

Abstract
Bacterial infection can lead to acidosis of the local microenvironment, which is believed to exacerbate disease pathogenesis; however, the mechanisms by which changes in pH alter disease progression are poorly understood. We test the hypothesis that acidosis enhances respiratory epithelial cell death in response to infection with Pseudomonas aeruginosa. Our findings support the idea that acidosis in the context of P. aeruginosa infection results in increased epithelial cell cytotoxicity due to ExoU intoxication. Importantly, enforced maintenance of neutral pH during P. aeruginosa infection demonstrates that cytotoxicity is dependent on the acidosis. Investigation of the underlying mechanisms revealed that host cell cytotoxicity correlated with increased bacterial survival during an acidic infection that was due to reduced bactericidal activity of host-derived antimicrobial peptides. These findings extend previous reports that the activities of antimicrobial peptides are pH-dependent and provide novel insights into the consequences of acidosis on infection-derived pathology. Therefore, this report provides the first evidence that physiological levels of acidosis increase the susceptibility of epithelial cells to acute Pseudomonas infection and demonstrates the benefit of maintaining pH homeostasis during a bacterial infection.
Keywords: Pseudomonas aeruginosa, acidosis, cell death, epithelial cells
the gram-negative bacterium Pseudomonas aeruginosa is an opportunistic pulmonary pathogen that establishes infection predominantly within immunocompromised individuals. P. aeruginosa is a leading cause of morbidity and mortality among hospital-acquired infections, as is the case with ventilator-associated pneumonia (28, 43), and within the lungs of cystic fibrosis (CF) patients (18, 32, 34). A preponderance of data from studies of CF supports previous findings that much of the pathogenesis and pathology result from bacterially derived damage to the lung epithelium, either through direct cytotoxicity or from associated inflammation (22, 35). Thus the progression and pathology of pulmonary diseases support the critical role of epithelial cells not only to maintain the structural integrity of the lung, but also to serve as key mediators of innate immunity. However, how the microenvironment alters the epithelial susceptibility to bacterial infection continues to be an emerging field of study.
Infection and inflammation promote acidification of the local microenvironment. This flux in local pH has been reported in a variety of inflammatory diseases, including CF, and is thought to be due to loss of bicarbonate transport and enhanced immune cell activity (13, 36, 38, 40, 50, 54, 56). Acidosis at these sites, with pH values below the physiological norm of 7.4, is thought to contribute to pathology and pathogenesis. Previous studies have demonstrated that low pH affects a variety of host responses, including regulation of the transcription factor NF-κB, neutrophil activation, and production of proinflammatory cytokines (7, 16, 30, 33, 37, 42, 44, 55, 57, 58), that can play an important role in the clearance of a P. aeruginosa infection. Furthermore, acidic pH impairs the bactericidal activity of antimicrobial peptides, which at neutral pH are capable of rapidly killing P. aeruginosa (1, 40). These antimicrobial peptides can be secreted by airway epithelial cells upon interaction with P. aeruginosa and contribute to protective antibacterial responses (4, 23, 25, 26, 60). However, how local acidosis affects epithelial cell responses and integrity during bacterial infection is not known.
The P. aeruginosa type III secretion system (T3SS) is a virulence factor that contributes to pathogenesis by inducing tissue damage and inflammation (27). Through the actions of its T3SS-secreted toxins, P. aeruginosa can disrupt the epithelium and, in severe cases, disseminate into the bloodstream, which can lead to septic shock. Accordingly, infection with P. aeruginosa expressing a functional T3SS is associated with severe disease and higher mortality rates in lung infections (17, 28, 46, 49). P. aeruginosa can inject four effector proteins into the eukaryotic host cell: ExoS, ExoT, ExoY, and ExoU. ExoS and ExoT activity interferes with bacterial internalization and wound healing (5), whereas ExoY induces cell rounding (15, 61). Even though all effector proteins contribute to virulence, ExoU is considered to be the most cytotoxic because of its phospholipase activity, which causes severe lung epithelial cell and tissue pathology (19, 28, 29, 39, 48). While the contributions of the P. aeruginosa exotoxins to tissue injury have previously been interrogated, the effects of environmental factors on T3SS effector-induced cytotoxicity are poorly understood.
In this study we have taken an integrated approach to determine the pH effects on Pseudomonas-induced cytotoxicity in epithelial cells. Bacterial binding, exotoxin activity, and epithelial cell responses were assessed to determine the mechanisms by which physiologically relevant levels of acidosis alter the pathogenicity of the bacterial infection. Our central finding is that infection within an acidic environment results in increased Pseudomonas-induced, ExoU-dependent cytotoxicity of respiratory epithelial cells. This is specifically due to impaired bactericidal activity by the epithelial cells at acidic pH, which supports previous observations that recombinant antimicrobial peptides lose activity at low pH (1, 40). Therefore, we establish the physiological effects of acidosis-impaired epithelial-derived antimicrobial peptides during a P. aeruginosa infection. These findings reveal how extracellular pH can regulate bactericidal responses by epithelial cells and can impact pulmonary pathology during infection.
MATERIALS AND METHODS
Reagents.
Hanks’ balanced salt solution (HBSS), Eagle’s minimum essential medium, and F-12K medium were purchased from Corning Cellgro (Manassas, VA), EGTA, HEPES, and Sephadex G-75 gel filtration medium from Sigma-Aldrich (St. Louis, MO), gentamicin from Lonza (Walkersville, MD), Luria broth (LB) agar from Genesee Scientific (San Diego, CA), and Ultrafree centrifugal filter units with a 3- or 5-kDa nominal molecular weight limit (NMWL) membrane from Millipore (Billerica, MA). CytoTox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI) and propidium iodide (PI; MP Biomedicals, Santa Ana, CA) were used to measure cytotoxicity.
Cell culture.
Human bronchial epithelial cells (CFBE41o−) expressing wild-type (WT) CFTR, referred to here as CFBE cells (6, 8), and A549 cells, a human pulmonary epithelial cell line, were obtained from B. Stanton (Geisel School of Medicine at Dartmouth). CFBE cells were cultured in Eagle’s minimum essential medium supplemented with 10% FBS, 100 U/ml penicillin-streptomycin, and 2 mM l-glutamine. A549 cells were maintained in F-12K medium with 10% FBS and 100 U/ml penicillin-streptomycin. Primary human bronchial epithelial (HBE) cells from three donors were provided by S. Randell (University of North Carolina, Chapel Hill, NC). HBE cells were grown on collagen-coated plates (Advanced Bio Matrix, Carlsbad, CA) in BronchiaLife basal medium (Lifeline Cell Technology, Frederick, MD) supplemented with the BronchiaLife B/T LifeFactors Kit (Lifeline Cell Technology), 10,000 U/ml penicillin, and 10,000 μg/ml streptomycin. For all experiments, CFBE, A549, and HBE cells were seeded in 24-well plates overnight in their respective culture medium at 37°C and 5% CO2. Before the cells were used in experiments, growth medium was removed, and preequilibrated pH medium at pH 6.7 or 7.3 was added to the wells. The pH medium consists of HBSS buffered with 25 mM HEPES to the desired pH and, where indicated, 1% FBS and is described elsewhere (58).
For pH neutralization experiments, HBSS without HEPES was used as the starting medium, and pH was adjusted to 7.3 by buffering with 25 mM HEPES at the time points indicated. For removal of bactericidal activity, seeded cells were washed with preequilibrated HBSS every hour for 3 h before use.
Bacteria.
P. aeruginosa strains on the PA14 background were obtained from G. O’Toole and D. Hogan (Geisel School of Medicine at Dartmouth). P. aeruginosa strains on the PA99 background were provided by A. Hauser (Northwestern University, Chicago, IL). The PA103 exoUT + pUCP-exoU-S142A strain was provided by D. Frank (Medical College of Wisconsin, Milwaukee, WI); all other PA103 strains were provided by B. Kazmierczak (Yale University, New Haven, CT). All strains have been previously used and published (21, 47, 48, 51, 58). Bacteria were cultured overnight at 37°C and subsequently subcultured for 2 h in LB. Bacterial concentrations were determined by optical density at 600 nm and subsequently validated and enumerated by colony-forming units (CFUs).
Lactate dehydrogenase assay.
Cells were infected with subcultured bacteria at the indicated multiplicity of infection (MOI) in preequilibrated pH medium or HBSS with 1% FBS for 4 h (A549) or 5 h (CFBE) at 37°C and 5% CO2. For infection of the CFBE cells with PA14 bacteria, 2.0 × 105 CFBE cells per well were used; for infection with PA103 or PA99 bacteria, 1.0 × 105 CFBE cells per well were used, unless otherwise indicated. For infections of A549 cells, 1.0 × 105 cells per well were infected with strain PA103 or PA99. Cytotoxicity, as measured by lactate dehydrogenase (LDH) release, was determined from cell-free supernatants using the CytoTox kit according to the manufacturer’s protocol.
PI staining assay.
A total of 2.0 × 105 CFBE or 1.0 × 105 A549 cells per well were infected with PA14 (MOI = 50) or PA103 (MOI = 10) in preequilibrated pH medium with 1% FBS at 37°C and 5% CO2. At 4 h postinfection, cells were stained with PI diluted 1:200 in HBSS and analyzed by microscopy. Images were acquired using an inverted microscope (Axio Observer.Z1, Carl Zeiss) at ×20 magnification.
Measurements of ExoU-mediated intoxication.
For the experiment in which the pH was switched, a total of 1.0 × 105 CFBE cells per well were infected with PA103 (MOI = 10) in preequilibrated pH medium with 1% FBS at 37°C and 5% CO2. At 3 h postinfection, the cells were treated with 100 μg/ml gentamicin for 20 min to kill the remaining bacteria. Culture medium was removed, and fresh preequilibrated pH medium with 1% FBS was added to the wells. Cell-free supernatants were collected 3 h after the medium was changed, and cytotoxicity was measured by LDH assay.
To determine ExoU-mediated cytotoxicity in the absence of bactericidal activity, a total of 2.0 × 105 CFBE cells per well were seeded and washed before infection with PA103 (MOI = 10). After addition of bacteria, cell contact to stimulate ExoU secretion was induced by centrifugation at 700 g for 10 min. At 3 h postinfection, the cells were treated with 100 μg/ml gentamicin to kill bacteria. Cell-free supernatants were collected 3 h after gentamicin treatment, and cytotoxicity was measured by LDH assay.
Immunoblotting.
To assess ExoU secretion at different pH values, equal amounts of subcultured exoUT + exoU-S142A PA103, which secretes noncytotoxic ExoU, and exoU PA103 were incubated for 3 h in preequilibrated pH medium with 2 mM EGTA at 37°C and 5% CO2. Supernatants were concentrated and subjected to Western blot analysis as previously described (19, 47). ExoU-S142A was detected with the monoclonal antibody U29F8, which was generously provided by D. Frank (Medical College of Wisconsin).
Association assay.
A total of 2.0 × 105 CFBE cells per well containing preequilibrated pH medium with 1% FBS were infected at MOI = 1 at 37°C and 5% CO2. At 1 or 2 h postinfection for the PA14 or PA103 strain, respectively, culture supernatants were removed and cells were washed with PBS to remove unattached bacteria. Cells were lysed with 0.1% Triton X-100 and then diluted and plated on LB agar, and recovered CFUs were counted. Bacterial association was determined by normalization of recovered CFUs to the input CFUs. Relative bacterial association is shown as percentage of pH 6.7 (normalized to 100%).
Plastic adhesion.
Attachment of bacteria to plastic was quantified using a modification of the protocol of Chen et al. (12). Briefly, subcultured bacteria were diluted 1:100 in HBSS, and 10 μl were added to 24-well plates containing 400 μl of preequilibrated pH medium, and the plates were incubated for 1 h at 37°C and 5% CO2. Images were taken using an inverted microscope (Axiovert 200, Carl Zeiss) with a ×63 objective.
Bacterial killing assay.
A total of 2.0 × 105 CFBE cells per well containing preequilibrated pH medium with 1% FBS were incubated for 1 h with or without bacteria (MOI = 1) at 37°C and 5% CO2. For experiments utilizing strain PA103, CFBE cells were incubated with bacteria for 3 h. Cell-free supernatants were collected and coincubated with 10 × 103 CFUs for 45 min (WT PA14) or 1.5 h (WT PA103) at 37°C and 5% CO2. After incubation, culture medium was plated on LB agar and CFUs were enumerated. Bacteria survival was determined by normalization of recovered CFUs to the input CFUs. Where indicated, relative bacteria survival is shown as percentage of pH 6.7 (normalized to 100%).
To remove bactericidal activity from the supernatant, cells were first incubated for 1 h in preequilibrated pH medium without serum at 37°C and 5% CO2. Cell-free supernatants were boiled for 15 min or filtered before incubation with bacteria. Supernatants were filtered through a 3- or 5-kDa NMWL membrane at 14,000 rpm for 20 min or through a Sephadex G-75 column that was preequilibrated with HBSS at pH 7.3. Boiled or filtered samples and paired untreated controls were utilized for bacterial killing assays with WT PA14.
For pH neutralization experiments, cells were incubated with preequilibrated pH medium or HBSS without serum, and pH values of cell-free supernatants were adjusted to neutral with 25 mM HEPES before incubation with WT PA14. Neutralized samples and paired controls were utilized for bacterial killing assays.
Statistical analyses.
Box-and-whisker plots (whiskers: minimum and maximum) or means ± SD from multiple independent experiments with technical duplicates are shown. Statistical analyses were performed using GraphPad Prism 7.
RESULTS
Epithelial cell death is increased by acidic conditions during P. aeruginosa infection.
To test the effect of an acidic microenvironment on epithelial cell viability in the context of P. aeruginosa infection, we infected CFBE cells at neutral pH (7.3) or physiologically acidic pH (6.7) with the PA14 strain of P. aeruginosa and assessed cytotoxicity by LDH release. CFBE cells infected at the acidic pH exhibited increased cytotoxicity compared with those infected at neutral pH, with a dose-dependent increase in cytotoxicity corresponding to the MOI (Fig. 1A). The enhanced cytotoxicity at the acidic pH was dependent on bacterial T3SS activity, since infection with the popB mutant of PA14, which lacks a functional T3SS translocon, did not elicit a response even at the highest MOI used in these studies (Fig. 1A). These results were validated with a complementary assay that measured cell death by PI staining. Infection at an acidic pH resulted in a higher percentage of PI-positive CFBE cells (Fig. 1B). The pH-dependent cell death phenotype is not specific to infection of CFBE cells by the PA14 strain, since the use of additional strains of P. aeruginosa, including PA103 and PA99, as well as the use of another lung epithelial cell line, A549, resulted in similar outcomes (Fig. 1, C and D). Consistent with Fig. 1A, infection with the exsA mutant of PA103, which is deficient in a functional T3SS, did not induce appreciable cell death in A549 cells (Fig. 1D). Importantly, infection of primary human bronchial epithelial cells with WT PA14 validated our aforementioned studies that used cultured cell lines: epithelial cell cytotoxicity was significantly higher at low pH following infection with PA14 (Fig. 1E). These data demonstrate that the epithelial cell death response is exacerbated at acidic pH and is dependent on a functional bacterial T3SS.
Fig. 1.

Cytotoxicity is exacerbated in epithelial cells infected with Pseudomonas aeruginosa at acidic pH. Human bronchial epithelial cells expressing wild-type (WT) CFTR (CFBE cells, A–C), A549 cells (D), or primary cultures of human bronchial epithelial cells (E) were infected with P. aeruginosa at pH 6.7 or 7.3. A lactate dehydrogenase (LDH) assay (A, C, D, and E) or propidium iodide (PI) staining (B) was used to determine epithelial cell death following infection. A: epithelial cells were infected with WT PA14 or the popB isogenic mutant at multiplicity of infection (MOI) = 100, 50, 10, 5, or 1 for 5 h. B: representative microscopy images and quantification of PI-positive cells at 4 h following infection with WT PA14 at MOI = 50. C: epithelial cells were infected with P. aeruginosa strain PA14, PA103, or PA99 at MOI = 50, 10, or 50, respectively, for 5 h. D: infection with WT PA103 or the exsA isogenic mutant at MOI = 25 for 4 h. E: epithelial cells from 3 individual donors were infected with WT PA14 at MOI = 50 for 5 h (n = 6). Data are derived from ≥2 independent experiments [n = 6 (A) and 4 (B–D)]. ***P ≤ 0.0005, **P ≤ 0.005, *P ≤ 0.05 vs. pH 7.3 (by unpaired Student’s t-test with Welch’s correction). #P ≤ 0.0001 vs. exsA [by 2-way analysis of variance (ANOVA)].
Epithelial cell death is due to ExoU-mediated cytotoxicity.
P. aeruginosa encodes four effector proteins that are translocated into host cells by the T3SS to mediate pathogenesis. These exotoxins can disrupt host defense mechanisms and induce cell necrosis (27). Since the enhanced cytotoxicity at acidic pH was dependent on the bacterial T3SS, we next investigated which effector protein was driving this response. We infected CFBE cells with isogenic strains of P. aeruginosa that express discrete T3SS effector proteins and subsequently quantified CFBE cytotoxicity by LDH release. As reported in Fig. 1, infection with WT PA103, which secretes ExoT and ExoU, led to increased epithelial cell cytotoxicity at low pH (Fig. 2A). Cell death after infection with the exoT mutant of PA103 was similar to that following infection by the WT bacteria (Fig. 2A). However, infection with exoU PA103 abrogated epithelial cell cytotoxicity at low pH, demonstrating that ExoU is the effector protein mediating epithelial cell death (Fig. 2A). These results were confirmed with the use of strain PA99, which secretes ExoS, ExoT, and ExoU (Fig. 2B). Loss of ExoU, but not ExoS and ExoT, reduced epithelial cell death to levels comparable to that induced by the T3SS-deficient pscJ mutant (Fig. 2B). These results were further validated with the use of A549 cells (Fig. 2, C and D) and by measurement of cell death by PI staining (Fig. 2E).
Fig. 2.

pH-dependent epithelial cell cytotoxicity induced by P. aeruginosa infection is due to intoxication by ExoU. Cytotoxicity of CFBE (A and B) or A549 (C–E) cells following infection with WT PA103 or isogenic mutants of P. aeruginosa strain PA103 (A, D, and E) or PA99 (B and C) at pH 6.7 and 7.3. Release of LDH into culture supernatants was measured at 5 h (A and B) or 4 h (C and D) postinfection. E: cytotoxicity was observed at 4 h postinfection by staining with PI (left) and quantified by counting PI-positive epithelial cells (right). MOI = 10 (A and E), 50 (B and C), and 25 (D). Data are derived from 2 independent experiments (n = 4). ***P ≤ 0.0001, **P ≤ 0.001 vs. pH 7.3 (by unpaired Student’s t-test with Welch’s correction). #P ≤ 0.0001 vs. WT bacteria; ns, not significant (by 2-way ANOVA).
The pH-dependent mechanism leading to increased cell death at acidic pH occurs before ExoU intoxication.
We next sought to determine the mechanism by which low pH leads to increased ExoU-dependent cell death. We previously showed that T3SS activity or expression of the T3SS effector genes is not regulated by pH (58). Furthermore, similar levels of ExoU-S142A were secreted by exoUT + exoU-S142A PA103 under T3SS-inducing conditions at acidic and neutral pH (Fig. 3A). Thus we decided to focus on the activity of ExoU. Upon injection, ExoU requires activation by host cell cofactors, whereupon it induces rapid cell lysis (2, 48, 59). Therefore, we tested the hypothesis that the differential epithelial cell death response was due to pH-dependent increased activation of ExoU following injection into the host cell. To test this, we conducted an experiment such that the pH during T3SS activation and the pH during ExoU delivery by the bacteria were different from the pH after epithelial cell intoxication with ExoU, thereby allowing us to determine if the pH-sensitive step occurred before or after ExoU intoxication. Briefly, we infected CFBE cells with WT PA103 at neutral or acidic pH for 3 h and then treated the infected cells with gentamicin to kill the bacteria. The 3-h duration of infection was chosen, since it enables ExoU delivery into cells while minimizing measurable cell death within this time frame (10–20% cell death) (47). Thereafter, we removed the medium and incubated the cells with fresh medium of the opposite pH for 3 h and then measured LDH release (Fig. 3B). As previously observed, infected epithelial cells that were maintained at an acidic pH throughout the study displayed higher cytotoxicity than cells kept at a neutral pH (Fig. 3C). Interestingly, epithelial cells that were infected with an initial pH of 6.7 and then switched to neutral pH following intoxication exhibited levels of cytotoxicity similar to those maintained at an acidic pH. In contrast, results from an epithelial cell infection at an initial pH of 7.3 and a final pH of 6.7 were similar to results from an infection maintained at neutral pH (Fig. 3C). This indicates that the pH at the time of ExoU delivery regulates the level of cytotoxicity. Together, these findings indicate that bacterial secretion of ExoU and ExoU activity is not pH-dependent and, therefore, likely not the cause of the difference in epithelial cell cytotoxicity. Instead, the enhanced cytotoxicity at low pH is dependent on an event preceding or concomitant with ExoU delivery into host cells.
Fig. 3.
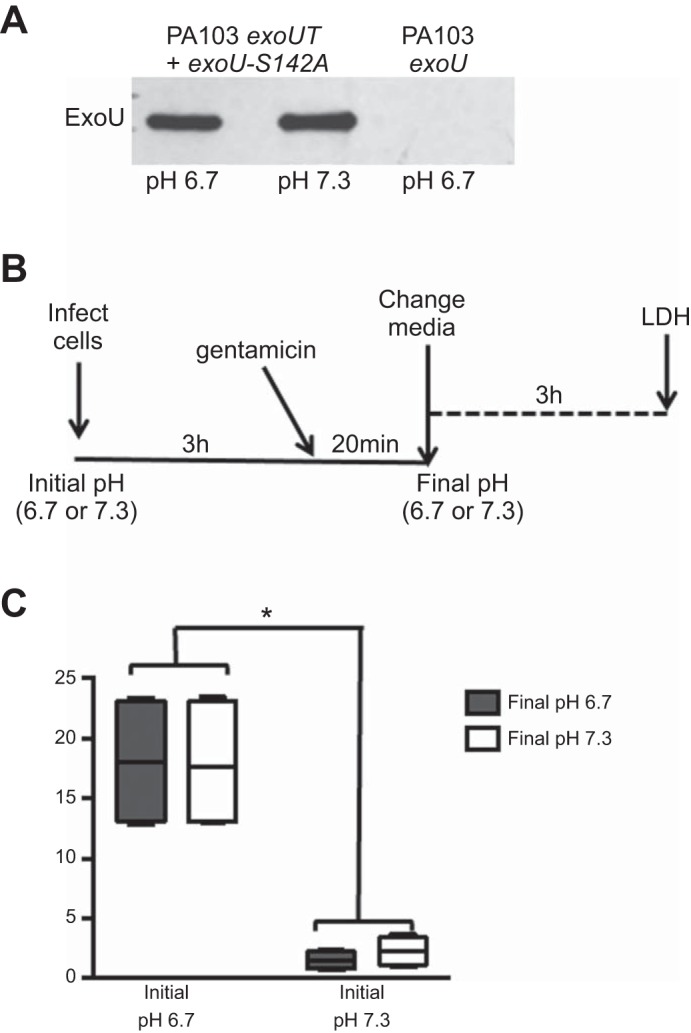
Acidosis enhances P. aeruginosa-induced cytotoxicity early during infection, rather than following ExoU intoxication. A: Western blot analyses of ExoU-S142A protein secreted by bacteria incubated under type III secretion system (T3SS)-inducing conditions for 3 h at pH 6.7 or 7.3. B: schematic of experimental design for pH switch. Briefly, CFBE cells were infected with WT PA103 (MOI = 10) at pH 6.7 or 7.3 (initial pH). At 3 h postinfection, gentamicin was added to kill the bacteria, the culture medium was removed, and fresh medium at pH 6.7 or 7.3 (final pH) was added. C: cytotoxicity was measured by an LDH assay 3 h following medium change. Data are derived from 2 independent experiments (n = 4). *P ≤ 0.01 vs. initial pH 7.3 (by 2-way ANOVA).
P. aeruginosa association with epithelial cells is enhanced during acidosis.
A fundamental step for the establishment of P. aeruginosa infection that occurs before T3SS activation and ExoU translocation is bacterial adherence to epithelial cells. Therefore, we hypothesized that the enhanced cytotoxicity under acidic conditions was due to increased bacterial association with epithelial cells. To address this, we infected CFBE cells with P. aeruginosa, removed unassociated bacteria, and then plated the cell lysates to count CFUs and determine relative bacterial association. Bacterial association at low pH was greater with the PA14 and PA103 strains (Fig. 4, A and B). As a control for binding specificity we employed the pilA mutant of PA14; the pili are required for efficient attachment to airway epithelium (Fig. 4A) (24). To analyze the kinetics of bacterial attachment at the respective pH values, bacterial association was measured every 15 min. It was surprising to find, after 15 min of infection, a similar number of CFUs associated with the epithelial cells in neutral and acidic conditions (Fig. 4C). However, at 30 min postinfection, a difference in bacterial attachment was observed (Fig. 4C). Bacterial association had increased at low pH and continued to increase thereafter, while, strikingly, it continually decreased at neutral pH (Fig. 4C). Since bacterial association with host cells is required for T3SS activation (27), these findings provide a critical insight into the lower level of epithelial cell cytotoxicity at neutral pH. Overall, these results demonstrate that the increased ExoU-dependent cytotoxicity at low pH is likely due to increased bacterial association with the epithelial cells. Additionally, our findings suggest that a pH-sensitive host- or bacterial-dependent mechanism contributes to bacterial attachment.
Fig. 4.
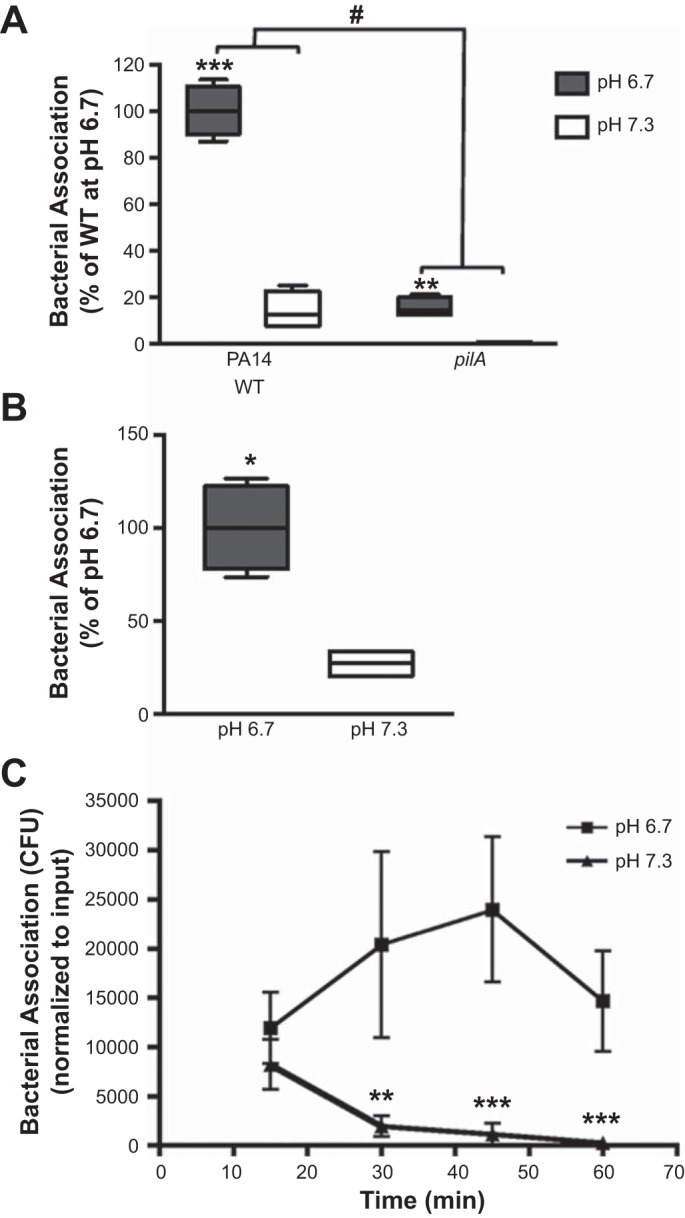
Infection at acidic pH leads to increased bacterial association with epithelial cells. Association of P. aeruginosa was assayed after addition of PA14 for 1 h (A and C) or WT PA103 at MOI = 1 for 2 h (B) to CFBE cells at pH 6.7 or 7.3. In A, the pilA mutant was employed as a control for specificity of the bacterial interaction with the epithelial cells. C: kinetics of P. aeruginosa association with epithelial cells at pH 6.7 and 7.3. CFU, colony-forming units. Data are derived from ≥2 independent experiments [n = 4 (A and B) and 6 (C)]. ***P ≤ 0.0005, **P ≤ 0.005, *P ≤ 0.05 vs. pH 7.3 (by unpaired Student’s t-test with Welch’s correction). #P ≤ 0.0001 vs. WT bacteria (by 2-way ANOVA).
Increased survival of P. aeruginosa under acidic conditions is due to impaired epithelial-derived bactericidal activity.
We next addressed why there was a pH-dependent difference in bacterial attachment to epithelial cells, as seen in Fig. 4C. Possible explanations for the continual reduction in bacterial attachment during a neutral infection are as follows: 1) P. aeruginosa is detaching from the epithelial cells, or 2) P. aeruginosa is killed at a faster rate during a neutral infection than during an acidic infection. To test these, we measured the total bacteria remaining after 1 h of incubation with CFBE cells. The amount of bacteria recovered was ∼10-fold greater at pH 6.7 than at neutral pH (Fig. 5A). Incubation of P. aeruginosa in neutral or acidic medium in the absence of host cells did not lead to a difference in bacterial growth or survival (data not shown). Therefore, we next assessed if the difference in bacterial survival was a consequence of an epithelial cell-derived factor. We collected the conditioned medium of CFBE cells infected with strain PA14 or PA103 at pH 6.7 or 7.3 and then tested the ability of this culture supernatant to kill the respective bacteria. Conditioned medium from PA14- or PA103-infected CFBE cells at neutral pH was able to efficiently kill bacteria, whereas medium collected in parallel under acidic conditions was not (Fig. 5, B and C). To assess whether the epithelial-derived bactericidal activity was elicited by ExoU intoxication, we infected CFBE cells with the exoU mutant of PA103 for 3 h and then tested the ability of the culture medium to kill WT PA103. Bactericidal activity within the conditioned medium was comparable to that elicited by infection with WT PA103 (Fig. 5C). Moreover, bacterial killing utilizing conditioned medium from uninfected CFBE cells mirrored that from infected cells (Fig. 5D). These observations indicate that an epithelial cell-secreted factor had bactericidal activity at neutral pH and that this factor was constitutively being secreted under our culture conditions, independent of an infection. This interpretation was validated with the methodology of Smith et al. (53), where efficient removal of the bactericidal activity at neutral pH was obtained through a series of washes of the epithelial cell surfaces (Fig. 5E). Consistent with the interpretation that epithelial-derived bactericidal activity is responsible for the differential in bacterial association, bacterial attachment to a plastic surface exhibited no differences between the experimental pH values tested (Fig. 5F).
Fig. 5.

Bacterial survival is reduced under neutral conditions due to epithelial cell-derived bactericidal activity. A: CFBE cells were infected with WT PA14 for 1 h at pH 6.7 or 7.3. Total recovered bacteria were determined by pooling culture media and cell lysates and plating for CFUs. B–E: CFBE cells were infected with WT PA14 for 1 h (B and E) or PA103 for 3 h (C) at MOI = 1 or remained uninfected (as indicated) for 1 h (D and E) at pH 6.7 and 7.3. Conditioned medium was then harvested and incubated with bacteria, and bacterial survival was assayed by CFUs. Survival of WT PA14 (B, D, and E) or WT PA103 (C) is shown. E: bacterial survival as described in B and D, except epithelial cells were washed before infection. F: quantification of WT PA14 attachment to plastic after 1 h of incubation in pH 6.7 or 7.3 medium. Data are derived from ≥2 independent experiments [n ≥ 5 (A), 4 (B, D, and E), and 6 (C and F)]. ***P ≤ 0.0005, **P ≤ 0.001; ns, not significant vs. pH 7.3 (by unpaired Student’s t-test with Welch’s correction). Not significant vs. WT bacteria (by 2-way ANOVA) in C.
We next sought to further characterize the epithelial-derived bactericidal factor. It has long been known that epithelial cells secrete antimicrobial peptides, and, in support of our observations, numerous studies have reported reduced antimicrobial peptide activity at low pH (1, 40, 53). Therefore, we ran a series of experiments to test the hypothesis that the epithelial-derived bactericidal activity originated from antimicrobial peptides. To confirm the low-molecular-mass origin of the bactericidal activity, we filtered uninfected supernatants through a 5-kDa cutoff filter. Recovery of WT PA14 following coincubation with the <5-kDa flow-through filter was comparable to that of unfiltered samples (Fig. 6A). This demonstrates that the epithelial-derived peptide with bactericidal activity is <5 kDa. Consistent with this, pH 7.3 supernatants filtered through a Sephadex G-75 gel filtration column exhibited a loss of bactericidal activity (Fig. 6B). To validate that the activity originated from peptides, rather than small molecules, boiling the pH 7.3 CFBE supernatant significantly reduced its bactericidal activity (Fig. 6C). Finally, to confirm that CBFE cells secreted the <5-kDa antimicrobial peptide at acidic pH, we filtered uninfected supernatant, adjusted the pH to neutral, and then ran a bacterial killing assay. Neutralizing the acidic supernatant restored its bactericidal activity (Fig. 6D). These data show that the bactericidal factor is secreted by epithelial cells at low pH but its activity is impaired and requires a neutral pH to effectively kill bacteria. Together, these data support the hypothesis that the majority of pH-dependent epithelial cell-derived bactericidal activity is due to antimicrobial peptides.
Fig. 6.

Antimicrobial peptides are responsible for bactericidal activity in neutral conditions. Culture media of uninfected CFBE cells incubated for 1 h at pH 7.3 or, where indicated, pH 6.7, were filtered through a 5-kDa (A) or 3-kDa (D) cutoff filter or a gel filtration column (B) or were boiled (C), and bacterial killing assays were performed with WT PA14. Bactericidal activity was lost after boiling (C) and after running culture medium through a Sephadex G-75 column (B). D: after culture medium was filtered, pH of the supernatant was neutralized with HEPES before incubation with the bacteria. Data are derived from ≥2 independent experiments [n = 4 (A, B, and D) and 6 (C)]. ***P ≤ 0.0005, **P ≤ 0.005 vs. paired control (by paired Student’s t-test with Welch’s correction). ns, Not significant (by 2-way ANOVA).
Reduced epithelial-derived antimicrobial activity is responsible for the enhanced cytotoxicity at low pH.
Since bactericidal activity is dependent on pH (Figs. 5 and 6), we hypothesized that our initial observation of decreased epithelial cell death at neutral pH was due to the bactericidal activity of antimicrobial peptides. Therefore, utilizing the methodology successfully employed for Fig. 5, we washed the epithelial cell surface to remove the bactericidal activity and measured bacterial association and epithelial cell cytotoxicity. Removal of antimicrobial peptides resulted in similar bacterial association between both pH values (Fig. 7A). To verify that similar bacterial association between pH values led to similar ExoU intoxication of cells in the absence of bactericidal activity, antimicrobial peptide activity was removed by washing the CFBE cell surface. The cells were then infected with WT and exoU PA103 for 3 h to allow for ExoU translocation into the cells, the bacteria were killed with gentamicin, and cytotoxicity was assessed 3 h later. Upon antimicrobial peptide removal, CFBE cells exhibited similar levels of ExoU-mediated cytotoxicity (Fig. 7B). Similar levels of epithelial cell cytotoxicity at both pH values were also observed following a 5-h infection with WT PA14 when the bactericidal activity was removed (Fig. 7C). These results suggest that, during P. aeruginosa infection, epithelial cells are able to control infection through action of antimicrobial peptides.
Fig. 7.
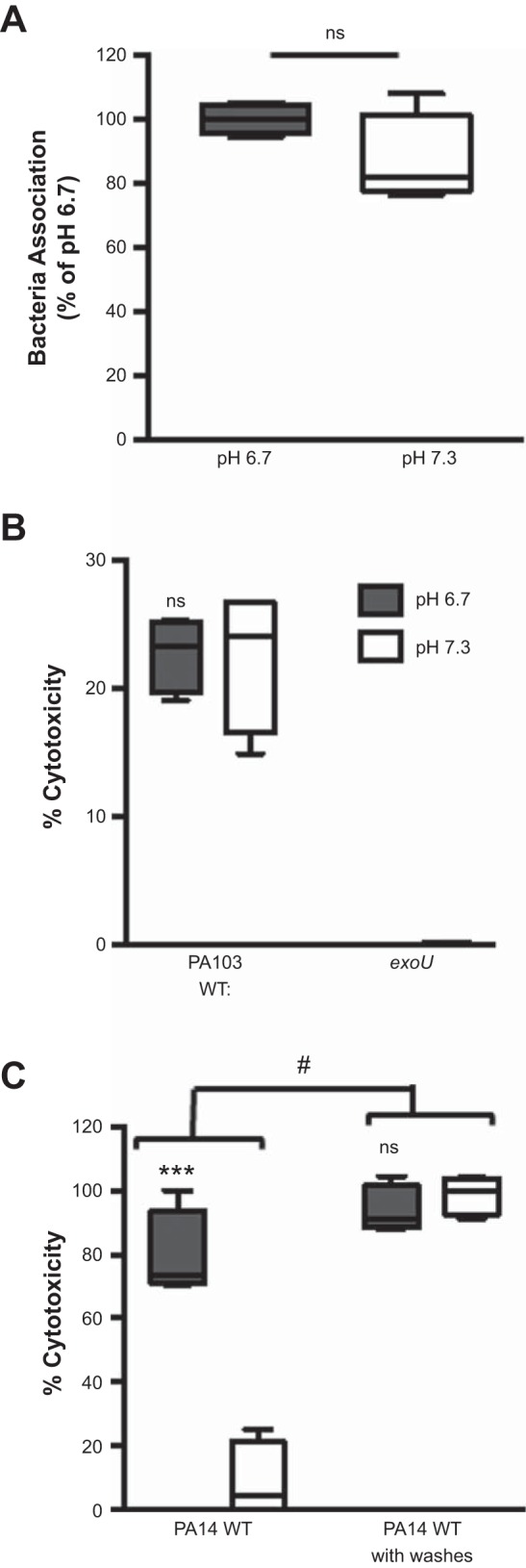
Removal of antimicrobial activity rescues bacterial-dependent killing of epithelial cells at neutral pH. CFBE cells were washed with preequilibrated pH medium to remove antimicrobial peptides, and association of WT PA14 (A) or epithelial cell cytotoxicity (B and C) was assessed at pH 6.7 and 7.3. A: association assay was performed 1 h postinfection at MOI = 1. B: WT PA103 or exoU at MOI = 10 was centrifuged onto CFBE cells to induce ExoU translocation. Gentamicin was added 3 h postinfection to kill the bacteria, and cytotoxicity was determined 3 h later by an LDH assay. C: CFBE cell cytotoxicity was determined by an LDH assay at 5 h postinfection with WT PA14 at MOI = 50. Data are derived from 2 independent experiments (n = 4). ***P ≤ 0.0005, not significant (ns) vs. pH 7.3 (by unpaired Student’s t-test with Welch’s correction). #P ≤ 0.0001 vs. infection with no washes (by 2-way ANOVA).
In an acidic environment, the antimicrobial activity is reduced, leading to increased bacterial association with the epithelial cells and subsequent ExoU-mediated epithelial cell death. Therefore, we reasoned that by returning the pH of the medium to neutral after, rather than before, infection, we could recover the bactericidal activity and reduce Pseudomonas-induced cytotoxicity. To test this, we infected CFBE cells with WT PA14 in the presence of HBSS without HEPES and assessed cytotoxicity after neutralizing the medium at 1, 2, and 3 h postinfection. Infection in HBSS without HEPES resulted in levels of cytotoxicity similar to those in epithelial cells infected at pH 6.7 (Fig. 8A). However, addition of HEPES to neutralize the medium significantly abrogated epithelial cell death (Fig. 8A). This was due to increased bactericidal activity of the host-secreted factor, because, in contrast to the HBSS control, neutralized supernatants were able to kill bacteria (Fig. 8B). Therefore, infection in an acidic environment leads to impairment of antimicrobial defenses of respiratory epithelial cells and enhances Pseudomonas-induced cytotoxicity; however, this can be prevented by forced maintenance of a neutral environment during infection.
Fig. 8.
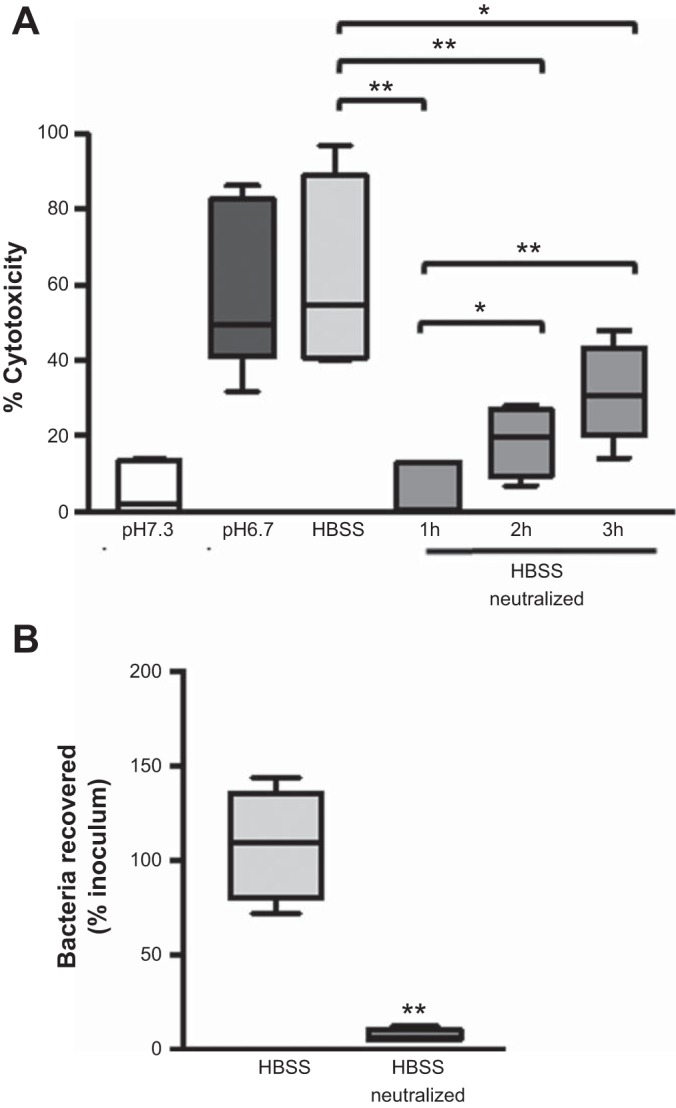
Neutralization of the pH of the medium following infection decreases P. aeruginosa-induced cytotoxicity of epithelial cells. A: CFBE cell cytotoxicity determined by an LDH assay at 5 h postinfection with WT PA14 at MOI = 10 in the indicated culture medium. B: culture media of uninfected CFBE cells incubated for 1 h at pH 7.3 were neutralized with HEPES, and bacterial killing assays were performed. Data are derived from ≥2 independent experiments [n = 6 (A) and 4 (B)]. **P ≤ 0.005, *P ≤ 0.05 (by unpaired Student’s t-test with Welch’s correction).
DISCUSSION
Recruitment and activation of immune cells, bacterial products, and hypoxia contribute to the reduced pH of the microenvironment at sites of infection and inflammation (9, 31, 33, 45, 52). Additionally, extracellular pH can also be influenced by intrinsic factors, as in CF, where the loss of bicarbonate transport by CFTR contributes to local acidosis (13, 20, 41, 54). Despite substantial evidence of acidification in inflammatory environments, the consequence of acidic pH on host responses and host cell integrity is poorly understood.
Previous studies have demonstrated alterations in a variety of host cellular responses during acidosis (1, 16, 40, 42, 44, 50, 55). Data from our laboratory and from others support this notion in the context of P. aeruginosa infection; specifically, we previously demonstrated an amplification of proinflammatory cytokine production by immune cells during acidosis (58). Based on this observation and the important role of epithelial cells during P. aeruginosa infection, we hypothesized that the epithelial cell response to P. aeruginosa may be altered during acidosis. To address this hypothesis, we assessed epithelial cell cytotoxicity upon P. aeruginosa infection in a neutral (pH 7.3) or physiologically acidic (pH 6.7) environment. The key finding from these studies is that respiratory epithelial cell cytotoxicity is pH-dependent, with increased cytotoxicity under acidic conditions. This is the first report that acidosis enhances the susceptibility of epithelial cells to Pseudomonas-induced cytotoxicity. To understand how an acidic environment impacts bacterial attachment and ensuing exotoxin-mediated cytotoxicity, we interrogated the component mechanisms of bacterial binding, exotoxin production and activity, and epithelial cell responses with regard to the pathogenicity and cytotoxicity resulting from the bacterial infection.
With the use of bacterial mutants deficient in a functional T3SS or its effector proteins, we determined that epithelial cell death at low pH was dependent on the function of the bacterial T3SS. Specifically, we identified the bacterial T3SS effector ExoU as the predominant mediator of cell death: isogenic P. aeruginosa strains deficient in ExoU did not measurably induce epithelial cell death under our experimental conditions. This finding supports previous reports that P. aeruginosa strains that express ExoU are more virulent, cytotoxic, and associated with increased disease severity (19, 27, 28, 39, 49, 51).
Our initial assays demonstrated that Pseudomonas-induced cytotoxicity of epithelial cells was pH-dependent; however, the fundamental basis for this could occur at several steps during the ExoU intoxication process, which have not been methodically tested for pH dependence. Our previous data indicate that T3SS expression and activity are not upregulated during acidosis (58). Ultimately, our present data demonstrate that although the cytotoxic activity of P. aeruginosa was pH-dependent, ExoU secretion and activity were independent of pH. Therefore, ExoU activity was the cause of epithelial cell cytotoxicity, but alterations in ExoU activity were not responsible for the pH-dependent differential in cell death. This directed us to examine additional steps during P. aeruginosa infection that could be altered by acidosis.
Since bacterial attachment to epithelial cells is one of the initial steps of infection and is required for efficient T3SS activation (27), we hypothesized that differential bacterial association could explain the pH dependence of epithelial cell cytotoxicity. Indeed, we identified a significant increase in bacterial attachment to the epithelial cells and a correlation between this increase in bacterial attachment and cytotoxicity. One possibility, not mutually exclusive with other pH-dependent steps in this pathway, was that the P. aeruginosa-epithelial cell interaction itself might be pH-sensitive through one of the variety of host receptors that have been proposed to mediate bacterial attachment to the epithelium (3, 10, 11, 14, 24). Consequently, we also studied bacterial attachment in a host cell-free system. However, similar numbers of bacteria associating with plastic were observed in both pH conditions, indicating that the pH-dependent differential in bacterial attachment required host cells.
Ultimately, in the course of determining the epithelial cell-specific component that contributed to the pH dependence, we demonstrated that the increased bacterial attachment and subsequent susceptibility of epithelial cells were due to decreased bactericidal activity derived from the epithelial cells at acidic pH. We were guided by studies that demonstrated sensitivity of airway antimicrobial peptides to the acidic pH in the airway surface liquid of CF patients (1, 40, 53). We show that, at both pH values tested, epithelial cells secrete factors and that these factors are likely to be antibacterial peptides. These peptides have bactericidal activity at neutral, but not low, pH, which is consistent with previous reports from studies of the CF lung (1, 40). The epithelial bactericidal activity derived from a low-molecular-weight molecule was constitutively secreted and partially heat-resistant. This is consistent with known properties of defensin-like molecules secreted by respiratory epithelial cells (23, 53). However, since boiling did diminish bactericidal activity of the epithelial cell supernatants, it is likely that other antimicrobial peptides might contribute as well (1, 4). Additionally, neutralization of acidic supernatants partially restored bactericidal activity. This demonstrates that the bactericidal activity is present but suppressed within the acidic pH medium and can be restored simply by a return to neutral pH. It is unclear whether the residual loss of activity not restored by the pH shift is due to inefficiencies in reviving the activity of the existing antimicrobial peptides or represents a difference in abundance or type of antimicrobial peptide being secreted by epithelial cells at acidic pH. Nonetheless, bulk removal of these peptides resulted in similar levels of bacterial survival and ExoU-mediated cytotoxicity of epithelial cells in neutral and acidic conditions, further establishing that the action of antimicrobial peptides significantly contributed to the outcome of epithelial cell cytotoxicity. Thus the pH during infection strongly impacts epithelial cell defenses and, consequently, their integrity.
Our findings are the first to demonstrate the functional consequence of reduced antimicrobial activity at low pH during a P. aeruginosa infection. First, reduced bacterial killing by antimicrobial peptides leads to increased bacterial association with epithelial cells at acidic pH. This increased interaction could lead to an enhanced establishment of infection and exacerbate P. aeruginosa pathogenesis in pulmonary infections. Second, with reduced antimicrobial activity and enhanced bacterial attachment, there is a direct consequence of significantly greater P. aeruginosa-induced cytotoxicity of epithelial cells. This suggests that during acidosis the epithelial cell response might be nullified and unable to control infection, which could affect subsequent host defenses and the inflammatory environment during P. aeruginosa infections. An interesting related observation is that an in vitro infection performed within normally buffered culture medium, for which we did not adjust or additionally stabilize the pH, follows the course of an infection within acidic-buffered medium. This suggests that epithelial cells are still able to produce bactericidal factors at low pH, however, with reduced activity. Moreover, this supports the idea that a bacterial infection lowers the pH of the microenvironment; therefore, the method used to manipulate and interpret in vitro culture assays should be taken into consideration. Our data support the concept of maintenance of pH homeostasis as a novel therapeutic approach. This dovetails well with a previous observation that increases in airway surface liquid pH in CF pigs increased killing of Staphylococcus aureus ex vivo (40). Therefore, our data further support the notion that neutralization of the pH in the pulmonary fluids may have therapeutic benefits to the host by increasing antimicrobial peptide activity and reducing P. aeruginosa-induced lung pathology.
In summary, we have demonstrated that physiological levels of acidosis enhance epithelial cell cytotoxicity during a P. aeruginosa infection. We have shown that the increased epithelial cytotoxicity during acidosis is due to reduced antimicrobial activity. In addition, we have established the pH dependence of epithelial-derived bactericidal activity, validating previous findings that employed recombinant antimicrobial peptides. Overall, these findings provide key insights into how changes in extracellular pH contribute to the pathogenesis of P. aeruginosa infections.
GRANTS
This work was supported by National Institutes of Health Grants P30 RR-032136-01, P30 GM-106394, R01 HL-074175, R21 AI-121820, and T32 AI-007363 (to I. M. Torres and B. Berwin), F32 ES-025082 (to B. C. Goodale), and R01 AI-104922 (to D. Frank) and Cystic Fibrosis Foundation Research Development Program grants STANTO19R0 and STANTO11R0 (to B. Berwin).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.M.T., S.D., and B.L.B. conceived and designed research; I.M.T., S.D., J.V., B.C.G., and B.L.B. performed experiments; I.M.T., S.D., J.V., and B.L.B. analyzed data; I.M.T., S.D., J.V., and B.L.B. interpreted results of experiments; I.M.T. prepared figures; I.M.T. drafted manuscript; I.M.T., S.D., and B.L.B. edited and revised manuscript; I.M.T., S.D., J.V., B.C.G., and B.L.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank George O’Toole, Deborah Hogan, and Bruce Stanton (Geisel School of Medicine at Dartmouth), Alan Hauser (Northwestern University, Chicago, IL), Dara Frank (Medical College of Wisconsin, Milwaukee, WI), Barbara Kazmierczak (Yale University, New Haven, CT), and Tim Yahr (University of Iowa, Iowa City, IA) for reagents and discussion. This work was facilitated by the Dartmouth Lung Biology Translational Research Core, the NCCC IML, and the Dartmouth ASURE Program.
REFERENCES
- 1.Abou Alaiwa MH, Reznikov LR, Gansemer ND, Sheets KA, Horswill AR, Stoltz DA, Zabner J, Welsh MJ. pH modulates the activity and synergism of the airway surface liquid antimicrobials β-defensin-3 and LL-37. Proc Natl Acad Sci USA 111: 18703–18708, 2014. doi: 10.1073/pnas.1422091112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson DM, Schmalzer KM, Sato H, Casey M, Terhune SS, Haas AL, Feix JB, Frank DW. Ubiquitin and ubiquitin-modified proteins activate the Pseudomonas aeruginosa T3SS cytotoxin, ExoU. Mol Microbiol 82: 1454–1467, 2011. doi: 10.1111/j.1365-2958.2011.07904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arora SK, Ritchings BW, Almira EC, Lory S, Ramphal R. The Pseudomonas aeruginosa flagellar cap protein, FliD, is responsible for mucin adhesion. Infect Immun 66: 1000–1007, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bals R, Wang X, Zasloff M, Wilson JM. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci USA 95: 9541–9546, 1998. doi: 10.1073/pnas.95.16.9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barbieri JT, Sun J. Pseudomonas aeruginosa ExoS and ExoT. Rev Physiol Biochem Pharmacol 152: 79–92, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Bebok Z, Collawn JF, Wakefield J, Parker W, Li Y, Varga K, Sorscher EJ, Clancy JP. Failure of cAMP agonists to activate rescued ΔF508 CFTR in CFBE41o- airway epithelial monolayers. J Physiol 569: 601–615, 2005. doi: 10.1113/jphysiol.2005.096669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellocq A, Suberville S, Philippe C, Bertrand F, Perez J, Fouqueray B, Cherqui G, Baud L. Low environmental pH is responsible for the induction of nitric-oxide synthase in macrophages. Evidence for involvement of nuclear factor-κB activation. J Biol Chem 273: 5086–5092, 1998. doi: 10.1074/jbc.273.9.5086. [DOI] [PubMed] [Google Scholar]
- 8.Bomberger JM, Maceachran DP, Coutermarsh BA, Ye S, O’Toole GA, Stanton BA. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog 5: e1000382, 2009. doi: 10.1371/journal.ppat.1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryant RE, Rashad AL, Mazza JA, Hammond D β-Lactamase activity in human pus. J Infect Dis 142: 594–601, 1980. doi: 10.1093/infdis/142.4.594. [DOI] [PubMed] [Google Scholar]
- 10.Bucior I, Mostov K, Engel JN. Pseudomonas aeruginosa-mediated damage requires distinct receptors at the apical and basolateral surfaces of the polarized epithelium. Infect Immun 78: 939–953, 2010. doi: 10.1128/IAI.01215-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bucior I, Pielage JF, Engel JN. Pseudomonas aeruginosa pili and flagella mediate distinct binding and signaling events at the apical and basolateral surface of airway epithelium. PLoS Pathog 8: e1002616, 2012. doi: 10.1371/journal.ppat.1002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen AI, Dolben EF, Okegbe C, Harty CE, Golub Y, Thao S, Ha DG, Willger SD, O’Toole GA, Harwood CS, Dietrich LE, Hogan DA. Candida albicans ethanol stimulates Pseudomonas aeruginosa WspR-controlled biofilm formation as part of a cyclic relationship involving phenazines. PLoS Pathog 10: e1004480, 2014. doi: 10.1371/journal.ppat.1004480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coakley RD, Grubb BR, Paradiso AM, Gatzy JT, Johnson LG, Kreda SM, O’Neal WK, Boucher RC. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci USA 100: 16083–16088, 2003. doi: 10.1073/pnas.2634339100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Comolli JC, Waite LL, Mostov KE, Engel JN. Pili binding to asialo-GM1 on epithelial cells can mediate cytotoxicity or bacterial internalization by Pseudomonas aeruginosa. Infect Immun 67: 3207–3214, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cowell BA, Evans DJ, Fleiszig SM. Actin cytoskeleton disruption by ExoY and its effects on Pseudomonas aeruginosa invasion. FEMS Microbiol Lett 250: 71–76, 2005. doi: 10.1016/j.femsle.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 16.Edye ME, Lopez-Castejon G, Allan SM, Brough D. Acidosis drives damage-associated molecular pattern (DAMP)-induced interleukin-1 secretion via a caspase-1-independent pathway. J Biol Chem 288: 30485–30494, 2013. doi: 10.1074/jbc.M113.478941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Solh AA, Hattemer A, Hauser AR, Alhajhusain A, Vora H. Clinical outcomes of type III Pseudomonas aeruginosa bacteremia. Crit Care Med 40: 1157–1163, 2012. doi: 10.1097/CCM.0b013e3182377906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fagon JY, Chastre J, Hance AJ, Montravers P, Novara A, Gibert C. Nosocomial pneumonia in ventilated patients: a cohort study evaluating attributable mortality and hospital stay. Am J Med 94: 281–288, 1993. doi: 10.1016/0002-9343(93)90060-3. [DOI] [PubMed] [Google Scholar]
- 19.Finck-Barbançon V, Goranson J, Zhu L, Sawa T, Wiener-Kronish JP, Fleiszig SM, Wu C, Mende-Mueller L, Frank DW. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol Microbiol 25: 547–557, 1997. doi: 10.1046/j.1365-2958.1997.4891851.x. [DOI] [PubMed] [Google Scholar]
- 20.Garland AL, Walton WG, Coakley RD, Tan CD, Gilmore RC, Hobbs CA, Tripathy A, Clunes LA, Bencharit S, Stutts MJ, Betts L, Redinbo MR, Tarran R. Molecular basis for pH-dependent mucosal dehydration in cystic fibrosis airways. Proc Natl Acad Sci USA 110: 15973–15978, 2013. doi: 10.1073/pnas.1311999110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garrity-Ryan L, Kazmierczak B, Kowal R, Comolli J, Hauser A, Engel JN. The arginine finger domain of ExoT contributes to actin cytoskeleton disruption and inhibition of internalization of Pseudomonas aeruginosa by epithelial cells and macrophages. Infect Immun 68: 7100–7113, 2000. doi: 10.1128/IAI.68.12.7100-7113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168: 918–951, 2003. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 23.Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. Human β-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 88: 553–560, 1997. doi: 10.1016/S0092-8674(00)81895-4. [DOI] [PubMed] [Google Scholar]
- 24.Hahn HP. The type-4 pilus is the major virulence-associated adhesin of Pseudomonas aeruginosa—a review. Gene 192: 99–108, 1997. doi: 10.1016/S0378-1119(97)00116-9. [DOI] [PubMed] [Google Scholar]
- 25.Hancock RE, Diamond G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol 8: 402–410, 2000. doi: 10.1016/S0966-842X(00)01823-0. [DOI] [PubMed] [Google Scholar]
- 26.Harder J, Meyer-Hoffert U, Teran LM, Schwichtenberg L, Bartels J, Maune S, Schröder JM. Mucoid Pseudomonas aeruginosa, TNF-α, and IL-1β, but not IL-6, induce human β-defensin-2 in respiratory epithelia. Am J Respir Cell Mol Biol 22: 714–721, 2000. doi: 10.1165/ajrcmb.22.6.4023. [DOI] [PubMed] [Google Scholar]
- 27.Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7: 654–665, 2009. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hauser AR, Cobb E, Bodi M, Mariscal D, Vallés J, Engel JN, Rello J. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Crit Care Med 30: 521–528, 2002. doi: 10.1097/00003246-200203000-00005. [DOI] [PubMed] [Google Scholar]
- 29.Hauser AR, Engel JN. Pseudomonas aeruginosa induces type-III-secretion-mediated apoptosis of macrophages and epithelial cells. Infect Immun 67: 5530–5537, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jancic CC, Cabrini M, Gabelloni ML, Rodríguez Rodrigues C, Salamone G, Trevani AS, Geffner J. Low extracellular pH stimulates the production of IL-1β by human monocytes. Cytokine 57: 258–268, 2012. doi: 10.1016/j.cyto.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 31.Kellum JA, Song M, Li J. Science review: extracellular acidosis and the immune response: clinical and physiologic implications. Crit Care 8: 331–336, 2004. doi: 10.1186/cc2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurahashi K, Kajikawa O, Sawa T, Ohara M, Gropper MA, Frank DW, Martin TR, Wiener-Kronish JP. Pathogenesis of septic shock in Pseudomonas aeruginosa pneumonia. J Clin Invest 104: 743–750, 1999. doi: 10.1172/JCI7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lardner A. The effects of extracellular pH on immune function. J Leukoc Biol 69: 522–530, 2001. [PubMed] [Google Scholar]
- 34.Lavoie EG, Wangdi T, Kazmierczak BI. Innate immune responses to Pseudomonas aeruginosa infection. Microbes Infect 13: 1133–1145, 2011. doi: 10.1016/j.micinf.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Machen TE. Innate immune response in CF airway epithelia: hyperinflammatory? Am J Physiol Cell Physiol 291: C218–C230, 2006. doi: 10.1152/ajpcell.00605.2005. [DOI] [PubMed] [Google Scholar]
- 36.Månsson B, Geborek P, Saxne T, Björnsson S. Cytidine deaminase activity in synovial fluid of patients with rheumatoid arthritis: relation to lactoferrin, acidosis, and cartilage proteoglycan release. Ann Rheum Dis 49: 594–597, 1990. doi: 10.1136/ard.49.8.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martínez D, Vermeulen M, Trevani A, Ceballos A, Sabatté J, Gamberale R, Alvarez ME, Salamone G, Tanos T, Coso OA, Geffner J. Extracellular acidosis induces neutrophil activation by a mechanism dependent on activation of phosphatidylinositol 3-kinase/Akt and ERK pathways. J Immunol 176: 1163–1171, 2006. doi: 10.4049/jimmunol.176.2.1163. [DOI] [PubMed] [Google Scholar]
- 38.Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 10: 767–777, 2011. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 39.Pankhaniya RR, Tamura M, Allmond LR, Moriyama K, Ajayi T, Wiener-Kronish JP, Sawa T. Pseudomonas aeruginosa causes acute lung injury via the catalytic activity of the patatin-like phospholipase domain of ExoU. Crit Care Med 32: 2293–2299, 2004. doi: 10.1097/01.CCM.0000145588.79063.07. [DOI] [PubMed] [Google Scholar]
- 40.Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Bánfi B, Horswill AR, Stoltz DA, McCray PB Jr, Welsh MJ, Zabner J. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487: 109–113, 2012. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poulsen JH, Fischer H, Illek B, Machen TE. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA 91: 5340–5344, 1994. doi: 10.1073/pnas.91.12.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajamäki K, Nordström T, Nurmi K, Åkerman KE, Kovanen PT, Öörni K, Eklund KK. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J Biol Chem 288: 13410–13419, 2013. doi: 10.1074/jbc.M112.426254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rello J, Díaz E, Rodríguez A. Etiology of ventilator-associated pneumonia. Clin Chest Med 26: 87–95, 2005. doi: 10.1016/j.ccm.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 44.Riemann A, Wußling H, Loppnow H, Fu H, Reime S, Thews O. Acidosis differently modulates the inflammatory program in monocytes and macrophages. Biochim Biophys Acta 1862: 72–81, 2016. doi: 10.1016/j.bbadis.2015.10.017. [DOI] [PubMed] [Google Scholar]
- 45.Rotstein OD, Nasmith PE, Grinstein S. The Bacteroides by-product succinic acid inhibits neutrophil respiratory burst by reducing intracellular pH. Infect Immun 55: 864–870, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J Infect Dis 183: 1767–1774, 2001. doi: 10.1086/320737. [DOI] [PubMed] [Google Scholar]
- 47.Sato H, Frank DW. Intoxication of host cells by the T3SS phospholipase ExoU: PI(4,5)P-2-associated, cytoskeletal collapse and late phase membrane blebbing. PLoS One 9: e103127, 2014. doi: 10.1371/journal.pone.0103127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sato H, Frank DW, Hillard CJ, Feix JB, Pankhaniya RR, Moriyama K, Finck-Barbançon V, Buchaklian A, Lei M, Long RM, Wiener-Kronish J, Sawa T. The mechanism of action of the Pseudomonas aeruginosa-encoded type III cytotoxin, ExoU. EMBO J 22: 2959–2969, 2003. doi: 10.1093/emboj/cdg290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schulert GS, Feltman H, Rabin SD, Martin CG, Battle SE, Rello J, Hauser AR. Secretion of the toxin ExoU is a marker for highly virulent Pseudomonas aeruginosa isolates obtained from patients with hospital-acquired pneumonia. J Infect Dis 188: 1695–1706, 2003. doi: 10.1086/379372. [DOI] [PubMed] [Google Scholar]
- 50.Shah VS, Meyerholz DK, Tang XX, Reznikov L, Abou Alaiwa M, Ernst SE, Karp PH, Wohlford-Lenane CL, Heilmann KP, Leidinger MR, Allen PD, Zabner J, McCray PB Jr, Ostedgaard LS, Stoltz DA, Randak CO, Welsh MJ. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science 351: 503–507, 2016. doi: 10.1126/science.aad5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaver CM, Hauser AR. Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infect Immun 72: 6969–6977, 2004. doi: 10.1128/IAI.72.12.6969-6977.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simmen HP, Blaser J. Analysis of pH and pO2 in abscesses, peritoneal fluid, and drainage fluid in the presence or absence of bacterial infection during and after abdominal surgery. Am J Surg 166: 24–27, 1993. doi: 10.1016/S0002-9610(05)80576-8. [DOI] [PubMed] [Google Scholar]
- 53.Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 85: 229–236, 1996. doi: 10.1016/S0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 54.Song Y, Salinas D, Nielson DW, Verkman AS. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am J Physiol Cell Physiol 290: C741–C749, 2006. doi: 10.1152/ajpcell.00379.2005. [DOI] [PubMed] [Google Scholar]
- 55.Takenouchi T, Iwamaru Y, Sugama S, Tsukimoto M, Fujita M, Sekigawa A, Sekiyama K, Sato M, Kojima S, Conti B, Hashimoto M, Kitani H. The activation of P2X7 receptor induces cathepsin D-dependent production of a 20-kDa form of IL-1β under acidic extracellular pH in LPS-primed microglial cells. J Neurochem 117: 712–723, 2011. [DOI] [PubMed] [Google Scholar]
- 56.Tang XX, Ostedgaard LS, Hoegger MJ, Moninger TO, Karp PH, McMenimen JD, Choudhury B, Varki A, Stoltz DA, Welsh MJ. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J Clin Invest 126: 879–891, 2016. doi: 10.1172/JCI83922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tong J, Wu WN, Kong X, Wu PF, Tian L, Du W, Fang M, Zheng F, Chen JG, Tan Z, Gong F. Acid-sensing ion channels contribute to the effect of acidosis on the function of dendritic cells. J Immunol 186: 3686–3692, 2011. doi: 10.4049/jimmunol.1001346. [DOI] [PubMed] [Google Scholar]
- 58.Torres IM, Patankar YR, Shabaneh TB, Dolben E, Hogan DA, Leib DA, Berwin BL. Acidosis potentiates the host proinflammatory interleukin-1β response to Pseudomonas aeruginosa infection. Infect Immun 82: 4689–4697, 2014. doi: 10.1128/IAI.02024-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tyson GH, Hauser AR. Phosphatidylinositol 4,5-bisphosphate is a novel coactivator of the Pseudomonas aeruginosa cytotoxin ExoU. Infect Immun 81: 2873–2881, 2013. doi: 10.1128/IAI.00414-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waterer GW. Airway defense mechanisms. Clin Chest Med 33: 199–209, 2012. doi: 10.1016/j.ccm.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 61.Yahr TL, Vallis AJ, Hancock MK, Barbieri JT, Frank DW. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc Natl Acad Sci USA 95: 13899–13904, 1998. doi: 10.1073/pnas.95.23.13899. [DOI] [PMC free article] [PubMed] [Google Scholar]