CKD: an update. Am J Kidney Dis 67: 307–317, 2016. doi: 10.1053/j.ajkd.2015.08.028. [DOI] [PubMed] [Google Scholar]
- 14.Lee HW, Verlander JW, Bishop JM, Igarashi P, Handlogten ME, Weiner ID. Collecting duct-specific Rh C glycoprotein deletion alters basal and acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 296: F1364–F1375, 2009. doi: 10.1152/ajprenal.90667.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HW, Osis G, Handlogten ME, Guo H, Verlander JW, Weiner ID. Effect of dietary protein restriction on renal ammonia metabolism. Am J Physiol Renal Physiol 308: F1463–F1473, 2015. doi: 10.1152/ajprenal.00077.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee HW, Osis G, Handlogten ME, Lamers WH, Chaudhry FA, Verlander JW, Weiner ID. Proximal tubule-specific glutamine synthetase deletion alters basal and acidosis-stimulated ammonia metabolism. Am J Physiol Renal Physiol 310: F1229–F1242, 2016. doi: 10.1152/ajprenal.00547.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levey AS, Greene T, Beck GJ, Caggiula AW, Kusek JW, Hunsicker LG, Klahr S. Dietary protein restriction and the progression of chronic renal disease: what have all of the results of the MDRD study shown? Modification of Diet in Renal Disease Study group. J Am Soc Nephrol 10: 2426–2439, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Mak DO, Dang B, Weiner ID, Foskett JK, Westhoff CM. Characterization of ammonia transport by the kidney Rh glycoproteins RhBG and RhCG. Am J Physiol Renal Physiol 290: F297–F305, 2006. doi: 10.1152/ajprenal.00147.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitch WW. Dietary protein restriction and progressive renal insufficiency. Am J Kidney Dis 30: 297–298, 1997. doi: 10.1016/S0272-6386(97)90069-X. [DOI] [PubMed] [Google Scholar]
- 20.Moorman AF, Vermeulen JL, Charles R, Lamers WH. Localization of ammonia-metabolizing enzymes in human liver: ontogenesis of heterogeneity. Hepatology 9: 367–372, 1989. doi: 10.1002/hep.1840090305. [DOI] [PubMed] [Google Scholar]
- 21.Moranne O, Froissart M, Rossert J, Gauci C, Boffa JJ, Haymann JP, M’rad MB, Jacquot C, Houillier P, Stengel B, Fouqueray B; NephroTest Study Group . Timing of onset of CKD-related metabolic complications. J Am Soc Nephrol 20: 164–171, 2009. doi: 10.1681/ASN.2008020159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Wehbe E, Raina R, Simon JF, Srinivas TR, Jain A, Schreiber MJ Jr, Nally JV Jr. Serum bicarbonate and mortality in stage 3 and stage 4 chronic kidney disease. Clin J Am Soc Nephrol 6: 2395–2402, 2011. doi: 10.2215/CJN.03730411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel YM, Yun JS, Liu J, McGrane MM, Hanson RW. An analysis of regulatory elements in the phosphoenolpyruvate carboxykinase (GTP) gene which are responsible for its tissue-specific expression and metabolic control in transgenic mice. J Biol Chem 269: 5619–5628, 1994. [PubMed] [Google Scholar]
- 24.Qvartskhava N, Lang PA, Görg B, Pozdeev VI, Ortiz MP, Lang KS, Bidmon HJ, Lang E, Leibrock CB, Herebian D, Bode JG, Lang F, Häussinger D. Hyperammonemia in gene-targeted mice lacking functional hepatic glutamine synthetase. Proc Natl Acad Sci USA 112: 5521–5526, 2015. doi: 10.1073/pnas.1423968112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Racine-Samson L, Scoazec JY, D’Errico A, Fiorentino M, Christa L, Moreau A, Roda C, Grigioni WF, Feldman G. The metabolic organization of the adult human liver: a comparative study of normal, fibrotic, and cirrhotic liver tissue. Hepatology 24: 104–113, 1996. doi: 10.1002/hep.510240118. [DOI] [PubMed] [Google Scholar]
- 26.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res 66: 2576–2583, 2006. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raphael KL, Murphy RA, Shlipak MG, Satterfield S, Huston HK, Sebastian A, Sellmeyer DE, Patel KV, Newman AB, Sarnak MJ, Ix JH, Fried LF; Health ABC Study . Bicarbonate concentration, acid-base status, and mortality in the health, aging, and body composition study. Clin J Am Soc Nephrol 11: 308–316, 2016. doi: 10.2215/CJN.06200615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Remer T, Manz F. Estimation of the renal net acid excretion by adults consuming diets containing variable amounts of protein. Am J Clin Nutr 59: 1356–1361, 1994. [DOI] [PubMed] [Google Scholar]
- 29.Schöls L, Mecke D, Gebhardt R. Reestablishment of the heterogeneous distribution of hepatic glutamine synthetase during regeneration after CCl4-intoxication. Histochemistry 94: 49–54, 1990. doi: 10.1007/BF00266789. [DOI] [PubMed] [Google Scholar]
- 30.Trilok G, Draper HH. Sources of protein-induced endogenous acid production and excretion by human adults. Calcif Tissue Int 44: 335–338, 1989. doi: 10.1007/BF02556313. [DOI] [PubMed] [Google Scholar]
- 31.Verlander JW, Miller RT, Frank AE, Royaux IE, Kim YH, Weiner ID. Localization of the ammonium transporter proteins, Rh B glycoprotein and Rh C glycoprotein, in the mouse kidney. Am J Physiol Renal 284: F323–F337, 2003. doi: 10.1152/ajprenal.00050.2002. [DOI] [PubMed] [Google Scholar]
- 32.Verlander JW, Chu D, Lee HW, Handlogten ME, Weiner ID. Expression of glutamine synthetase in the mouse kidney: localization in multiple epithelial cell types and differential regulation by hypokalemia. Am J Physiol Renal Physiol 305: F701–F713, 2013. doi: 10.1152/ajprenal.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weiner ID. Roles of renal ammonia metabolism other than in acid-base homeostasis. Pediatr Nephrol, 2016. doi: 10.1007/s00467-016-3401-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiner ID, Hamm LL. Molecular mechanisms of renal ammonia transport. Annu Rev Physiol 69: 317–340, 2007. doi: 10.1146/annurev.physiol.69.040705.142215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weiner ID, Miller RT, Verlander JW. Localization of the ammonium transporters, Rh B glycoprotein and Rh C glycoprotein, in the mouse liver. Gastroenterology 124: 1432–1440, 2003. doi: 10.1016/S0016-5085(03)00277-4. [DOI] [PubMed] [Google Scholar]
- 36.Weiner ID, Mitch WE, Sands JM. Urea and ammonia metabolism and the control of renal nitrogen excretion. Clin J Am Soc Nephrol 10: 1444–1458, 2015. doi: 10.2215/CJN.10311013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weiner ID, Verlander JW. Role of NH3 and NH4+ transporters in renal acid-base transport. Am J Physiol Renal Physiol 300: F11–F23, 2011. doi: 10.1152/ajprenal.00554.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weiner ID, Verlander JW. Recent advances in understanding renal ammonia metabolism and transport. Curr Opin Nephrol Hypertens 25: 436–443, 2016. doi: 10.1097/MNH.0000000000000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiner ID, Verlander JW. Ammonia transporters and their role in acid-base balance. Physiol Rev 97: 465–494, 2017. doi: 10.1152/physrev.00011.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiner ID, Verlander JW. Renal ammonia metabolism and transport. Compr Physiol 3: 201–220, 2013. doi: 10.1002/cphy.c120010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weiner ID, Verlander JW. Ammonia transport in the kidney by Rhesus glycoproteins. Am J Physiol Renal Physiol 306: F1107–F1120, 2014. doi: 10.1152/ajprenal.00013.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue Y, Liao SF, Son KW, Greenwood SL, McBride BW, Boling JA, Matthews JC. Metabolic acidosis in sheep alters expression of renal and skeletal muscle amino acid enzymes and transporters. J Anim Sci 88: 707–717, 2010. doi: 10.2527/jas.2009-2101. [DOI] [PubMed] [Google Scholar]