

Abstract
Protozoans of the genus Plasmodium are the causative agents of malaria; they have a complex life cycle involving vertebrate and arthropod hosts and have three distinct invasive stages. Although the invasive stages probably invade cells using similar mechanisms, each stage has a different host cell specificity and utilizes different receptors to enter cells.
Of the many forms the malaria parasite takes during its complex life cycle, three stages must invade host cells; the ookinete, the sporozoite and the merozoite (see Fig. 1 for an overview of the life cycle of the malaria parasite). In this review, we will discuss sporozoite invasion of hepatocytes and erythrocyte invasion by merozoites (see Box 1 for a glossary of terms used). Unlike most viral and bacterial intracellular pathogens, which trigger their own uptake by the host cell, Plasmodium invasion of cells appears to be an active process in which the parasite utilizes an actin-based motility system to enter the target cell (Ref. 1 and reviewed in Ref. 2). Although the invasive stages of the parasite are morphologically and biochemically different from one another, they share a highly conserved structural organization and apical organelles called micronemes and rhoptries. The function of these conserved apical structures has not been fully elucidated but they are likely to be required for host cell invasion. Although these common structures may confer a similar overall pattern of target cell invasion, the details differ. Each invasive stage of the parasite has a different target cell specificity, probably governed by specific receptor–ligand interactions. Much of the work on the sporozoite stage of the parasite has centered around the challenge faced by the parasite in finding its target. In contrast, merozoites are usually released in close proximity to a potential host cell and most of the work in this field has focused on the process of invasion after initial attachment of the parasite to the erythrocyte.
Fig. 1.
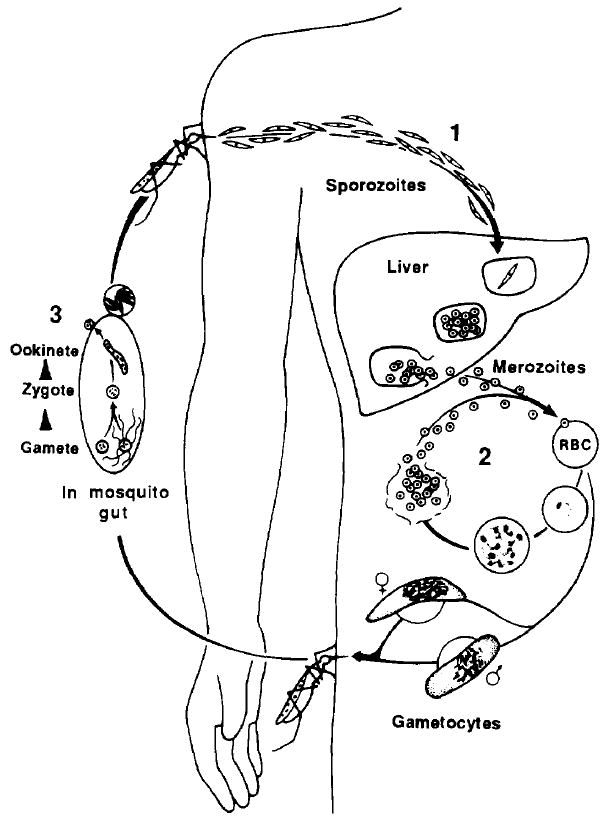
Life cycle of the malaria parasite. (1) Sporozoites are injected into a vertebrate host during the blood meal of a female anopheline mosquito and rapidly invade hepatocytes. One sporozoite can develop into 20 000 merozoites, which rupture from the hepatocyte, enter the bloodstream and invade erythrocytes. (2) In the asexual erythrocytic cycle, merozoites invade erythrocytes and mature from ring stages to schizonts within these cells in 48–72 h, the time varying with the species of malaria parasite. (3) Some erythrocytic stages differentiate into gametocytes, which are infective for mosquitoes. Fertilization occurs in the mosquito midgut, and within 24 h zygotes transform into ookinetes, which penetrate the midgut and form oocysts. Sporozoites rupture from these oocysts and invade the salivary glands of the mosquito from where they are injected into a vertebrate host.
Box 1. Glossary.
CS: Circumsporozoite protein.
Duffy blood group antigens: A group of antigens on the erythrocyte surface used for blood typing.
EBA-175: 175-kDa erythrocyte-binding antigen of Plasmodium falciparum.
EEF: Exoerythrocytic forms of Plasmodium.
GAGs: Glycosaminoglycan chains of HSPG.
HepG2 cells: A hepatoma cell line permissive for Plasmodium berghei sporozoite invasion and development.
HSPGs: Heparan sulfate proteoglycans.
Kupffer cells: Actively phagocytic cells that line the liver sinusoids.
MSP-I: Merozoite surface protein 1.
PVM: Parasitophorous vacuole membrane.
Region II: Receptor-binding domain of erythrocyte-binding proteins of merozoites.
Region II-plus: A cell-adhesive motif that occurs in both CS and SSP2/TRAP with homology to the type I thrombospondin repeats and which binds to sulfated glycoconjugates.
RESA: 155-kDa ring-infected erythrocyte surface antigen.
Space of Disse: The space between hepatocytes and the sinusoid lining that is continuous with the sinusoid lumen.
SSP2/TRAP: Sporozoite surface protein 2 or thrombospondin-related adhesion protein.
Hepatocyte invasion by sporozoites
Injection of two to ten Plasmodium sporozoites can initiate infection3,4. Although it is not known how many parasites are injected by a mosquito in the field, laboratory studies suggest that the median number of injected parasites during a blood meal is 15 (Ref. 5). In addition to being efficient, sporozoite invasion of hepatocytes is a rapid process, occurring minutes after intravenous injection6. The speed and efficiency of hepatocyte infection by sporozoites suggest that the parasites go to the liver directly.
The Kupffer cell hypothesi
The mechanism by which the parasites are arrested in the liver is not known. The endothelial cell lining of the liver sinusoids is unique in that it has open fenestrations, but the diameter of the fenestrae (0.1μm)7 is smaller than that of a sporozoite (1 μm). To circumvent this difficulty, it was thought that the parasites initially interact with Kupffer cells (which are found in the sinusoids) before invading hepatocytes.
Morphological studies performed to address the role of Kupffer cells are difficult to interpret because parasites are found in hepatocytes, sinusoids and Kupffer cells minutes after injection6,8. When Kupffer cells are eliminated using liposome-encapsulated dichloromethylene diphosphonate, there are 4–5-fold more exoerythrocytic forms (EEF) developing in the liver compared with controls, indicating that macrophages are not required for sporozoite invasion of hepatocytes9. Studies in vitro show that sporozoites can interact with, invade and sometimes kill macrophages1,10. It is possible, therefore, that sporozoites may interact with Kupffer cells in vivo, although the nature of this interaction requires further investigation.
An alternative theory is that sporozoites bind to and pass through hepatic endothelial cells11. Although there are no data to demonstrate the transendothelial passage of sporozoites, the electron microscopic techniques used to localize parasites in the liver may not be able to distinguish between the presence of parasites in liver sinusoids and the binding of parasites to hepatic endothelia. The use of new microscopic techniques, as well as increased knowledge of organ-specific endothelial cell markers, may help to answer this question.
Hepatocyte-adhesive domains on sporozoite surface proteins
Alternatively, sporozoites may invade hepatocytes directly, without initially interacting with Kupffer cells or endothelial cells. This hypothesis is supported by recent studies demonstrating the specific binding of two sporozoite surface proteins, the circumsporozoite protein (CS; reviewed in Ref. 12) and the sporozoite surface protein 2 (SSP2; Ref. 13) or thrombospondin-related adhesion protein (TRAP; Ref. 14), to the basolateral domain of hepatocytes15,16. Immunoelectron microscopy of liver sections incubated with either CS or SSP2/TRAP shows that both proteins bind to hepatocyte microvilli within the space of Disse, the portion of the cell exposed to the circulation. Neither protein binds to Kupffer ceils nor to endothelial cells. In addition, antibodies to either CS or SSP2/TRAP inhibit sporozoite invasion of HepG2 cells15,17,18, a hepatoma cell line permissive for Plasmodium berghei sporozoite invasion and development in vitro.
CS and SSP2/TRAP have only one region of identity: a known cell-adhesive motif with a high degree of homology to the type I thrombospondin repeats14. In CS, this motif is called region II-plus19,20; it is conserved in all CS and SSP2/TRAP proteins sequenced to date (Table 1) and it is required for binding of CS and SSP2/TRAP to hepatocytes15,17.
Table 1.
Region II-plus sequences of CS and homologous sequences of SSP2/TRAP from several species of malaria parasitesa
| Malaria parasite | Region II-plus sequencesb |
|---|---|
| Circumsporozoite protein | |
| Plasmodium falciparum | EWSPCSVTCGNGIQVRIK |
| Plasmodium vivax | EWTPCSVTCGVGVRVRRR |
| Plasmodium malariae | EWSPCSVTCGSGIRARRK |
| Plasmodium knowlesi | EWTPCSVTCGNGVRIRRK |
| Plasmodium cynomolgi | EWSPCSVTCGKRVRMRRK |
| Plasmodium brasilianum | EWSPCSVTCGKRVRMRRK |
| Plasmodium berghei | EWSPCSVTCGSGIRARRK |
| Plasmodium yoelii | EWSQCNVTCGSGIRVRKR |
| SSP2/TRAP | |
| Plasmodium falciparum | EWSPCSVTCGKGTRSRKR |
| Plasmodium yoelii | EWSECSTTCDEGRKIRRR |
Abbreviations: CS, circumsporozoite protein; SSP2, sporozoite surface protein 2; TRAP, thrombospondin-related adhesion protein.
The highly conserved cysteines and the downstream basic residues that are essential for binding to heparan sulfate proteoglycans are marked in bold.
Initial studies have shown that CS and SSP2FFRAP bind to sulfated glycoconjugates in a region II-plus- dependent fashion17,21. Subsequent experiments have demonstrated that CS binds to heparan sulfate proteoglycans (HSPGs)22. Although the identity of the cell-surface sulfated glycoconjugate that binds to SSP2/TRAP is not known, it may also be an HSPG because SSP2FFRAP binding to liver sections is eliminated after treatment with heparitinase16.
Structural studies on the parasite ligand have shown that the downstream, positively charged residues, as well as the interspersed hydrophobic amino acids of region II-plus, are required for binding to HSPGs (Ref. 20). In addition, only multimers of CS, or of peptides representing region II-plus, bind in a stable fashion to the hepatocyte receptors. It is likely that the lysines and arginines of region II-plus form ionic bonds with the negatively charged sulfate molecules of the HSPG glycosaminoglycan chains (GAGs).
It is not clear why the parasite should have two surface proteins with the same cell-adhesive motif. Perhaps the fine specificity of the receptor for each protein differs because heparan sulfate consists of repeating disaccharides that undergo secondary modifications, giving rise to an enormous amount of structural diversity throughout the length of each GAG chain. CS and SSP2/TRAP, therefore, may have different binding specificities based on conservative changes in region II-plus and/or amino acid residues in neighboring regions. Alternatively, CS and SSP2/TRAP may function at different stages of hepatocyte invasion. A different functional role for each protein is suggested by their different patterns of expression on the surface of the parasite: CS forms a dense coat whereas SSP2FFRAP gives a punctate staining pattern by immunofluorescence and immunoelectron microscopy18,23.
A role for CS binding to hepatic HSPGs in vivo
The density and structure of CS on the parasite surface make it an ideal ligand for trapping the parasite in the liver. When radiolabeled CS is intravenously injected into mice, it is rapidly cleared from the blood circulation and found in the liver24. Competition experiments with the physiological ligands for hepatic HSPGs, namely lipoprotein remnant particles and lactoferrin (reviewed in Ref. 25), show that CS clearance is delayed in mice preinjected with lactoferrin and in mice with high circulating levels of remnant lipoproteins26.
To determine whether or not sporozoites are captured in the liver by the same mechanism as CS, mice with high circulating levels of remnant lipoproteins were used for experiments in vivo. Forty hours after injection with sporozoites, mice with high remnant lipoprotein levels had 8–10-fold fewer parasites developing in the liver than control mice26, suggesting that the binding between the abundant hepatocyte HSPGs and the dense CS coat of the parasite is critical for the arrest of the parasite in the liver.
As mentioned earlier, one confounding issue is that the hepatocyte microvilli are within the space of Disse behind the fenestrated endothelial ceils. Perhaps the long GAG chains of the HSPGs protrude through the endothelial fenestrae into the sinusoids and capture circulating particles, such as lipoprotein remnants and Plasmodium sporozoites (Fig. 2). Lyon and Gallagher have shown that the bulk of hepatic HSPG sulfation is along the distal third of the molecule, the part that would be in the sinusoidal lumen if this hypothesis is correct27. An alternative possibility is that the fenestrae are not rigid structures and their diameter can increase during blood circulation, allowing for direct contact between sporozoites and hepatocyte microvilli.
Flg. 2.
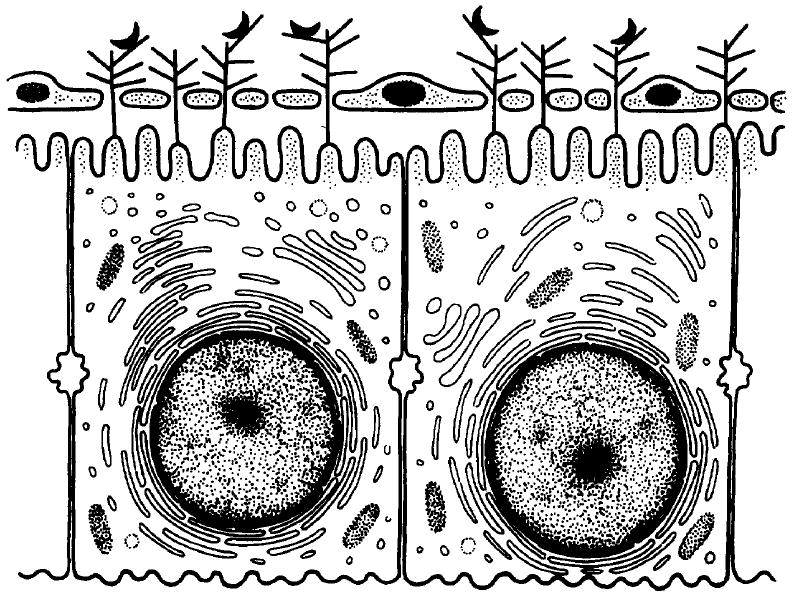
Diagrammatic representation of direct interaction between sporozoites and hepatocytes. Heparan sulfate proteoglycans (HSPGs) are shown as branched structures on the hepatocyte membrane, protruding through the open fenestrae of the endothelial cells into the sinusoidal lumen. Sporozoites, shown as black half-moon structures, are captured by these HSPGs and then invade hepatocytes.
Other sporozoite surface proteins
Sporozoite arrest in the liver is the first step of hepatocyte invasion; once in the liver, sporozoites must enter hepatocytes and develop into EEF. In contrast to what is known about merozoite invasion of red cells, little is known about how sporozoites invade hepatocytes. Thus far, only one other sporozoite surface protein has been cloned and sequenced: the 78-kDa sporozoite threonine- and asparagine-rich protein STARP (Ref. 28). STARP has a high degree of structural conservation in different Plasmodium falciparum isolates, as well as a high degree of homology with the gene from the closely related Plasmodium reichenowi, suggesting that it is important in the life cycle of the parasite29. Whether this protein is involved in sporozoite invasion of hepatocytes and, if so, what role it plays is not yet known.
Erythrocyte invasion by Plasmodium
When merozoites are released from erythrocytes they are in close proximity to their target cells: uninfected erythrocytes. If the target erythrocyte is in direct contact with the red cell from which the merozoite emerges, the parasite may only be extracellular briefly. Finding host cells is more difficult for Plasmodium vivax merozoites, which primarily, if not exclusively, invade reticulocytes30-32. However, even for P. vivax, the extra-cellular journey of a merozoite is much less complex than that of a sporozoite, which must travel from the site of inoculation in the skin to the hepatocyte.
Erythrocyte invasion by malaria parasites is a multistep process that involves attachment and reorientation, apical end binding and junction formation, parasitophorous vacuole membrane (PVM) formation and internalization (reviewed in Ref. 2) (Fig. 3). The apical organelles (micronemes and rhoptries), as well as other organdies called dense granules, seem to be involved in several of these steps. How this process is controlled is not understood, but it may occur as a defined sequence, in which one event occurs as a consequence of the previous step, or as a cascade catalyzed by a single trigger.
Fig. 3.
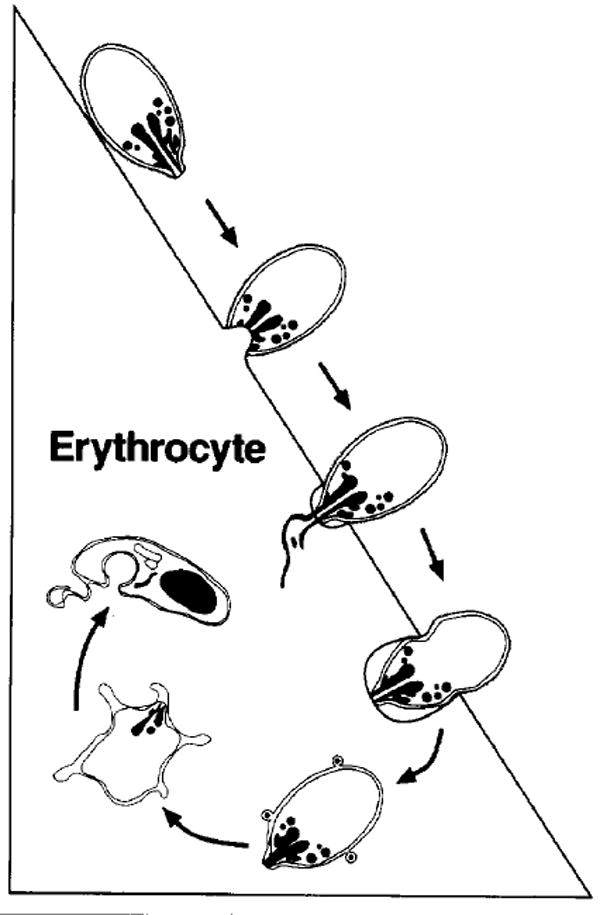
Schematic representation of the major morphological events of merozoite invasion. On top, the merozoite is depicted in the attachment and orientation phase, followed by binding, junction formation and rhoptry discharge. The merozoite is then shown entering the parasitophorous vacuole as the junction between the merozoite and the erythrocyte moves towards the posterior end of the merozoite. The parasite is then completely intracellular and flattens with its cytoplasm remaining thick only at the edges, giving the appearance of a ‘ring’.
Attachment and orientation
After release from ruptured schizonts, merozoites come into contact with erythrocytes and reorient so that ligands expressed at the apical end of the parasite are able to interact with erythrocyte receptors. Reorientation appears to involve attachment, dissociation and reattachment as the merozoite appositions itself in the correct orientation on the erythrocyte membrane (reviewed in Ref. 2). The interaction between the erythrocyte membrane and the merozoite surface must be of low affinity because the attachment is reversible. The specific receptors and ligands that mediate this interaction have not been identified; however, it seems likely that the re-orientation process involves an abundant surface protein of merozoites, and one possible candidate is merozoite surface protein 1 (MSP-1)33,34. Full-length MSP-1 from supernatants of P. falciparum erythrocytic stage cultures binds specifically to the sialic acid residues of glycophorins on erythrocytes, but this binding is of low affinity35. However, others have found that MSP-1 is processed at about the time of merozoite release from erythrocytes and a 19-kDa cysteine-rich glycosyl-phosphatidylinositol (GPI)-linked glycosylated fragment is left on the merozoite surface as it enters the erythrocyte36. How then does full- length MSP-1 participate in invasion? Possibly, sequential binding and proteolysis produce a gradient of contact that facilitates directionally oriented movement towards the apical pole37. Although it is not clear if MSP-1 is involved in attachment and orientation of the merozoite to the erythrocyte, antibodies raised against this protein inhibit merozoite invasion of erythrocytes36 and confer protection38,39, suggesting that MSP-1 participates in the invasion process.
Apical end binding and junction formation
To date, all of the ligands involved in apical end binding of the merozoite to the erythrocyte have been localized to the micronemes. These ligands are the 175-kDa erythrocyte-binding antigen (EBA-175) of P. falciparum40-42 and the Duffy binding proteins of P. vivax43,44 and Plasmodium knowlesi45,46. Another group of proteins, the reticulocyte-binding proteins of P. vivax47, is seen at the apical end of the merozoite but its precise localization in the merozoite has not yet been described.
Interestingly, the CS and SSP2/TRAP proteins of sporozoites, which both bind to hepatocytes, have been localized to the micronemes of sporozoites18,48,49. Although there are no obvious similarities between the gene structure of the merozoite and sporozoite ligands, the co-localization of these binding proteins in the micronemes suggests a common subcellular trafficking and sequestration route. As both CS and SSP2/TRAP are found on the surface of the sporozoite, the micronemes may function as a constitutive intermediate compartment in the trafficking of surface proteins. However, neither EBA-175 nor the Duffy binding proteins have been visualized on the surface of merozoites. Visualizing these ligands on the surface of P. falciparum and P. vivax merozoites may not be easy because trafficking to the surface may depend on the viability of merozoites and the isolation of viable merozoites of these species is not yet feasible. However, the Duffy binding protein of P. knowlesi, whose merozoites can be isolated in a viable state, has not been visualized on the merozoite surface. Another possibility is that the micronemes function as a regulated secretory granule, releasing their contents at a specific time during invasion. Both CS and SSP2/TRAP are found on the surface of sporozoites before they contact target cells, in contrast to EBA-175 and the Duffy binding proteins, which are not found on the merozoite surface and may only be secreted after contact with target cells. Perhaps the signals regulating protein secretion by the micronemes vary depending on the stage of the parasite.
The binding of the merozoite ligand to the erythrocyte receptor leads to the formation of a tight junction that can be visualized as an electron-dense zone beneath the erythrocyte membrane (Ref. 50 and reviewed in Ref. 2). The formation of a tight junction appears to be the committed step in the invasion process. The tight junction starts as a localized patch and transforms into a circumferential interaction between the apical end of the merozoite and the invaginated erythrocyte membrane. This moves as a band around the merozoite and fuses as the posterior end of the merozoite enters the erythrocyte, closing the orifice not unlike an iris diaphragm. The merozoite is now within the erythrocyte surrounded by the PVM (Fig. 3).
Parasitophorous vacuole formation
As mature erythrocytes are incapable of receptor- mediated endocytosis51, the driving force behind PVM formation must be provided by the merozoites. During PVM formation, the contents of the apical organelles are released onto the erythrocyte membrane. Rhoptries are a pair of electron-dense, teardrop-shaped organelles connected by ducts leading to the apical end of the parasite. Several rhoptry proteins have been identified and characterized52-55, and one of these, a 110-kDa protein, is part of a rhoptry protein complex that is secreted into the erythrocyte membrane during invasion56. However, it is not known whether the PVM is formed as a result of the insertion of apical organelle proteins into the erythrocyte membrane, which could lead to its deformation, or if the membrane is deformed by the active invasion of the merozoite, which then leads to the release of rhoptry contents.
The protein composition of the PVM is distinct from that of the erythrocyte membrane. Rhoptry proteins are inserted into the PVM (Ref. 56) and several major erythrocyte proteins are excluded from the PVM (Refs 57,58). However, in experiments using fluorescent lipophilic tracers, it has been found that the lipid composition of the PVM is indistinguishable from that of the normal erythrocyte membrane2,59.
After rhoptry discharge, dense granules from the apical and lateral surfaces of the merozoite discharge their contents into the PVM (Refs 50,60), and the trailing end of the invaginated erythrocyte membrane fuses. Dense granules are spherical organelles that are larger than a microneme but smaller than the rhoptry60. A parasite protein that has been localized to the dense granules is the 155-kDa ring-infected erythrocyte surface antigen (RESA). RESA is released from the dense granules49,61 and then, by as yet undefined mechanisms, is transported across the PVM and binds spectrin in the erythrocyte cytoskeleton62,63.
Parasite proteases are thought to play a role at various steps of the invasion process64-66, perhaps by local disruption of the erythrocyte membrane. Elegant studies by Braun Breton and co-workers have shown that in Plasmodium chabaudi, the parasite p68 serine protease cleaves band 3 protein in the erythrocyte membrane, leading to successful invasion67. Cleavage of band 3 may modify the erythrocyte cytoskeleton so that it can deform during merozoite entry.
Receptor and ligands
The important historical observations and data that have implicated particular proteins as receptors and ligands for invasion will not be described here. The initial observation that P. knowlesi and P. vivax do not invade Duffy-negative erythrocytes has led to the identification of the Duffy blood group antigen as a putative invasion receptor for these parasites (reviewed in Ref. 2). In the case of P. falciparum, invasion studies with mutant and enzymatically treated erythrocytes in vitro have demonstrated that the sialic acid residues of glycophorins are receptors for invasion68-70.
More recently, parasite ligands for the Duffy blood group antigens and glycophorin A have been discovered using an erythrocyte-binding assay40,45 and supernatants from labeled parasite cultures. The 140-kDa and 135-kDa Duffy binding proteins of P. vivax43 and P. knowlesi45, respectively, and EBA-175 of P. falciparum40 were identified in this way. There are striking similarities between the gene structures of the Duffy binding proteins and EBA-175 and they have been proposed to be members of an erythrocyte-binding family of proteins71. They all have three exons encoding transmembrane and cytoplasmic domains and they share 5′ and 3′ cysteine-rich domains that contain conserved cysteine motifs.
The 5′ cysteine-rich region of these erythrocyte- binding proteins, designated region II, has been identified as the receptor-binding domain42,72. Within region II, the cysteines and some aromatic residues are strictly conserved and probably constitute the erythrocyte-binding motif, while the intervening sequences determine the specificity of the receptor binding71. In the case of EBA-175, binding is specific for sialic acids linked 2,3 on O-linked tetrasaccharides73 and requires both sialic acids and the protein backbone of glycophorin A for binding to erythrocytes42.
Conservation of functional domains in evolutionarily distant species was previously described for the CS protein, in which the only regions conserved among species were those shown to bind hepatocytes15,74. As the Duffy binding proteins of P. knowlesi appear to be required for junction formation during invasion, it is likely that erythrocyte-binding proteins in other Plasmodium species have the same role.
The specificities of the erythrocyte-binding proteins do not account for all of the host cell preferences observed in vivo. P. vivax merozoites preferentially invade reticulocytes30,31,47, even though the Duffy antigen is expressed on both reticulocytes and mature erythrocytes. The reasons for this preferential invasion of reticulocytes is not understood; however, two erythrocyte-binding proteins of 250 kDa and 280 kDa that preferentially bind reticulocytes have recently been identified 47. These proteins are expressed at the invasive, apical end of merozoites and bind both Duffy-positive and Duffy- negative erythrocytes. Whether these proteins are the parasite ligands responsible for the preferential invasion of reticulocytes remains to be determined.
There are undoubtedly many more, as yet unidentified, parasite proteins involved in the invasion process. In addition, there is evidence that these parasites use alternative pathways to invade erythrocytes. For example, the P. vivax Duffy binding protein does not bind to Saimiri monkey erythrocytes, even though these erythrocytes are invaded by P. vivax merozoites43. P. falciparum can also use alternative pathways for entering erythrocytes. All strains of P. falciparum studied to date primarily use sialic acid residues on glycophorin A as receptors because neuraminidase treatment of erythrocytes greatly affects the invasion efficiencies of these strains in vitro68. However, certain strains of P. falciparum can be selected to invade and develop in neuraminidase-treated erythrocytes vs and it has been observed that some strains of P. falciparum (Dd2 and FCR3/A2) can invade after trypsin treatment of erythrocytes76; trypsin cleaves glycophorin A. These alternative pathways of invasion possibly indicate a redundancy required to give a selective advantage to parasites for survival in a polymorphic human population.
Conclusions
Invasion of host cells by malaria parasites is a tightly controlled multistep process that occurs over a short period of time in specific host compartments (i.e. blood-stream, liver sinusoids and mosquito midgut). In the case of erythrocyte invasion by merozoites, this process has been visualized and involves a defined sequence of events, which includes recognition and attachment, reorientation, junction formation, PVM formation and internalization. Although sporozoite entry into hepatocytes has not been visualized in this detail, the process may involve some of the same steps77.
Microscopic observations, as well as biochemical studies of merozoite entry into host cells, have shown that the micronemes contain proteins required for invasion, and that the contents of the rhoptries and dense granules are released during invasion. However, there are no known apical organelle proteins that are identical, or even similar, in different stages of the same Plasmodium species. Perhaps this strategy enables the parasite to better avoid the immune response of the host. Despite these differences, within a given stage of the parasite, but among evolutionarily distant species, there is conservation of protein domains that have been shown to function during cell invasion, for example, region II-plus of CS and SSP2/TRAP and region II of the erythrocyte-binding proteins of P. vivax , P. knowlesi and P. falciparum. Identifying the receptors and ligands involved in parasite invasion is important; however, experiments aimed at understanding the overall mechanisms of invasion are also of interest and perhaps of wider significance. It is hoped that future work will elucidate some of the common mechanisms utilized by apicomplexan parasites for entry into host cells.
Questions for future research.
What are the signaling events that trigger release of the contents of apical organelles during invasion?
Once in the circulation of the vertebrate host, the sporozoite is probably cleared by hepatocytes via binding of region II-plus motifs in CS (and perhaps SSP2/TRAP) to hepatocyte HSPGs. However, sporozoites are normally injected intradermally, but not intravenously, by mosquitoes. How do the sporozoites get from the site of injection into the circulation?
Although blood smears from malarious patients and Plasmodium falciparum cultures in vitro frequently reveal several multiply infected erythrocytes, different stages of the parasite are never seen in the same erythrocytes. This suggests that a merozoite can not invade an already infected erythrocyte. In what way has the intracellular parasite changed its host erythrocyte, such that a merozoite can no longer enter?
Sporozoites and merozoites have surface molecules with transmembrane domains and GPI-anchored surface molecules. Do these differently anchored proteins on the surface of the parasite have distinct roles in invasion and motility?
In what way is the membrane of the parasitophorous vacuole different from the host cell membrane, and how do these differences ensure the survival of the intracellular parasite?
Acknowledgments
P.S. is supported by an NIH Physician Scientist Award (Kll AI-01175).
Contributor Information
Photini Sinnis, Dept of Medical and Molecular Parasitology, New York University Medical Center, New York, NY 10016, USA.
B. Kim Lee Sim, EntreMed, 9610 Medical Center Drive, Rockville, MD 20850, USA.
References
- 1.Vanderberg J, Chew S, Stewart MJ. J Protozool. 1990;37:528–536. doi: 10.1111/j.1550-7408.1990.tb01260.x. [DOI] [PubMed] [Google Scholar]
- 2.Ward G, Chitnis CE, Miller LH. In: Strategtes for Intracellular Survival of Microbes. Russell D, editor. Saunders; 1994. pp. 155–190. [Google Scholar]
- 3.Khusmith S, Sedegah M, Hoffman SL. Infect Immun. 1994;62:2979–2983. doi: 10.1128/iai.62.7.2979-2983.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ungureanu E, et al. Trans R Soc Trop Med Hyg. 1976;70:482–483. doi: 10.1016/0035-9203(76)90133-4. [DOI] [PubMed] [Google Scholar]
- 5.Ponnudurai T, et al. Trans R Soc Trop Med Hyg. 1991;85:175–180. doi: 10.1016/0035-9203(91)90012-n. [DOI] [PubMed] [Google Scholar]
- 6.Shin SCJ, Vanderberg JP, Terzakis JA. J Protozool. 1982;29:448–454. doi: 10.1111/j.1550-7408.1982.tb05431.x. [DOI] [PubMed] [Google Scholar]
- 7.Wisse E, et al. Hepatology. 1985;5:683–692. doi: 10.1002/hep.1840050427. [DOI] [PubMed] [Google Scholar]
- 8.Meis JFGM, et al. Parasitology. 1983;86:231–242. doi: 10.1017/s003118200005040x. [DOI] [PubMed] [Google Scholar]
- 9.Vreden SGS, et al. Infect lmmun. 1993;61:1936–1939. [Google Scholar]
- 10.Seguin MC, Ballou WR, Nacy CA. Immunol. 1989;143:1716–1722. [PubMed] [Google Scholar]
- 11.Vanderberg JP. Parasitol Today. 1995;11:24. doi: 10.1016/0169-4758(91)90213-8. [DOI] [PubMed] [Google Scholar]
- 12.Nussenzweig V, Nussenzweig RS. In: Advances in Immunology. Dixon FJ, editor. Academic Press; 1989. pp. 283–334. [DOI] [PubMed] [Google Scholar]
- 13.Hedstrom RC, et al. Bull WHO. 1990;68:152–157. [PMC free article] [PubMed] [Google Scholar]
- 14.Robson KJH, et al. Nature. 1988;335:79–82. doi: 10.1038/335079a0. [DOI] [PubMed] [Google Scholar]
- 15.Cerami C, et al. Cell. 1992;70:1021–1035. doi: 10.1016/0092-8674(92)90251-7. [DOI] [PubMed] [Google Scholar]
- 16.Robson KJH, et al. EMBO J. 1995;14:3883–3894. doi: 10.1002/j.1460-2075.1995.tb00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller HM, et al. EMBO J. 1993;12:2881–2889. doi: 10.1002/j.1460-2075.1993.tb05950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers WO, et al. Proc Natl Acad Sci U S A. 1992;89:9176–9180. doi: 10.1073/pnas.89.19.9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dame JB, et al. Science. 1984;225:593–599. doi: 10.1126/science.6204383. [DOI] [PubMed] [Google Scholar]
- 20.Sinnis P, et al. J Exp Med. 1994;180:297–306. doi: 10.1084/jem.180.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pancake SJ, et al. J Cell Biol. 1992;6:1351–1357. doi: 10.1083/jcb.117.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frevert U, et al. J Exp Med. 1993;177:1287–1298. doi: 10.1084/jem.177.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cowan G, et al. Lancet. 1992;339:1412–1413. doi: 10.1016/0140-6736(92)91229-2. [DOI] [PubMed] [Google Scholar]
- 24.Cerami C, et al. J Exp Med. 1994;179:695–701. doi: 10.1084/jem.179.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahley RW, et al. In: Biology of α2-Macroglobulin, its Receptor, and Related Proteins. Borth W, et al., editors. Vol. 737. New York Academy of Sciences; 1994. pp. 39–52. [Google Scholar]
- 26.Sinnis P, et al. J Exp Med. 1996;184:945–954. doi: 10.1084/jem.184.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyon M, Deakin JA, Gallagher JT. J Biol Chem. 1994;269:11208–11215. [PubMed] [Google Scholar]
- 28.Fidock DA, et al. Mol Biochem Parasitol. 1994;64:219–232. doi: 10.1016/0166-6851(94)00012-3. [DOI] [PubMed] [Google Scholar]
- 29.Fidock DA, et al. Mol Biochem Parasitol. 1994;67:255–267. doi: 10.1016/0166-6851(94)00138-3. [DOI] [PubMed] [Google Scholar]
- 30.Kitchen SF. Am J Trop Med Hyg. 1938;18:347–359. [Google Scholar]
- 31.Mons B, et al. Exp Parasitol. 1988;66:183–188. doi: 10.1016/0014-4894(88)90089-6. [DOI] [PubMed] [Google Scholar]
- 32.Barnwell JW, et al. J Exp Med. 1989;169:162–167. doi: 10.1084/jem.169.5.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holder AA, et al. Mol Biochem Parasitol. 1985;14:293–303. doi: 10.1016/0166-6851(85)90057-x. [DOI] [PubMed] [Google Scholar]
- 34.Miller LH, et al. Mol Biochem Parasitol. 1993;59:1–14. doi: 10.1016/0166-6851(93)90002-f. [DOI] [PubMed] [Google Scholar]
- 35.Perkins ME, Rocco LJ. J Immunol. 1988;141:3190–3196. [PubMed] [Google Scholar]
- 36.Blackman MJ, et al. Exp Med. 1990;172:379–382. doi: 10.1084/jem.172.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barnwell JW, Galinski MR. Res Immunol. 1991;142:666–671. doi: 10.1016/0923-2494(91)90147-b. [DOI] [PubMed] [Google Scholar]
- 38.Siddiqui WA, et al. Proc Natl Acad Sci U S A. 1987;84:3014–3018. doi: 10.1073/pnas.84.9.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Etlinger HM, et al. Infect Immun. 1991;59:3498–3503. doi: 10.1128/iai.59.10.3498-3503.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Camus D, Hadley TJ. Science. 1985;230:553–556. doi: 10.1126/science.3901257. [DOI] [PubMed] [Google Scholar]
- 41.Sim BKL, et al. J Cell Biol. 1990;111:1877–1884. doi: 10.1083/jcb.111.5.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sim BKL, et al. Science. 1994;264:1941–1944. [Google Scholar]
- 43.Werthheimer SP, Barnwell JW. Exp Parasitol. 1989;69:340–350. doi: 10.1016/0014-4894(89)90083-0. [DOI] [PubMed] [Google Scholar]
- 44.Fang X, et al. Mol Biochem ParasitoI. 1991;44:125–132. doi: 10.1016/0166-6851(91)90228-x. [DOI] [PubMed] [Google Scholar]
- 45.Haynes JD, et al. Exp Med. 1988;367:1873–1881. doi: 10.1084/jem.167.6.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams JH, et al. Cell. 1990;63:141–153. doi: 10.1016/0092-8674(90)90295-p. [DOI] [PubMed] [Google Scholar]
- 47.Galinski M, et al. Cell. 1992;69:1213–1226. doi: 10.1016/0092-8674(92)90642-p. [DOI] [PubMed] [Google Scholar]
- 48.Fine E, et al. Am J Trop Med Hyg. 1984;30:220–226. doi: 10.4269/ajtmh.1984.33.220. [DOI] [PubMed] [Google Scholar]
- 49.Aikawa M, et al. Bull WHO. 1990;68:165–171. [Google Scholar]
- 50.Bannister LH, et al. Parasitology. 1975;71:483–491. doi: 10.1017/s0031182000047247. [DOI] [PubMed] [Google Scholar]
- 51.Zweig S, Singer SJ. J Cell Biol. 1979;80:487–491. doi: 10.1083/jcb.80.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petersen MG, et al. Mol Cell Biol. 1989;89:3151–3154. doi: 10.1128/mcb.9.7.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ridley RG, et al. Mol Biochem Parasitol. 1990;41:125–134. doi: 10.1016/0166-6851(90)90103-s. [DOI] [PubMed] [Google Scholar]
- 54.Brown HJ, Coppel RL. Mol Biochem Parasitol. 1991;49:99–110. doi: 10.1016/0166-6851(91)90133-q. [DOI] [PubMed] [Google Scholar]
- 55.Saul A, et al. Mol Biochem Parasitol. 1992;50:139–150. doi: 10.1016/0166-6851(92)90251-e. [DOI] [PubMed] [Google Scholar]
- 56.Sam-Yellowe TY, Shio H, Perkins ME. J Cell Biol. 1988;106:1507–1513. doi: 10.1083/jcb.106.5.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Atkinson CT, et al. J Cell Biol. 1987;45:192–199. [Google Scholar]
- 58.Dluzewski AR, et al. J Cell Sci. 1989;92:691–699. doi: 10.1242/jcs.92.4.691. [DOI] [PubMed] [Google Scholar]
- 59.Ward GE, Miller LH, Dvorak JA. J Cell Sci. 1993;106:237–248. doi: 10.1242/jcs.106.1.237. [DOI] [PubMed] [Google Scholar]
- 60.Torii M, et al. Infect Immun. 1989;57:3230–3233. doi: 10.1128/iai.57.10.3230-3233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Culvenor JG, Day KP, Anders RF. Infect Immun. 1991;59:1183–1187. doi: 10.1128/iai.59.3.1183-1187.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Foley M, et al. Mol Biochem Parasitol. 1991;46:137–148. doi: 10.1016/0166-6851(91)90207-m. [DOI] [PubMed] [Google Scholar]
- 63.Ruangjirachuport W, et al. Exp Parasitol. 1991;73:62–72. doi: 10.1016/0014-4894(91)90008-k. [DOI] [PubMed] [Google Scholar]
- 64.Barale JC, et al. Res Immunol. 1991;142:672–681. doi: 10.1016/0923-2494(91)90148-c. [DOI] [PubMed] [Google Scholar]
- 65.Braun Breton C, Pereira da Silva LH. Parasitol Today. 1993;9:9296–9300. doi: 10.1016/0169-4758(93)90212-x. [DOI] [PubMed] [Google Scholar]
- 66.McKerrow JH, et al. Annu Rev Microbiol. 1993;47:821–853. doi: 10.1146/annurev.mi.47.100193.004133. [DOI] [PubMed] [Google Scholar]
- 67.Braun Breton C, et al. Proc Natl Acad 5ci U S A. 1992;89:9647–9651. doi: 10.1073/pnas.89.20.9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pasvol G. Philos Trans R Soc London. 1984;307:189–200. doi: 10.1098/rstb.1984.0119. [DOI] [PubMed] [Google Scholar]
- 69.Hadley TJ, Miller LH. In: Protein Blood Group Antigens of the Human Red Cell. Agree PC, Cartron JC, editors. Johns Hopkins University Press; 1992. pp. 228–245. [Google Scholar]
- 70.Haynes JD. In: Current Opinion in HaematoIogy. Adamson JW, editor. Current Science; 1993. pp. 79–89. [Google Scholar]
- 71.Adams JH, et al. Proc Natl Acad Sci U S A. 1992;89:7085–7089. doi: 10.1073/pnas.89.15.7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chitnis CE, Miller LH. J Exp Med. 1994;180:497–506. doi: 10.1084/jem.180.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Orlandi PA, et al. J Cell Biol. 1992;116:901–911. doi: 10.1083/jcb.116.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aley SB, et al. J Exp Med. 1986;164:1915–1921. doi: 10.1084/jem.164.6.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dolan SA, Miller LH, Wellems TE. J Clin Invest. 1990;86:618–624. doi: 10.1172/JCI114753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dolan SA, et al. Mol Biochem Parasitol. 1994;64:55–63. doi: 10.1016/0166-6851(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 77.Russell DG. Parasitology. 1983;87:199–209. doi: 10.1017/s0031182000052562. [DOI] [PubMed] [Google Scholar]