

This novel study systematically examined the power-generating capacity of cardiac myofilaments during the progression from hypertension to heart disease. Previously undiscovered changes in myofibrillar power output were found and were associated with alterations in myofilament proteins, providing potential new targets to exploit for improved ventricular pump function in heart failure.
Keywords: cardiac myocyte, force development, force velocity, power output, hypertensive heart failure, spontaneous hypertensive heart failure
Abstract
Heart failure arises, in part, from a constellation of changes in cardiac myocytes including remodeling, energetics, Ca2+ handling, and myofibrillar function. However, little is known about the changes in myofibrillar contractile properties during the progression from hypertension to decompensated heart failure. The aim of the present study was to provide a comprehensive assessment of myofibrillar functional properties from health to heart disease. A rodent model of uncontrolled hypertension was used to test the hypothesis that myocytes in compensated hearts exhibit increased force, higher rates of force development, faster loaded shortening, and greater power output; however, with progression to overt heart failure, we predicted marked depression in these contractile properties. We assessed contractile properties in skinned cardiac myocyte preparations from left ventricles of Wistar-Kyoto control rats and spontaneous hypertensive heart failure (SHHF) rats at ~3, ~12, and >20 mo of age to evaluate the time course of myofilament properties associated with normal aging processes compared with myofilaments from rats with a predisposition to heart failure. In control rats, the myofilament contractile properties were virtually unchanged throughout the aging process. Conversely, in SHHF rats, the rate of force development, loaded shortening velocity, and power all increased at ~12 mo and then significantly fell at the >20-mo time point, which coincided with a decrease in left ventricular fractional shortening. Furthermore, these changes occurred independent of changes in β-myosin heavy chain but were associated with depressed phosphorylation of myofibrillar proteins, and the fall in loaded shortening and peak power output corresponded with the onset of clinical signs of heart failure.
NEW & NOTEWORTHY This novel study systematically examined the power-generating capacity of cardiac myofilaments during the progression from hypertension to heart disease. Previously undiscovered changes in myofibrillar power output were found and were associated with alterations in myofilament proteins, providing potential new targets to exploit for improved ventricular pump function in heart failure.
heart failure is characterized by the inability of the contractile pump to adequately match blood supply with metabolic demands of the tissues. Heart failure is thought to initiate from a variety of changes in cardiac myocyte biology, including structural changes, disrupted energetics, altered Ca2+ handling, and modifications in myofibrillar function. The progression of heart failure typically involves a compensatory period whereby physiological neurohumoral signals increase hemodynamic loads that elevate ventricular filling pressures, which augment the inherent pumping capacity of the heart by stretching cardiac myocytes (i.e., myocytes/ventricles ascend up the Frank-Starling relationship to a new working set point). Unfortunately, upregulation of these physiological processes can lead to maladaptive changes that often spiral into decompensation; these maladaptive changes include inflammation, fluid retention, labored breathing, fibrosis, remodeling, diastolic dysfunction, and exacerbation of systolic dysfunction (3). Decompensation often results in dire clinical outcomes, as evident by the fact that there is a 5-yr mortality rate of ~50% in the reported ~6 million heart failure cases in the United States. The current clinical approach to heart failure primarily involves treatment of signs and symptoms, i.e., mitigating congestion and labored breathing by reducing both hemodynamic loads and ventricular wall stress with the combined use of diuretics, angiotensin-converting enzyme inhibitors, angiotensin/aldosterone receptor antagonists, β-adrenergic receptor blockade, and Ca2+ channel blockers. Although these treatments have improved survival outcomes and quality of life for patients with heart failure, all these treatments compromise ventricular contractile reserve and offer minimal hope for a healthy and active lifestyle. In fact, there are very few applicable therapies directed to prevent and/or reverse adverse changes in myocyte contractile properties, which are the convergent end point for numerous heart disease-related cellular signaling processes and thus an eventual cause of heart failure. One major barrier toward more precise therapy is the large knowledge gap of how myofibrillar function changes during the progression from healthy hearts to heart disease. With this in mind, we undertook a longitudinal study to determine myofibrillar functional properties during the progression from hypertension to decompensated heart failure.
For this study, we used the lean male spontaneously hypertensive heart failure (SHHF) rat model, which was selected for three primary reasons: 1) it provides a congenital, multifactorial model of hypertensive heart disease that transitions into dilated cardiomyopathy, which mimics a highly prevalent human condition (24); 2) the SHHF rat model has been well characterized with regard to hemodynamics, hypertrophy, inflammation, fibrosis, and function by echocardiography (13, 23, 24); and 3) the model exhibits distinct sex variations with regard to the onset of heart failure (23). We chose to investigate male rats in this study because the onset of overt heart failure is more rapid and tends to be more pronounced than in female rats (23, 24). A final factor for selecting this model is that our laboratory has extensive experience with contractile property measurements using rat skinned cardiac myocyte preparations (10, 17, 18, 25). The purpose of this study was to test the hypothesis that permeabilized cardiac myocyte preparations from ~12-mo-old SHHF rat hearts presenting with compensation and concentric remodeling will exhibit increased force, higher rate of force development, faster loaded shortening velocity, and greater power output; however, with progression to overt heart failure, we predicted a marked depression in all these contractile properties.
METHODS
Animals
Male Wistar-Kyoto (WK) control and SHHF rat strains were obtained from Charles River Laboratory (Wilmington, MA) at 6 wk of age and assigned an age end point of either ~3, ~12, or >20 mo for functional analysis. SHHF rats assigned to the >20-mo time point underwent intermittent echocardiography to monitor cardiac performance, and these rats were killed when fractional shortening was substantially depressed and/or exhibited overt signs and symptoms of congestive heart failure. The average age of >20-mo-old rats was 22.48 ± 0.80 mo of age for control WK rats and 21.79 ± 0.62 mo of age for SHHF rats (P = 0.504). All animals were housed pairwise with a 12:12-h light-dark cycle at room temperature of ~72°F and provided water and rat chow ad libitum. Animals were monitored daily for health, and all procedures involving animal use were performed according to the Animal Care and Use Committee of the University of Missouri.
In Vivo Assessment of Cardiovascular Function
Blood pressure measurements.
Nonanesthetized rat blood pressure was measured in the tail of the rats using the volume-pressure recording (VPR) sensor technology system from Kent Scientific (CODA 2 Animal System, Torrington, CT).
Echocardiography.
Transthoracic echocardiography was performed with a 12-MHz pediatric transducer using a GE Vivid I Ultrasound system to assess in vivo cardiac morphology and diastolic and systolic function under 0.5–1.0% inhaled isofluorane anesthesia as previously described (4).
Permeabilized cardiac myocyte functional measurements.
Rats were weighed and then anesthetized by 20% isofluorane in olive oil (vol/vol), and their hearts were quickly removed and placed in ice-cold relaxing solution (2 mM EGTA, 5 mM MgCl2, 4 mM ATP, 10 mM imidazole, and 100 mM KCl at pH 7.0 with the addition of Protease Inhibitor Cocktail Set 1 from Calbiochem, San Diego, CA). The heart was weighed, and the left ventricle was isolated and permeabilized; cardiac myocytes were prepared as previously described (25). Permeabilized cardiac myocyte preparations were attached by monofilament nylon suture between a force transducer and motor, which was then mounted on the stage of an inverted microscope (model IX-70, Olympus Instrument, Tokyo, Japan). Mechanical measurements were performed using a capacitance-gauge transducer plus a 10× amplifier (Aurora Scientific, Aurora, ON, Canada). Length changes were introduced using a direct current torque motor (model 308, Aurora Scientific) driven by voltage commands from a personal computer via a 12- or 16-bit digital-to-analog converter (AT-MIO-16E-1, National Instruments, Austin, TX). Force and length signals were digitized at 1 kHz and stored on a personal computer using LabView for Windows (National Instruments). Sarcomere length was monitored using an IonOptix SarcLen system (IonOptix, Milton, MA), which used a fast Fourier transform algorithm of the video image of the myocyte. After attachment, the relaxed, permeabilized cardiac myocyte preparation was adjusted to a sarcomere length of ~2.30 µm. Cardiac myocyte preparation force, rates of force development, and force-velocity and power-load measurements were made at 13 ± 1°C in maximal Ca2+ activating solution (7.0 mM EGTA, 1.0 mM free Mg2+, 20.0 mM imidazole, 4.0 mM MgATP, 14.5 mM creatinine phosphate, and a Ca2+ concentration of 10-4.5 M, pH 7.0). For force measurements, steady-state tension was allowed to develop, and the cell was rapidly slackened to determine total tension. The amount of active tension was calculated as the difference between total tension and passive tension, which was assessed by slackening the preparation in the relaxed state (pCa 9.0 solution). The rates of force development were obtained using a procedure previously described for skinned cardiac myocyte preparations (10, 15, 19). Force-velocity and power-load measurements were performed as previously described (25). The cardiac myocyte preparation was kept in maximal Ca2+-activating solution for 2–3 min, during which 10–15 force clamps were performed. If force fell below 70% of initial force during the force clamp cycle, data from that myocyte were discarded.
Biochemical Analysis of Ventricular Myocyte Preparations
The relative expression of α- and β-myosin heavy chain (MyHC) was determined in ventricular myocytes from each rat heart using SDS-PAGE, ultrasensitive silver staining, and visualization of the relative intensity for each MyHC band (11, 17). For Western blot analysis of myofibrillar proteins, samples containing ventricular myocyte preparations were resolved with 12% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were then blocked in 5% milk in PBS (pH 7.4) and incubated with primary antibodies overnight. Immunoreactivity was detected with peroxidase-labeled secondary antibodies and supersignal WestPico chemiluminescence (Pierce Chemical, Rockford, IL). Western blot assessment of cMyBP-C phosphorylation levels was carried out using a mixture of polyclonal antibodies specific to Ser273, Ser282, and Ser302. Phospho-specific and total cMyBP-C antibodies (clone C2-14) were a gift from Dr. Sakthivel Sadayappan. Western blot assessment of cardiac troponin I (cTnI) was carried out using Invitrogen phospho-Ser23 and phospho-Ser24 cardiac TnI antibody (Thermo Scientific, Waltham, MA), and total TnI was detected by monoclonal antibody to cTnI (MAB1691, EMD Millipore, Billerica, MA). For two-dimensional (2-D) electrophoresis (Kendrick Laboratories, Madison, WI), ventricular myocyte pellets were lysed in osmotic lysis buffer [100 mM Tris (pH 7.0), 50 mM MgCl2, 500 µg/ml RNase, 1,000 µg/ml DNase, ribonuclease A from bovine pancreas type IIIA, deoxyribonuclease I, type II from the bovine pancreas, and protease inhibitors] and then diluted 1:1 in SDS boiling buffer (per liter: 100.0 g glycerol, 50.0 ml 2-mercaptoethanol, 50.0 g SDS, and 62.5 mmol Tris; pH 6.8). Samples were pulled through a small-bore syringe, sonicated 5 min, and vortexed with washed glass beads. Samples were heated in a boiling water bath for 5 min, and protein concentration was then determined by BCA assay (Pierce Chemical). Samples were lyophilized and redissolved 1:1 in SDS boiling buffer-urea sample buffer. 2-D electrophoresis was performed using 2% pH 3–10 ampholyte isoelectric focusing, resulting in a gradient of ~pH 5–8.5 followed by SDS 10% acrylamide slab gel electrophoresis. Gels were dried between sheets of cellophane, and individual protein spots were computer analyzed for comparison between samples from each time point for both WK control and SHHF rats. Protein spots that were greater than twofold different were putatively identified in silico using ExPasy TagIdent NIH software incorporating the parameters of taxID Rattus, pI range of 0.05 and 1% molecular weight range.
Data and Statistical Analysis
Force redevelopment after a slack-restretch maneuver was fit by the following single-exponential equation: F = Fmax(1 − ), where F is tension at time t, Fmax is maximal tension, and ktr is the rate constant of force development. Myocyte length traces, force-velocity curves, and power-load curves were analyzed as previously described (25). Myocyte length and sarcomere length traces during loaded shortening were fit to the following single decaying exponential equation: L = Ae−kt + C, where L is cell length at time t, A and C are constants with dimensions of length, and k is the rate constant of shortening (kshortening). Velocity of shortening at any given time t was determined as the slope of the tangent to the fitted curve at that time point. In this study, velocities of shortening were calculated by extrapolation of the fitted curve to the onset of the force clamp (i.e., t = 0). Hyperbolic force-velocity curves were fit to the relative force-velocity data using the following Hill equation (14): (P + a)(V + b) = (Po + a)b, where P is force during shortening at velocity V, Po is the peak isometric force, and a and b are constants with dimensions of force and velocity, respectively. Power-load curves were obtained by multiplying force by velocity at each load on the force-velocity curve. Curve fitting was performed using a customized program written in Qbasic as well as commercial software (SigmaPlot, Systat, San Jose, CA).
Two-way ANOVA was used to compare main effects and interactions between age and rat strain on blood pressure, in vivo echocardiography measurements, organ morphology, and permeabilized cardiac myocyte preparation structure and functional dependent variables. For biochemical analysis, two-way ANOVA was used to compare main effects and interactions between age and strain of cMyBP-C and cTnI phosphorylation level at putative PKA sites from Western blots. A Student-Newman-Keuls test was used for all pairwise comparisons among the different groups. P < 0.05 was accepted as a statistically significant difference. N is the number of animals, and n is the sample number of observations. Values are expressed as means ± SE.
RESULTS
In Vivo Hemodynamic and Echocardiographic Analysis of Systolic and Diastolic Function
There was a significant age × strain interaction in both systolic and diastolic blood pressures. Specifically, in WK control rats, systolic blood pressure increased at ~12 and >20 mo of age compared with ~3 mo of age. In SHHF rats, both systolic and diastolic blood pressures were similar between ~3 and ~12 mo of age and then dropped significantly at >20 mo (Fig. 1A). Between rat strain comparison indicated that both systolic and diastolic blood pressures were markedly higher in SHHF rats than WK control rats at ~3 and ~12 mo of age before dropping to lower pressures compared with WK control rats at >20 mo of age (Fig. 1A). Echocardiography indicated that diastolic left ventricular free wall thickness was increased, but left ventricular internal diastolic diameter and fractional shortening were similar in SHHF rats at ~3 mo and ~12 mo of age compared with WK control rats, indicative of compensated concentric hypertrophic remodeling (Table 1). However, at >20 mo of age, SHHF rat left ventricles were dilated and exhibited depressed fractional shortening compared with all other groups (Table 1 and Fig. 1, B and C). The time course of in vivo hemodynamics and echocardiography was consistent with previous characterizations of the SHHF rat model (13, 20, 24). Furthermore, the decline in ventricular function assessed by echocardiography, in most cases, occurred in conjunction with overt signs of heart failure, including dyspnea, ascites, and edema.
Fig. 1.

Time course of in vivo hemodynamic, functional properties and cardiac hypertrophy from Wistar-Kyoto (WK) control and spontaneous hypertensive heart failure (SHHF) rats. A: both systolic (closed symbols and solid lines) and diastolic (shaded symbols and dashed lines) blood pressures were significantly different in SHHF rats compared with age-matched control rats at all time points. B: representative in vivo M-mode echocardiography of WK control and SHHF rats at ~12 and >20 mo of age. LVAWs, left ventricular (LV) systolic anterior wall thickness; LVAWd, LV diastolic anterior wall thickness; LVPWs, LV systolic posterior wall thickness; LVPWd, LV diastolic posterior wall thickness; LVIDs, LV systolic internal diameter; LVIDd, LV diastolic internal diameter. C: LVIDd was significantly increased in >20-mo-old SHHF rats. D: the heart weight-to-tibia length ratio was significantly increased at >20 mo of age in both WK control and SHHF rats compared with the other time points, and the heart weight-to-tibia length ratio was significantly increased in >20-mo-old SHHF rats compared with WK control rats. n values for each group are shown in Table 1 or 2. *Different from ~12 and >20 mo; #different from ~3 and ~12 mo; †different from 3 and >20 mo; §different between age-matched WK control and SHHF groups.
Table 1.
In vivo hemodynamic and echocardiography analysis of 3-, 12-, and >20-mo-old WK control and SHHF rats
| WK Control, 3 mo old | SHHF, 3 mo old | WK Control, 12 mo old | SHHF, 12 mo old | WK Control, >20 mo old | SHHF, >20 mo old | |
|---|---|---|---|---|---|---|
| Tail-cuff blood pressure | ||||||
| n | 13 | 17 | 27 | 27 | 25 | 18 |
| Systolic bloodp pressure, mmHg | 133.0 ± 6.0b | 198.0 ± 6.0a | 155.0 ± 4.0d | 207.0 ± 4.0a | 169.0 ± 5.0c | 87.0 ± 5.0a,c |
| Diastolic blood pressure, mmHg | 93.0 ± 5.0 | 144.0 ± 5.0a | 101.0 ± 3.0 | 159.0 ± 6.0a | 107.0 ± 6.0 | 67.0 ± 2.0a,c |
| Systolic function | ||||||
| n | 4 | 4 | 4 | 4 | 4 | 4 |
| Heart rate, beats/min | 325.0 ± 9.0 | 344.0 ± 18.0 | 304.0 ± 10.0 | 344.0 ± 10.0a | 303.0 ± 6.0 | 279.0 ± 23.0c |
| Fractional shortening, % | 51.0 ± 3.0 | 45.0 ± 3.0 | 48.0 ± 3.0 | 49.0 ± 4.0 | 53.0 ± 3.0 | 22.0 ± 3.0a,c |
| LV stroke volume, µl | 266.0 ± 41.0b | 378.0 ± 50.0 | 524.0 ± 40.0 | 529.0 ± 48.0 | 448.0 ± 51.0 | 389.0 ± 53.0e |
| LV systolic anterior wall thickness, mm | 3.3 ± 0.2 | 3.9 ± 0.2 | 3.4 ± 0.1 | 4.3 ± 0.2a | 4.2 ± 0.2 | 4.1 ± 0.5 |
| LV systolic posterior wall thickness, mm | 2.8 ± 0.2 | 3.2 ± 0.1 | 2.9 ± 0.2 | 3.4 ± 0.3 | 2.7 ± 0.2 | 2.3 ± 0.1c |
| LV internal systolic diameter, mm | 3.3 ± 0.3 | 3.6 ± 0.2 | 3.7 ± 0.3 | 3.3 ± 0.2 | 3.4 ± 0.3 | 7.1 ± 0.3a,c |
| Diastolic function | ||||||
| n | 4 | 4 | 4 | 4 | 4 | 4 |
| LV diastolic anterior wall thickness, mm | 2.0 ± 0.1 | 2.7 ± 0.1a | 2.1 ± 0.1 | 3.1 ± 0.1a | 2.7 ± 0.2 | 2.9 ± 0.3 |
| LV diastolic posterior wall thickness, mm | 1.9 ± 0.2 | 2.3 ± 0.2 | 1.8 ± 0.2 | 2.6 ± 0.2a | 1.7 ± 0.2 | 1.8 ± 0.1c |
| LV internal diastolic diameter, mm | 6.7 ± 0.4 | 6.6 ± 0.1 | 7.0 ± 0.3 | 6.5 ± 0.2 | 7.0 ± 0.2 | 9.2 ± 0.4a,c |
| Isovolumic relaxation time, ms | 15.1 ± 1.2 | 22.8 ± 2.7a | 21.6 ± 1.6 | 25.5 ± 0.9 | 21.4 ± 0.9 | 27.6 ± 3.6a |
Values are means ± SE. WK, Wistar-Kyoto; SHHF, spontaneously hypertensive heart failure; LV, left ventricular.
Different from age-matched control rats. Within the same group:
different from 12- and 20-mo-old rats,
different from 3- and 12-mo-old rats,
different from 3- and >20-mo-old rats,
different from 12-mo-old rats. P < 0.05.
Time Course of Heart Size and Permeabilized Cardiac Myocyte Preparation Mechanical Properties During the Progression of Hypertension to Heart Failure
Isolated heart weights normalized to tibia length (HW/TL) were greater in SHHF rats compared with WK control rats, indicative of cardiac hypertrophy and ventricular remodeling in SHHF rats (Table 2 and Fig. 1D). In addition, HW/TL was greater at >20 mo of age compared with all other time points (Table 2 and Fig. 1D). The characteristics of the permeabilized cardiac myocyte preparations are also shown in Table 2. There were no statistical differences in the length, width, and passive force of the permeabilized cardiac myocyte preparations between groups. Regarding contractile properties, a significant age × strain interaction indicated that changes in maximal tension (in kN/m2) with age were dependent on rat strain. Specifically, maximal tension was lower in the >20-mo-old SHHF group compared with both the ~3-mo SHHF group and >20-mo WK control group (Fig. 2A). We also assessed the rate of force development in permeabilized cardiac myocyte preparations. There was a significant age × strain interaction in the rate of force development, which was quantified by the rate constant ktr. Specifically, ktr was statistically similar at all time points in WK control rats (Fig. 2B). In contrast, ktr significantly increased from ~3 to ~12 mo in SHHF rats before falling at >20 mo of age. Furthermore, at >20 mo of age, ktr was significantly lower in SHHF rats compared with >20-mo WK control rats. Both absolute and normalized force-velocity and power-load relationships were also characterized in cardiac myocyte preparations for the two groups at each time point (Table 2). Interestingly, peak normalized power output (PNPO) demonstrated the same age × strain interaction pattern as ktr, whereby PNPO was statistically unchanged at all time points in WK control rats but increased from ~3 to ~12 mo in SHHF rats before falling at >20 mo of age (Fig. 2C). Also, as observed with ktr, at >20 mo of age, PNPO was significantly lower in SHHF rats compared with >20-mo-old WK control rats (Fig. 2C). Figure 2D shows representative force-velocity and power-load curves from permeabilized myocyte preparations from a 12- and >20-mo-old SHHF rat, respectively, illustrating the marked depression in power output over this time frame. A similar trend was observed for absolute power-generating capacity normalized for the size of permeabilized cardiac myocyte preparations (in µW/mg), whereby, in the SHHF group, absolute power increased at ~12 mo (compared with ~3 mo) and then fell significantly at >20 mo, which coincided with lower left ventricular fractional shortening and overt signs and symptoms of clinical heart failure. Additional characteristics of the force-velocity relationships and power output are shown in Table 2. For example, the curvature of the force-velocity relationship is indexed by the a-to-Po ratio and is related to the relative force of optimal power (Fopt). Both these parameters exhibited a significant change in the >20-mo-old age group compared with the ~3- and ~12-mo time points in SHHF rats. Also, both the velocity at optimal power (Vopt) and absolute power (in pW) displayed a main effect of age, where both parameters were greater at ~12 mo compared with >20 mo in SHHF rats.
Table 2.
Characteristics of animals, hearts, and permeabilized cardiac myocyte preparations from 3-, 12-, and >20-mo-old WK control and SHHF rats
| WK Control, 3 mo old | SHHF, 3 mo old | WK Control, 12 mo old | SHHF, 12 mo old | WK Control, >20 mo old | SHHF, >20 mo old | |
|---|---|---|---|---|---|---|
| Animal weight and organ morphology | ||||||
| n | 6 | 6 | 4 | 4 | 7 | 7 |
| Body weight, g | 292.000 ± 24.000b | 381.000 ± 13.000a,b | 448.000 ± 4.000 | 477.000 ± 10.000 | 476.000 ± 11.000 | 467.000 ± 18.000 |
| Heart weight, g | 1.020 ± 0.070b | 1.400 ± 0.040a | 1.480 ± 0.030 | 1.700 ± 0.050 | 1.830 ± 0.110 | 2.740 ± 0.200a,c |
| Tibia length, cm | 3.510 ± 0.170 | 3.350 ± 0.130b | 3.850 ± 0.050 | 3.730 ± 0.100 | 3.810 ± 0.110 | 3.890 ± 0.050 |
| Heart weight/tibia length, g/cm | 0.291 ± 0.020 | 0.420 ± 0.017a | 0.385 ± 0.009 | 0.458 ± 0.022 | 0.482 ± 0.030c | 0.703 ± 0.046a,c |
| Skinned cardiac myocyte preparations | ||||||
| n | 9 | 10 | 10 | 8 | 12 | 15 |
| Length, µm | 145.000 ± 8.000 | 127.000 ± 9.000 | 141.000 ± 9.000 | 156.000 ± 10.000 | 131.000 ± 9.000 | 137.000 ± 7.000 |
| Width, µm | 22.000 ± 2.000 | 23.000 ± 1.000 | 25.000 ± 2.000 | 23.000 ± 1.000 | 21.000 ± 2.000 | 24.000 ± 1.000 |
| Resting sarcomere length, µm | 2.280 ± 0.010 | 2.280 ± 0.010 | 2.240 ± 0.010 | 2.260 ± 0.020 | 2.240 ± 0.020 | 2.230 ± 0.020 |
| Passive force, µN | 0.327 ± 0.035 | 0.419 ± 0.56 | 0.417 ± 0.052 | 0.398 ± 0.045 | 0.496 ± 0.057 | 0.472 ± 0.041 |
| Maximum force, µN | 16.800 ± 1.300 | 23.900 ± 2.700 | 23.100 ± 2.700 | 19.500 ± 2.600 | 19.800 ± 2.200 | 19.700 ± 2.300 |
| Maximum tension, kN/m2 | 68.100 ± 11.500 | 82.400 ± 10.200f | 65.100 ± 4.600 | 66.400 ± 5.800 | 77.800 ± 4.800 | 58.200 ± 4.700a |
| Peak normalized power output, P/P0⋅ml⋅s−1 | 0.092 ± 0.009 | 0.090 ± 0.009b | 0.109 ± 0.007 | 0.120 ± 0.010d | 0.089 ± 0.007 | 0.060 ± 0.005a,c |
| Peak power output, pW | 192.000 ± 48.000 | 207.000 ± 49.000 | 327.000 ± 42.000 | 305.000 ± 29.000f | 209.000 ± 52.000 | 136.000 ± 27.000 |
| Peak power output, µW/mg | 4.070 ± 0.580e | 5.740 ± 0.900 | 6.710 ± 0.990 | 6.530 ± 0.380 | 5.620 ± 0.550 | 2.770 ± 0.320a,c |
| a/P0 | 0.170 ± 0.030 | 0.160 ± 0.030 | 0.190 ± 0.020 | 0.200 ± 0.020 | 0.150 ± 0.020 | 0.080 ± 0.020a,c |
| Optimal power, P/P0 | 0.270 ± 0.020 | 0.260 ± 0.010 | 0.280 ± 0.010 | 0.280 ± 0.010 | 0.260 ± 0.020 | 0.200 ± 0.020a,c |
| Optimal power, ml/s | 0.340 ± 0.030 | 0.330 ± 0.020 | 0.400 ± 0.040 | 0.410 ± 0.030f | 0.330 ± 0.020 | 0.300 ± 0.010 |
| Rate constant of force development, per s | 5.730 ± 0.350 | 5.740 ± 0.560b | 6.710 ± 0.240 | 7.650 ± 0.550d | 5.460 ± 0.410 | 3.790 ± 0.270a,c |
Values are means ± SE.
Different from age-matched control rats. Within the same group:
different from 12- and 20-mo-old rats,
different from 3- and 12-mo-old rats,
different from 3- and >20-mo-old rats,
different from 12-mo-old rats, and
different from >20 mo. P < 0.05.
Fig. 2.

Time course of myofibrillar properties in permeabilized cardiac myocyte preparations from WK control and SHHF rats. A: maximal tension was significantly reduced in myocyte preparations from >20-mo-old SHHF rats. B, right: force development rates increased at ~12 mo and then decreased at >20 mo in SHHF rats. Left, simultaneous length and relative force traces. C: peak normalized power output increased at ~12 mo and then decreased at >20 mo in SHHF rats. Inset: time course of in vivo fractional shortening, which decreased at >20 mo in SHHF rats. D, left: force-velocity and power-load curves in a myocyte preparation from ~12- and >20-mo-old SHHF rats. Right, force clamps and length traces during maximal Ca2+ activation. The n value for each group is shown in Table 2. ¥Different vs. >20 mo of age; *different from ~12 and >20 mo; #different from ~3 and ~12 mo; †different from 3 and >20 mo; §different between age-matched WK control and SHHF rats.
Relation Between Skinned Cardiac Myocyte Preparation Mechanical Properties and Cardiac Myofibrillar Proteome During the Progression From Hypertension to Heart Failure
In an attempt to examine molecular mechanisms for the alterations in cardiac myofilament mechanical properties, we first assessed MyHC isoform content, which has been shown to be a canonical regulator of rates of force development, force-velocity relationships, and power-load curves (6, 11, 17, 18, 29). Figure 3A shows the percentage of β-MyHC isoform content in permeabilized cardiac myocyte preparations from all groups. There was a significant increase in β-MyHC content with age for both WK controls and SHHF rats; however, at each respective time point, there lacked any interstrain differences in β-MyHC isoform content. To further evaluate the relationship between the slower β-MyHC isoform and kinetic contractile properties with age and during the progression from hypertension to heart failure, we plotted PNPO as a function of β-MyHC isoform (Fig. 3B). Interestingly, in the SHHF group, PNPO increased despite higher amounts of slower β-MyHC isoform content between ~3 and ~12 mo. Furthermore, at >20 mo, there was a significant decrease in PNPO in SHHF rats compared with WK control rats; however, the depression in this contractile property occurred even though the β-MyHC content was similar between the two rat strain groups. A similar relationship was observed between β-MyHC content and ktr. Overall, these results implicate myofibrillar alterations, independent of and/or in addition to β-MyHC, contribute to the changes in cardiac myofibrillar contractile properties during the progression from hypertension to heart failure.
Fig. 3.

Relationship between myosin heavy chain (MyHC) and myofilament power output. A: time course of changes in the β-MyHC content of myofibrillar samples from WK control and SHHF hearts and a representative SDS-PAGE showing α- and β-MyHCs in permeabilized myocyte preparations (n = 9–23 for each data point). B: peak normalized power output of skinned cardiac myocytes from WK control and SHHF rats plotted against β-MyHC content. Power output increased at ~12 mo, although β-MyHC increased in both WK control and SHHF groups. In SHHF rats, myocyte power output was significantly depressed compared with >20-mo-old WK control rats even though β-MyHC content was similar. †Different from ~3 and >20 mo; #different from ~3 and ~12 mo.
Myofilament Phosphorylation During the Progression From Hypertension to Heart Failure
Previous work from our laboratory and others has found that PKA-mediated phosphorylation of sarcomeric proteins modulates force, rates of force development, loaded shortening, power output, and length dependence of force (8–10, 12, 27, 31, 32). Because PKA has been shown to increase maximal force and power in permeabilized cardiac muscle preparations (12), we hypothesized that there would be reduced PKA-mediated phosphorylation in permeabilized cardiac myocyte preparations from heart failure rats. Western blot analysis using a phosphoserine antibody to putative PKA phosphorylation sites on cMyBP-C (i.e., Ser273, Ser282, and Ser302) and cTnI (i.e., Ser22 and Ser23) indicated reduced levels of phosphorylation of both cMyBP-C and cTnI in myofilaments from >20-mo-old SHHF hearts (Fig. 4, A and B), which may contribute, at least in part, to depressed force and power associated with decompensated heart failure. In addition, cMyBP-C phosphorylation was increased between ~3 and ~12 mo in cardiac myofilaments from SHHF rats (Fig. 4, A and B). For a more comprehensive investigation into the cardiac myofibrillar proteomic changes throughout the progression from hypertension to heart failure, we used 2-D gel electrophoretic analysis. Blinded computerized spot algorithm analysis yielded over 1,117 spot changes via pairwise assessment of all groups. Figure 5 shows spot differences between silver-stained 2-D gels of permeabilized cardiac myofilaments from SHHF versus WK control rats at ~3, ~12, and >20 mo of age. To further investigate changes within just SHHF rats during the progression from hypertension to heart failure, 2-D gel difference images were analyzed between ~3 and ~12 mo and between ~12 and >20 mo (Fig. 6). Polypeptide spots were putatively identified in silico when spot intensity differed by greater than twofold, and these are shown in Table 3. A considerable number of the identified protein changes involved signaling molecules involved in hypertrophic pathways, actin regulatory molecules, and A-kinase-anchoring proteins (AKAPs), the latter of which are consistent with the pattern of our biochemical assessment of phosphorylation of myofilament proteins during the transition from compensated, hypertensive hearts to decompensated heart failure.
Fig. 4.

Assessment of the phosphorylation status of cMyBP-C and cardiac troponin I (cTnI) in permeabilized cardiac myocytes from WK control (Con) and SHHF rats at ~3, ~12, and >20 mo of age. A: Western blots using phosphoserine antibody to PKA phosphorylation sites on cMyBP-C (i.e., Ser273, Ser282, and Ser302) (top) and total cMyBP-C (middle). Bottom, line plot showing the time course of phosphoserine cMyBP-C normalized to total cMyBP-C (n = 5 for each data point). B: Western blots using phosphoserine antibody to PKA phosphorylation sites on cTnI (i.e., Ser22 and Ser23) (top) and total cTnI (middle). Bottom, line plot showing the time course of phosphoserine cTnI normalized to total cTnI (n = 6 for each data point). §Different between age-matched WK control and SHHF rats; †different from ~3 and >20 mo; #different from ~3 and ~12 mo of age.
Fig. 5.

Two-dimensional (2-D) gel difference images of silver-stained gels of permeabilized cardiac myofilaments from SHHF versus WK control rats at ~3 mo (A), ~12 mo (B), and >20 mo (C) of age. Polypeptide spots that increased in SHHF samples compared with WK control samples are outlined in blue, whereas spots that decreased in SHHF versus WK control samples are outlined in red.
Fig. 6.
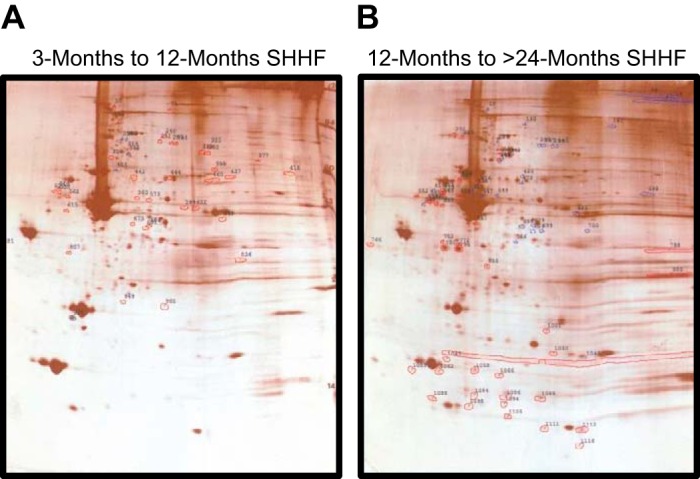
2-D gel difference images of silver-stained gels of cardiac myofilaments from SHHF rats at different ages. A: overlay of 2-D gels of myofilaments from an ~3-mo-old SHHF rat compared with an ~12-mo-old SHHF rat. Polypeptide spots that increased from ~3 to ~12 mo are outlined in red, whereas spots that decreased are outlined in blue. B: overlay of 2-D gels of myofilaments from an ~12-mo-old SHHF rat compared with a >20-mo-old SHHF rat. Polypeptide spots that increased from ~12 to >20 mo are outlined in red, whereas spots that decreased are outlined in blue.
Table 3.
Two-dimensional gel difference polypeptide spot identification
| Spot No. | PI | Molecular Weight, Da | Fold Change | Putative Protein |
|---|---|---|---|---|
| 12-mo-old SHHF vs. 12-mo-old WK control | ||||
| 142 | 6.0 | 104,967 | +5.00 | Dual 3′5′-cAMP and GMP phosphodiesterase |
| 238 | 6.1 | 88,740 | +4.80 | Rho-GTPase-activating protein 44 |
| cAMP and cAMP-inhibited cGMP phosphodiesterase | ||||
| 272 | 6.5 | 85,214 | +4.75 | Serine/threonine protein kinase |
| ARF GTPase-activating protein | ||||
| 418 | 6.6 | 62,087 | +4.80 | Dihydropyrimidase-related proteins 1 and 5 |
| 599 | 6.8 | 42,519 | +5.50 | Ca2+/calmodulin-dependent protein kinase 1G |
| 656 | 6.4 | 40,234 | +5.00 | Tyrosine protein phosphatase nonreceptor type 7 |
| 689 | 6.6 | 39,193 | +26.50 | Dual-specificity protein phosphatase 1 |
| 691 | 6.3 | 38,991 | +7.70 | Isoform-γ of A-kinase anchor protein 7 |
| PDZ and LIM domain protein 3 | ||||
| 930 | 5.8 | 27,979 | +7.00 | PKC-δ-binding protein |
| 716 | 6.7 | 38,184 | −12.00 | Isoform 2 of transcriptional coactivator YAP1 |
| >20-mo-old SHHF vs. >20-mo-old WK control | ||||
| 570 | 6.6 | 46,335 | +55.30 | MAPK-8 |
| 667 | 5.9 | 40,136 | +5.75 | Isoform 2 of focal adhesion kinase 1 |
| 689 | 6.6 | 39,193 | +13.00 | Dual-specificity protein phosphatase |
| 865 | 5.3 | 31,811 | +4.00 | Microtubule-associated protein RP/EB family member |
| 930 | 5.8 | 27,979 | +6.00 | PKC-δ-binding protein |
| 38 | 5.9 | 183,196 | −7.00 | PH domain leucine-rich repeat protein phosphatase |
| 225 | 6.0 | 123,058 | −5.10 | Protooncogene tyrosine protein kinase receptor |
| 3-mo-old SHHF vs. 12-mo-old SHHF | ||||
| 238 | 6.1 | 88,740 | +12.80 | Rho-GTPase-activating protein 44 |
| cAMP and cAMP-inhibited cGMP phosphodiesterase | ||||
| 272 | 6.5 | 85,214 | +6.30 | Serine/threonine protein kinase |
| 442 | 6.2 | 60,870 | +10.60 | Tyrosine protein kinase yes |
| Tyrosine protein kinase Fyn | ||||
| Inositol 1,4,5-trisphosphate receptor-interacting protein | ||||
| 449 | 6.3 | 60,473 | +5.00 | Nonmuscle caldesmon |
| Phosphatidylinositol 4-phosphate 5-kinase type 1β | ||||
| 488 | 5.9 | 54,810 | +4.30 | Voltage-dependent L-type Ca2+ channel-β |
| 562 | 5.6 | 48,137 | +6.00 | MAPK-9 |
| 599 | 6.8 | 42,519 | +14.80 | Isoform 2 of Ca2+/calmodulin-dependent protein kinase 1G |
| 675 | 6.2 | 39,730 | +21.00 | Twinfilin-1 |
| 684 | 6.4 | 39,361 | +8.50 | Phosphotriesterase-related protein A-kinase anchor protein 7 isoform-δ and isoform-γ |
| 691 | 6.3 | 38,991 | +7.70 | Isoform-γ of A-kinase anchor protein 7 PDZ and LIM domain protein 3 |
| 12-mo-old SHHF vs. >20-mo-old SHHF | ||||
| 335 | 6.1 | 77,458 | +5.70 | Isoform 2 of dual 3′5′-cAMP and GMP phosphodiesterase |
| 552 | 5.3 | 4,900 | +9.50 | Angiotensinogen, gap junction α8 protein |
| 561 | 5.5 | 48,137 | +7.00 | Serine/threonine protein kinase DCLK1 |
| 1,062 | 5.5 | 17,414 | +7.80 | Caveolin 3 |
| 1,088 | 5.5 | 13,761 | +7.40 | Cyclin-dependent kinase 4 inhibitor B |
| 480 | 7.2 | 55,221 | −17.90 | Protein phosphatase 1J |
| 488 | 5.9 | 54,810 | −4.30 | Voltage dependent L-type Ca2+ channel-β |
| 526 | 5.8 | 51,103 | −13.90 | Serine/threonine protein phosphatase 2A |
| 621 | 7.1 | 41,272 | −5.40 | Isoform 3 of receptor type tyrosine protein phosphatase |
| 679 | 6.5 | 39,562 | −8.00 | Dual-specificity protein phosphatase 1 |
| 684 | 6.4 | 39,361 | −17.00 | A-kinase anchor protein 7 isoform-δ and isoform-γ |
| 699 | 6.6 | 38,655 | −6.00 | Isoform 2 of coxsackievirus and adenoviral receptor homolog |
A total of 1,117 polypeptide spot differences were detected by two-dimensional differences. Putative in silico protein identification was carried out using Expasy TagIdent software with the parameters of taxID 10116 Rattus, PI range of 0.05, and 1% molecular weight range. Shown are the identifiable proteins with a fold change of ≥2.0.
DISCUSSION
This study examined the changes in myofilament contractile properties during the clinically relevant progression from hypertension to heart failure in the rat SHHF model. In SHHF rats, the cardiac myofibrillar dynamic contractile properties increased between ~3 to ~12 mo of age, and these changes in contractile kinetics occurred independent of changes in cardiac MyHC because there was actually greater expression of the slower β-MyHC isoform over this timeframe. Subsequently, during the progression from hypertension (~12 mo of age) to decompensated heart failure (>20 mo of age), there was a significant decrease in all myofibrillar contractile properties, which seemed to arise from factors in addition to greater β-MyHC content, which include, at least in part, decreased phosphorylation of myofibrillar proteins.
Congestive heart failure is a multifaceted disease characterized by the inability of ventricles to sustain normal cardiac output or to elevate output to meet higher peripheral demand. The onset of heart failure is triggered by a range of possible factors, such as coronary artery disease, myocarditis, genetic abnormalities, chronic hypertension, or valvular dysfunction. If the primary disease state remains untreated, heart failure often follows and gives rise to symptoms, including exercise intolerance, pulmonary and peripheral edema, dyspnea, and fatigue. Present treatments for heart failure mainly help reduce wall stress and circulatory loads, and, although such therapeutic regimens may improve quality of life, the prognosis following the initial diagnosis of heart failure has remained stagnant. Unfortunately, there are very few applicable therapies that are targeted to prevent or reverse adverse changes in myocyte contractile properties, which likely are the convergent end point for numerous heart disease-related cellular signaling processes and a major cause of heart failure (2, 16, 26, 33). A critical barrier toward more precise therapy is the significant knowledge gap in the changes in myofibrillar function that occur during the progression of heart failure, which was the main focus of this study.
Although there are a vast number of animal heart failure models, we selected the SHHF rat model to study the time course of myofibrillar contractile properties for a variety of reasons, one of which is its clinical relevance because a large number of human congestive heart failure cases are preceded by hypertension. In addition, previous studies have characterized several clinically relevant variables in SHHF rats. For instance, lean male SHHF rats develop hypertension at ~3 mo of age and spontaneously develop overt congestive heart failure (including signs of dyspnea, orthopnea, cyanosis, subcutaneous edema, and ascites) at ~20 mo of age (23, 24). From the standpoint of cardiac structure, the progression of hypertension to heart failure in the SHHF model is associated with wall thickening, chamber dilation, atrial enlargement, altered ventricular MyHC isoform content, increased fibrosis, inflammatory markers, and depressed ventricular fractional shortening (5, 13, 23, 24). Although ventricular pump performance depends on several factors including adequate filling times, ventricular architecture, afterload against which the ventricles work, and the contractile state of the myocardium, the capacity of the myocardium to pump blood is ultimately determined by the power-generating capacity of individual myocytes. From a mechanical standpoint, in vivo myocyte power output is determined by three myofibrillar properties: 1) amount of force developed, 2) rate of force development, and 3) velocity of shortening against afterloads. The purpose of this study was to systematically examine the time course of changes that occur in the power-generating capacity of cardiac myofilaments during the progression from hypertension to heart disease. There have been no studies to our knowledge that have systematically studied the time course of changes in these specific properties in the SHHF rat model. Conrad et al. (1) did, however, characterize force-velocity properties of intact papillary preparations in the spontaneous hypertensive rat (SHR) model. Force-velocity relationships were depressed in papillary preparations from failing hearts compared with myocardial preparations from age-matched SHRs without signs of heart failure. It remains unclear whether these changes arose from altered myofibrillar function, per se, or alterations in Ca2+ handling or extracellular mechanical properties. Along these lines, Perez et al. (28) examined Ca2+ handling and force in intact trabecular preparations from failing SHHF rats. Ca2+ transients were unchanged, whereas force was markedly depressed, suggesting a central role of myofilaments in altered myocardial function in SHHF failing hearts (28). We extended these studies to address the time course of changes of both steady-state isometric force and dynamic contractile properties (e.g., rate of force development and loaded shortening) in myofilaments from SHHF rats and aged-matched control rats. We found that the rate of force development, loaded shortening, and normalized power output of permeabilized cardiac myocyte preparations were all significantly increased from ~3 to ~12 mo in SHHF rats. Conversely, the transition to overt heart failure coincided with marked depression in maximal force-generating capacity, rate of force development, loaded shortening, normalized power output, and absolute power output in SHHF rats, implicating detriments in myofilament function as a causative factor in depressed pump function in heart failure.
Deciphering the molecular underpinnings for these functional changes is a formidable task, as there are near infinite numbers of combinatorial changes in contractile protein isoforms, regulatory proteins, and posttranslational modifications. To address this question, we undertook a dual-based proteomic approach: one focused on targets known to alter myofibrillar contractile properties and the second a more broad-based approach that could be compared with other more high-throughput studies (20). We first examined the relationship between contractile properties and cardiac MyHC isoform content, which has been largely reported to be a major regulator of dynamic functional properties of myofilaments and cardiac myocytes (6, 11, 17, 18, 29). Although cardiac β-MyHC content increased to a similar extent in both groups over time, the functional ramifications of this shift were different in the SHHF group. We found that, between ~3 and ~12 mo in the SHHF group, rates of force development, loaded shortening velocities, and normalized power output were significantly elevated despite increased β-MyHC content, which tends to slow both these properties. This finding has the potential to be leveraged to either preserve cardiac function with age/disease and/or be used to help restore power and ventricular performance in failing hearts. The discovery of a molecule(s) involved in enhanced cardiac myofibrillar force development rates, loaded shortening, and power would be highly significant in this regard. In addition, at >20 mo, these same contractile properties were all decreased in SHHF rats versus WK control rats, and these variations occurred despite similar α:β MyHC ratios between rat strain groups. Overall, these findings implicate an additional molecular mechanism(s) beyond α-to-β-MyHC ratios underlying contractile function changes during the progression from hypertension to heart failure.
Because PKA-mediated phosphorylation of cMyBP-C and cTnI have been previously shown to alter force, rate of force development, and power output (8–10, 12, 27, 31, 32), we addressed whether phosphorylation at the putative PKA sites differed between groups. We observed increased phosphorylation of MyBP-C between ~3 and ~12 mo, which may, at least in part, involve augmenting myofibrillar contractile properties (12, 27, 31, 32). Furthermore, in SHHF rats, phosphorylation of both cMyBP-C and cTnI appeared markedly depressed at >20 mo of age compared with WK control rats. Whether or not these changes in phosphorylation of cMyBP-C and cTnI are causative in depressed ventricular function remains uncertain, but because these residue-specific alterations clearly modify force and power, they certainly gain priority as targets worthy of more investigation as a means to restore contractile properties and ventricular performance in failing hearts. Another potential myofibrillar target for altered phosphorylation during the progression from hypertension to failure is the phosphorylation of myosin regulatory light chain. We did not examine the phosphorylation profile of myosin regulatory light chain in the present study. However, a previous paper (20) found a significant decrease in myosin regulatory light chain phosphorylation in 18-mo-old SHHF male rats, and additional studies have implicated phosphorylation of myosin regulatory light chain in the development (30) and prevention (34) of cardiomyopathy.
We also undertook a high-throughput, data-intensive assessment of protein modification during the progression of hypertension to heart failure. There were a few putative pathways that changed throughout the time course. In particular, AKAP-7γδ appeared to increase over eightfold from 3 to 12 mo before declining ~95% between 12 and >20 mo in cardiac myofibrillar samples from SHHF rats (Table 3). Interestingly, AKAP-7γδ has been found to interact with phospholamban (21) and may coordinate cAMP signaling events by anchoring PKA and associated phosphatases. However, AKAP-7γδ has not been shown to regulate PKA-mediated phosphorylation of myofibrillar proteins. 2-D gel electrophoresis also yielded significant increases in additional putative signaling molecules (e.g., MAPK-9, isoform 2 of Ca2+/calmodulin-dependent protein kinase 1G, and serine/threonine protein) that play a role in the MAPK/ERK stress hypertrophy pathway between ~3 and ~12 mo, but these molecules remained elevated at the >20-mo time point. In addition, molecules involved in cytoskeleton and actin organization such as twinfilin-1, PDZ, LIM domain protein 3, and caldesmon also appeared to significantly increase between ~3 and ~12 mo in cardiac myofilaments from SHHF rats. The implications that these molecules have in altering thick and thin filament properties and modulation of myofibrillar contractile properties are presently unknown but may provide some new avenues of exploration regarding their therapeutic value for treating heart failure.
In conclusion, a direct therapeutic approach that optimizes myofibrillar power output could be paramount toward improving cardiac pump function. As an example, a novel systolic pump activator, omecamtiv mecarbil, has been found to improve systolic function in patients with late-stage heart failure (22). A goal for the design of omecamtiv mecarbil was to improve myofilament power output by accelerating inorganic phosphate release from cycling myosin cross bridges (15). Although the drug has limitations (e.g., suboptimal diastolic times), it demonstrates feasibility for tuning cardiac myocyte power output as a discovery framework to impact patient quality of life and long-term outcomes. In fact, a recent proof-of-concept study (7) used small-molecule therapy to tune myocyte power output, and, excitingly, this therapy suppressed hypertrophic cardiomyopathy in mice. Overall, our present study fills a gap about how myofibrillar power output is altered during the progression from hypertension to decompensated heart failure and identifies several molecules associated with these changes, thus providing potential new targets to exploit for improved ventricular pump function in patients with heart failure.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant R01-HL-57852 (to K. S. McDonald).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
L.M.H. and K.S.M. conceived and designed research; L.M.H., C.A.E., and K.S.M. performed experiments; L.M.H., C.A.E., and K.S.M. analyzed data; L.M.H., C.A.E., and K.S.M. interpreted results of experiments; L.M.H., C.A.E., and K.S.M. prepared figures; L.M.H. and K.S.M. drafted manuscript; L.M.H., C.A.E., and K.S.M. edited and revised manuscript; L.M.H., C.A.E., and K.S.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge Dr. Vincent G. DeMarco, Terry L. Carmack, and Lisa D. Watkinson for technical assistance with the in vivo assessment of cardiovascular function and Sakthivel Sadayappan for providing cMyBP-C polyclonal antibodies. We also thank Dr. Timothy L. Domeier and Dr. Maike Krenz for reviewing previous versions of the manuscript.
REFERENCES
- 1.Conrad CH, Brooks WW, Robinson KG, Bing OH. Impaired myocardial function in spontaneously hypertensive rats with heart failure. Am J Physiol Heart Circ Physiol 260: H136–H145, 1991. [DOI] [PubMed] [Google Scholar]
- 2.de Tombe PP. Altered contractile function in heart failure. Cardiovasc Res 37: 367–380, 1998. doi: 10.1016/S0008-6363(97)00275-7. [DOI] [PubMed] [Google Scholar]
- 3.Eichhorn EJ, Bristow MR. Medical therapy can improve the biological properties of the chronically failing heart. A new era in the treatment of heart failure. Circulation 94: 2285–2296, 1996. doi: 10.1161/01.CIR.94.9.2285. [DOI] [PubMed] [Google Scholar]
- 4.Emter CA, Baines CP. Low-intensity aerobic interval training attenuates pathological left ventricular remodeling and mitochondrial dysfunction in aortic-banded miniature swine. Am J Physiol Heart Circ Physiol 299: H1348–H1356, 2010. doi: 10.1152/ajpheart.00578.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emter CA, McCune SA, Sparagna GC, Radin MJ, Moore RL. Low-intensity exercise training delays onset of decompensated heart failure in spontaneously hypertensive heart failure rats. Am J Physiol Heart Circ Physiol 289: H2030–H2038, 2005. doi: 10.1152/ajpheart.00526.2005. [DOI] [PubMed] [Google Scholar]
- 6.Fitzsimons DP, Patel JR, Moss RL. Role of myosin heavy chain composition in kinetics of force development and relaxation in rat myocardium. J Physiol 513: 171–183, 1998. doi: 10.1111/j.1469-7793.1998.171by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG, Seidman CE. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351: 617–621, 2016. doi: 10.1126/science.aad3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanft LM, Cornell TD, McDonald CA, Rovetto MJ, Emter CA, McDonald KS. Molecule specific effects of PKA-mediated phosphorylation on rat isolated heart and cardiac myofibrillar function. Arch Biochem Biophys 601: 22–31, 2016. doi: 10.1016/j.abb.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanft LM, McDonald KS. Length dependence of force generation exhibit similarities between rat cardiac myocytes and skeletal muscle fibres. J Physiol 588: 2891–2903, 2010. doi: 10.1113/jphysiol.2010.190504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanft LM, McDonald KS. Sarcomere length dependence of power output is increased after PKA treatment in rat cardiac myocytes. Am J Physiol Heart Circ Physiol 296: H1524–H1531, 2009. doi: 10.1152/ajpheart.00864.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herron TJ, Korte FS, McDonald KS. Loaded shortening and power output in cardiac myocytes are dependent on myosin heavy chain isoform expression. Am J Physiol Heart Circ Physiol 281: H1217–H1222, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Herron TJ, Korte FS, McDonald KS. Power output is increased after phosphorylation of myofibrillar proteins in rat skinned cardiac myocytes. Circ Res 89: 1184–1190, 2001. doi: 10.1161/hh2401.101908. [DOI] [PubMed] [Google Scholar]
- 13.Heyen JRR, Blasi ER, Nikula K, Rocha R, Daust HA, Frierdich G, Van Vleet JF, De Ciechi P, McMahon EG, Rudolph AE. Structural, functional, and molecular characterization of the SHHF model of heart failure. Am J Physiol Heart Circ Physiol 283: H1775–H1784, 2002. doi: 10.1152/ajpheart.00305.2002. [DOI] [PubMed] [Google Scholar]
- 14.Hill AV. The heat of shortening and the dynamic constants of muscle. Proc R Soc Lond B Biol Sci 126: 136–195, 1938. doi: 10.1098/rspb.1938.0050. [DOI] [PubMed] [Google Scholar]
- 15.Hinken AC, McDonald KS. Inorganic phosphate speeds loaded shortening in rat skinned cardiac myocytes. Am J Physiol Cell Physiol 287: C500–C507, 2004. doi: 10.1152/ajpcell.00049.2004. [DOI] [PubMed] [Google Scholar]
- 16.Hinken AC, Solaro RJ. A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiology (Bethesda) 22: 73–80, 2007. doi: 10.1152/physiol.00043.2006. [DOI] [PubMed] [Google Scholar]
- 17.Korte FS, Herron TJ, Rovetto MJ, McDonald KS. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am J Physiol Heart Circ Physiol 289: H801–H812, 2005. doi: 10.1152/ajpheart.01227.2004. [DOI] [PubMed] [Google Scholar]
- 18.Korte FS, McDonald KS. Sarcomere length dependence of rat skinned cardiac myocyte mechanical properties: dependence on myosin heavy chain. J Physiol 581: 725–739, 2007. doi: 10.1113/jphysiol.2007.128199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korte FS, McDonald KS, Harris SP, Moss RL. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ Res 93: 752–758, 2003. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 20.Kotlo K, Johnson KR, Grillon JM, Geenen DL, deTombe P, Danziger RS. Phosphoprotein abundance changes in hypertensive cardiac remodeling. J Proteomics 77: 1–13, 2012. doi: 10.1016/j.jprot.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lygren B, Carlson CR, Santamaria K, Lissandron V, McSorley T, Litzenberg J, Lorenz D, Wiesner B, Rosenthal W, Zaccolo M, Taskén K, Klussmann E. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep 8: 1061–1067, 2007. doi: 10.1038/sj.embor.7401081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, Anderson RL, Sueoka SH, Lee KH, Finer JT, Sakowicz R, Baliga R, Cox DR, Garard M, Godinez G, Kawas R, Kraynack E, Lenzi D, Lu PP, Muci A, Niu C, Qian X, Pierce DW, Pokrovskii M, Suehiro I, Sylvester S, Tochimoto T, Valdez C, Wang W, Katori T, Kass DA, Shen Y-T, Vatner SF, Morgans DJ. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science 331: 1439–1443, 2011. doi: 10.1126/science.1200113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCune S, Radin MJ, Jenkins JE, Chu Y-Y, Park S, Peterson RG. SHHF-Mcc-facap rat model: effects of gender and genotype on age of expression of metabolic complications and congestive heart failure and on response to drug therapy. In: Lessons from Animal Diabetes, edited by Shafrir E. Great Britan: Smith-Gordon, 1995, p. 255–270. [Google Scholar]
- 24.McCune SA, Park S, Radin MJ, Jurin RR. SHHF/Mcc-facp rat model: a genetic model of congestive heart failure. In: Mechanisms of Heart Failure, edited by Singal PK, Dixon IM, Beamish RE, Dhalla NS. New York, NY: Kluwer Academic, 1995, p. 91–106. doi: 10.1007/978-1-4615-2003-0_8. [DOI] [Google Scholar]
- 25.McDonald KS. Ca2+ dependence of loaded shortening in rat skinned cardiac myocytes and skeletal muscle fibres. J Physiol 525: 169–181, 2000. doi: 10.1111/j.1469-7793.2000.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moss RL, Razumova M, Fitzsimons DP. Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circ Res 94: 1290–1300, 2004. doi: 10.1161/01.RES.0000127125.61647.4F. [DOI] [PubMed] [Google Scholar]
- 27.Patel JR, Fitzsimons DP, Buck SH, Muthuchamy M, Wieczorek DF, Moss RL. PKA accelerates rate of force development in murine skinned myocardium expressing α- or β-tropomyosin. Am J Physiol Heart Circ Physiol 280: H2732–H2739, 2001. [DOI] [PubMed] [Google Scholar]
- 28.Pérez NG, Hashimoto K, McCune S, Altschuld RA, Marbán E. Origin of contractile dysfunction in heart failure: calcium cycling versus myofilaments. Circulation 99: 1077–1083, 1999. doi: 10.1161/01.CIR.99.8.1077. [DOI] [PubMed] [Google Scholar]
- 29.Rundell VLM, Manaves V, Martin AF, de Tombe PP. Impact of β-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Heart Circ Physiol 288: H896–H903, 2005. doi: 10.1152/ajpheart.00407.2004. [DOI] [PubMed] [Google Scholar]
- 30.Sheikh F, Ouyang K, Campbell SG, Lyon RC, Chuang J, Fitzsimons D, Tangney J, Hidalgo CG, Chung CS, Cheng H, Dalton ND, Gu Y, Kasahara H, Ghassemian M, Omens JH, Peterson KL, Granzier HL, Moss RL, McCulloch AD, Chen J. Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease. J Clin Invest 122: 1209–1221, 2012. doi: 10.1172/JCI61134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ Res 99: 884–890, 2006. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 32.Strang KT, Sweitzer NK, Greaser ML, Moss RL. Beta-adrenergic receptor stimulation increases unloaded shortening velocity of skinned single ventricular myocytes from rats. Circ Res 74: 542–549, 1994. doi: 10.1161/01.RES.74.3.542. [DOI] [PubMed] [Google Scholar]
- 33.Yar S, Monasky MM, Solaro RJ. Maladaptive modifications in myofilament proteins and triggers in the progression to heart failure and sudden death. Pflugers Arch 466: 1189–1197, 2014. doi: 10.1007/s00424-014-1457-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan CC, Muthu P, Kazmierczak K, Liang J, Huang W, Irving TC, Kanashiro-Takeuchi RM, Hare JM, Szczesna-Cordary D. Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice. Proc Natl Acad Sci USA 112: E4138–E4146, 2015. doi: 10.1073/pnas.1505819112. [DOI] [PMC free article] [PubMed] [Google Scholar]